

Abstract
Tombusviruses, such as Carnation Italian ringspot virus (CIRV), encode a protein homodimer called p19 that is capable of suppressing RNA silencing in their infected hosts by binding to and sequestering short-interfering RNA (siRNA) away from the RNA silencing pathway. P19 binding stability has been shown to be sensitive to changes in pH but the specific amino acid residues involved have remained unclear. Using constant pH molecular dynamics simulations, we have identified key pH-dependent residues that affect CIRV p19–siRNA binding stability at various pH ranges based on calculated changes in the free energy contribution from each titratable residue. At high pH, the deprotonation of Lys60, Lys67, Lys71, and Cys134 has the largest effect on the binding stability. Similarly, deprotonation of several acidic residues (Asp9, Glu12, Asp20, Glu35, and/or Glu41) at low pH results in a decrease in binding stability. At neutral pH, residues Glu17 and His132 provide a small increase in the binding stability and we find that the optimal pH range for siRNA binding is between 7.0 and 10.0. Overall, our findings further inform recent experiments and are in excellent agreement with data on the pH-dependent binding profile.
Keywords: pH-dependence, CIRV p19, Constant pH molecular dynamics simulations, protein–RNA interactions
Introduction
RNA silencing (or RNA interference [RNAi])1–3 is an evolutionarily conserved gene inactivation pathway in eukaryotes that involves the conversion of long double-stranded RNA (dsRNA) into 21–24 nucleotide-long short-interfering RNA (siRNA) or microRNA (miRNA) by DICER, an enzyme that is a part of the endoribonuclease family of proteins.4 These small RNAs are then separated into individual strands, incorporated into a multiprotein complex called RNA-induced silencing complex (RISC),5 and ultimately used to target the degradation of reasonably complementary messenger RNA (mRNA). In plants, RNA silencing has evolved into a mechanism that can respond to both endogenous and exogenous dsRNA, the latter of which helps to defend against transgenes, transposons, and infection by RNA viruses. As a result, Tombusviruses such as Tomato bushy stunt virus (TBSV) and Carnation Italian ringspot virus (CIRV) have evolved an elegant 19 kDa protein, aptly named p19, which is capable of suppressing RNA silencing in its host by binding to and sequestering siRNA from a RISC.6, 7
X-ray structures of p19 bound to a 21 nucleotide (nt) siRNA have been crystallized from TBSV8 and CIRV.9 In both structures, p19 exists as a homodimer and is composed of five α-helices and four β-strands in each monomer (Fig. 1). Key tryptophan residues, shown experimentally and computationally to be crucial for siRNA recognition, form end-capping calipers around the RNA by stacking with the exposed terminal base pairs.8–10 Three conserved lysine residues (Lys60, Lys67, and Lys71) found in the β-sheet–RNA interface form important sequence-independent interactions with the siRNA phosphate backbone.8, 9 Mutations of Lys60 and Lys71 to alanine in TBSV have displayed decreases in the lethal necrosis phenotype.11, 12 Recent experiments have also identified two cysteine residues that appear to be responsible for maintaining the overall structural integrity of the p19 protein as modifications of these cysteines (Cys110 and Cys134 in CIRV) resulted in a reduction of siRNA binding activity.13, 14
Figure 1.
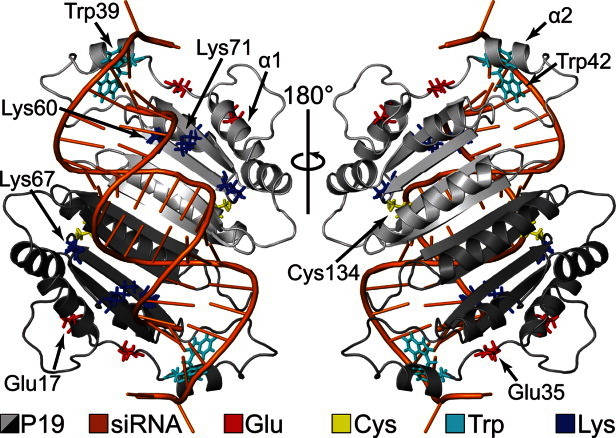
CIRV p19–siRNA complex. The complex has been rotated by 180° (PDBID: 1RPU).
Over the past few years, p19 has been used in several systems to suppress RNAi15–22 and has also emerged as a valuable tool for characterizing small RNAs.8,9,23–26 Furthermore, as environmental factors (e.g., acidity/alkalinity, salt concentration, water levels, etc.) can vary significantly across different plant hosts, it has become increasingly important to understand how the protein environment can affect the function of p19. Recent investigations using fluorescence detection assays have revealed that CIRV p19 has the most significant affinity for 21-nt siRNA in the pH range from 6.0 to 9.0.27 More specifically, it was shown that p19–siRNA binding is dependent on three apparent pKa values, 7.1, 8.0, and 10.6, that were hypothesized to correspond to one or more histidine, cysteine, and lysine residues, respectively. However, due to the limited resolution of the experiment, the identity of these ionizable residues has remained unknown. Therefore, it is necessary to consider alternative approaches.
Computational methods using molecular dynamics (MD) simulations an/or Monte Carlo (MC) sampling have been developed with considerable success for predicting protein pKa values (see reviews28–30). Often referred to as constant pH MD (CPHMD) simulations, the titration coordinate is typically implemented in either a discrete manner31–43 where protonation states are modified with an MC step at some regular MD interval or using a continuous function44–46 that describes the protonation state via the λ dynamics method developed by Brooks and coworkers.47–49 Recent studies have shown that CPHMD is a reliable and robust method that is capable of predicting pKa values in a variety of biomolecular systems.50–56 Thus, to uncover the pH-dependent residues in the CIRV p19 protein involved in siRNA binding stability, we have carried out CPHMD simulations44–46 of the p19 protein dimer in both holo (siRNA-bound) and apo (siRNA-free) forms and determined the pKa values for all titratable residues. These results were then used to calculate the pH-dependent siRNA binding stability profile and corresponding pH-dissociation constant profile. Details of the conformational dynamics for important titratable residues at different pH conditions were also investigated and the results were compared with experiment.
Results
CPHMD simulation stability
CPHMD simulations ranging from pH 1 to 14 were performed for both apo and holo systems and the Cα-root-mean-square deviation (RMSD) relative to the crystal structure at different pHs is shown in Supporting Information Figure S1. In general, the apo simulations demonstrated larger average Cα-RMSDs than the holo simulations. The largest Cα-RMSD was ∼5.3 Å among all apo simulations (at pH 2) and ∼3.7 Å from all holo simulations (at pH 14). Visual inspection of the protein structure from the apo simulations revealed that the p19 core and RNA-binding interface (residues 55–152) were very stable. Instead, the elevated p19 Cα-RMSDs in the apo systems came from the increased dynamics of the α2-helix (residues 39–45) from both monomers (Fig. 1 and Supporting Information Fig. S2). In the holo system, α2 is connected to the N-terminal α1-helix (residues 9–17) by a long flexible linker (residues 18–38) and contains important tryptophan residues (Trp39 and Trp42) that form end-capping calipers around the terminal base pairs of the siRNA.9 In the apo simulations, these base stacking interactions are lost due to the absence of RNA which resulted in an increase in α2 dynamics and, to a lesser extent, α1 dynamics.
pKa values calculated from holo and apo simulations
The pKa values for each Glu, Asp, His, Cys, and Lys residue were obtained by fitting their simulated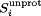 values (combined from both monomers) to the Henderson–Hasselbalch equation (see Materials and Methods section). The calculated results are summarized in Table I and Supporting Information Table S1.
values (combined from both monomers) to the Henderson–Hasselbalch equation (see Materials and Methods section). The calculated results are summarized in Table I and Supporting Information Table S1.
