

Abstract
Brain death is associated with dramatic and serious pathophysiologic changes that adversely affect both the quantity and quality of organs available for transplant. To fully optimise the donor pool necessitates a more complete understanding of the underlying pathophysiology of organ dysfunction associated with transplantation. These injurious processes are initially triggered by catastrophic brain injury and are further enhanced during both brain death and graft transplantation. The activated inflammatory systems then contribute to graft dysfunction in the recipient. Inflammatory mediators drive this process in concert with the innate and adaptive immune systems. Activation of deleterious immunological pathways in organ grafts occurs, priming them for further inflammation after engraftment. Finally, posttransplantation ischaemia reperfusion injury leads to further generation of inflammatory mediators and consequent activation of the recipient's immune system. Ongoing research has identified key mediators that contribute to the inflammatory milieu inherent in brain dead organ donation. This has seen the development of novel therapies that directly target the inflammatory cascade.
1. Introduction
Organ transplantation is the gold standard treatment for patients with end stage solid organ failure. An ever increasing disparity between available organs and potential recipients is the cause of avoidable morbidity and mortality [1–4]. Ongoing efforts are being made to increase the quantity and quality of organs available for transplant. Although outcomes from non-heart-beating donors have become increasingly successful [5], the majority of organs are still donated from donors after brain death (BD). Significant brain injury of any aetiology will cause a systemic response [6], creating a proinflammatory environment prior to the occurrence of brain death itself. BD then also creates a variety of inflammatory, haemodynamic and endocrine effects, which induce adverse sequelae in distant organs [7–10]. Finally, ischaemia-reperfusion injury (IRI), inherent in transplantation, generates reactive oxygen species (ROS), activates complement, and independently drives cytokine release and inflammation [11, 12]. Every transplanted organ from a BD donor will face these stages of potential injury. Consequently, donor management must consider each step from donor to recipient in order to maximise recipient outcomes. The purpose of this paper is to explore the current understanding of the three main contributors to injury that an organ will travel through from donor to recipient. Additionally, donor management and organ preservation strategies that are currently being investigated will be briefly considered.
2. Stage Zero of Potential Organ Injury: Current Concepts in Immunological Signalling
Inflammation, secondary to brain injury, BD, and IRI, is driven by both the innate and adaptive immune systems. The complexity of these systems means that our understanding continues to evolve at a rapid pace (Figure 1). Prior to reviewing the specific inflammatory responses at each major step of the donor organ journey, it is important to discuss current concepts in the normally functioning immune system.
Figure 1.
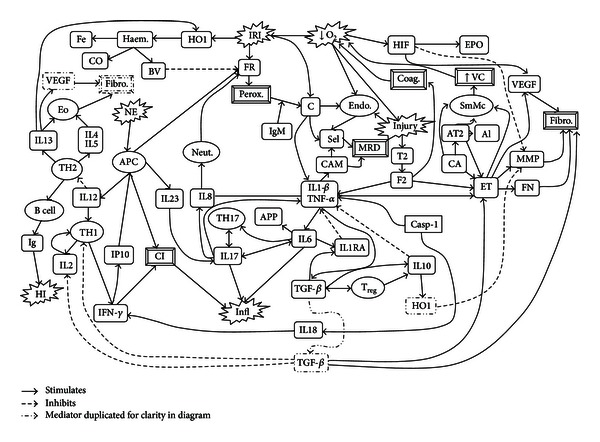
Primary mediators of peri-transplant related inflammation. Al: aldosterone, APC: antigen presenting cell, APP: acute phase proteins, AT2: angiotensin II, BV: biliverdin, C: complement, CA: catecholamines, CAM: cellular adhesion molecule, Casp-1: caspase 1, CI: cellular inflammation, CO: carbon monoxide, Coag: coagulation, Endo: endothelial cells, Eo: eosinophils, EPO: erythropoietin, ET: endothelin, F2: factor II (Thrombin), Fe: iron, Fibro: fibrosis, FN: fibronectin, FR: free radicals, HI: humoral immunity, HIF: hypoxia inducible factor, HO1: heme oxygenase 1, IFN: interferon, Ig: immunoglobulin, IL: interleukin, IL1RA: interleukin 1 receptor antagonist, Infl: inflammation, IP: interferon-γ-induced protein, IRI: ischaemia reperfusion injury, MMP: matrixmetalloproteinases, MRD: margination/rolling/diapedesis, NE: new antigens/neoepitopes, Neut: neutrophils, O2: oxygen, Perox: peroxidation, Sel: selectin, SmMc: smooth muscle contraction, TF: tissue factor, TGF: transforming growth factor, TH1: type 1 helper T-cell, TH17: type 17 helper T-cell, TH2: type 2 helper T-cell, TNF: tumour necrosis factor, Treg: regulatory T-cell, VC: vasoconstriction, VEGF: vascular endothelial growth factor.
Traditionally, T-cell responses are grouped according to the TH1/TH2 paradigm. TH1 lymphocytes (CD4+) are responsible for cell-mediated immunity through activation of killer CD8+ T cells and cytotoxic macrophages [13, 14]. TH2 cells are responsible for the control of humoral immunity through antibody producing B cells. Additionally, they regulate eosinophil and basophil functions. Recent work has identified TH17 and T-regulatory (Treg) subsets. TH17 cells have been implicated in autoimmunity [13, 14]. Treg cells are related to TH17 cells and function to regulate immunological reactions and prevent uncontrolled inflammation. Each of these T cells plays a specific role in inflammation and their actions can be identified by certain inflammatory mediators. Although cytokines may interact with multiple T-cell subsets, previous authors have classified the major cytokines into “types” reflecting the major T-cell subtype to which they are related [15–18]. This convention will be used in the current paper.
2.1. TH1-Cell-Related Cytokines
Communicating via tumour necrosis factor (TNF)-α, interleukin (IL)-1, IL-2, IL-12, and IFN-γ [19–21], TH1 cells play a fundamental role in acute rejection. These type 1 cytokines are upregulated early in the inflammatory process. After their release, IL-1β and TNF-α support the inflammatory response via activation of endothelial cells [22]. These cytokines act early in the inflammatory cascade, stimulating generation of cellular adhesion molecules, innate immune defence mechanisms, and participating in cross-talk between the various inflammatory pathways [23, 24]. IL-2 plays an essential role in resting T-cell activation and proliferation, contributing to T-cell maturation [25]. After T-cell induction via IL-2, IL-12 directs cellular maturation towards TH1, leading to a cell-mediated immune response [26]. IFN-γ influences both the innate and adaptive immune systems and is integral in the antigen presenting cell (APC) controlled balance between effector and suppressor T cells [27]. IFN-γ not only acts as the primary effector cytokine of IL-12 as part of cellular immunity, but also provides negative feedback control of IL-12 and indoleamine dioxygenase-mediated T-cell inhibition, under the control of APC's [27].
2.2. TH2-Cell-Related Cytokines
TH2 cell-related-cytokines include IL-4, IL-5, IL-10, and IL-13 [14, 28]. Type 2 cytokines are generally considered anti-inflammatory when associated with brain injury and BD, and in the early transplant period [29–31]. IL-4 inhibits formation of TH1 cells and encourages development of TH2 cells [29]. It also plays an essential role in B-cell generation of IgE [32]. IL-4 may activate macrophages via an alternative pathway that reduces inflammation through sequestration and metabolism of arginine, an essential requirement for nitric oxide generation by inflammatory IFN-γ-activated macrophages [33]. IL-4 has been postulated to depress T-cell activity through the production of indoleamine dioxygenase; Wang et al. demonstrated increased indoleamine dioxygenase produced by natural killer cells in IL-4-treated rat livers [33]. IL-13 is best known for its role in allergy. Through interaction with its receptor, IL-13 stimulates inflammatory cells as well as epithelial and smooth muscle cells [34]. This may contribute to smooth muscle hypertrophy and pulmonary hypertension in various lung diseases [34]. IL-13 inhibits cell-mediated immunity through downregulation of E-selectin, reduction of neutrophil recruitment, and macrophage inhibition [35]. IL-5 is essential for the development, recruitment and activation of eosinophils [36]. Once these cells are recruited to inflammatory sites, IL-5 is a potent costimulator of eosinophil degranulation and maintains their presence through inhibition of apoptosis [36, 37]. IL-5 also acts as a key mediator for generation of antigen-specific IgE. Furthermore, it is important for terminal B-cell differentiation, including the switch to mature IgM and IgG1 secreting plasma cells [38].
IL-10 acts to inhibit the production of inflammatory cytokines and upregulate inhibitors of IL-1 and TNF [15]. It may also specifically block the production of IL-1 and TNF [39]. Direct activity on inflammatory cells impairs or reverses the effects of proinflammatory mediators [39]. While IL-10 is classified as a type 2 cytokine, it is also able to be produced by TH1 cells under the influence of transforming growth factor (TGF)-β [19].
2.3. TH17-Cell-Related Cytokines
The TH17 cells are identified by their association with IL-6, IL-17, IL-21, IL-22, and IL-23 [14, 26, 40]. IL-17 and IL-23 direct TH17 cell differentiation, proliferation, and maturation [26]. Apart from directing TH17 development, IL-17 functions to stimulate production of chemokines, IL-1β, TNF-α, IL-6, and IL-8 [19, 41]. Its production is reinforced by IL-6, IL-23, and TGF-β [19, 41]. IL-8-related neutrophil attraction and activation may contribute partly to the inflammatory action of IL-17 [42]. IL-23 is an important upstream regulator of IL-17 expression [26, 43]. Generation of IL-17 by γδ-T cells is directly activated by IL-23, and these cells are an important source of IL-17 [43]. Furthermore, IL-23 induces IL-17 production from natural killer T cells [41]. IL-21 stimulates natural killer T cells, CD40 dependent B-cell proliferation, and T-cell expansion [44]. Hagn et al. recently demonstrated that incompletely activated CD4+ T cells, through expression of IL-21 and CD40 ligand, stimulate B cells to differentiate into Granzyme B generating cytotoxic B cells [45].
IL-6 has been extensively investigated in many conditions. It's pro- and anti-inflammatory effects have recently been comprehensively reviewed [46]. Briefly, it is a pro-inflammatory agent which has been classified as a type 17 cytokine [47], although some authors may include it as a type 1 [48]. IL-6, the prototypical member of its family, acts through receptor complex formation with glycoprotein gp130 on the cell surface [46]. The IL-6 receptor molecule is present on the surface of hepatocytes, neutrophils, monocytes, and macrophages [46, 49]. Direct activation of these receptors is associated with an inflammatory response [49]. Other cells may also respond to IL-6 through a process termed trans-signalling [49]. Free soluble IL-6 receptor binds circulating IL-6 and then interacts with the ubiquitous cell surface protein, gp130, to affect cell signalling [46]. The dual roles of IL-6 may be partly explained by the differing signalling mechanisms. Soluble IL-6 receptor generated from apoptotic neutrophils in areas of inflammation activates signalling pathways after interaction with epithelial gp130, attracting regulatory monocytes and macrophages and contributing to resolution of inflammation [46].
2.4. Treg Cells and Related Cytokines
Named due to their ability to downregulate inflammatory processes, Treg cells are another important source of the anti-inflammatory IL-10. Treg are closely related to TH17 cells; both lineages are derived from the same naïve T-cell precursor in a similar fashion to TH1/TH2 cells [14, 20]. Deknuydt et al. recently highlighted the fluidity of the TH17/Treg balance by demonstrating that Treg cells can be stimulated to become TH17 cells under the influence of IL-1β and IL-2 [14]. TGF-β also directs the differentiation of T-cell populations in inflammatory conditions and is important in the TH17/Treg balance. TGF-β modulates the effects of IL-2, reducing expansion of inflammatory T-cell populations [19]. When acting synergistically with IL-2, TGF-β is able to direct naïve T cells to become Treg cells [50]. Selective inhibition of TH1 producing mediators by TGF-β further contributes to the diversion from inflammatory T cells to Treg cells, mediating the inflammatory response [19]. However, costimulation of TGF-β by IL-6 directs T-cell differentiation towards TH17 cells and production of type 17 cytokines [20]. Treg cells are immunosuppressive through production of IL-10 and TGF-β, cellular anergy and direct contact with inflammatory cells [51].
3. Stage One of Potential Organ Injury: Brain Injury
Most brain dead donors suffer from three main causes of BD: cerebrovascular injury, anoxia, or traumatic brain injury (TBI) [52, 53]. Donor cause of death can significantly influence recipient survival rates, though this varies according to the organ. Renal transplant outcomes are adversely affected by cerebrovascular causes of BD [54, 55]. Lung transplant is unaffected by donor cause of death [56], while heart transplant outcomes remain controversial [57, 58]. For this reason, it is important to consider the pathophysiologic responses to severe central nervous system injury, and their systemic sequelae, prior to brain death.
3.1. Systemic Inflammatory Response Secondary to Brain Injury
Central nervous system (CNS) injury is associated with the systemic inflammatory response syndrome (SIRS). This can occur with an intact blood brain barrier (BBB), indicating an additional mechanism distinct from CNS-derived cytokine release [59, 60]. The link between the brain and SIRS has been comprehensively reviewed recently by Lu et al. [61]. Briefly, SIRS is associated with leukocyte mobilisation and recruitment to major organs, activation and release of inflammatory mediators, generation of ROS, increased vascular permeability, and organ dysfunction [62, 63]. Brain intraparenchymal injection of TNF-α recruits and activates systemic monocytes while IL-1β activates and recruits neutrophils via release of chemokines from the liver [59, 64].
TNF-α is released from the spleen in the early stages of brain injury to augment the peripheral inflammatory response [6, 65]. Lee et al. demonstrated upregulation of TNF-α, IL-1β, IL-4, and IL-6 in the spleens of rats with subarachnoid haemorrhage (SAH) [65]. Intravenous administration of neural stem cells attenuated the inflammatory response via a chaperone mechanism which was localised to the spleen and reversed on splenectomy [65]. Splenic inflammation may also be directly downregulated via vagal messages from the brain [6]. The SIRS response activates gut-derived inflammatory mediators, resulting in leaky gut wall [66]. This contributes to global inflammation through cytokine generation and systemic endotoxin exposure, worsening pulmonary inflammation and impairing oxygenation [66–68]. Similar to the spleen, gut generation of cytokines is also modulated by the CNS through vagal input [67].
