

Abstract
Low hemoglobin oxygen saturation (SpO2) is common in Sickle Cell Anemia (SCA) and associated with complications including stroke, although determinants remain unknown. We investigated potential hematological, genetic, and nutritional predictors of daytime SpO2 in Tanzanian children with SCA and compared them with non-SCA controls. Steady-state resting pulse oximetry, full blood count, transferrin saturation, and clinical chemistry were measured. Median daytime SpO2 was 97% (IQ range 94–99%) in SCA (N = 458), lower (P < 0.0001) than non-SCA (median 99%, IQ range 98–100%; N = 394). Within SCA, associations with SpO2 were observed for hematological variables, transferrin saturation, body-mass-index z-score, hemoglobin F (HbF%), genotypes, and hemolytic markers; mean cell hemoglobin (MCH) explained most variability (P < 0.001, Adj r 2 = 0.09). In non-SCA only age correlated with SpO2. α-thalassemia 3.7 deletion highly correlated with decreased MCH (Pearson correlation coefficient −0.60, P < 0.0001). In multivariable models, lower SpO2 correlated with higher MCH (β-coefficient −0.32, P < 0.001) or with decreased copies of α-thalassemia 3.7 deletion (β-coefficient 1.1, P < 0.001), and independently in both models with lower HbF% (β-coefficient 0.15, P < 0.001) and Glucose-6-Phosphate Dehydrogenase genotype (β-coefficient −1.12, P = 0.012). This study provides evidence to support the hypothesis that effects on red cell rheology are important in determining SpO2 in children with SCA. Potential mechanisms and implications are discussed.
1. Introduction
Hemoglobin oxygen desaturation in the absence of acute illness is common in children with Sickle Cell Anemia (SCA), and is associated with higher cerebral blood flow velocities [1, 2], and with risk of complications including stroke [3]. The underlying mechanisms of hemoglobin oxygen desaturation in SCA are poorly understood but may involve the severity of anemia [4] as well as differences in hemoglobin oxygen affinity compared to hemoglobin A (HbA), with increased expression of 2.3 DPG in hemoglobin S (HbS) resulting in a right-shifted hemoglobin oxygen affinity curve and other differences in red cell physiology [5]. Other potential causes include a history of acute chest syndrome and reduced pulmonary [6] and cardiac function [7]. Coinheritance of alpha-thalassemia deletions and glucose-6-phosphate deficiency (G6PD) may affect the degree of anemia [8, 9] whilst alpha-thalassemia status modifies red cell indices [10–12] and rheology [13], as can iron status [14]. We therefore investigated potential hematological, genetic, and nutritional predictors of daytime hemoglobin oxygen saturation in Tanzanian pediatric patients homozygous for HbS (SCA) and in non-SCA local controls.
2. Patients and Methods
Ethical permission was granted by the Muhimbili University of Health and Allied Sciences Ethics Committee (MU/RP/AECNoI.XII/77). Written informed consent was obtained from parents or guardians in their own language.
2.1. Patients and Clinical Procedures
Children (less than 17 years) with SCA (HbSS genotype) were enrolled in the SCD cohort study at Muhimbili National Hospital, Dar-es-Salaam [15]. Resting pulse oximetry data (Masimo Radical, Masimo Corporation, USA) and blood samples were collected at routine outpatient clinic visits between November 2007 and December 2008. Analysis was limited to data collected at a single steady-state time point. A strict definition of steady state was employed (temperature <37.5°C, no malaria parasitaemia, no reported pain, no blood transfusion within 90 days or hospital admission within 30 days on either side of the selected time point) and determined to be clinically well by the attending doctor. All cohort children are routinely prescribed folate supplementation (5 mg/day).
Non-SCA children were those who presented for sickle testing between October 2004 and December 2008 but who had HbAA or HbAS by hemoglobin electrophoresis. None of the children had malaria parasitaemia or fever (temperature >37.4°C) and all were clinically well.
2.2. Laboratory Procedures
Blood samples were collected between 8 and 10 am. Full blood counts were performed using an automated cell counter (Pentra 60, Horiba ABX, Kyoto, Japan). Serum iron and total iron binding capacity were measured in serum samples stored at −80°C (Architect C8000, Abbott, New York, USA). Transferrin saturation was calculated from serum iron and total iron binding capacity. Lactate dehydrogenase (LDH) and bilirubin (total and conjugated) were measured in fresh serum samples (Architect C8000, Abbott, New York) by Muhimbili Central Pathology Laboratory.
Children attending screening for sickle status were typed for HbS by alkaline Hb electrophoresis (Helena, Sunderland, Tyne and Wear, UK). Children enrolled in the Muhimbili Sickle cohort also had hemoglobin fractions, including HbF, quantified by HPLC using the β-thalassemia Short Programme on the Variant analyzer (BioRad, Hercules, CA, USA). In addition, children enrolled in the Muhimbili Sickle Cohort had HbSS status confirmed by genotype and were genotyped for the 3.7 alpha-thalassemia deletion using a PCR-based method and agarose gel visualization as per published methods [16] and the 202- and 376-single nucleotide polymorphisms (SNPs) for glucose 6-phosphate dehydrogenase deficiency (G6PD) and HbS using multiplex Sequenom [17].