Table I.
pKaValuesaCalculated from CPHMD for the Holo and Apo States
| Residue | 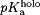 |
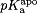 |
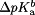 |
|---|---|---|---|
| Asp9 | 4.17 | 3.51 | 0.66 |
| Glu12 | 5.29 | 4.30 | 1.00 |
| Glu17 | 4.94 | 6.15 | -1.20 |
| Asp20 | 5.54 | 4.48 | 1.06 |
| Asp34 | 3.56 | 2.41 | 1.15 |
| Glu35 | 5.97 | 5.20 | 0.77 |
| Glu41 | 4.75 | 4.24 | 0.51 |
| Lys60 | 11.11 | 8.94 | 2.17 |
| Lys67 | 11.43 | 10.26 | 1.18 |
| Lys71 | 12.00 | 9.93 | 2.06 |
| His132 | 5.76 | 6.39 | -0.63 |
| Cys134 | 10.93 | 10.05 | 0.88 |
| Glu151 | 5.15 | 4.59 | 0.56 |
Only pKa values with 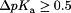 are displayed. A full list pKa values for all titratable residues is provided in Supporting Information Table S1.
are displayed. A full list pKa values for all titratable residues is provided in Supporting Information Table S1.
b
 .
.
Effects of pH on p19–siRNA binding
The net charge of both holo and apo p19 as a function of pH was determined from the computed pKa values [Fig. 2(A)]. Overall, both systems became progressively more negative as the pH was increased until all 26 titratable residues were completely deprotonated. The apo form of the protein was found to be more negatively charged than the holo form except between pH 6 and 8 where the total charge of both forms of the protein was nearly the same. Substituting the pKa values into the Wyman–Tanford linkage equation, we calculated the pH-dependent changes in the total binding stability (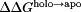 ) as well as the individual contributions from each titratable residue [Figs. 2(B) and 3]. At pH > 9, the deprotonation of several conserved lysine residues (Lys60, Lys67, and Lys71) and a nonconserved cysteine residue (Cys134) led to a large destabilization in siRNA binding by about 14.0 kcal/mol. Upon deprotonation at 4 < pH < 6, several acidic residues (Asp9, Glu12, Asp20, Glu35, and Glu41) destabilized the protein–siRNA complex by ∼7 kcal/mol. Changes in the binding stability were smallest (<1.0 kcal/mol) between pH 6 and 8, which is consistent with the lack of difference in net charge between both apo and holo systems in the same pH range. Only deprotonation of Glu17 and His132 contributed significantly to stabilizing the siRNA-bound complex in this pH range.
) as well as the individual contributions from each titratable residue [Figs. 2(B) and 3]. At pH > 9, the deprotonation of several conserved lysine residues (Lys60, Lys67, and Lys71) and a nonconserved cysteine residue (Cys134) led to a large destabilization in siRNA binding by about 14.0 kcal/mol. Upon deprotonation at 4 < pH < 6, several acidic residues (Asp9, Glu12, Asp20, Glu35, and Glu41) destabilized the protein–siRNA complex by ∼7 kcal/mol. Changes in the binding stability were smallest (<1.0 kcal/mol) between pH 6 and 8, which is consistent with the lack of difference in net charge between both apo and holo systems in the same pH range. Only deprotonation of Glu17 and His132 contributed significantly to stabilizing the siRNA-bound complex in this pH range.
Figure 2.
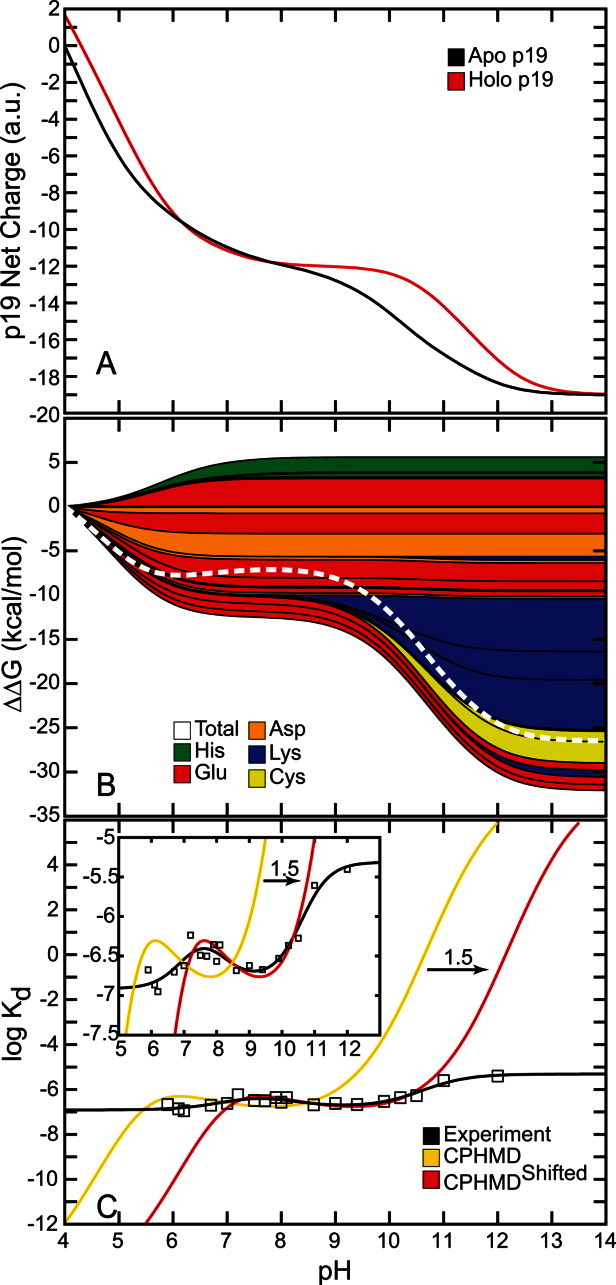
Effects of pH on p19–siRNA binding. (A) Net charge of the holo and apo p19. (B) Total binding stability (white) and per residue contributions to binding stability (colors). Positive  values increase binding stability (i.e., favors holo form) while negative
values increase binding stability (i.e., favors holo form) while negative  values destabilizes binding (i.e., favors apo form). (C) pH-dependent dissociation constant profiles compared with experiment. CPHMDshifted (red) is identical to CPHMD (yellow) except that it is shifted to the right by 1.5 pH units for comparison with experiment (black).
values destabilizes binding (i.e., favors apo form). (C) pH-dependent dissociation constant profiles compared with experiment. CPHMDshifted (red) is identical to CPHMD (yellow) except that it is shifted to the right by 1.5 pH units for comparison with experiment (black).