3.2. Localised Response to Brain Injury and Loss of Blood Brain Barrier Function
Local responses to severe brain injury can be classified into two phases [69]. The primary phase is due to the insult itself and includes cellular death, direct BBB disruption, and cerebral oedema [69]. The secondary phase of injury is caused by elevated intracranial pressure (ICP), global brain ischaemia, excitotoxicity, metabolic derangements, and haemodynamic instability [69–71]. Whatever the cause of brain death, a cytokinaemia secondary to brain injury occurs prior to brain death itself [72–75].
Local inflammation, and the direct effect of the insult itself, causes the highly selective BBB to become disrupted [74, 76]. Matrix metalloproteinases (MMP), especially MMP-9, act to break down extracellular proteins, including basal lamina and endothelial tight junctions [77]. In a rat model of closed head trauma, Higashida et al. investigated the role of MMP-9 and hypoxia inducible factor (HIF) in cerebral oedema resulting from lost BBB integrity [77]. Inhibition of MMP-9 in this model significantly reduced the amount of brain oedema observed after 24 hours. Additionally, inhibition of HIF (an upstream regulator of protein expression associated with hypoxia) also significantly reduced the expression of MMP-9 and brain oedema [77]. This observation was confirmed in an intracranial haemorrhage model in rats; Wu et al. showed that MMP-9 is upregulated early after injury and is associated with brain oedema [78]. A postmortem study of intracranial haemorrhage confirmed these findings in humans [79].
The effect of the loss of BBB integrity is to allow bidirectional access of inflammatory cells and mediators [76, 80–84]. CNS-derived cytokines are then free to interact at receptors within the systemic tissues, inducing local inflammation and “priming” organs for further injury [81, 85]. The importance of brain injury-derived cytokinaemia was recently demonstrated by Graetz et al., who reviewed compartmental levels of IL-6 in SAH and found that elevated plasma IL-6 is associated with increased mortality [72]. This provides further evidence that isolated brain injury causes a systemic inflammatory response and upregulates the peripheral immune system [67, 83].
3.3. Type 1 Cytokines
Type 1 cytokines are upregulated in the brain after injury and contribute to BBB breakdown, vasospasm, and secondary injury [70, 86, 87]. The general roles of these and other inflammatory mediators have been previously reviewed [88]. Briefly, IL-1β is a pleiotropic proinflammatory mediator that stimulates multiple pathways of inflammation after brain injury [88]. TNF-α acts as a proinflammatory agent early in the inflammatory process in the CNS [89]. Microdialysis techniques have confirmed the presence of IL-1β and TNF-α in extracellular fluid after TBI and SAH [72, 87, 90]. Both of these cytokines are also released peripherally as part of a systemic acute phase response (APR) [6, 59, 91, 92]. IL-1β and TNF-α can be detected in blood within as little as one hour after brain ischaemia, even before significant neuronal cell death can be demonstrated [83, 93]. Quantitative systemic levels of type 1 cytokines may be affected by the type of brain insult; these were decreased in a middle cerebral artery occlusion model in mice, partially explaining the mechanism of the observed shift from TH1- to TH2-driven immunity after-stroke [16].
The soluble TNF Receptors (TNFR), p55 and p75, also contribute to the inflammatory process in traumatic brain injury, though the specifics of their involvement are not currently clear [84]. They act as anti-inflammatory agents through free TNF scavenging, although TNFR levels are more closely correlated to mortality in potential donors than TNF itself [84, 89]. This observation may actually reflect an imbalance in pro- and anti-inflammatory mechanisms or simply be due to the very short half-life of TNF [84].
3.4. Type 2 Cytokines
A recent study of stroke in IL-4 knockout mice showed that IL-4 reduces the TH1 : TH2 cell ratio and infarct volume, and improves neurological outcome [29]. Studies in humans have shown that brain-derived IL-4 can be detected in the jugular vein in patients with serious head injury [94]. A post-mortem study of TBI patients confirmed elevated IL-4 in brain tissue [70]. IL-13 has been less studied in brain injury. One in vitro study of IL-13 and IL-4 did show that these mediators induced apoptosis of activated microglia, which may account for part of the observed anti-inflammatory effect [95]. IL-13 is not significantly elevated in the plasma after TBI [31].
IL-10, a type 2 cytokine with anti-inflammatory properties [29, 30], plays a protective role in the CNS, reducing infarct size in stroke patients [39, 96]. Analysis of post-mortem TBI brains confirmed the presence of IL-10, though levels were more modest than similarly identified pro-inflammatory cytokines [70]. This was consistent with intraparenchymal levels measured by microdialysis in TBI and SAH patients [90]. Overflow of IL-10 into the cerebrospinal fluid (CSF) after TBI has also been demonstrated [97]. Systemic IL-10 levels peak early in TBI patients, declining to baseline within 48 hours [84]. Although IL-10 decreases inflammation through its immunomodulatory action, it also increases susceptibility to infection through immune system downregulation [96].
3.5. Type 17 Cytokines
In the CNS, IL-6 plays a dichotomous role through modulation of glial responses and neuronal survival, contributing to the early inflammatory response, but modulating later inflammatory pathways to assist with brain recovery [89, 98–100]. While its proinflammatory role is well known, it has also been shown to protect against excitotoxicity in vitro and brain ischaemic or excitotoxic states in vivo [98]. The specifics of how this balance are achieved are less clear [98]. One suggestion is that the role of IL-6 depends on the amount of neuronal cell damage and is concentration dependent, but it is also probably subject to negative feedback inhibition via crosstalk between NMDA and IL-6 receptors [98]. It may also downregulate inflammation through stimulating IL-1 receptor antagonist [90]. Microdialysis techniques have confirmed that IL-6 is acutely increased after brain injury [72, 73]. Furthermore, Graetz et al. demonstrated that IL-6 is released from the brain parenchyma into the systemic circulation after brain injury, particularly in the presence of high ICP [72]. Previous studies have also shown that IL-6 interferes with BBB integrity [72, 80]. Similar to IL-4, IL-6 is detectable in jugular blood, and the transcranial gradient correlates with poor outcome in TBI [72, 80]. The APR is stimulated by circulating IL-6 [101] and this may provide a link between central injury and the peripheral immune response seen with intracranial injury [83].
The roles of IL-17 and IL-23 in acute brain injury remain to be fully elucidated. While a role has been established in central autoimmune disorders including experimental models of multiple sclerosis [13], less has been published on acute CNS injury. Murine models demonstrate that both of these interleukins are locally upregulated after stroke [102–104]. Currently, there are no published data on their peripheral release after acute brain injury.
3.6. The Endothelin Axis in Brain Injury
Endothelin-1 (ET-1) is the most active member of a family of small polypeptides which are potent vasoconstrictors, mitogens of smooth muscle cells, and stimulators of fibroblasts (Table 1) [105–109]. ET-1 is an important mediator in TBI, stroke, and SAH [110–113]. In acute brain injury, ET-1 leads to constriction of large vessels, altering the normal balance between vascular relaxation and constriction, resulting in impaired cerebral blood flow [113]. This alteration of blood flow has been targeted in studies of SAH [114]. Clazosentan, an ET receptor A antagonist, reduces large cerebral artery vasospasm in murine models, but this did not reduce other mechanisms of secondary brain injury [114]. Salonia et al. analysed CSF levels of ET-1 in paediatric head trauma [113]. They found that ET-1 is significantly elevated after injury and remains so for up to 5 days. Central production of ET-1 in adult TBI was confirmed by Chatfield et al. [111]. Their analysis of the juguloarterial gradient showed that ET-1 is produced intracranially and spills over into the systemic circulation.
Table 1.
Properties of endothelin.
| Endothelin | |
|---|---|
| Endothelin subtypes [105, 106, 115] | |
| ET-1 ET-2 ET-3 |
|
| Sites of production [105] | |
| Smooth muscle cells Cardiomyocytes Leukocytes Macrophages Mesangial cells Airway epithelium Alveolar epithelial cells |
|
| Receptor subtypes [105, 107, 108, 116, 117] | |
| Endothelin receptor A Endothelin receptor B |
|
| Action | |
| Endothelin receptor A [105, 107, 108, 116, 117] Smooth muscle contraction Fibrogenesis Endothelin receptor B [105, 107, 108, 116, 117] Smooth muscle contraction Smooth muscle relaxation ET-1 clearance |
|
| Localisation of receptors [105, 107, 115] | |
| Heart Endocardium Conducting system Coronary vessels Lung Kidneys CNS Liver Neutrophils |
|
| Stimulators of release [105] | |
| Endothelial shear stress Thrombin AT2 Cytokines Free radicals Catecholamines |
|
| Inhibitors of release [105] | |
| NO ANP Prostacyclin |
ET: endothelin, AT2: angiotensin 2, NO: nitric oxide, ANP: atrial natriuretic peptide, CNS: central nervous system.
4. Stage Two of Potential Organ Injury: Brain Death
Serious brain injury, augmented by local inflammation, may eventually lead to an irretrievable state of impaired brain function and brain death. BD then further causes a massive autonomic storm and cytokinaemia which increases the inflammatory state of the individual [126, 140, 141]. A complex interplay of immunologic [142], coagulopathic [143], autonomic, haemodynamic, and endocrine [144, 145] dysregulation drives inflammation through local and global cytokine release, cellular activation, organ priming, IRI, and secondary ischaemic insult (Figure 2).
Figure 2.
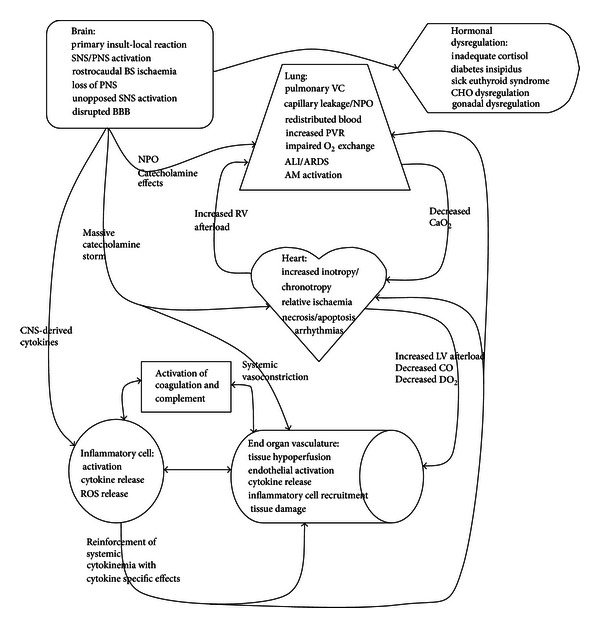
Interaction of homeostatic mechanisms after BD. ALI: acute lung injury, AM: alveolar macrophage, ARDS: acute respiratory distress syndrome, CaO2: arterial oxygen content, BBB: blood brain barrier, BS: brain stem, CHO: carbohydrate, CNS: central nervous system, CO: cardiac output, DO2: oxygen delivery, NPO: neurogenic pulmonary oedema, PNS: parasympathetic nervous system, PVR: pulmonary vascular resistance, ROS: reactive oxygen species, VC: vasoconstriction.
4.1. The Autonomic Nervous System during Brain Death
Brain stem dysfunction is associated with extreme physiological perturbations due to its “master control” function [146–148]. Brain stem failure secondary to high ICP occurs in a rostrocaudal direction, with initial hypertension and bradycardia (classically known as Cushing's reflex [149, 150]), followed by an intense “sympathetic storm” which remains unopposed due to ischaemia of the parasympathetic vagal nucleus [151, 152]. This storm results from an overwhelming release of catecholamines in an attempt to perfuse the brain by increasing the mean arterial pressure (MAP) to overcome the elevated ICP [153, 154]. Such changes in autonomic outflow can be detected prior to the occurrence of brain death [155]. The initial massive upsurge in sympathetic tone results in widespread vasoconstriction and microthrombus formation, impairing organ and tissue perfusion [146].
As the ICP outpaces the MAP, ischaemia progresses down the brain stem, sympathetic centres become necrotic, vascular and myocardial sympathetic stimulation drops and a second phase of hypotension ensues [146, 156, 157]. The resulting uncontrolled hypotension further impairs the already tenuous end organ perfusion that resulted during the sympathetic storm [156].
While the effects of the sympathetic nervous system are the most obvious clinically during and after BD, inflammatory and haemodynamic responses are also influenced by the parasympathetic nervous system (PNS). The effect of BD is to inhibit PNS-mediated anti-inflammatory responses by direct destruction of vagal centres in the brain stem [67]. Under normal conditions, vagal stimulation directly decreases inflammation via cholinergic receptors on inflammatory cells [158, 159]. Central activation of vagal efferent pathways downregulates inflammation in the brain, gut, and spleen [6, 67]. Balance is normally achieved through negative feedback by the innate immune system interacting with the PNS via IL-1 receptors in the parasympathetic paraganglia [67].
4.2. Cytokine Upregulation after Brain Death
4.2.1. Type 1-Associated Cytokines
Cytokine upregulation after BD has been recognised for many years [66, 160]. Animal models have shown that serum levels of IL-1β and TNF-α may be influenced by the rate of induction of brain death [22, 161]. Avlonitis et al. reported that explosive brain death induced a rapid increase in IL-1β, with significantly elevated levels detectable within one hour, remaining so throughout the duration of the study [161]. TNF-α levels initially rose and then decreased by five hours, though it remained above baseline [161]. Zhu et al. showed that gradual induction of brain death leads to steady elevation of IL-1β over 24 hours in a pig model [162]. Conversely, Damman and colleagues, utilising gradual BD induction in a rat model, showed that IL-1β and TNF-α did not change significantly over the four hours of their study [101]. Interestingly, this group also analysed serum cytokine levels in human BD donors and showed that they were not significantly elevated [101]. Cypel and colleagues recently reported that TNF-α and IL-1β mRNA are significantly elevated in lungs rejected for transplant, highlighting the clinical importance of these proinflammatory cytokines [163].
4.2.2. Type 2-Associated Cytokines Including IL-10
Early studies of cytokine upregulation after BD suggested that type 2 cytokines are not significant contributors to BD-induced inflammation [160]. Takada et al. did not show upregulation of IL-4 in rat kidneys, hearts, livers, or lungs after BD [160]. Weiss et al. studied cytokine expression at various timepoints during the liver transplantation process [91]. This group reported that IL-4 expression is increased after brain death [91]. IL-10 is elevated in the plasma of human BD donors [66, 123, 126]. Additionally, IL-10 has been shown to be upregulated in human livers [91] and kidneys [164]. Work undertaken by Li et al. suggested that IL-10 expression after BD may be important in stimulating apoptosis of graft infiltrating lymphocytes through activation of the Fas/Fas Ligand pathway [165]. There is little published in the literature investigating the role of IL-5 and IL-13 during brain death. This may be an area for future research.