Figure 2(C) shows the dissociation constant profile and a modified profile in comparison with the experimentally determined profile from Koukiekolo et al.27 The unmodified log Kd profile showed minimal change over the last 5 ns of production simulation (see Supporting Information Fig. S3) and was in close agreement with the experimental profile. A much better match with experiment was achieved in the modified profile (CPHMDshifted), which was shifted by 1.5 pH units relative to the unmodified profile [see Fig. 2(C) and inset].
pH-dependent conformational dynamics
Various pH-dependent intraprotein and protein–RNA interactions were assessed from the holo simulations (Fig. 4). Deprotonation of Lys60 and Lys67 showed a drastic decrease in Lys–RNA salt bridge formation [Supporting Information Fig. S2 and Fig. 4(A, B)]. However, Lys71, in its neutral form, only displayed a moderate decrease in Lys–RNA interactions [Fig. 4(C)]. Next, measuring the solvent accessible surface area (SASA) for the Cys134 sulfur atom revealed that its side chain was essentially buried when in its reduced form and was much more solvent exposed when negatively charged [Fig. 4(D)]. Upon ionization, Glu17 and Glu35 formed more stable salt bridges with Arg72 and Arg85, respectively [Supporting Information Fig. S2 and Fig. 4(E, F)]. Formation of the Glu35–Arg85 salt bridge also appears to stabilize the Trp39–RNA base stacking interactions but has little to no effect on Trp42–RNA interactions [Supporting Information Fig. S2 and Fig. 4(G, H)].
Figure 4.
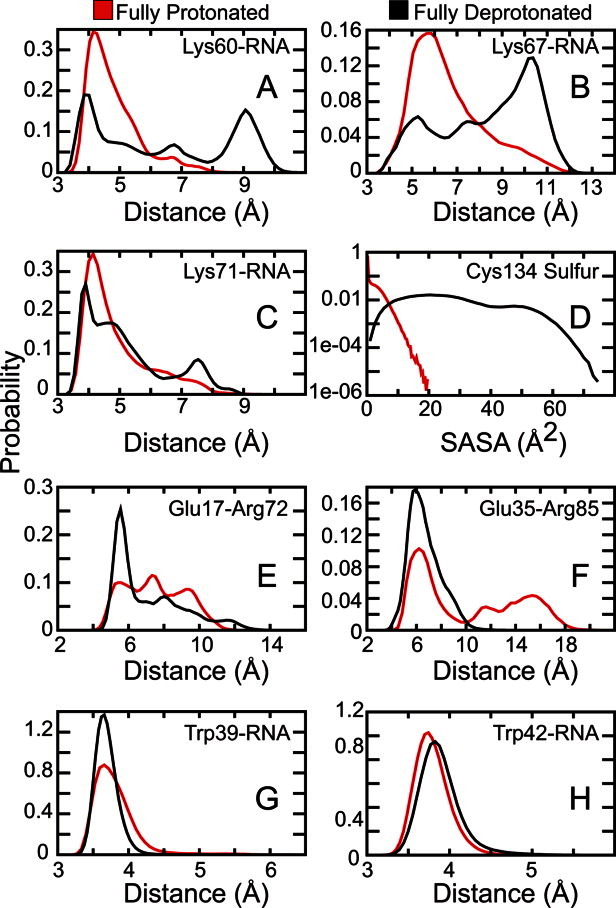
Conformational dynamics of key intraprotein and protein–RNA interactions. Lys–RNA and Glu–Arg distances correspond to salt bridge interactions. Trp–RNA distances and Cys SASA correspond to end-capping interactions and the solvent accessible surface area (SASA) for the cysteine sulfur atom, respectively. See Materials and Methods section for more detail.
Discussion
The primary goal of this study was to assess the overall p19–siRNA binding stability and to identify the important pH-sensitive residues that affect siRNA binding. Previously, Koukiekolo et al. hypothesized that p19–siRNA binding is dependent on the ionization of one or more histidine, cysteine, and lysine residues.27 They determined this by fitting fluorescence data to an equation that represents the titration of three apparent pKa values (found to be 7.1, 8.0, and 10.6) and then attributed these numbers to a particular type of residue based on each residue's reference pKa value. However, the detailed resolution needed to pinpoint the residues associated with these experimental pKa values is well beyond the capacity of their assays. Thus, using atomic-level resolution CPHMD simulations, we have computed pKa values for 26 titratable residues from the p19 protein dimer in both holo and apo systems and compared our results with the current literature. As pointed out previously, the observed changes in the binding stability are likely the result of the deprotonation/protonation of titratable residues that interact with the siRNA and/or the result of local pH-sensitive changes that affect the structural integrity of the p19 protein.27
Overall, the 14 holo and 14 apo CPHMD simulations showed remarkable stability in the p19 structure as demonstrated by their Cα-RMSD in different pH environments (Supporting Information Fig. S1). This structural stability is consistent with traditional explicit solvent MD simulations of CIRV p19 with fixed protonation states.10 The holo simulations displayed slightly lower Cα-RMSD values compared with the apo systems due to the presence of the siRNA. Visual inspection of the apo p19 simulations showed that the differences in RMSD were caused by the loss of Trp39/Trp42–RNA end-capping interactions which resulted in a significant increase in the α2-helix dynamics along with added mobility in the α1-helix (Fig. 1 and Supporting Information Fig. S2). The flexibility found in the N-terminal region of p19 is in line with a model where a negatively charged siRNA first binds to the positively charged p19 β-sheet surface and then the tryptophan residues act as calipers to measure the length of the bound dsRNA by stacking with the terminal base pairs.9, 10
Using the pKa values calculated from all 26 titratable residues in the holo and apo p19 simulations (Table I and Supporting Information Table S1), we computed the net charge of both p19 systems as well as the pH-dependent binding stability (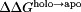 ) of the entire system and for each titratable residue [Fig. 2(A, B)].
) of the entire system and for each titratable residue [Fig. 2(A, B)]. 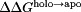 > 0 increases binding stability while
> 0 increases binding stability while 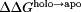 < 0 decreases binding stability. Overall, both systems became more negative as each titratable residue was deprotonated due to an increase in pH and, in general, the net charge of the holo system was more positive than the apo system [Fig. 2(A)]. This was expected as a more negative net charge would result in siRNA dissociation due to the charge–charge repulsion between the negatively charged siRNA and the protein in the holo system. Between pH 6 and pH 8, the total charge for both systems was nearly identical. It is also interesting to note that, with the exception of Cys134 (not conserved) and His132 (charged conserved as arginine), 11 of the 13 titratable residues found to contribute significantly to the binding stability (Table I) are well conserved across the Tombusvirus p19 family9 which generally implies some level of functional importance.
< 0 decreases binding stability. Overall, both systems became more negative as each titratable residue was deprotonated due to an increase in pH and, in general, the net charge of the holo system was more positive than the apo system [Fig. 2(A)]. This was expected as a more negative net charge would result in siRNA dissociation due to the charge–charge repulsion between the negatively charged siRNA and the protein in the holo system. Between pH 6 and pH 8, the total charge for both systems was nearly identical. It is also interesting to note that, with the exception of Cys134 (not conserved) and His132 (charged conserved as arginine), 11 of the 13 titratable residues found to contribute significantly to the binding stability (Table I) are well conserved across the Tombusvirus p19 family9 which generally implies some level of functional importance.
At pH > 9, the side chains of Lys60, Lys67, and Lys71 become neutralized and, as a result, their direct interactions with the negatively charged siRNA backbone are reduced [Figs. 3 and 4(A–C)]. Deprotonation of these three conserved residues has the largest destabilizing effect on siRNA binding as reflected in the 14.0 kcal/mol drop in free energy [Fig. 2(B)]. This is consistent with past mutations of Lys60 and Lys71 that resulted in decreases in the lethal necrosis phenotype.11, 12 Thus, we hypothesize that mutating one or more of these lysine residues to arginine, which has a much higher pKa and therefore would remain positively charged, may help to increase the binding stability at higher pH ranges. In addition, Cys134 was found to be highly solvent exposed when it is deprotonated [Figs 3 and 4(D)]. Koukiekolo et al. suggested that the role of the cysteine amino acid is to preserve the structural integrity of the protein and that the deprotonation of cysteine (or other titratable residues) could lead to structural changes that could either destabilize the p19 dimer or the p19–siRNA complex.13,14,27 From our simulations, we suggest that Cys134 becomes more solvent exposed to prevent having a buried charge that could affect the stability of the local p19 structure. Therefore, we proffer that mutation of Cys134 to its isosteric equivalent, serine, may be beneficial to the CIRV p19 binding affinity.
Figure 3.
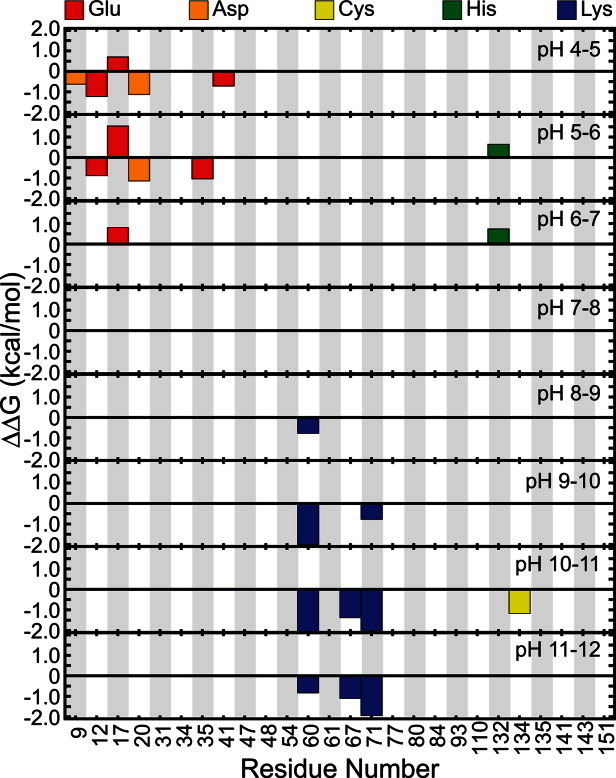
Individual contributions to binding stability at different pH ranges. Negative and positive contributions to binding stability will have 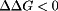 and
and 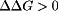 , respectively, for a given titratable residue. Only residues with
, respectively, for a given titratable residue. Only residues with 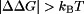 are shown where
are shown where 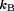 is the Boltzmann constant and T is the temperature (298 K).
is the Boltzmann constant and T is the temperature (298 K).
At 4 < pH < 6, deprotonation of Glu35 not only facilitates the formation of a salt bridge with Arg85 [Figs 3 and 4(F)] but it also leads to an increase in base stacking interactions between the nearby Trp39 and terminal RNA base pair [Fig. 4(G)]. Surprisingly, the Trp42–RNA base stacking interactions were not affected [Fig. 4(H)]. Glu12 and Asp20 are located near the p19–siRNA binding surface and likely destabilize the complex once they are deprotonated by conferring strong electrostatic repulsion with the siRNA (Fig. 3). Based on this observation, we hypothesize that replacing Glu12 and/or Asp20 to neutral glutamine and asparagine, respectively, would increase the overall binding stability.
At neutral pH, there was a small increase in 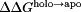 that was caused by the deprotonation of Glu17 and His132 [Figs. 2(B) and 3]. In the crystal structure, His132 is positioned beside a buried Arg117 which is expected to be structurally less stable when both residues are protonated.9 We speculate that the neutralization of His132 reduces the local concentration of positive charges and ultimately stabilizes the p19 structure. In contrast, ionized Glu17 appears to facilitate the positioning of key lysine residues (Lys60, Lys67, and Lys71) along the protein–RNA interface by forming salt bridge interactions with Arg72 [Supporting Information Fig. S2 and Fig. 4(E)]. However, detailed correlation analyses revealed that the Glu17–Arg72 salt bridge formation and the Lys–RNA interactions are basically uncorrelated (with correlation coefficients, R, between −0.04 and 0.22). Alternatively, it has also been proposed that the Glu17–Arg72 salt bridge (and possibly Glu35–Arg85 and Glu41–Arg75 salt bridges) may play a role in positioning the end-capping tryptophan residues.9, 10 The importance of this specific salt bridge is further illustrated by previous studies that swapped Arg72 for glycine and found that the activity of p19 was severely compromised.11,12,57,58
that was caused by the deprotonation of Glu17 and His132 [Figs. 2(B) and 3]. In the crystal structure, His132 is positioned beside a buried Arg117 which is expected to be structurally less stable when both residues are protonated.9 We speculate that the neutralization of His132 reduces the local concentration of positive charges and ultimately stabilizes the p19 structure. In contrast, ionized Glu17 appears to facilitate the positioning of key lysine residues (Lys60, Lys67, and Lys71) along the protein–RNA interface by forming salt bridge interactions with Arg72 [Supporting Information Fig. S2 and Fig. 4(E)]. However, detailed correlation analyses revealed that the Glu17–Arg72 salt bridge formation and the Lys–RNA interactions are basically uncorrelated (with correlation coefficients, R, between −0.04 and 0.22). Alternatively, it has also been proposed that the Glu17–Arg72 salt bridge (and possibly Glu35–Arg85 and Glu41–Arg75 salt bridges) may play a role in positioning the end-capping tryptophan residues.9, 10 The importance of this specific salt bridge is further illustrated by previous studies that swapped Arg72 for glycine and found that the activity of p19 was severely compromised.11,12,57,58
The computed dissociation constant profile [Fig. 2(C)] showed little change after the first 15 ns of production simulation time (Supporting Information Fig. S3) and was in good agreement with experiment.13 This suggests that the results from the CPHMD simulations were converged. Also, a much better correspondence with experiment was obtained when we shifted the dissociation constant profile horizontally by 1.5 pH units [see “CPHMDshifted” in Fig. 2(C) and inset]. The shift in the log Kd profile can be attributed to an underestimation of the desolvation energy and has been shown to lead to a systematic underestimation of the pKa values (from which the dissociation constant profiles are derived).59 Henceforth, discussions of the computed dissociation constant profile will be in reference to the shifted profile (CPHMDshifted).