4.2.3. Type 17-Associated Cytokines
IL-6 is heavily implicated in BD-related inflammation [127, 128], where it is an important instigator of the generalised APR [101]. The levels increase in human brain dead donors up until the time of organ retrieval [101]. Systemic venous and CNS-derived IL-6 is significantly higher at brain death than at admission to the intensive care unit (ICU) in TBI patients that progress to BD [80]. Brain death induces the production of IL-6 in multiple organs, including the kidney [166], heart [167], liver [168], and lung [141]. IL-6 signalling induces nitric oxide synthase in cardiac myocytes [167] and contributes to early haemodynamic compromise in the donor via direct negative inotropy [121, 127]. IL-6 mRNA and protein are elevated in nonstructural donor heart dysfunction [167].
Damman et al. recently investigated IL-6-related renal acute phase protein synthesis in rats [101]. As expected, IL-6 was upregulated after brain death. This correlated with an increase in renal acute phase proteins, notably complement 3 (C3), fibrinogen, α2-macroglobulin, and haptoglobin [101]. Furthermore, in vitro analysis indicated that renal production of C3 is directly related to IL-6 exposure [101].
Overall, elevated plasma levels of IL-6 are associated with poorer transplantation outcomes [91, 168]. Murugan and colleagues demonstrated an inverse relationship between donor plasma IL-6 levels and recipient six-month hospital-free survival [126]. Kaneda et al. also showed that higher donor IL-6 levels increased the risk of recipient death within 30 days of lung transplant [169].
TH17 cells, through production of IL-17, stimulate inflammation in donor organs [170]. Pretransplant renal biopsies from deceased donors showed little elevation of IL-17 positive cells, though few graft infiltrating cells were demonstrated in the biopsy samples [170]. Although a number of authors have studied IL-17 in the context of chronic rejection, the role of BD donor IL-17 currently remains unexplored.
4.2.4. Treg-Associated Cytokines
TGF-β is upregulated in heart and lung tissue in animal models [81]. Elevated TGF-β mRNA has been identified in renal and liver biopsies from brain dead donors [91, 164]. Weiss et al. showed that the greatest stimulus for TGF-β expression in liver grafts is BD itself [91]. A slight decrease in expression occurred prior to cold storage and to reperfusion. TGF-β mRNA expression increased by one hour after implantation and reperfusion but did not exceed levels measured before surgical manipulation (i.e., after BD alone) [91]. Skrabal et al. also demonstrated that TGF-β mRNA transcription is increased in donor heart and lungs in a porcine model of brain death [81]. The role of TGF-β in acute organ injury may relate to its role in the TH17/Treg balance [20, 50]; however, increased expression prior to transplantation may start fibrotic processes through activation of MMP's and tissue inhibitor of metalloproteinases (TIMP's). MMP-2, -9, TIMP-1, and -2 expression is increased after BD in pulmonary tissue [115].
4.2.5. Interleukin 8
IL-8 is a chemokine which attracts and activates neutrophils [132, 171]. Similar to other cytokines, IL-8 is produced peripherally after BD where it stimulates neutrophil-driven angiogenesis and fibroproliferation [132, 172]. IL-8-induced neovascularisation, alveolar-capillary disruption, and extracellular matrix deposition contribute to the development of acute lung injury after brain death [132]. In lung donors, bronchoalveolar lavage fluid IL-8 levels are positively correlated with neutrophil infiltration in pretransplant lung tissue, contributing to early graft dysfunction [132].
4.2.6. The Endothelin Axis
ET-1 release from endothelium is stimulated by noradrenaline, thrombin, and TGF-β [139]. Animal experiments have shown that ET-1 is upregulated in serum and in donor lung after BD and that this is related to MMP activation [115, 173]. Salama et al. demonstrated a correlation between donor ET-1 and primary graft dysfunction (PGD) [174]. In this study, ET-1 upregulation (as measured by donor lung mRNA and donor serum levels) adversely affected recipients after transplantation, contributing to the development of PGD.
5. Stage Three of Potential Organ Injury: Ischaemia Reperfusion Injury
Ischaemia reperfusion injury is implicated in early and late stage transplant complications [175]. IRI leads to organ dysfunction through induction of cytokines, generation of free radicals, and activation of immunocompetent cells [175, 176]. Endothelial cell dysfunction secondary to IRI is a key contributor to chronic allograft dysfunction in hearts [177], lungs [11], livers [178], and kidneys [179]. Early injury to cells occurs as a direct result of ischaemia, with impaired oxygen delivery, altered energy metabolism, and accumulation of waste products. Cell death occurs through necrosis and apoptosis, the latter through caspase signalling [66, 180]. Further injury occurs upon reperfusion, with recruitment of inflammatory cells, interaction between local and systemic cytokine signalling systems, and generation of ROS [127, 181, 182].
APC's of the innate immune system play a key role providing antigens and costimulatory molecules to activate the adaptive immune system, contributing to IRI and early graft dysfunction. Activation of cellular immunity can be classified as direct or indirect [181]. Direct activation occurs due to the transfer of donor APC's in the allograft, which activate recipient TH1 cells [181]. Atkinson et al. recently demonstrated that passenger leukocytes are recruited to donor hearts after BD in a murine model [22]. This finding was also confirmed in lung [183] and renal allografts [184]. Gelman et al. also demonstrated that recipient T cells interact with donor APC's and that this is sufficient to activate an inflammatory response [183]. Alternatively, the indirect pathway results from the interaction of recipient APC's with native T cells to stimulate inflammation.
5.1. Contribution of Preservation Strategies to Cytokine Expression
Hypothermic preservation strategies are widely used to decrease inflammation, depress the metabolic rate of cells, and reduce the effects of ischaemia [185]. However, cold storage does cause cell death via both apoptosis and necrosis [15]. BD donor organs predominantly display the latter mechanism [178]. The duration and type (warm or cold) of ischaemic time may also directly influence cytokine production. A correlation was recently identified between cold ischaemia time and levels of IL-1 and IL-8 in human liver transplants [186]. Warm ischaemia time correlated with IL-6 and IL-10 in the same study. Significantly, the authors found that the excess cytokines generated by hepatic graft warm ischaemia time resulted in systemic adverse effects, most notably increased intraoperative pulmonary shunt [186]. Another study found that, while cold ischaemic time per se did not adversely affect liver function, the associated graft-generated IL-8 did correlate with PGD [187]. Weiss et al., in a study of transplanted human livers, showed that IL-4 was increased in BD donors prior to explantation, but cold ischaemia and reperfusion did not result in further increases in the cytokine [91]. Indeed, while it was elevated compared to living donors, it failed to reach statistical significance at time points other than immediately after laparotomy. IL-10 was highly expressed prior to organ preservation, but cold ischaemia and reperfusion did not result in further elevation of this cytokine [91]. Livers from living donors showed a relatively greater increase in expression of IL-10 one hour after reperfusion than BD organs, which may partially contribute to better outcomes with organs from these donors [91].
Delayed graft function in transplanted kidneys has been shown to be dependent on cold ischaemic time [170]. Kaminska et al. showed that while cytokine upregulation occurred, associated with brain death, mRNA expression did not increase further after cold ischaemia and prior to reperfusion [164]. In keeping with this, de Vries et al. were unable to detect an arteriovenous difference across human BD donor kidneys for multiple cytokines, including IL-4, IL-5, IL-10, and IL-13 [184]. Cold ischaemia and reperfusion do not induce excess TGF-β mRNA production, indicating that the primary stimulus for this mediator is brain death itself [164].
5.2. Other Mediators of Ischaemia Reperfusion Injury
The combination of BD and IRI activates allografts greater than either insult alone. Kusaka et al. studied rat renal isografts to analyse gene activation after BD, IRI, or combined BD/IRI [175]. They found that BD primarily upregulated cytokines, chemokines and adhesion molecules while IRI tended to upregulate transcription factors. Combined BD/IRI was synergistic in enhancing upregulation of these genes. More recent work has maintained these findings. Inhibition of JNK, a phosphorylator of the transcription factor c-Jun, decreases IRI-induced renal damage in rats [179]. In humans, de Vries et al. demonstrated that reperfusion of BD kidneys generates higher cytokine levels than living donor allografts (i.e., those that only underwent IRI) [184].
Complement interacts with, and reinforces, the inflammatory process of IRI by increasing TNF-α and IL-1 [22]. C3a and C5a, potent anaphylatoxins generated by the complement cascade, activate mast cells and neutrophils [182]. While the specific mechanism of complement activation in BD is unknown, it is postulated that ischaemia leads to defects in cell membranes, uncovering neoepitopes via exposure of internal cellular components to the humoral immune system, which leads to interaction with natural IgM and activation of the classical complement pathway [182]. ROS generated during infarction and IRI may lead to lipid peroxidation and alteration of cellular cytoskeletal structure providing further neoepitopes for IgM [182].
The importance of toll-like receptors (TLR) in IRI is currently being investigated. It was previously noted that low levels of lipopolysaccharide (LPS) may precondition and therefore protect the lung from IRI [188]. Merry et al. demonstrated that low-dose preconditioning with LPS in rat lung ischaemia reduced injury [188]. The authors postulated that this may be due to LPS activating TLR-4 via an alternative pathway that results in protective interferon and IL-10 generation. Unfortunately, they did not measure IL-10 protein or mRNA to confirm this hypothesis. The role of TLR's in renal IRI has recently been reviewed elsewhere [12]. TLRs may also contribute to inflammation through interaction with T cells via cytokine signalling. APC TLR activation leads to generation of cytokines, including IL-6, which may decrease the sensitivity of TH1 cells to the immunosuppressive effects of Treg cells [189]. Additionally, TLR on Treg cells may directly inhibit their immunosuppressive effects [189].
ET-1 contributes to IRI through activated neutrophils, leading to endothelial injury, neutrophil superoxide production and generation of ROS [115]. Both ET-1 and its receptors are upregulated in the lungs after brain death [115]. Alveolar macrophages have been demonstrated to increase expression of endothelin receptors in the donor lung in animal models [115]. This may then prime passenger macrophages for further activation by recipient ET-1, which is generated in the pro-inflammatory environment of chronic lung disease, surgery, and the posttransplant course [115, 190].
Heme-oxygenase-1 (HO-1) is essential for the metabolism of heme to carbon monoxide, free iron, and biliverdin [25]. Its ability to reduce injury secondary to IRI, with resulting better recipient outcomes after transplantation, has been the subject of much research. HO-1 exerts its beneficial effects through antioxidant, antiapoptotic, and anti-inflammatory mechanisms [25, 191–193]. Carbon monoxide contributes to these beneficial effects through inhibiting T-cell proliferation and IL-2 secretion [25]. Zhou et al., in studying a rat model of BD, demonstrated improved lung function and decreased lung injury when carbon monoxide was administered at 250 ppm [194]. Carbon monoxide decreased myeloperoxidase activity, TNF-α, and IL-6 [194]. More recently, the same group demonstrated that both carbon monoxide and biliverdin reduce myeloperoxidase activity and cytokine signalling while improving respiratory mechanics in rat lung after BD [195]. HO-1 has also been linked to anti-inflammatory cytokine generation. IL-10 production secondary to HO-1 is increased in both BD [192] and non-BD models [196]. HO-1 may also be an important mediator of IL-13's anti-inflammatory effect [191, 197].
6. Management Implications and Potential Future Directions
Recipients of organs from brain dead donors continue to have poorer outcomes than those that receive living donor organs. Aggressive donor management (ADM) improves both quality and quantity of organs available for transplant [198]. Current ADM recommendations include early identification of potential donors, ICU admission, pulmonary artery catheterisation, aggressive fluid management, vasopressors, hormonal resuscitation therapy, pulmonary toilet, and bronchoscopy [148, 199–202].
Even with ADM, up to 25% of potential donors are lost due to haemodynamic instability [148]. Studies comparing use of noradrenaline and vasopressin as pressor agents demonstrate differing effects on transplantable organs. Although no studies directly comparing these agents in humans have been published, animal models suggest that both agents decrease lung inflammation and serum cytokine release [68]. While a similar effect is seen in the kidney, hepatic inflammation is increased by both agents [203]. Dopamine decreased monocyte kidney graft infiltration and markers of inflammation in a rat model of BD [193]. Additionally, dopamine increased the expression of HO-1 [193]. Regardless of the agents used, the evidence supports that aggressive haemodynamic monitoring and management do convert marginal donors to acceptable donors [141, 145].
The inflammatory cascade may be downregulated by ADM. Currently, there are no standard interventions specifically directed at individual cytokines, though many are being investigated (Table 2). Steroid administration, as part of hormonal resuscitation, is now commonplace in the management of organ donors and, in addition to addressing a relatively inadequate adrenal response, reduces inflammatory cytokines to levels similar to living donors [141, 151, 168].
Table 2.