The optimal range for siRNA binding appears to be between pH 7 and 10 where the binding stability fluctuates between −7.0 ≤ log Kd ≤ −6.5 [Fig. 2(C)]. From our simulations, we can attribute the highest experimentally observed pKa of 10.627 to residues Lys60, Lys67, Lys71, and Cys134 (Fig. 3). These residues demonstrated the largest reduction in binding stability at high pH [Fig. 2(B)] and are excellent candidates for further mutational studies (see discussion above). Similarly, the lowest experimental pKa value, 7.1, can be assigned to multiple residues (i.e., Asp9, Glu12, Asp20, Glu35, and/or Glu41) (Fig. 3). Ionization of all these residues appeared to have a synergistic destabilizing effect on the p19–siRNA complex. Finally, the last observed apparent pKa of 8.0 corresponded to residues Glu17 and His132. These two residues were the only groups that were beneficial for significantly increasing the binding affinity upon deprotonation and we found them to be important for maintaining the structural integrity of p19.
Conclusions
In summary, we presented CPHMD simulations of a large protein–RNA complex in implicit solvent. Overall, the results agree well with experiment. We identified several titratable residues that are highly pH-dependent and that could be assigned to experimentally observed pKa values. Lys60, Lys67, Lys71, and Cys134 appear to affect binding stability at pH > 9 while several glutamic and aspartic acids destabilize the complex at pH < 6. These residues were found to be important for maintaining the stability of the protein structure and/or for siRNA binding. The optimal pH range for siRNA binding is from about 7.0 to 10.0 and is largely stabilized by Glu17 and His132.
The CPHMD method has developed into an accurate and powerful tool for predicting protein pKa values30 and for generating pH-dependent binding stability curves that can be directly compared with experiment. Ultimately, identifying the key pH-sensitive residues using the CPHMD approach would allow us to design p19 proteins that have a higher affinity for siRNA which could be used to characterize RNA silencing complexes, to manage cellular levels of siRNA levels, and for discriminating between single-stranded RNA and dsRNA, and so forth.13,23,26,60 Furthermore, understanding the pH-dependence of the viral protein could enable us to engineer plants that can survive outside of the virulent pH range and avoid infection altogether. This study clearly illustrates the value of complementing experiment with theoretical techniques and offers results that can be further validated.
Materials and Methods
Simulation setup
The CIRV p19 X-ray crystal structure bound to a 21-nt siRNA (PDBID: 1RPU)9 was used as our model. The unresolved short linker (residues 49–54) in each protein monomer was constructed using MODELLER61, 62 and the loop modeling facility in the MMTSB Tool Set.63 Missing hydrogen atoms were added using the HBUILD algorithm in the CHARMM simulation package.64 The holo (siRNA-bound) and apo (siRNA-free) systems contain 5889 and 4551 atoms, respectively. All simulations were performed in implicit solvent using the generalized born with a simple switching (GBSW) model65, 66 implemented in CHARMM along with the CHARMM27 protein–nucleic acid force field67, 68 and an energy correction map (CMAP).69 A 50 ps−1 friction coefficient was used for Langevin dynamics and the experimental salt concentration of 0.1 M27 was simulated using the Debye–Hückel model for screening charge–charge interactions.70 Consistent with previous GBSW simulations, the nonpolar surface tension coefficient was set to 0.005 kcal/mol/Å253,65,71,72 and a switching cutoff that reduces the electrostatic solvation and van der Waals contributions to zero beginning from 20 Å to 24 Å was used. Optimized atomic radii for proteins65, 73 and nucleic acids74 were used in place of the standard van der Waals radii for the GBSW calculations. The siRNA was harmonically restrained to its initial starting position using a 2 kcal/mol/Å2 force constant to prevent large structural changes in the RNA. Both holo and apo systems were energy minimized and heated up slowly to a final temperature of 298 K followed by 500 ps of equilibration using a 1 fs simulation time step. All molecular images were created using PyMOL.75
Constant pH MD simulations and pKa calculations
A total of 28 independent simulations (14 for siRNA-bound and 14 for siRNA-free) ranging from pH 1 to 14 were conducted using the CPHMD methodology.44–46 Each simulation was 25 ns long and resulted in a cumulative simulation time of 0.7 μs. The first 5 ns of each simulation were discarded in the analysis so that all 28 production simulations were each 20 ns long. Atomic charges for protonated and unprotonated states of aspartic acid, glutamic acid, histidine, lysine, and cysteine have been described previously.44,68,76,77 Arginines, whose model pKa value is typically ∼12,78 were kept permanently in its protonated form because CIRV p19 was found experimentally to be unstable at pH > 1227 All titratable residues were simulated following the CPHMD method originally developed in the Brooks research group where a continuous titration coordinate, 0 > λi ≤ 1, controls the protonation state for the ith titratable residue.44–46 In that model, λi = 1 and λi = 0 correspond to the fully unprotonated and fully protonated states, respectively, and N(λi) is the number of simulation snapshots with protonation state λ. However, to increase the number of times that a titratable residue is considered to be fully protonated (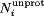 ) or fully unprotonated (
) or fully unprotonated (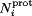 ), we have defined a more generous cutoff for λ:
), we have defined a more generous cutoff for λ:
| (1) |
Thus, the fraction of unprotonated states, 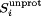 , is given by:
, is given by:
| (2) |
and the pKa of the ith titratable residue can be calculated by fitting a set of Sunprot (at different pH values) to the standard Hendersen–Hasselbalch equation:
| (3) |
where n represents the Hill coefficient. It has been discussed previously that small deviations in the Hill coefficient away from 1 have a negligible effect on the free energy53, 54 and, indeed, we find only small differences in n during the curve fitting process. Thus, we have set n = 1 for all our calculations. Finally, due to the fact that the experimentally determined pKa values were extracted from a homodimer, we have combined the data from both monomers to effectively double the sampling for calculating 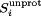 and its corresponding pKa.
and its corresponding pKa.
pH-dependent binding stability and pH-dissociation constant profiles
The pH-dependent binding stability profile was calculated using the Wyman–Tanford linkage equation79, 80:
| (4) |
where 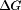 is the dissociation free energy, R is the gas constant, and T is the temperature in Kelvins.
is the dissociation free energy, R is the gas constant, and T is the temperature in Kelvins.  is the difference in the net charge between the holo and apo states at a particular pH and is calculated from:
is the difference in the net charge between the holo and apo states at a particular pH and is calculated from:
| (5) |
The average charge of the system, 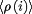 , is obtained from:
, is obtained from:
| (6) |
where 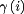 , defined previously,81 is equal to 1 or −1 for a basic and acidic group, respectively. Integrating Eq. (4) after substituting in Eqs. (5) and (6) gives the dissociation free energy at a given pH relative to a reference pH (pHref):
, defined previously,81 is equal to 1 or −1 for a basic and acidic group, respectively. Integrating Eq. (4) after substituting in Eqs. (5) and (6) gives the dissociation free energy at a given pH relative to a reference pH (pHref):
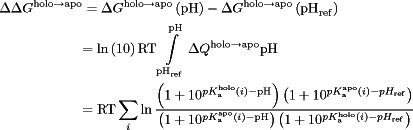 |
(7) |
where 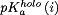 and
and 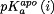 are the pKas for the holo and apo states, respectively. The final summation in Eq. (7) allows the binding stability to be decomposed into contributions from each titratable residue. The final dissociation constant profile was then computed by applying the basic relation:
are the pKas for the holo and apo states, respectively. The final summation in Eq. (7) allows the binding stability to be decomposed into contributions from each titratable residue. The final dissociation constant profile was then computed by applying the basic relation:
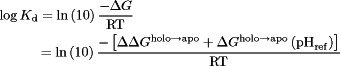 |
(8) |
where 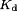 is the dissociation constant. However, as Eq. (7) gives us
is the dissociation constant. However, as Eq. (7) gives us 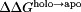 , then the resulting
, then the resulting 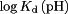 in Eq. (8) at any given pH depends on
in Eq. (8) at any given pH depends on 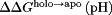 and
and 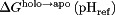 , the latter of which is a constant. Thus,
, the latter of which is a constant. Thus, 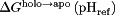 was chosen to allow the best match of
was chosen to allow the best match of 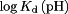 to experiment.
to experiment.
Side chain conformational dynamics
Motivated by previous discussions,8,9,27 the conformational dynamics of several different intraprotein and protein–RNA interactions were analyzed (from the holo simulations) by comparing the normalized probability of an interaction when a particular titratable side chain is either fully protonated (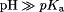 ) or fully deprotonated (
) or fully deprotonated (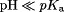 ). Glu–Arg salt bridge distances were measured from the Glu-Cδ atom to the Arg-Cζ atom. Trp–RNA base stacking distances were measured from the center-of-mass of the Trp side chain (not titrated) to the center-of-mass of the closest RNA base. Lys–RNA salt bridges were measured from the Lys-Nζ atom to the closest RNA backbone phosphorus atom. The SASA was measured for the Cys-Sγ atom.
). Glu–Arg salt bridge distances were measured from the Glu-Cδ atom to the Arg-Cζ atom. Trp–RNA base stacking distances were measured from the center-of-mass of the Trp side chain (not titrated) to the center-of-mass of the closest RNA base. Lys–RNA salt bridges were measured from the Lys-Nζ atom to the closest RNA backbone phosphorus atom. The SASA was measured for the Cys-Sγ atom.
Acknowledgments
The authors would like to thank Garrett B. Goh for many valuable scientific discussions.
Glossary
Abbreviations
- CIRV
carnation Italian ringspot virus
- CPHMD
constant pH molecular dynamics
- dsRNA
double-stranded RNA
- MC
Monte Carlo
- MD
molecular dynamics
- miRNA
microRNA
- RISC
RNA-induced silencing complex
- RMSD
root mean squared deviation
- RNAi
RNA interference
- Sunprot
fraction of unprotonated states
- siRNA
short-interfering RNA
- TBSV
tomato bushy stunt virus
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Fire A, Xu SQ, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 2.Hannon GJ. Rna interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 3.Voinnet O. RNA silencing as a plant immune system against viruses. Trends Genet. 2001;17:449–459. doi: 10.1016/s0168-9525(01)02367-8. [DOI] [PubMed] [Google Scholar]
- 4.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 5.Hutvagner G, Simard MJ. Argonaute proteins: key players in RNA silencing. Nat Rev Mol Cell Bio. 2008;9:22–32. doi: 10.1038/nrm2321. [DOI] [PubMed] [Google Scholar]
- 6.Voinnet O, Pinto YM, Baulcombe DC. Suppression of gene silencing: a general strategy used by diverse DNA and RNA viruses of plants. Proc Natl Acad Sci USA. 1999;96:14147–14152. doi: 10.1073/pnas.96.24.14147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silhavy D, Molnar A, Lucioli A, Szittya G, Hornyik C, Tavazza M, Burgyan J. A viral protein suppresses RNA silencing and binds silencing-generated, 21-to 25-nucleotide double-stranded RNAs. EMBO J. 2002;21:3070–3080. doi: 10.1093/emboj/cdf312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ye KQ, Malinina L, Patel DJ. Recognition of small interfering RNA by a viral suppressor of RNA silencing. Nature. 2003;426:874–878. doi: 10.1038/nature02213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vargason JM, Szittya G, Burgyan J, Hall TMT. Size selective recognition of siRNA by an RNA silencing suppressor. Cell. 2003;115:799–811. doi: 10.1016/s0092-8674(03)00984-x. [DOI] [PubMed] [Google Scholar]
- 10.Xia Z, Zhu ZH, Zhu J, Zhou RH. Recognition mechanism of siRNA by viral p19 suppressor of RNA silencing: a molecular dynamics study. Biophys J. 2009;96:1761–1769. doi: 10.1016/j.bpj.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu M, Desvoyes B, Turina M, Noad R, Scholthof HB. Genetic dissection of tomato bushy stunt virus p19-protein-mediated host-dependent symptom induction and systemic invasion. Virology. 2000;266:79–87. doi: 10.1006/viro.1999.0071. [DOI] [PubMed] [Google Scholar]
- 12.Turina M, Omarov R, Murphy JF, Bazaldua-Hernandez C, Desvoyes B, Scholthof HB. A newly identified role for Tomato bushy stunt virus P19 in short distance spread. Mol Plant Pathol. 2003;4:67–72. doi: 10.1046/j.1364-3703.2003.00151.x. [DOI] [PubMed] [Google Scholar]
- 13.Sagan SM, Koukiekolo R, Rodgers E, Goto NK, Pezacki JP. Inhibition of siRNA binding to a p19 viral suppressor of RNA silencing by cysteine alkylation. Angew Chem Int Ed Engl. 2007;46:2005–2009. doi: 10.1002/anie.200603284. [DOI] [PubMed] [Google Scholar]