Major cytokines associated with brain injury and BD.
| Cytokine/chemokine | Organs/cells upregulated in BD/CNS injury | Stimulation in BD/TBI | Action | Potential therapeutic agents in brain injury |
|---|---|---|---|---|
| TNF-α | CNS—astrocytes, microglia, and neurons [70, 118] Endothelial cells [118] Lungs [119] Splenocytes, macrophages [6] |
Infection, TBI, SAH [118] | Endothelial cell detachment/apoptosis, activation caspase-3, disruption of BBB [118] Induction of CAM's, and other inflammatory cytokines [120] Impairment of cardiac function [121] |
IFN-β [119], NNZ-2566 [122], etanercept, and IFN inhibitors [60] Haemoadsorption [123] |
|
| ||||
| IL-1β | CNS—neurons, microglia, and infiltrating macrophages [124] Endothelial cells [118] |
Neuroexcitation, infection, and trauma [124] SAH [118] |
Synaptic modulation, central regulation of systemic inflammatory response [124] Proinflammatory, activation of NFκB and SAPK with upregulation of E-selectin/ICAM/VCAM [118] |
IL-1RA [124] and NNZ-2566 [122] Haemoadsorption [123] |
|
| ||||
| IL-6 | CNS—microglia [125] Kidney, liver, spleen, and heart [80, 121, 126–128] Macrophages [121] |
IL-1β [118] TNF-α [121] Sepsis, major surgery, heart failure, multitrauma, and burns [80, 84, 127, 129, 130] |
Regulator of inflammation—inhibition of TNF and upregulation of control of glial responses and neuronal survival [98–100] IL-1RA in CNS, induction of NGF [122] Disruption of BBB [118] Inducer of acute phase reaction [70, 131] Cardiac dysfunction, fibroblast activation [127] |
Haemoadsorption [123] |
|
| ||||
| IL-8/CXCL-8/MIP-2 | Microglia [125] Lung—alveolar macrophages, endothelial cells [132, 133] |
Trauma, ischaemia, SAH, ET-1 [118] TNF-α, IL-1β [120] |
Disruption of BBB [118, 120] CXC chemokine—neutrophil migration and activation [120] Induces ROS by neutrophils [131] |
Haemoadsorption [123] |
|
| ||||
| IL-10 | Macrophages, microglia [125] Splenocytes [83] |
TBI [97] Burns, MT, surgery, and infection [131] |
Anti-inflammatory—downregulates TNF-α, IL-1β, and IFN-γ, upregulates antagonists [39, 134] Reverses effect of proinflammatory cytokines directly on cells [39] |
Haemoadsorption [123] |
|
| ||||
| E-Selectin | Endothelial cells in multiple organs [119] | IL-1β [118] TNF-α [121] TBI [122] SAH [118] |
Essential for neutrophil rolling, margination, and diapedesis [118] | |
|
| ||||
| ICAM | Endothelial cells in multiple organs [119] | IL-1β [118] TNF-α [121] SAH [118] |
Essential for neutrophil rolling, margination, and diapedesis [118] | Monoclonal antibodies [118] and IFN-β [119] |
|
| ||||
| VCAM | Endothelial cells in multiple organs [119] | IL-1β [118] TNF-α [121] SAH [118] |
Essential for neutrophil rolling, margination, and diapedesis [118] | Monoclonal antibodies [118] and IFN-β [119] |
|
| ||||
| TGF-β | Macrophages, microglia, astrocytes, and neurons [135, 136] Platelets, choroid epithelium [137] |
Constitutively expressed by microglia [134] SAH [137] |
Anti-inflammatory, may block activation by IL-1β [125] Regulates T-cell survival and function [136] Suppresses IFN-γ-induced macrophage upregulation, cytokine and chemokine generation [136] Downregulation of adhesion molecules [136] Reduces COX-2 production in microglia [125] ECM component generation [138] Angiogenesis [137] ET-1 generation [139] |
Haemoadsorption [123] |
|
| ||||
| IFN-γ | Microglia [125] Macrophages [122] |
TBI, SAH [122, 125] | Upregulation of CAM's, chemokines, and innate immune system cells [122] | IFN inhibitors [60] |
|
| ||||
| COX-2 | CNS—Microglia, endothelial cells [125] | Inflammatory mediators including IL-1β, TNF-α, and IL-6 [125] | Production of prostaglandins, reinforcement of inflammation [125] |
COX inhibitors [125] |
BD: brain stem death, TBI: traumatic brain injury, CNS: central nervous system, SAH: subarachnoid haemorrhage, BBB: blood brain barrier, MT: multitrauma, ECM: extracellular matrix, COX: cyclooxygenase, IL: interleukin, TNF: tumour necrosis factor, CAM: cellular adhesion molecule (ICAM: intercellular adhesion molecule/VCAM: vascular cellular adhesion molecule), NFκB: nuclear factor κ B, SAPK: stress-activated protein kinases, MIP-2: macrophage inflammatory protein 2, TGF: transforming growth factor, IFN: interferon, NGF: nerve growth factor.
Other methods directly addressing anti-inflammatory mechanisms are currently being investigated. Gene transfer of IL-10 holds great promise. Manning and colleagues investigated viral IL-10 (virIL-10) transfer into a rat model of lung IRI using mesenchymal stem cells [11]. This study showed that virIL-10 was detectable in the lungs and that presence of this cytokine was related to improved lung function, less microscopic pathology, and decreased lung oedema at four hours after injury. Gene transfer pretreatment of rat liver grafts to generate recombinant human IL-10 significantly decreases IRI and markers of apoptosis, with upregulation of HO-1 and the antiapoptotic agent, Bcl-2 [15]. HO-1 may then act as a downstream regulator of protective mechanisms in IRI [15].
HO-1 or its metabolites (carbon monoxide and biliverdin) may offer potential therapeutic benefits [25, 192, 194, 195]. Overexpression of HO-1, through adeno-associated virus gene transfer, was associated with a beneficial increase in HO-1 expression [192]. This resulted in downregulation of IL-2 and TNF-α, decreased infiltration of cytotoxic and helper T-cells, and an increase in IL-10, TGF-β, and Treg infiltration in transplanted rat livers [192]. IL-13 gene transfer in rat livers increased HO-1 expression with reduced evidence of IRI [197]. Inhibition of HO-1 activity reversed this effect, suggesting that part of IL-13's anti-inflammatory properties in IRI is mediated by HO-1 [197].
Hypothermic ischaemic storage prior to transplantation does not allow sufficient metabolic activity for gene transfer to be beneficial [204]. Cypel et al. therefore trialled an ex vivo lung perfusion (EVLP) model to transfer recombinant human IL-10 genes into porcine lungs [204]. Perfusate IL-10 was increased while IL-6 decreased. This effect was maintained after transplantation and four hours of reperfusion. Lung function, as assessed by PaO2 : FiO2 ratio, was significantly improved in the transfected lungs. When transfection was trialled in human lungs rejected for transplantation, Cypel and colleagues found similar results including improved gas exchange and pulmonary vascular resistance [204].
Lung conditioning using EVLP is able to improve the function of lungs initially rejected for transplant [205]. Sadaria et al. have established a baseline cytokine profile of human lungs undergoing EVLP [205]. Cytokine analysis during 12 hours of EVLP showed an upregulation in IL-6, IL-8, G-CSF, and MCP-1 [205]. IL-1β, IL-4, IL-7, IL-12, and TNF-α were detectable but remained unchanged [205]. IL-17 was undetectable, as were IL-10 and IL-13 [205]. Kakishita et al. also investigated the cytokine profile of EVLP in pigs [206]. Inflammatory cytokines were similarly elevated. Interestingly, based on a previously published concept of haemoadsorption of cytokines [123], Kakishita investigated the benefit removing perfusate cytokines within the circuit. Cytokine levels were significantly reduced with haemoadsorption, but oxygenation, pulmonary vascular resistance, peak airway pressure, and myeloperoxidase activity (as a marker of neutrophil accumulation) were not statistically different [206].
Numerous other agents have been investigated as part of organ protection and preservation strategies. Donor simvastatin may reduce IRI in cardiac allografts [177]. This agent appears to work through multiple mechanisms and provides a lasting effect after a single dose to the donor prior to graft removal [177]. Organ donors in this animal model were not brain dead; therefore, simvastatin's effects seem to be related to downregulation of ischaemia reperfusion injury. A study of N-acetylcysteine after pig non-BD lung transplantation demonstrated increased glutathione and downregulation of the inflammatory transcription factor NFκB in tissue samples [171]. IL-6 and IL-8 levels were also reduced. Lung function was improved despite extended cold ischaemia and reperfusion [171]. Intraoperative administration of N-acetylcysteine to human liver transplant recipients significantly increased the transhepatic gradient of IL-4 and IL-10 around the time of reperfusion, but not at other measured time points [207]. The authors theorised that the presence of these anti-inflammatory cytokines at reperfusion may benefit recipients through downregulation of inflammation. Unfortunately, although the agent was administered as a continuous infusion for 24 hours, no further information is given about levels of cytokines later than the first hour of transplant, nor any information about hepatic biochemistry and patient outcomes.
In renal transplantation, carbamylated erythropoietin (EPO) downregulated renal IL-1β and IL-6 in a rat model of brain death [166]. This agent retains the protective effects of EPO without stimulating haematopoiesis [166]. Utilising an isolated perfused kidney circuit, Nijboer and colleagues demonstrated that carbamylated EPO downregulated IL-1β and IL-6, reduced neutrophil infiltration, and reversed brain death-induced renal impairment. Of note, other authors are also investigating EPO in preventing brain IRI [208]. Such use in pre-BD conditions may eventually spill over to benefit the recipients of organs from these patients in the case of nonsurvival.
Further research is required into the impact of pre-BD management of organ donors. There are substantial data examining the management of TBI or SAH patients which specifically addresses inflammatory/anti-inflammatory interventions and long-term recovery. The impact of such management on the transplanted organs of those that fail treatment and become BD organ donors may reveal interesting results.
7. Conclusion
Engrafted organs undergo significant pathophysiological challenges as they are transplanted from the donor to the recipient. Brain injury, brain death, ischaemia, and reperfusion all contribute to inflammation and injury. As has been discussed, a vast amount of research is ongoing at each of these steps of transplant. Understanding the molecular inflammatory responses and utilising interventions that can reduce haemodynamic instability, inflammation, and IRI is the key to further advancing donor management. With time and more successful interventions, it may be possible to further address the ongoing shortage of donor organs and decrease the number of patients who die whilst waiting for a transplant.
Disclosure
Appropriate terms were searched on Medline, Pubmed, and Embase on November 2011. This was repeated on March 2012. A full list of search terms is available on request.
Acknowledgment
The authors declare no funding sources for the development of this paper. Professor J. Fraser is supported by the Health Research Fellowship funded by Queensland Health.
Abbreviations
- ADM:
Aggressive donor management
- APC:
Antigen presenting cell
- APR:
Acute phase response
- BBB:
Blood brain barrier
- BD:
Brain death
- C:
Complement
- CD:
Cluster of differentiation
- CNS:
Central nervous system
- CSF:
Cerebrospinal fluid
- EPO:
Erythropoietin
- ET:
Endothelin
- EVLP:
Ex vivo lung perfusion
- G-CSF:
Granulocyte colony stimulating factor
- HIF:
Hypoxia inducible factor
- HO:
Heme oxygenase
- ICP:
Intracranial pressure
- ICU:
Intensive care unit
- IFN:
Interferon
- IL:
Interleukin
- IRI:
Ischaemia reperfusion injury
- LPS:
Lipopolysaccharide
- MAP:
Mean arterial pressure
- MCP:
Monocyte chemoattractant protein
- MMP:
Matrix metalloproteinase
- mRNA:
Messenger ribonucleic acid
- NFKB:
Nuclear factor kappa B
- NMDA:
N-Methyl D-aspartic acid
- PGD:
Primary graft dysfunction
- PNS:
Parasympathetic nervous system
- ROS:
Reactive oxygen species
- SAH:
Subarachnoid haemorrhage
- SIRS:
Systemic inflammatory response syndrome
- SNS:
Sympathetic nervous system
- TBI:
Traumatic brain injury
- TGF:
Transforming growth factor
- TH:
Helper T cell
- TIMP:
Tissue inhibitor of metalloproteinases
- TLR:
Toll-like receptor
- TNF:
Tumour necrosis factor
- TNFR:
Tumour necrosis factor receptor
- Treg:
Regulatory T cell.
References
- 1.Organ. Procurement and Transplantation Network. Organ by Status, 2011, http://optn.transplant.hrsa.gov/
- 2.Fink MA, Berry SR, Gow PJ, et al. Risk factors for liver transplantation waiting list mortality. Journal of Gastroenterology and Hepatology. 2007;22(1):119–124. doi: 10.1111/j.1440-1746.2006.04422.x. [DOI] [PubMed] [Google Scholar]
- 3.Banner NR, Rogers CA, Bonser RS, et al. Effect of heart transplantation on survival in ambulatory and decompensated heart failure. Transplantation. 2008;96(11, article 8) doi: 10.1097/TP.0b013e31818b3328. [DOI] [PubMed] [Google Scholar]
- 4.Reed A, Snell GI, McLean C, Williams TJ. Outcomes of patients with interstitial lung disease referred for lung transplant assessment. Internal Medicine Journal. 2006;36(7):423–430. doi: 10.1111/j.1445-5994.2006.01103.x. [DOI] [PubMed] [Google Scholar]
- 5.De Vleeschauwer SI, Wauters S, Dupont LJ, et al. Medium-term outcome after lung transplantation is comparable between brain-dead and cardiac-dead donors. Journal of Heart and Lung Transplantation. 2011;30(9):975–981. doi: 10.1016/j.healun.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 6.Lee S-T, Chu K, Jung KH, et al. Cholinergic anti-inflammatory pathway in intracerebral hemorrhage. Brain Research. 2010;1309:164–171. doi: 10.1016/j.brainres.2009.10.076. [DOI] [PubMed] [Google Scholar]
- 7.Nijboer WN, Schuurs TA, Van Der Hoeven JAB, et al. Effect of brain death on gene expression and tissue activation in human donor kidneys. Transplantation. 2004;78(7):978–986. doi: 10.1097/01.tp.0000135565.49535.60. [DOI] [PubMed] [Google Scholar]
- 8.Naderi GH, Mehraban D, Kazemeyni SM, Darvishi M, Latif AH. Living or deceased donor kidney transplantation: a comparison of results and survival rates among Iranian patients. Transplantation Proceedings. 2009;41(7):2772–2774. doi: 10.1016/j.transproceed.2009.07.041. [DOI] [PubMed] [Google Scholar]
- 9.Anyanwu AC, Banner NR, Radley-Smith R, Khaghani A, Yacoub MH. Long-term results of cardiac transplantation from live donors: the domino heart transplant. Journal of Heart and Lung Transplantation. 2002;21(9):971–975. doi: 10.1016/s1053-2498(02)00406-0. [DOI] [PubMed] [Google Scholar]
- 10.Khaghani A, Birks EJ, Anyanwu AC, Banner NR. Heart transplantation from live donors: ‘Domino procedure’. Journal of Heart and Lung Transplantation. 2004;23(9, supplement 1):S257–S259. doi: 10.1016/j.healun.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 11.Manning E, Pham S, Li S, et al. Interleukin-10 delivery via mesenchymal stem cells: a novel gene therapy approach to prevent lung ischemia-reperfusion injury. Human Gene Therapy. 2010;21(6):713–727. doi: 10.1089/hum.2009.147. [DOI] [PubMed] [Google Scholar]
- 12.Damman J, Daha MR, Van Son WJ, Leuvenink HG, Ploeg RJ, Seelen MA. Crosstalk between complement and toll-like receptor activation in relation to donor brain death and renal ischemia-reperfusion injury. American Journal of Transplantation. 2011;11(4):660–669. doi: 10.1111/j.1600-6143.2011.03475.x. [DOI] [PubMed] [Google Scholar]
- 13.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24(6):677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 14.Deknuydt F, Bioley G, Valmori D, Ayyoub M. IL-1β and IL-2 convert human Treg into TH17 cells. Clinical Immunology. 2009;131(2):298–307. doi: 10.1016/j.clim.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 15.Li J-Q, Qi HZ, He ZJ, et al. Cytoprotective effects of human interleukin-10 gene transfer against necrosis and apoptosis induced by hepatic cold ischemia/reperfusion injury. Journal of Surgical Research. 2009;157(1):e71–e78. doi: 10.1016/j.jss.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Wong CHY, Jenne CN, Lee WY, Léger C, Kubes P. Functional innervation of hepatic iNKT cells is immunosuppressive following stroke. Science. 2011;334(6052):101–105. doi: 10.1126/science.1210301. [DOI] [PubMed] [Google Scholar]
- 17.Wadia PP, Tambur AR. Yin and yan of cytokine regulation in solid organ graft rejection and tolerance. Clinics in Laboratory Medicine. 2008;28(3):469–479. doi: 10.1016/j.cll.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 18.Nakajima T, Palchevsky V, Perkins DL, Belperio JA, Finn PW. Lung transplantation: infection, inflammation, and the microbiome. Seminars in Immunopathology. 2011;33(2):135–156. doi: 10.1007/s00281-011-0249-9. [DOI] [PubMed] [Google Scholar]
- 19.Huss DJ, Winger RC, Peng H, Yang Y, Racke MK, Lovett-Racke AE. TGF-β enhances effector Th1 cell activation but promotes self-regulation via IL-10. Journal of Immunology. 2010;184(10):5628–5636. doi: 10.4049/jimmunol.1000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Itoh S, Kimura N, Axtell RC, et al. Interleukin-17 accelerates allograft rejection by suppressing regulatory T cell expansion. Circulation. 2011;124(supplement 11):S187–S196. doi: 10.1161/CIRCULATIONAHA.110.014852. [DOI] [PubMed] [Google Scholar]
- 21.Kastelijn EA, Rijkers GT, Van Moorsel CH, et al. Systemic and exhaled cytokine and chemokine profiles are associated with the development of bronchiolitis obliterans syndrome. The Journal of Heart and Lung Transplantation. 2010;29(9):997–1008. doi: 10.1016/j.healun.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 22.Atkinson C, Varela JC, Tomlinson S. Complement-dependent inflammation and injury in a murine model of brain dead donor hearts. Circulation Research. 2009;105(11):1094–1101. doi: 10.1161/CIRCRESAHA.109.194977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshida N, Yoshikawa T, Nakamura Y, et al. Interactions of neutrophils and endothelial cells under low flow conditions in vitro . Shock. 1997;8(2):125–130. doi: 10.1097/00024382-199708000-00011. [DOI] [PubMed] [Google Scholar]
- 24.Lattmann T, Hein M, Horber S, et al. Activation of pro-inflammatory and anti-inflammatory cytokines in host organs during chronic allograft rejection: role of endothelin receptor signaling. American Journal of Transplantation. 2005;5(5):1042–1049. doi: 10.1111/j.1600-6143.2005.00807.x. [DOI] [PubMed] [Google Scholar]
- 25.Pae H-O, Oh GS, Choi BM, et al. Carbon monoxide produced by heme oxygenase-1 suppresses T cell proliferation via inhibition of IL-2 production. Journal of Immunology. 2004;172(8):4744–4751. doi: 10.4049/jimmunol.172.8.4744. [DOI] [PubMed] [Google Scholar]
- 26.Ishizaki M, Akimoto T, Muromoto R, et al. Involvement of tyrosine kinase-2 in both the IL-12/Th1 and IL-23/Th17 axes in vivo . Journal of Immunology. 2011;187(1):181–189. doi: 10.4049/jimmunol.1003244. [DOI] [PubMed] [Google Scholar]
- 27.Harden JL, Gu T, Kilinc MO, Rowswell-Turner RB, Virtuoso LP, Egilmez NK. Dichotomous effects of IFN-γ on dendritic cell function determine the extent of IL-12 - driven antitumor T cell immunity. Journal of Immunology. 2011;187(1):126–132. doi: 10.4049/jimmunol.1100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ueno T, Yamada A, Ito T, et al. Role of nuclear factor of activated T cell (NFAT) transcription factors in skin and vascularized cardiac allograft rejection. Transplantation. 2011;92(5):e26–e27. doi: 10.1097/TP.0b013e318228061c. [DOI] [PubMed] [Google Scholar]
- 29.Xiong X, Barreto GE, Xu L, Ouyang YB, Xie X, Giffard RG. Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke. 2011;42(7):2026–2032. doi: 10.1161/STROKEAHA.110.593772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Theodorou GL, Marousi S, Ellul J, et al. T helper 1 (Th1)/Th2 cytokine expression shift of peripheral blood CD4+ and CD8+ T cells in patients at the post-acute phase of stroke. Clinical and Experimental Immunology. 2008;152(3):456–463. doi: 10.1111/j.1365-2249.2008.03650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hensler T, Sauerland S, Riess P, et al. The effect of additional brain injury on systemic interleukin (IL)-10 and IL-13 levels in trauma patients. Inflammation Research. 2000;49(10):524–528. doi: 10.1007/s000110050626. [DOI] [PubMed] [Google Scholar]
- 32.Finkelman FD, Katona IM, Urban JF, Jr., et al. IL-4 is required to generate and sustain in vivo IgE responses. Journal of Immunology. 1988;141(7):2335–2341. [PubMed] [Google Scholar]
- 33.Wang C, Tay SS, Tran GT, et al. Donor IL-4-treatment induces alternatively activated liver macrophages and IDO-expressing NK cells and promotes rat liver allograft acceptance. Transplant Immunology. 2010;22(3-4):172–178. doi: 10.1016/j.trim.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 34.Bansal G, Wong CM, Liu L, Suzuki YJ, et al. Oxidant signaling for interleukin-13 gene expression in lung smooth muscle cells. Free Radical Biology and Medicine. 2012;52(9):1552–1559. doi: 10.1016/j.freeradbiomed.2012.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandovici M, Deelman LE, van Goor H, Helfrich W, de Zeeuw D, Henning RH. Adenovirus-mediated interleukin-13 gene therapy attenuates acute kidney allograft injury. The Journal of Gene Medicine. 2007;9(12):1024–1032. doi: 10.1002/jgm.1106. [DOI] [PubMed] [Google Scholar]
- 36.Corren J. Inhibition of interleukin-5 for the treatment of eosinophilic diseases. Discovery Medicine. 2012;13(71):305–312. [PubMed] [Google Scholar]
- 37.Fujisawa T, Abu-Ghazaleh R, Kita H, Sanderson CJ, Gleich GJ. Regulatory effect of cytokines on eosinophil degranulation. Journal of Immunology. 1990;144(2):642–646. [PubMed] [Google Scholar]
- 38.Grund LZ, Komegae EN, Lopes-Ferreira M, Lima C. IL-5 and IL-17A are critical for the chronic IgE response and differentiation of long-lived antibody-secreting cells in inflamed tissues. Cytokine. 2012;59(2):335–351. doi: 10.1016/j.cyto.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 39.Knoblach SM, Faden AI. Interleukin-10 improves outcome and alters proinflammatory cytokine expression after experimental traumatic brain injury. Experimental Neurology. 1998;153(1):143–151. doi: 10.1006/exnr.1998.6877. [DOI] [PubMed] [Google Scholar]
- 40.Nath DS, Ilias Basha H, Tiriveedhi V, et al. Characterization of immune responses to cardiac self-antigens myosin and vimentin in human cardiac allograft recipients with antibody-mediated rejection and cardiac allograft vasculopathy. Journal of Heart and Lung Transplantation. 2010;29(11):1277–1285. doi: 10.1016/j.healun.2010.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu XC, Zhai A, Li JQ, Qi HZ. Interleukin-23 promotes natural killer T-cell production of IL-17 during rat liver transplantation. Transplantation Proceedings. 2011;43(5):1962–1966. doi: 10.1016/j.transproceed.2011.01.175. [DOI] [PubMed] [Google Scholar]
- 42.Laan M, Cui ZH, Hoshino H, et al. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. Journal of Immunology. 1999;162(4):2347–2352. [PubMed] [Google Scholar]
- 43.Shichita T, Sugiyama Y, Ooboshi H, et al. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nature Medicine. 2009;15(8):946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- 44.Parrish-Novak J, Dillon SR, Nelson A, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature. 2000;408(6808):57–63. doi: 10.1038/35040504. [DOI] [PubMed] [Google Scholar]
- 45.Hagn M, Sontheimer K, Dahlke K, et al. Human B cells differentiate into granzyme B-secreting cytotoxic B lymphocytes upon incomplete T-cell help. Immunology and Cell Biology. 2012;90(4):457–467. doi: 10.1038/icb.2011.64. [DOI] [PubMed] [Google Scholar]
- 46.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochimica et Biophysica Acta. 2011;1813(5):878–888. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 47.Akdis M. The cellular orchestra in skin allergy; Are differences to lung and nose relevant? Current Opinion in Allergy and Clinical Immunology. 2010;10(5):443–451. doi: 10.1097/ACI.0b013e32833d7d48. [DOI] [PubMed] [Google Scholar]
- 48.Chen Y, Li D, Tsang JYS, et al. PPAR-γ signaling and IL-5 inhibition together prevent chronic rejection of MHC Class IImismatched cardiac grafts. Journal of Heart and Lung Transplantation. 2011;30(6):698–706. doi: 10.1016/j.healun.2011.01.704. [DOI] [PubMed] [Google Scholar]
- 49.Eulenfeld R, et al. Interleukin-6 signalling: more than Jaks and STATs. European Journal of Cell Biology. 2012;91(6-7):486–495. doi: 10.1016/j.ejcb.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 50.Wang G, Zhong A, Wang S, Dong N, Sun Z, Xia J. Retinoic acid attenuates acute heart rejection by increasing regulatory t cell and repressing differentiation of th17 cell in the presence of TGF-β . Transplant International. 2010;23(10):986–997. doi: 10.1111/j.1432-2277.2010.01080.x. [DOI] [PubMed] [Google Scholar]
- 51.Neujahr DC, Larsen CP. Regulatory T cells in lung transplantation-an emerging concept. Seminars in Immunopathology. 2011;33(2):117–127. doi: 10.1007/s00281-011-0253-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Organ Procurement and Transplantation Network, Deceased Donors Recovered in the U.S. by Cause of Death, 2011, http://optn.transplant.hrsa.gov/latestData/rptData.asp.