- 14.Cheng J, Koukiekolo R, Kieliszkiewicz K, Sagan SM, Pezacki JP. Cysteine residues of Carnation Italian Ringspot virus p19 suppressor of RNA silencing maintain global structural integrity and stability for siRNA binding. Biochim Biophys Acta. 2009;1794:1197–1203. doi: 10.1016/j.bbapap.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 15.Papp I, Mette MF, Aufsatz W, Daxinger L, Schauer SE, Ray A, van der Winden J, Matzke M, Matzke AJM. Evidence for nuclear processing of plant micro RNA and short interfering RNA precursors. Plant Physiol. 2003;132:1382–1390. doi: 10.1104/pp.103.021980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lakatos L, Szittya G, Silhavy D, Burgyan J. Molecular mechanism of RNA silencing suppression mediated by p19 protein of tombusviruses. EMBO J. 2004;23:876–884. doi: 10.1038/sj.emboj.7600096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chapman EJ, Prokhnevsky AI, Gopinath K, Dolja VV, Carrington JC. Viral RNA silencing suppressors inhibit the microRNA pathway at an intermediate step. Gene Dev. 2004;18:1179–1186. doi: 10.1101/gad.1201204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunoyer P, Lecellier CH, Parizotto EA, Himber C, Voinnet O. Probing the microRNA and small interfering RNA pathways with virus-encoded suppressors of RNA silencing. Plant Cell. 2004;16:1235–1250. doi: 10.1105/tpc.020719. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Li WX, Li HW, Lu R, Li F, Dus M, Atkinson P, Brydon EWA, Johnson KL, Garcia-Sastre A, Ball LA, Palese P, Ding SW. Interferon antagonist proteins of influenza and vaccinia viruses are suppressors of RNA silencing. Proc Natl Acad Sci USA. 2004;101:1350–1355. doi: 10.1073/pnas.0308308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lecellier CH, Dunoyer P, Arar K, Lehmann-Che J, Eyquem S, Himber C, Saib A, Voinnet O. A cellular MicroRNA mediates antiviral defense in human cells. Science. 2005;308:557–560. doi: 10.1126/science.1108784. [DOI] [PubMed] [Google Scholar]
- 21.Lu R, Maduro M, Li F, Li HW, Broitman-Maduro G, Li WX, Ding SW. Animal virus replication and RNAi-mediated antiviral silencing in Caenorhabditis elegans. Nature. 2005;436:1040–1043. doi: 10.1038/nature03870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Voinnet O, Rivas S, Mestre P, Baulcombe D. An enhanced transient expression system in plants based on suppression of gene silencing by the p19 protein of tomato bushy stunt virus. Plant J. 2003;33:949–956. doi: 10.1046/j.1365-313x.2003.01676.x. [DOI] [PubMed] [Google Scholar]
- 23.Cheng J, Sagan SM, Assem N, Koukiekolo R, Goto NK, Pezacki JP. Stabilized recombinant suppressors of RNA silencing: functional effects of linking monomers of Carnation Italian Ringspot virus p19. Biochim Biophys Acta. 2007;1774:1528–1535. doi: 10.1016/j.bbapap.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 24.Calabrese JM, Sharp PA. Characterization of the short RNAs bound by the P19 suppressor of RNA silencing in mouse embryonic stem cells. RNA. 2006;12:2092–2102. doi: 10.1261/rna.224606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin JM, Cid M, Poole CB, McReynolds LA. Protein mediated miRNA detection and siRNA enrichment using p19. Biotechniques. 2010;48:Xvii–Xxiii. doi: 10.2144/000113364. [DOI] [PubMed] [Google Scholar]
- 26.Koukiekolo R, Jakubek ZJ, Cheng J, Sagan SM, Pezacki JP. Studies of a viral suppressor of RNA silencing p19-CFP fusion protein: a FRET-based probe for sensing double-stranded fluorophore tagged small RNAs. Biophys Chem. 2009;143:166–169. doi: 10.1016/j.bpc.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 27.Koukiekolo R, Sagan SM, Pezacki JP. Effects of pH and salt concentration on the siRNA binding activity of the RNA silencing suppressor protein p19. FEBS Lett. 2007;581:3051–3056. doi: 10.1016/j.febslet.2007.05.064. [DOI] [PubMed] [Google Scholar]
- 28.Alexov E, Mehler EL, Baker N, Baptista AM, Huang Y, Milletti F, Nielsen JE, Farrell D, Carstensen T, Olsson MHM, Shen JK, Warwicker J, Williams S, Word JM. Progress in the prediction of pK(a) values in proteins. Proteins. 2011;79:3260–3275. doi: 10.1002/prot.23189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mongan J, Case DA. Biomolecular simulations at constant pH. Curr Opin Struct Biol. 2005;15:157–163. doi: 10.1016/j.sbi.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Wallace JA, Shen JK. Predicting pKa values with continuous constant pH molecular dynamics. Methods Enzymol. 2009;466:455–475. doi: 10.1016/S0076-6879(09)66019-5. [DOI] [PubMed] [Google Scholar]
- 31.Burgi R, Kollman PA, van Gunsteren WF. Simulating proteins at constant pH: an approach combining molecular dynamics and Monte Carlo simulation. Proteins. 2002;47:469–480. doi: 10.1002/prot.10046. [DOI] [PubMed] [Google Scholar]
- 32.Baptista AM, Teixeira VH, Soares CM. Constant-pH molecular dynamics using stochastic titration. J Chem Phys. 2002;117:4184–4200. [Google Scholar]
- 33.Machuqueiro M, Baptista AM. Constant-pH molecular dynamics with ionic strength effects: protonation-conformation coupling in decalysine. J Phys Chem B. 2006;110:2927–2933. doi: 10.1021/jp056456q. [DOI] [PubMed] [Google Scholar]
- 34.Machuqueiro M, Baptista AM. Acidic range titration of HEWL using a constant-pH molecular dynamics method. Proteins. 2008;72:289–298. doi: 10.1002/prot.21923. [DOI] [PubMed] [Google Scholar]
- 35.Machuqueiro M, Baptista AM. Molecular dynamics at constant pH and reduction potential: application to cytochrome c(3. J Am Chem Soc. 2009;131:12586–12594. doi: 10.1021/ja808463e. [DOI] [PubMed] [Google Scholar]
- 36.Dlugosz M, Antosiewicz JM. Constant-pH molecular dynamics simulations: a test case of succinic acid. Chem Phys. 2004;302:161–170. [Google Scholar]
- 37.Dlugosz M, Antosiewicz JM, Robertson AD. Constant-pH molecular dynamics study of protonation-structure relationship in a heptapeptide derived from ovomucoid third domain. Phys Rev E. 2004;69:021915. doi: 10.1103/PhysRevE.69.021915. [DOI] [PubMed] [Google Scholar]
- 38.Mongan J, Case DA, McCammon JA. Constant pH molecular dynamics in generalized born implicit solvent. J Comput Chem. 2004;25:2038–2048. doi: 10.1002/jcc.20139. [DOI] [PubMed] [Google Scholar]
- 39.Mongan J, Case DA, McCammon J. Discrete-state constant pH molecular dynamics in generalized born implicit solvent. Protein Sci. 2004;13:219. doi: 10.1002/jcc.20139. [DOI] [PubMed] [Google Scholar]
- 40.Meng YL, Roitberg AE. Constant pH replica exchange molecular dynamics in biomolecules using a discrete protonation model. J Chem Theory Comput. 2010;6:1401–1412. doi: 10.1021/ct900676b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams SL, de Oliveira CAF, McCammon JA. Coupling constant pH molecular dynamics with accelerated molecular dynamics. J Chem Theory Comput. 2010;6:560–568. doi: 10.1021/ct9005294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Messer BM, Roca M, Chu ZT, Vicatos S, Kilshtain AV, Warshel A. Multiscale simulations of protein landscapes: using coarse-grained models as reference potentials to full explicit models. Proteins. 2010;78:1212–1227. doi: 10.1002/prot.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aqvist J, Warshel A. Simulation of enzyme-reactions using valence-bond force-fields and other hybrid quantum-classical approaches. Chem Rev. 1993;93:2523–2544. [Google Scholar]