- 53.Inaba K, Branco BC, Lam L, et al. Organ donation and time to procurement: late is not too late. Journal of Trauma. 2010;68(6):1362–1366. doi: 10.1097/TA.0b013e3181db30d3. [DOI] [PubMed] [Google Scholar]
- 54.Lee S, Shin M, Kim E, et al. Donor characteristics associated with reduced survival of transplanted kidney grafts in Korea. Transplantation Proceedings. 2010;42(3):778–781. doi: 10.1016/j.transproceed.2010.02.060. [DOI] [PubMed] [Google Scholar]
- 55.Zukowski M, Bohatyrewicz R, Biernawska J, et al. Cause of death in multiorgan donors and its relation to the function of transplanted kidneys. Transplantation Proceedings. 2009;41(8):2972–2974. doi: 10.1016/j.transproceed.2009.07.081. [DOI] [PubMed] [Google Scholar]
- 56.Wauters S, Verleden GM, Belmans A, et al. Donor cause of brain death and related time intervals: does it affect outcome after lung transplantation? European Journal of Cardio-thoracic Surgery. 2011;39(4):e68–e76. doi: 10.1016/j.ejcts.2010.11.049. [DOI] [PubMed] [Google Scholar]
- 57.Ganesh JS, Rogers CA, Banner NR, Bonser RS. Donor cause of death and medium-term survival after heart transplantation: a United Kingdom national study. Journal of Thoracic and Cardiovascular Surgery. 2005;129(5):1153–1159. doi: 10.1016/j.jtcvs.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 58.Godino M, Lander M, Cacciatore A, Perez-Protto S, Mizraji R. Ventricular dysfunction associated with brain trauma is cause for exclusion of young heart donors. Transplantation Proceedings. 2010;42(5):1507–1509. doi: 10.1016/j.transproceed.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 59.Campbell SJ, Perry VH, Pitossi FJ, et al. Central nervous system injury triggers Hepatic CC and CXC chemokine expression that is associated with leukocyte mobilization and recruitment to both the central nervous system and the liver. American Journal of Pathology. 2005;166(5):1487–1497. doi: 10.1016/S0002-9440(10)62365-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Campbell SJ, Jiang Y, Davis AEM, et al. Immunomodulatory effects of etanercept in a model of brain injury act through attenuation of the acute-phase response. Journal of Neurochemistry. 2007;103(6):2245–2255. doi: 10.1111/j.1471-4159.2007.04928.x. [DOI] [PubMed] [Google Scholar]
- 61.Lu J, Goh SJ, Tng PYL, Deng YY, Ling EA, Moochhala S. Systemic inflammatory response following acute traumatic brain injury. Frontiers in Bioscience. 2009;14(10):3795–3813. doi: 10.2741/3489. [DOI] [PubMed] [Google Scholar]
- 62.Kitamura Y, Nomura M, Shima H, et al. Acute lung injury associated with systemic inflammatory response syndrome following subarachnoid hemorrhage: a survey by the shonan neurosurgical association. Neurologia Medico-Chirurgica. 2010;50(6):456–460. doi: 10.2176/nmc.50.456. [DOI] [PubMed] [Google Scholar]
- 63.Tam AKH, Ilodigwe D, Mocco J, et al. Impact of systemic inflammatory response syndrome on vasospasm, cerebral infarction, and outcome after subarachnoid hemorrhage: exploratory analysis of CONSCIOUS-1 database. Neurocritical Care. 2010;13(2):182–189. doi: 10.1007/s12028-010-9402-x. [DOI] [PubMed] [Google Scholar]
- 64.Campbell SJ, Hughes PM, Iredale JP, et al. CINC-1 is an acute-phase protein induced by focal brain injury causing leukocyte mobilization and liver injury. The FASEB Journal. 2003;17(9):1168–1170. doi: 10.1096/fj.02-0757fje. [DOI] [PubMed] [Google Scholar]
- 65.Lee S-T, Chu K, Jung KH, et al. Anti-inflammatory mechanism of intravascular neural stem cell transplantation in haemorrhagic stroke. Brain. 2008;131(3):616–629. doi: 10.1093/brain/awm306. [DOI] [PubMed] [Google Scholar]
- 66.Adrie C, Monchi M, Fulgencio JP, et al. Immune status and apoptosis activation during brain death. Shock. 2010;33(4):353–362. doi: 10.1097/SHK.0b013e3181b65b99. [DOI] [PubMed] [Google Scholar]
- 67.Hoeger S, Bergstraesser C, Selhorst J, et al. Modulation of brain dead induced inflammation by vagus nerve stimulation. American Journal of Transplantation. 2010;10(3):477–489. doi: 10.1111/j.1600-6143.2009.02951.x. [DOI] [PubMed] [Google Scholar]
- 68.Rostron AJ, Avlonitis VS, Cork DM, Grenade DS, Kirby JA, Dark JH. Hemodynamic resuscitation with arginine vasopressin reduces lung injury after brain death in the transplant donor. Transplantation. 2008;85(4):597–606. doi: 10.1097/TP.0b013e31816398dd. [DOI] [PubMed] [Google Scholar]
- 69.Guo Z-D, Sun XC, Zhang JH. Mechanisms of early brain injury after SAH: matrix metalloproteinase 9. Acta Neurochirurgica. Supplement. 2011;110(1):63–65. doi: 10.1007/978-3-7091-0353-1_11. [DOI] [PubMed] [Google Scholar]
- 70.Frugier T, Morganti-Kossmann MC, O’Reilly D, McLean CA. In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. Journal of Neurotrauma. 2010;27(3):497–507. doi: 10.1089/neu.2009.1120. [DOI] [PubMed] [Google Scholar]
- 71.Jeremitsky E, Omert L, Dunham CM, Protetch J, Rodriguez A. Harbingers of poor outcome the day after severe brain injury: hypothermia, hypoxia, and hypoperfusion. Journal of Trauma. 2003;54(2):312–319. doi: 10.1097/01.TA.0000037876.37236.D6. [DOI] [PubMed] [Google Scholar]
- 72.Graetz D, Nagel A, Schlenk F, Sakowitz O, Vajkoczy P, Sarrafzadeh A. High ICP as trigger of proinflammatory IL-6 cytokine activation in aneurysmal subarachnoid hemorrhage. Neurological Research. 2010;32(7):728–735. doi: 10.1179/016164109X12464612122650. [DOI] [PubMed] [Google Scholar]
- 73.Perez-Barcena J, Ibáñez J, Brell M, et al. Lack of correlation among intracerebral cytokines, intracranial pressure, and brain tissue oxygenation in patients with traumatic brain injury and diffuse lesions. Critical Care Medicine. 2011;39(3):533–540. doi: 10.1097/CCM.0b013e318205c7a4. [DOI] [PubMed] [Google Scholar]
- 74.Hergenroeder GW, Redell JB, Moore AN, Dash PK. Biomarkers in the clinical diagnosis and management of traumatic brain injury. Molecular Diagnosis and Therapy. 2008;12(6):345–358. doi: 10.1007/BF03256301. [DOI] [PubMed] [Google Scholar]
- 75.Rhind SG, Crnko NT, Baker AJ, et al. Prehospital resuscitation with hypertonic saline-dextran modulates inflammatory, coagulation and endothelial activation marker profiles in severe traumatic brain injured patients. Journal of Neuroinflammation. 2010;7(1, article 5) doi: 10.1186/1742-2094-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nature Reviews Neurology. 2010;6(7):393–403. doi: 10.1038/nrneurol.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Higashida T, Kreipke CW, Rafols JA, et al. The role of hypoxia-inducible factor-1α, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury: laboratory investigation. Journal of Neurosurgery. 2011;114(1):92–101. doi: 10.3171/2010.6.JNS10207. [DOI] [PubMed] [Google Scholar]
- 78.Wu H, Zhang Z, Li Y, et al. Time course of upregulation of inflammatory mediators in the hemorrhagic brain in rats: correlation with brain edema. Neurochemistry International. 2010;57(3):248–253. doi: 10.1016/j.neuint.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu H, Zhang Z, Hu X, et al. Dynamic changes of inflammatory markers in brain after hemorrhagic stroke in humans: a postmortem study. Brain Research. 2010;1342:111–117. doi: 10.1016/j.brainres.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Miñambres E, Cemborain A, Sánchez-Velasco P, et al. Correlation between transcranial interleukin-6 gradient and outcome in patients with acute brain injury. Critical Care Medicine. 2003;31(3):933–938. doi: 10.1097/01.CCM.0000055370.66389.59. [DOI] [PubMed] [Google Scholar]
- 81.Skrabal CA, Thompson LO, Potapov EV, et al. Organ-specific regulation of pro-inflammatory molecules in heart, lung, and kidney following brain death. Journal of Surgical Research. 2005;123(1):118–125. doi: 10.1016/j.jss.2004.07.245. [DOI] [PubMed] [Google Scholar]
- 82.Amado JA, Lopez-Espadas F, Vazquez-Barquero A, et al. Blood levels of cytokines in brain-dead patients: relationship with circulating hormones and acute-phase reactants. Metabolism. 1995;44(6):812–816. doi: 10.1016/0026-0495(95)90198-1. [DOI] [PubMed] [Google Scholar]
- 83.Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. Journal of Cerebral Blood Flow and Metabolism. 2006;26(5):654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- 84.Maier B, Lefering R, Lehnert M, et al. Early versus late onset of multiple organ failure is associated with differing patterns of plasma cytokine biomarker expression and outcome after severe trauma. Shock. 2007;28(6):668–674. [PubMed] [Google Scholar]
- 85.Koudstaal LG, 'T Hart NA, Ottens PJ, et al. Brain death induces inflammation in the donor intestine. Transplantation. 2008;86(1):148–154. doi: 10.1097/TP.0b013e31817ba53a. [DOI] [PubMed] [Google Scholar]
- 86.Jedrzejowska-Szypulka H, Straszak G, Larysz-Brysz M, et al. Interleukin-1β plays a role in the activation of peripheral leukocytes after blood-brain barrier rupture in the course of subarachnoid hemorrhage. Current Neurovascular Research. 2010;7(1):39–48. doi: 10.2174/156720210790820226. [DOI] [PubMed] [Google Scholar]
- 87.Hanafy KA, Grobelny B, Fernandez L, et al. Brain interstitial fluid TNF-α after subarachnoid hemorrhage. Journal of the Neurological Sciences. 2010;291(1-2):69–73. doi: 10.1016/j.jns.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. British Journal of Pharmacology. 2006;147(supplement 1):S232–S240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Maier B, Lehnert M, Laurer HL, Mautes AE, Steudel WI, Marzi I. Delayed elevation of soluble tumor necrosis factor receptors p75 and p55 in cerebrospinal fluid and plasma after traumatic brain injury. Shock. 2006;26(2):122–127. doi: 10.1097/01.shk.0000223127.41641.f4. [DOI] [PubMed] [Google Scholar]
- 90.Mellergård P, Åneman O, Sjögren F, Säberg C, Hillman J. Differences in cerebral extracellular response of interleukin-1β, interleukin-6, and interleukin-10 after subarachnoid hemorrhage or severe head trauma in humans. Neurosurgery. 2011;68(1):12–19. doi: 10.1227/NEU.0b013e3181ef2a40. [DOI] [PubMed] [Google Scholar]
- 91.Weiss S, Kotsch K, Francuski M, et al. Brain death activates donor organs and is associated with a worse I/R injury after liver transplantation. American Journal of Transplantation. 2007;7(6):1584–1593. doi: 10.1111/j.1600-6143.2007.01799.x. [DOI] [PubMed] [Google Scholar]
- 92.Campbell SJ, Zahid I, Losey P, et al. Liver Kupffer cells control the magnitude of the inflammatory response in the injured brain and spinal cord. Neuropharmacology. 2008;55(5):780–787. doi: 10.1016/j.neuropharm.2008.06.074. [DOI] [PubMed] [Google Scholar]
- 93.McLean KM, Duffy JY, Pandalai PK, et al. Glucocorticoids alter the balance between pro- and anti-inflammatory mediators in the myocardium in a porcine model of brain death. Journal of Heart and Lung Transplantation. 2007;26(1):78–84. doi: 10.1016/j.healun.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 94.Katsuno M, Yokota H, Yamamoto Y, Teramoto A. Increased regional interleukin-4 during the acute stage of severe intracranial disorders. Neurologia Medico-Chirurgica. 2006;46(10):471–474. doi: 10.2176/nmc.46.471. [DOI] [PubMed] [Google Scholar]
- 95.Yang MS, Park EJ, Sohn S, et al. Interleukin-13 and -4 induce death of activated microglia. GLIA. 2002;38(4):273–280. doi: 10.1002/glia.10057. [DOI] [PubMed] [Google Scholar]
- 96.Planas AM, Gorina R, Chamorro A. Signalling pathways mediating inflammatory responses in brain ischaemia. Biochemical Society Transactions. 2006;34(6):1267–1270. doi: 10.1042/BST0341267. [DOI] [PubMed] [Google Scholar]
- 97.Hayakata T, Shiozaki T, Tasaki O, et al. Changes in CSF S100B and cytokine concentrations in early-phase severe traumatic brain injury. Shock. 2004;22(2):102–107. doi: 10.1097/01.shk.0000131193.80038.f1. [DOI] [PubMed] [Google Scholar]
- 98.Wang X-Q, Peng YP, Lu JH, Cao BB, Qiu YH. Neuroprotection of interleukin-6 against NMDA attack and its signal transduction by JAK and MAPK. Neuroscience Letters. 2009;450(2):122–126. doi: 10.1016/j.neulet.2008.11.051. [DOI] [PubMed] [Google Scholar]
- 99.Dimopoulou I, Korfias S, Dafni U, et al. Protein S-100b serum levels in trauma-induced brain death. Neurology. 2003;60(6):947–951. doi: 10.1212/01.wnl.0000049931.77887.7f. [DOI] [PubMed] [Google Scholar]
- 100.Quintana A, Giralt M, Molinero A, Campbell IL, Penkowa M, Hidalgo J. Analysis of the cerebral transcriptome in mice subjected to traumatic brain injury: importance of IL-6. NeuroImmunoModulation. 2007;14(3-4):139–143. doi: 10.1159/000110637. [DOI] [PubMed] [Google Scholar]
- 101.Damman J, Nijboer WN, Schuurs TA, et al. Local renal complement C3 induction by donor brain death is associated with reduced renal allograft function after transplantation. Nephrology Dialysis Transplantation. 2011;26(7):2345–2354. doi: 10.1093/ndt/gfq717. [DOI] [PubMed] [Google Scholar]
- 102.Lv M, Liu Y, Zhang J, et al. Roles of inflammation response in microglia cell through Toll-like receptors 2/interleukin-23/interleukin-17 pathway in cerebral ischemia/reperfusion injury. Neuroscience. 2011;176:162–172. doi: 10.1016/j.neuroscience.2010.11.066. [DOI] [PubMed] [Google Scholar]
- 103.Konoeda F, Shichita T, Yoshida H, et al. Therapeutic effect of IL-12/23 and their signaling pathway blockade on brain ischemia model. Biochemical and Biophysical Research Communications. 2010;402(3):500–506. doi: 10.1016/j.bbrc.2010.10.058. [DOI] [PubMed] [Google Scholar]
- 104.Wang D-D, Zhao YF, Wang GY, et al. IL-17 potentiates neuronal injury induced by oxygen-glucose deprivation and affects neuronal IL-17 receptor expression. Journal of Neuroimmunology. 2009;212(1-2):17–25. doi: 10.1016/j.jneuroim.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 105.Comellas AP, Briva A. Role of endothelin-1 in acute lung injury. Translational Research. 2009;153(6):263–271. doi: 10.1016/j.trsl.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Persson BP, Rossi P, Weitzberg E, Oldner A. Inhaled tezosentan reduces pulmonary hypertension in endotoxin-induced lung injury. Shock. 2009;32(4):427–434. doi: 10.1097/SHK.0b013e31819e2cbb. [DOI] [PubMed] [Google Scholar]
- 107.Konrad D, Haney M, Johansson G, Wanecek M, Weitzberg E, Oldner A. Cardiac effects of endothelin receptor antagonism in endotoxemic pigs. American Journal of Physiology. 2007;293(2):H988–H996. doi: 10.1152/ajpheart.01023.2006. [DOI] [PubMed] [Google Scholar]
- 108.Leuchte HH, Meis T, El-Nounou M, Michalek J, Behr J. Inhalation of endothelin receptor blockers in pulmonary hypertension. American Journal of Physiology. 2008;294(4):L772–L777. doi: 10.1152/ajplung.00405.2007. [DOI] [PubMed] [Google Scholar]
- 109.Kuklin VN, Kirov MY, Evgenov OV, et al. Novel endothelin receptor antagonist attenuates endotoxin-induced lung injury in sheep. Critical Care Medicine. 2004;32(3):766–773. doi: 10.1097/01.ccm.0000114575.08269.f6. [DOI] [PubMed] [Google Scholar]
- 110.Kallakuri S, Kreipke CW, Schafer PC, Schafer SM, Rafols JA. Brain cellular localization of endothelin receptors A and B in a rodent model of diffuse traumatic brain injury. Neuroscience. 2010;168(3):820–830. doi: 10.1016/j.neuroscience.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 111.Chatfield DA, Brahmbhatt DH, Sharp T, Perkes IE, Outrim JG, Menon DK. Juguloarterial endothelin-1 gradients after severe traumatic brain injury. Neurocritical Care. 2011;14(1):55–60. doi: 10.1007/s12028-010-9413-7. [DOI] [PubMed] [Google Scholar]
- 112.Vatter H, Konczalla J, Seifert V. Endothelin related pathophysiology in cerebral vasospasm: what happens to the cerebral vessels? Acta Neurochirurgica. Supplement. 2011;110(1):177–180. doi: 10.1007/978-3-7091-0353-1_31. [DOI] [PubMed] [Google Scholar]
- 113.Salonia R, Empey PE, Poloyac SM, et al. Endothelin-1 is increased in cerebrospinal fluid and associated with unfavorable outcomes in children after severe traumatic brain injury. Journal of Neurotrauma. 2010;27(10):1819–1825. doi: 10.1089/neu.2010.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sabri M, Ai J, MacDonald RL. Dissociation of vasospasm and secondary effects of experimental subarachnoid hemorrhage by clazosentan. Stroke. 2011;42(5):1454–1460. doi: 10.1161/STROKEAHA.110.604728. [DOI] [PubMed] [Google Scholar]
- 115.Sutherland AJ, Ware RS, Winterford C, Fraser JF. The endothelin axis and gelatinase activity in alveolar macrophages after brain-stem death injury: a pilot study. Journal of Heart and Lung Transplantation. 2007;26(10):1040–1047. doi: 10.1016/j.healun.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 116.Reel B, Oktay G, Ozkal S, et al. MMP-2 and MMP-9 alteration in response to collaring in rabbits: the effects of endothelin receptor antagonism. Journal of Cardiovascular Pharmacology and Therapeutics. 2009;14(4):292–301. doi: 10.1177/1074248409343690. [DOI] [PubMed] [Google Scholar]
- 117.Rossi P, Persson B, Boels PJM, Arner A, Weitzberg E, Oldner A. Endotoxemic pulmonary hypertension is largely mediated by endothelin-induced venous constriction. Intensive Care Medicine. 2008;34(5):873–880. doi: 10.1007/s00134-007-0980-9. [DOI] [PubMed] [Google Scholar]
- 118.Kimura H, Gules I, Meguro T, Zhang JH. Cytotoxicity of cytokines in cerebral microvascular endothelial cell. Brain Research. 2003;990(1-2):148–156. doi: 10.1016/s0006-8993(03)03450-4. [DOI] [PubMed] [Google Scholar]
- 119.Cobelens PM, Tiebosch IACW, Dijkhuizen RM, et al. Interferon-β attenuates lung inflammation following experimental subarachnoid hemorrhage. Critical Care. 2010;14(4, article R157) doi: 10.1186/cc9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Otto VI, Heinzel-Pleines UE, Gloor SM, Trentz O, Kossmann T. Morganti-Kossmann MCsICAM-1 and TNF-α induce MIP-2 with distinct kinetics in astrocytes and brain microvascular endothelial cells. Journal of Neuroscience Research. 2000;60(6):733–742. doi: 10.1002/1097-4547(20000615)60:6<733::AID-JNR5>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 121.Birks EJ, Burton PBJ, Owen V, et al. Elevated tumor necrosis factor-α and interleukin-6 in myocardium and serum of malfunctioning donor hearts. Circulation. 2000;102(19):III352–III358. doi: 10.1161/01.cir.102.suppl_3.iii-352. [DOI] [PubMed] [Google Scholar]
- 122.Wei HH, Lu XCM, Shear DA, et al. NNZ-2566 treatment inhibits neuroinflammation and pro-inflammatory cytokine expression induced by experimental penetrating ballistic-like brain injury in rats. Journal of Neuroinflammation. 2009;6(1, article 19) doi: 10.1186/1742-2094-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kellum JA, Venkataraman R, Powner D, Elder M, Hergenroeder G, Carter M. Feasibility study of cytokine removal by hemoadsorption in brain-dead humans. Critical Care Medicine. 2008;36(1):268–272. doi: 10.1097/01.CCM.0000291646.34815.BB. [DOI] [PubMed] [Google Scholar]
- 124.Bartfai T, Sanchez-Alavez M, Andell-Jonsson S, et al. Interleukin-1 system in CNS stress: seizures, fever, and neurotrauma. Annals of the New York Academy of Sciences. 2007;1113(1):173–177. doi: 10.1196/annals.1391.022. [DOI] [PubMed] [Google Scholar]
- 125.Basu A, Krady JK, Enterline JR, Levison SW. Transforming growth factor β1 prevents IL-1β-induced microglial activation, whereas TNFα- and IL-6-stimulated activation are not antagonized. GLIA. 2002;40(1):109–120. doi: 10.1002/glia.10118. [DOI] [PubMed] [Google Scholar]
- 126.Murugan R, Venkataraman R, Wahed AS, et al. Increased plasma interleukin-6 in donors is associated with lower recipient hospital-free survival after cadaveric organ transplantation. Critical Care Medicine. 2008;36(6):1810–1816. doi: 10.1097/CCM.0b013e318174d89f. [DOI] [PubMed] [Google Scholar]
- 127.Plenz G, Eschert H, Erren M, et al. The interleukin-6/interleukin-6-receptor system is activated in donor hearts. Journal of the American College of Cardiology. 2002;39(9):1508–1512. doi: 10.1016/s0735-1097(02)01791-6. [DOI] [PubMed] [Google Scholar]
- 128.Murugan R, Venkataraman R, Wahed AS, et al. Preload responsiveness is associated with increased interleukin-6 and lower organ yield from brain-dead donors. Critical Care Medicine. 2009;37(8):2387–2393. doi: 10.1097/CCM.0b013e3181a960d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gregoric P, Sijacki A, Stankovic S, et al. SIRS score on admission and initial concentration of IL-6 as severe acute pancreatitis outcome predictors. Hepato-Gastroenterology. 2011;58(105):p. 263. [PubMed] [Google Scholar]
- 130.Wang L, Quan J, Johnston WE, et al. Age-dependent differences of interleukin-6 activity in cardiac function after burn complicated by sepsis. Burns. 2010;36(2):232–238. doi: 10.1016/j.burns.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Maier B, Schwerdtfeger K, Mautes A, et al. Differential release of interleukines 6, 8, and 10 in cerebrospinal fluid and plasma after traumatic brain injury. Shock. 2001;15(6):421–426. doi: 10.1097/00024382-200115060-00002. [DOI] [PubMed] [Google Scholar]
- 132.Fisher AJ, Donnelly SC, Hirani N, et al. Elevated levels of interleukin-8 in donor lungs is associated with early graft failure after lung transplantation. American Journal of Respiratory and Critical Care Medicine. 2001;163(1):259–265. doi: 10.1164/ajrccm.163.1.2005093. [DOI] [PubMed] [Google Scholar]
- 133.Donnelly SC, Strieter RM, Kunkel SL, et al. Interleukin-8 and development of adult respiratory distress syndrome in at-risk patient groups. The Lancet. 1993;341(8846):643–647. doi: 10.1016/0140-6736(93)90416-e. [DOI] [PubMed] [Google Scholar]
- 134.Csuka E, Morganti-Kossmann MC, Lenzlinger PM, Joller H, Trentz O, Kossmann T. IL-10 levels in cerebrospinal fluid and serum of patients with severe traumatic brain injury: relationship to IL-6, TNF-α, TGF-β1 and blood-brain barrier function. Journal of Neuroimmunology. 1999;101(2):211–221. doi: 10.1016/s0165-5728(99)00148-4. [DOI] [PubMed] [Google Scholar]
- 135.Morganti-Kossman MC, Lenzlinger PM, Hans V, et al. Production of cytokines following brain injury: beneficial and deleterious for the damaged tissue. Molecular Psychiatry. 1997;2(2):133–136. doi: 10.1038/sj.mp.4000227. [DOI] [PubMed] [Google Scholar]
- 136.Malipiero U, Koedel U, Pfister W, Fontana A. Bacterial meningitis: the role of transforming growth factor-beta in innate immunity and secondary brain damage. Neurodegenerative Diseases. 2007;4(1):43–50. doi: 10.1159/000100358. [DOI] [PubMed] [Google Scholar]
- 137.Douglas MR, Daniel M, Lagord C, et al. High CSF transforming growth factor b levels after subarachnoid haemorrhage: association with chronic communicating hydrocephalus. Journal of Neurology, Neurosurgery and Psychiatry. 2009;80(5):545–550. doi: 10.1136/jnnp.2008.155671. [DOI] [PubMed] [Google Scholar]
- 138.Okamoto T, Takahashi S, Nakamura E, Nagaya K, Hayashi T, Fujieda K. Transforming growth factor-β1 induces matrix metalloproteinase-9 expression in human meningeal cells via ERK and Smad pathways. Biochemical and Biophysical Research Communications. 2009;383(4):475–479. doi: 10.1016/j.bbrc.2009.04.038. [DOI] [PubMed] [Google Scholar]
- 139.Oishi Y, Nishimura Y, Tanoue Y, et al. Endothelin-1 receptor antagonist prevents deterioration of left ventricular function and coronary flow reserve in brain-dead canine heart. Journal of Heart and Lung Transplantation. 2005;24(9):1354–1361. doi: 10.1016/j.healun.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 140.Ferrera R, Hadour G, Tamion F, et al. Brain death provokes very acute alteration in myocardial morphology detected by echocardiography: preventive effect of beta-blockers. Transplant International. 2011;24(3):300–306. doi: 10.1111/j.1432-2277.2010.01184.x. [DOI] [PubMed] [Google Scholar]
- 141.Venkateswaran RV, Dronavalli V, Lambert PA, et al. The proinflammatory environment in potential heart and lung donors: prevalence and impact of donor management and hormonal therapy. Transplantation. 2009;88(4):582–588. doi: 10.1097/TP.0b013e3181b11e5d. [DOI] [PubMed] [Google Scholar]
- 142.Catania A, Lonati C, Sordi A, Gatti S. Detrimental consequences of brain injury on peripheral cells. Brain, Behavior, and Immunity. 2009;23(7):877–884. doi: 10.1016/j.bbi.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 143.Valdivia M, Chamorro C, Romera MA, Balandín B, Pérez M. Effect of posttraumatic donor's disseminated intravascular coagulation in intrathoracic organ donation and transplantation. Transplantation Proceedings. 2007;39(7):2427–2428. doi: 10.1016/j.transproceed.2007.07.052. [DOI] [PubMed] [Google Scholar]
- 144.James SR, Ranasinghe AM, Venkateswaran R, McCabe CJ, Franklyn JA, Bonser RS. The effects of acute triiodothyronine therapy on myocardial gene expression in brain stem dead cardiac donors. Journal of Clinical Endocrinology and Metabolism. 2010;95(3):1338–1343. doi: 10.1210/jc.2009-1659. [DOI] [PubMed] [Google Scholar]
- 145.Venkateswaran RV, Steeds RP, Quinn DW, et al. The haemodynamic effects of adjunctive hormone therapy in potential heart donors: a prospective randomized double-blind factorially designed controlled trial. European Heart Journal. 2009;30(14):1771–1780. doi: 10.1093/eurheartj/ehp086. [DOI] [PubMed] [Google Scholar]
- 146.Avlonitis VS, Wigfield CH, Kirby JA, Dark JH. The hemodynamic mechanisms of lung injury and systemic inflammatory response following brain death in the transplant donor. American Journal of Transplantation. 2005;5(4 I):684–693. doi: 10.1111/j.1600-6143.2005.00755.x. [DOI] [PubMed] [Google Scholar]
- 147.Salim A, Martin M, Brown C, Belzberg H, Rhee P, Demetriades D. Complications of brain death: frequency and impact on organ retrieval. American Surgeon. 2006;72(5):377–381. doi: 10.1177/000313480607200502. [DOI] [PubMed] [Google Scholar]
- 148.Salim A, Velmahos GC, Brown C, Belzberg H, Demetriades D. Aggressive organ donor management significantly increases the number of organs available for transplantation. Journal of Trauma. 2005;58(5):991–994. doi: 10.1097/01.ta.0000168708.78049.32. [DOI] [PubMed] [Google Scholar]
- 149.Cushing H. Concerning a definite regulatory mechanism of the vaso-motor centre which controls blood pressure during cerebral compression. The Johns Hopkins Hospital Bulletin. 1901;12:290–292. [Google Scholar]
- 150.Cushing H. Some experimental and clinical observations concerning states of increased intracranial tension. The Mütter Lecture for 1901. American Journal of Medical Sciences. 1902;124:375–400. [Google Scholar]
- 151.Dictus C, Vienenkoetter B, Esmaeilzadeh M, Unterberg A, Ahmadi R. Critical care management of potential organ donors: our current standard. Clinical Transplantation. 2009;23(21):2–9. doi: 10.1111/j.1399-0012.2009.01102.x. [DOI] [PubMed] [Google Scholar]
- 152.Schrader H, Hall C, Zwetnow NN. Effects of prolonged supratentorial mass expansion on regional blood flow and cardiovascular parameters during the Cushing response. Acta Neurologica Scandinavica. 1985;72(3):283–294. doi: 10.1111/j.1600-0404.1985.tb00872.x. [DOI] [PubMed] [Google Scholar]
- 153.Gao C, Liu X, Shi H, et al. Relationship between sympathetic nervous activity and inflammatory response after subarachnoid hemorrhage in a perforating canine model. Autonomic Neuroscience. 2009;147(1-2):70–74. doi: 10.1016/j.autneu.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 154.Ferrera R, Ovize M, Claustrat B, Hadour G. Stable myocardial function and endocrine dysfunction during experimental brain death. Journal of Heart and Lung Transplantation. 2005;24(7):921–927. doi: 10.1016/j.healun.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 155.Marthol H, Intravooth T, Bardutzky J, De Fina P, Schwab S, Hilz MJ. Sympathetic cardiovascular hyperactivity precedes brain death. Clinical Autonomic Research. 2010;20(6):363–369. doi: 10.1007/s10286-010-0072-8. [DOI] [PubMed] [Google Scholar]
- 156.Chamorro C, Falcón JA, Michelena JC. Controversial points in organ donor management. Transplantation Proceedings. 2009;41(8):3473–3475. doi: 10.1016/j.transproceed.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 157.Herijgers P, Borgers M, Flameng W. The effect of brain death on cardiovascular function in rats. Part I. Is the heart damaged? Cardiovascular Research. 1998;38(1):98–106. doi: 10.1016/s0008-6363(97)00285-x. [DOI] [PubMed] [Google Scholar]