- 44.Lee MS, Salsbury FR, Brooks CL., III Constant-pH molecular dynamics using continuous titration coordinates. Proteins. 2004;56:738–752. doi: 10.1002/prot.20128. [DOI] [PubMed] [Google Scholar]
- 45.Khandogin J, Brooks CL., III Constant pH molecular dynamics with proton tautomerism. Biophys J. 2005;89:141–157. doi: 10.1529/biophysj.105.061341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khandogin J, Brooks CL., III Toward the accurate first-principles prediction of ionization equilibria in proteins. Biochemistry. 2006;45:9363–9373. doi: 10.1021/bi060706r. [DOI] [PubMed] [Google Scholar]
- 47.Guo Z, Brooks CL, III, Kong X. Efficient and flexible algorithm for free energy calculations using the lambda-dynamics approach. J Phys Chem B. 1998;102:2032–2036. [Google Scholar]
- 48.Knight JL, Brooks CL., III lambda-dynamics free energy simulation methods. J Comput Chem. 2009;30:1692–1700. doi: 10.1002/jcc.21295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kong XJ, Brooks CL., III lambda-Dynamics: a new approach to free energy calculations. J Chem Phys. 1996;105:2414–2423. [Google Scholar]
- 50.Wallace JA, Shen JK. Continuous constant pH molecular dynamics in explicit solvent with pH-based replica exchange. J Chem Theory Comput. 2011;7:2617–2629. doi: 10.1021/ct200146j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goh GB, Knight JL, Brooks CL., III Constant pH molecular dynamics simulations of nucleic acids in explicit solvent. J Chem Theory Comput. 2012;8:36–46. doi: 10.1021/ct2006314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang BW, Brunetti L, Brooks CL., III Probing pH-dependent dissociation of HdeA dimers. J Am Chem Soc. 2011;133:19393–19398. doi: 10.1021/ja2060066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shen JK. Uncovering specific electrostatic interactions in the denatured states of proteins. Biophys J. 2010;99:924–932. doi: 10.1016/j.bpj.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shen JK. A method to determine residue-specific unfolded-state pK(a) values from analysis of stability changes in single mutant cycles. J Am Chem Soc. 2010;132:7258. doi: 10.1021/ja101761m. [DOI] [PubMed] [Google Scholar]
- 55.Wallace JA, Shen JK. Unraveling a trap-and-trigger mechanism in the pH-sensitive self-assembly of spider silk proteins. J Phys Chem Lett. 2012;3:658–662. doi: 10.1021/jz2016846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arthur EJ, Yesselman JD, Brooks CL., III Predicting extreme pK(a) shifts in staphylococcal nuclease mutants with constant pH molecular dynamics. Proteins. 2011;79:3276–3286. doi: 10.1002/prot.23195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uhrig JF, Canto T, Marshall D, MacFarlane SA. Relocalization of nuclear ALY proteins to the cytoplasm by the tomato bushy stunt virus P19 pathogenicity protein. Plant Physiol. 2004;135:2411–2423. doi: 10.1104/pp.104.046086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Park JW, Faure-Rabasse S, Robinson MA, Desvoyes B, Scholthof HB. The multifunctional plant viral suppressor of gene silencing P19 interacts with itself and an RNA binding host protein. Virology. 2004;323:49–58. doi: 10.1016/j.virol.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 59.Wallace JA, Wang YH, Shi CY, Pastoor KJ, Nguyen BL, Xia K, Shen JK. Toward accurate prediction of pK(a) values for internal protein residues: the importance of conformational relaxation and desolvation energy. Proteins. 2011;79:3364–3373. doi: 10.1002/prot.23080. [DOI] [PubMed] [Google Scholar]
- 60.Scholthof HB. Timeline—the Tombusvirus-encoded P19: from irrelevance to elegance. Nat Rev Microbiol. 2006;4:405–411. doi: 10.1038/nrmicro1395. [DOI] [PubMed] [Google Scholar]
- 61.Fiser A, Do RKG, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 63.Feig M, Karanicolas J, Brooks CL., III MMTSB tool set: enhanced sampling and multiscale modeling methods for applications in structural biology. J Mol Graphics Model. 2004;22:377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 64.Brooks BR, Brooks CL, III, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen JH, Im WP, Brooks CL., III Balancing solvation and intramolecular interactions: toward a consistent generalized born force field. J Am Chem Soc. 2006;128:3728–3736. doi: 10.1021/ja057216r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Im WP, Lee MS, Brooks CL., III Generalized born model with a simple smoothing function. J Comput Chem. 2003;24:1691–1702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 67.Foloppe N, MacKerell AD. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J Comput Chem. 2000;21:86–104. [Google Scholar]
- 68.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 69.MacKerell AD, Feig M, Brooks CL., III Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 70.Srinivasan J, Trevathan MW, Beroza P, Case DA. Application of a pairwise generalized Born model to proteins and nucleic acids: inclusion of salt effects. Theor Chem Acc. 1999;101:426–434. [Google Scholar]
- 71.Khandogin J, Chen JH, Brooks CL., III Exploring atomistic details of pH-dependent peptide folding. Proc Natl Acad Sci USA. 2006;103:18546–18550. doi: 10.1073/pnas.0605216103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khandogin J, Brooks CL., III Linking folding with aggregation in Alzheimer's beta-amyloid peptides. Proc Natl Acad Sci USA. 2007;104:16880–16885. doi: 10.1073/pnas.0703832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nina M, Beglov D, Roux B. Atomic radii for continuum electrostatics calculations based on molecular dynamics free energy simulations. J Phys Chem B. 1997;101:5239–5248. [Google Scholar]
- 74.Banavali NK, Roux B. Atomic radii for continuum electrostatics calculations on nucleic acids. J Phys Chem B. 2002;106:11026–11035. [Google Scholar]
- 75.Schrödinger L. The PyMOL Molecular Graphics System, Version 1.3. Schrödinger, LLC; [Google Scholar]
- 76.Foloppe N, Sagemark J, Nordstrand K, Berndt KD, Nilsson L. Structure, dynamics and electrostatics of the active site of glutaredoxin 3 from Escherichia coli: comparison with functionally related proteins. J Mol Biol. 2001;310:449–470. doi: 10.1006/jmbi.2001.4767. [DOI] [PubMed] [Google Scholar]
- 77.Foloppe N, Nilsson L. The glutaredoxin -C-P-Y-C- motif: influence of peripheral residues. Structure. 2004;12:289–300. doi: 10.1016/j.str.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 78.Nozaki Y, Tanford C. Examination of titration behavior. In: Hirs CHW, editor. Methods enzymol. Vol. 11. New York: Academic Press; 1967. pp. 715–734. [Google Scholar]
- 79.Tanford C. Protein denaturation: part C. Theoretical models for the mechanism of denaturation. Adv Prot Chem. 1970;24:1–95. [PubMed] [Google Scholar]
- 80.Wyman J. Linked functions and reciprocal effects in hemoglobin—a 2nd look. Adv Prot Chem. 1964;19:223–286. doi: 10.1016/s0065-3233(08)60190-4. [DOI] [PubMed] [Google Scholar]
- 81.Yang AS, Honig B. On the pH-dependence of protein stability. J Mol Biol. 1993;231:459–474. doi: 10.1006/jmbi.1993.1294. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.