- 158.Borovikova LV, Ivanova S, Zhang M, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405(6785):458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 159.Flierl MA, Rittirsch D, Nadeau BA, et al. Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature. 2007;449(7163):721–725. doi: 10.1038/nature06185. [DOI] [PubMed] [Google Scholar]
- 160.Takada M, Nadeau KC, Hancock WW, et al. Effects of explosive brain death on cytokine activation of peripheral organs in the rat. Transplantation. 1998;65(12):1533–1542. doi: 10.1097/00007890-199806270-00001. [DOI] [PubMed] [Google Scholar]
- 161.Avlonitis VS, Wigfield CH, Kirby JA, Dark JH. Treatment of the brain-dead lung donor with aprotinin and nitric oxide. Journal of Heart and Lung Transplantation. 2010;29(10):1177–1184. doi: 10.1016/j.healun.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 162.Zhu C, Li J, Zhang G, et al. Brain death disrupts structure and function of pig liver. Transplantation Proceedings. 2010;42(3):733–736. doi: 10.1016/j.transproceed.2010.02.028. [DOI] [PubMed] [Google Scholar]
- 163.Cypel M, Kaneda H, Yeung JC, et al. Increased levels of interleukin-1β and tumor necrosis factor-α in donor lungs rejected for transplantation. Journal of Heart and Lung Transplantation. 2011;30(4):452–459. doi: 10.1016/j.healun.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 164.Kaminska D, Tyran B, Mazanowska O, et al. Cytokine gene expression in kidney allograft biopsies after donor brain death and ischemia-reperfusion injury using in situ reverse-transcription polymerase chain reaction analysis. Transplantation. 2007;84(9):1118–1124. doi: 10.1097/01.tp.0000287190.86654.74. [DOI] [PubMed] [Google Scholar]
- 165.Li JQ, Qi HZ, He ZJ, Hu W, Si ZZ, Li YN. Induction of lymphocyte apoptosis in rat liver allograft by adenoviral gene transfection of human interleukin-10. European Surgical Research. 2010;44(3-4):133–141. doi: 10.1159/000276989. [DOI] [PubMed] [Google Scholar]
- 166.Nijboer WN, Ottens PJ, Van Dijk A, Van Goor H, Ploeg RJ, Leuvenink HGD. Donor pretreatment with carbamylated erythropoietin in a brain death model reduces inflammation more effectively than erythropoietin while preserving renal function. Critical Care Medicine. 2010;38(4):1155–1161. doi: 10.1097/CCM.0b013e3181cf6e78. [DOI] [PubMed] [Google Scholar]
- 167.Bulcao CF, D'Souza KM, Malhotra R, et al. Activation of JAK-STAT and nitric oxide signaling as a mechanism for donor heart dysfunction. Journal of Heart and Lung Transplantation. 2010;29(3):346–351. doi: 10.1016/j.healun.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kotsch KP, Ulrich F, Reutzel-Selke A, et al. Methylprednisolone therapy in deceased donors reduces inflammation in the donor liver and improves outcome after liver transplantation a prospective randomized controlled trial. Annals of Surgery. 2008;248(6):1042–1049. doi: 10.1097/SLA.0b013e318190e70c. [DOI] [PubMed] [Google Scholar]
- 169.Kaneda H, Waddell TK, De Perrot M, et al. Pre-implantation multiple cytokine mRNA expression analysis of donor lung grafts predicts survival after lung transplantation in humans. American Journal of Transplantation. 2006;6(3):544–551. doi: 10.1111/j.1600-6143.2005.01204.x. [DOI] [PubMed] [Google Scholar]
- 170.Loverre A, Divella C, Castellano G, et al. T helper 1, 2 and 17 cell subsets in renal transplant patients with delayed graft function. Transplant International. 2011;24(3):233–242. doi: 10.1111/j.1432-2277.2010.01157.x. [DOI] [PubMed] [Google Scholar]
- 171.Inci I, Erne B, Arni S, et al. Prevention of primary graft dysfunction in lung transplantation by N-acetylcysteine after prolonged cold ischemia. Journal of Heart and Lung Transplantation. 2010;29(11):1293–1301. doi: 10.1016/j.healun.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 172.Lascano EC, Bertolotti A, Gómez CB, et al. Failure of IL-8 to assess early reperfusion injury following lung transplantation of cardiac death donor pigs. Transplant International. 2009;22(5):574–582. doi: 10.1111/j.1432-2277.2008.00833.x. [DOI] [PubMed] [Google Scholar]
- 173.Zhang SJ, Chen S. The influence of brain death on liver in rats. Transplantation Proceedings. 2004;36(7):1925–1927. doi: 10.1016/j.transproceed.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 174.Salama M, Andrukhova O, Hoda MA, et al. Concomitant endothelin-1 overexpression in lung transplant donors and recipients predicts primary graft dysfunction. American Journal of Transplantation. 2010;10(3):628–636. doi: 10.1111/j.1600-6143.2009.02957.x. [DOI] [PubMed] [Google Scholar]
- 175.Kusaka M, Kuroyanagi Y, Kowa H, et al. Genomewide expression profiles of rat model renal isografts from brain dead donors. Transplantation. 2007;83(1):62–70. doi: 10.1097/01.tp.0000250485.53865.b8. [DOI] [PubMed] [Google Scholar]
- 176.Shiotani S, Shimada M, Suehiro T, et al. Involvement of Rho-kinase in cold ischemia-reperfusion injury after liver transplantation in rats. Transplantation. 2004;78(3):375–382. doi: 10.1097/01.tp.0000128618.41619.e7. [DOI] [PubMed] [Google Scholar]
- 177.Tuuminen R, Syrjälä S, Krebs R, et al. Donor simvastatin treatment abolishes rat cardiac allograft ischemia/reperfusion injury and chronic rejection through microvascular protection. Circulation. 2011;124(10):1138–1150. doi: 10.1161/CIRCULATIONAHA.110.005249. [DOI] [PubMed] [Google Scholar]
- 178.Ulukaya S, Ulukaya E, Alper I, Yilmaztepe-Oral A, Kilic M. Soluble cytokeratin 18 biomarkers may provide information on the type of cell death during early ischemia and reperfusion periods of liver transplantation. Clinical Transplantation. 2010;24(6):848–854. doi: 10.1111/j.1399-0012.2009.01177.x. [DOI] [PubMed] [Google Scholar]
- 179.Xu YF, Liu M, Peng B, et al. Protective effects of SP600125 on renal ischemia-reperfusion injury in rats. Journal of Surgical Research. 2011;169(1):e77–e84. doi: 10.1016/j.jss.2011.02.021. [DOI] [PubMed] [Google Scholar]
- 180.Blagonravov ML, Azova MM, Onufriev MV, Frolov VA. Activities of some caspase cascade enzymes and myocardial contractility in experimental left ventricular focal ischemia. Bulletin of Experimental Biology and Medicine. 2011;150(6):672–675. doi: 10.1007/s10517-011-1219-x. [DOI] [PubMed] [Google Scholar]
- 181.Ballet C, Renaudin K, Degauque N, et al. Indirect CD4+ TH1 response, antidonor antibodies and diffuse C4d graft deposits in long-term recipients conditioned by donor antigens priming. American Journal of Transplantation. 2009;9(4):697–708. doi: 10.1111/j.1600-6143.2009.02556.x. [DOI] [PubMed] [Google Scholar]
- 182.Zhang M, Michael LH, Grosjean SA, Kelly RA, Carroll MC, Entman ML. The role of natural IgM in myocardial ischemia-reperfusion injury. Journal of Molecular and Cellular Cardiology. 2006;41(1):62–67. doi: 10.1016/j.yjmcc.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 183.Gelman AE, Okazaki M, Sugimoto S, et al. CCR2 regulates monocyte recruitment as well as CD4+ T h1 allorecognition after lung transplantation. American Journal of Transplantation. 2010;10(5):1189–1199. doi: 10.1111/j.1600-6143.2010.03101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.de Vries DK, Lindeman JHN, Ringers J, Reinders MEJ, Rabelink TJ, Schaapherder AFM. Donor brain death predisposes human kidney grafts to a proinflammatory reaction after transplantation. American Journal of Transplantation. 2011;11(5):1064–1070. doi: 10.1111/j.1600-6143.2011.03466.x. [DOI] [PubMed] [Google Scholar]
- 185.Hosgood SA, Mohamed IH, Bagul A, Nicholson ML. Hypothermic machine perfusion after static cold storage does not improve the preservation condition in an experimental porcine kidney model. British Journal of Surgery. 2011;98(7):943–950. doi: 10.1002/bjs.7481. [DOI] [PubMed] [Google Scholar]
- 186.Duran JA, González AA, García DD, et al. Variation in the levels of inflammatory cytokines depending on ischemic time: effects on respiratory variables. Transplantation Proceedings. 2009;41(3):980–982. doi: 10.1016/j.transproceed.2009.01.045. [DOI] [PubMed] [Google Scholar]
- 187.Ilmakunnas M, Höckerstedt K, Mäkisalo H, Siitonen S, Repo H, Pesonen EJ. Hepatic IL-8 release during graft procurement is associated with impaired graft function after human liver transplantation. Clinical Transplantation. 2010;24(1):29–35. doi: 10.1111/j.1399-0012.2009.00975.x. [DOI] [PubMed] [Google Scholar]
- 188.Merry HE, Wolf PS, Fitzsullivan E, Keech JC, Mulligan MS. Lipopolysaccharide pre-conditioning is protective in lung ischemia-reperfusion injury. Journal of Heart and Lung Transplantation. 2010;29(4):471–478. doi: 10.1016/j.healun.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Benson A, Murray S, Divakar P, et al. Microbial infection-induced expansion of effector T cells overcomes the suppressive effects of regulatory T cells via an IL-2 deprivation mechanism. The Journal of Immunology. 2012;188(2):800–810. doi: 10.4049/jimmunol.1100769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Jonigk D, Merk M, Hussein K, et al. Obliterative airway remodeling: molecular evidence for shared pathways in transplanted and native lungs. American Journal of Pathology. 2011;178(2):599–608. doi: 10.1016/j.ajpath.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ke B, Shen XD, Lassman CR, Gao F, Busuttil RW, Kupiec-Weglinski JW. Cytoprotective and antiapoptotic effects of IL-13 in hepatic cold ischemia/reperfusion injury are heme oxygenase-1 dependent. American Journal of Transplantation. 2003;3(9):1076–1082. doi: 10.1034/j.1600-6143.2003.00147.x. [DOI] [PubMed] [Google Scholar]
- 192.Sun L, Shi T, Qiao H, et al. Hepatic overexpression of heme oxygenase-1 improves liver allograft survival by expanding t regulatory cells. Journal of Surgical Research. 2011;166(2):e187–e194. doi: 10.1016/j.jss.2010.11.917. [DOI] [PubMed] [Google Scholar]
- 193.Schaub M, Ploetz CJ, Gerbaulet D, et al. Effect of dopamine on inflammatory status in kidneys of brain-dead rats. Transplantation. 2004;77(9):1333–1340. doi: 10.1097/01.tp.0000119164.47302.49. [DOI] [PubMed] [Google Scholar]
- 194.Zhou H, Liu J, Pan P, Jin D, Ding W, Li W. Carbon monoxide inhalation decreased lung injury via anti-inflammatory and anti-apoptotic effects in brain death rats. Experimental Biology and Medicine. 2010;235(10):1236–1243. doi: 10.1258/ebm.2010.010147. [DOI] [PubMed] [Google Scholar]
- 195.Zhou H, Qian H, Liu J, et al. Protection against lung graft injury from brain-dead donors with carbon monoxide, biliverdin, or both. Journal of Heart and Lung Transplantation. 2011;30(4):460–466. doi: 10.1016/j.healun.2010.11.020. [DOI] [PubMed] [Google Scholar]
- 196.Sierra-Filardi E, Vega MA, Sánchez-Mateos P, Corbí AL, Puig-Kröger A. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology. 2010;215(9-10):788–795. doi: 10.1016/j.imbio.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 197.Ke B, Shen XD, Zhai Y, et al. Heme oxygenase 1 mediates the immunomodulatory and antiapoptotic effects of interleukin 13 gene therapy in vivo and in vitro . Human Gene Therapy. 2002;13(15):1845–1857. doi: 10.1089/104303402760372945. [DOI] [PubMed] [Google Scholar]
- 198.Selck FW, Deb P, Grossman EB. Deceased organ donor characteristics and clinical interventions associated with organ yield. American Journal of Transplantation. 2008;8(5):965–974. doi: 10.1111/j.1600-6143.2008.02205.x. [DOI] [PubMed] [Google Scholar]
- 199.Salim A, Martin M, Brown C, Rhee P, Demetriades D, Belzberg H. The effect of a protocol of aggressive donor management: implications for the national organ donor shortage. Journal of Trauma. 2006;61(2):429–432. doi: 10.1097/01.ta.0000228968.63652.c1. [DOI] [PubMed] [Google Scholar]
- 200.Rosendale JD, Kauffman HM, McBride MA, et al. Aggressive pharmacologic donor management results in more transplanted organs. Transplantation. 2003;75(4):482–487. doi: 10.1097/01.TP.0000045683.85282.93. [DOI] [PubMed] [Google Scholar]
- 201. Critical Pathway for the Organ Donor, United Network for Organ Sharing, 2002, http://www.unos.org/docs/Critical_Pathway.pdf.
- 202.Dare A, Bartlett A, Fraser J. Critical care of the potential organ donor. Current Neurology and Neuroscience Reports. 2012;12(4):456–465. doi: 10.1007/s11910-012-0272-9. [DOI] [PubMed] [Google Scholar]
- 203.Lewis AJM, Rostron AJ, Cork DMW, Kirby JA, Dark JH. Norepinephrine and arginine vasopressin increase hepatic but not renal inflammatory activation during hemodynamic resuscitation in a rodent model of brain-dead donors. Experimental and Clinical Transplantation. 2009;7(2):119–123. [PubMed] [Google Scholar]
- 204.Cypel M, Liu M, Rubacha M, et al. Functional repair of human donor lungs by IL-10 gene therapy. Science Translational Medicine. 2009;1(4, article 4ra9) doi: 10.1126/scitranslmed.3000266. [DOI] [PubMed] [Google Scholar]
- 205.Sadaria MR, Smith PD, Fullerton DA, et al. Cytokine expression profile in human lungs undergoing normothermic ex-vivo lung perfusion. Annals of Thoracic Surgery. 2011;92(2):478–484. doi: 10.1016/j.athoracsur.2011.04.027. [DOI] [PubMed] [Google Scholar]
- 206.Kakishita T, Oto T, Hori S, et al. Suppression of inflammatory cytokines during ex vivo lung perfusion with an adsorbent membrane. Annals of Thoracic Surgery. 2010;89(6):1773–1779. doi: 10.1016/j.athoracsur.2010.02.077. [DOI] [PubMed] [Google Scholar]
- 207.Santiago FM, Bueno P, Olmedo C, et al. Effect of N-acetylcysteine administration on intraoperative plasma levels of interleukin-4 and interleukin-10 in liver transplant recipients. Transplantation Proceedings. 2008;40(9):2978–2980. doi: 10.1016/j.transproceed.2008.08.103. [DOI] [PubMed] [Google Scholar]
- 208.Brissaud O, Villega F, Konsman JP, et al. Short-term effect of erythropoietin on brain lesions and aquaporin-4 expression in a hypoxic-ischemic neonatal rat model assessed by magnetic resonance diffusion weighted imaging and immunohistochemistry. Pediatric Research. 2010;68(2):123–127. doi: 10.1203/PDR.0b013e3181e67d02. [DOI] [PubMed] [Google Scholar]