2.3. Data Analysis
Data were analyzed using STATA 11-IC (StataCorp, College Station, TX, USA). Daytime SpO2 is not normally distributed due to an excess of observations having the maximum possible value of 100% and a skewed distribution towards the lower values. The data are not normalized by the usual log transformations. Thus we investigated the use of negative binomial regression of count data using a new variable of SpO2-100 compared to zero-inflated negative binomial regression. Vuong tests [18] indicated no strong consistent preference for either models across the explanatory variables tested. As we had no prior hypotheses that mechanisms to predict 100% versus <100% SpO2 may differ from mechanisms underlying the degree of desaturation, we selected the negative binomial regression model. We next compared the results of these models to those from simple linear regression of the nontransformed data and observed no major differences in the results. Whilst analysis of the residuals from the linear regression models indicated some skew, this was not judged to be sufficient to render the results invalid, which were in agreement with those from the better fitting negative binomial regression models. Thus for ease of presentation and interpretation the results of the linear regression models are presented. P values <0.05 were considered significant.
3. Results
Complete hematological, genetic, and iron status (transferrin saturation) data were available for 458 SCA children. None of the children were receiving hydroxyurea or routine blood transfusions. None of the children had received more than 4 blood transfusions in their lifetime. In addition, complete hematological and pulse oximetry data were available for 394 non-SCA children, although transferrin saturation data were only available in a small subset of 62 children and genotyping was not conducted in these controls. Pulse oximetry, hematological and iron status data for the two groups of children are summarized in Table 1. Of the 458 SCA children, 239 were boys (52%) with a mean age of 9.7 y (SD 4.3 y) compared to 212 boys (54%) in the 394 non-SCA children with a mean age 7.0 y (SD 4.7 y). In the SCA children median daytime SpO2 was 97% (IQ range 94–99%) (Table 1), significantly lower (Wilcoxon rank sum test; P < 0.0001) than in the non-SCA children (median 99%; IQ range 98–100%) (Table 1 and Figure 1). Hemoglobin concentration, red cell indices (red blood cell count (RBC), mean cell hemoglobin (MCH), and mean cell hemoglobin concentration (MCHC), mean cell volume (MCV)), and markers of hemolysis (LDH and unconjugated bilirubin) were significantly different between the SCA and non-SCA groups (Table 1). The proportion of SCA children with low transferrin saturation (<16%) indicating probable iron deficiency, was 28% (126/458) but was lower than that in non-SCA Tanzanian control children (32/62, 52%) (Table 1). Only 9 (2%) of the SCA and none of the non-SCA patients had high transferrin saturation (>55%) which might indicate iron over-load. Forty-one percent of the SCA children (192/458) were heterozygous for the 3.7 alpha-thalassemia deletion and 17% were homozygous (78/458). Eleven percent of SCA children were heterozygous females for both the −202 and −376 G6PD single nucleotide polymorphisms (SNPs) which results in a moderate phenotype [19], whilst a further 12% (54/458) of SCA children were affected homozygous females or affected males.
Table 1.
Daytime pulse oximetry data, haematological and haemolytic indices and iron status in Tanzanian SCA and non-SCA children.
| Variable | SCA children with daytime oximetry (N = 458) | Non-SCA children with daytime oximetry (N = 394) |
|---|---|---|
| Pulse oximetry | ||
| Median daytime SpO2 (%) | 97 (IQ range 94–99) | 99 (IQ range 98–100)*** |
| Haematology | ||
| Mean hemoglobin* (g/dL) | 7.4 (SD 1.17) | 10.9 (SD 2.2)*** |
| Mean red cell count × 10−9 | 2.89 (SD 0.66) | 4.71 (SD 0.86)*** |
| Mean cell hemoglobin concentration (g/dL) | 31.9 (SD 1.52) | 32.3 (SD 1.60)** |
| Mean cell hemoglobin (pg/cell) | 26.1 (SD 3.31) | 23.6 (SD 3.86)*** |
| Mean cell volume* (fL) | 79.5 (SD 15.28) | 73.1 (SD 9.21)*** |
| Median reticulocyte % (N = 440/335) | 12.8 (IQ range 9.1–16.4) | 2.4 (IQ range 0.8–6.7)*** |
| Median hemoglobin F (Hb%) (N = 316/60)† | 4.7 (IQ range 2.8–7.9) | 0.6 (IQ range 0.2–1.0)*** |
| Haemolytic markers | ||
| Median lactate dehydrogenase (IU) (N = 427/340) | 762 (533–1121) | 498 (367–671)*** |
| Median unconjugated bilirubin (μmol/L) (N = 417/181) | 33.3 (IQ range 18.2–61.1) | 7.2 (IQ range 3.5–12.5)*** |
| Iron status | ||
| Median transferrin saturation (%) (N = 458/62) | 20.9 (IQ range 15.4–27.7) | 14.9 (IQ range 9.1–25.3)** |
| Median serum iron (μmol/L) (N = 458/62) | 11.5 (SD 5.7) (range 8.6–14.9) | 10.0 (SD 8.5) (range 5.8–17.2) |
| Mean serum TIBC (μmol/L) (N = 458/62) | 55.3 (SD 10.9) (range 47.7–62.75) | 70.5 (SD 22.3) (range 50.4–84.3)*** |
| Iron deficiency transferrin saturation < 16% | 126/458 (27.5%) | 32/62 (51.6%)*** |
†Within the SCA children, HbF% was not assessed at the same time point as the hematology and SpO2 data and was limited to children older than 5 years at the time of measurement of HbF% & SpO2. HbF% was available for some non-SCA children and was measured at the same time point as the SpO2.
*P < 0.05, **P < 0.01, ***P < 0.001.
Figure 1.
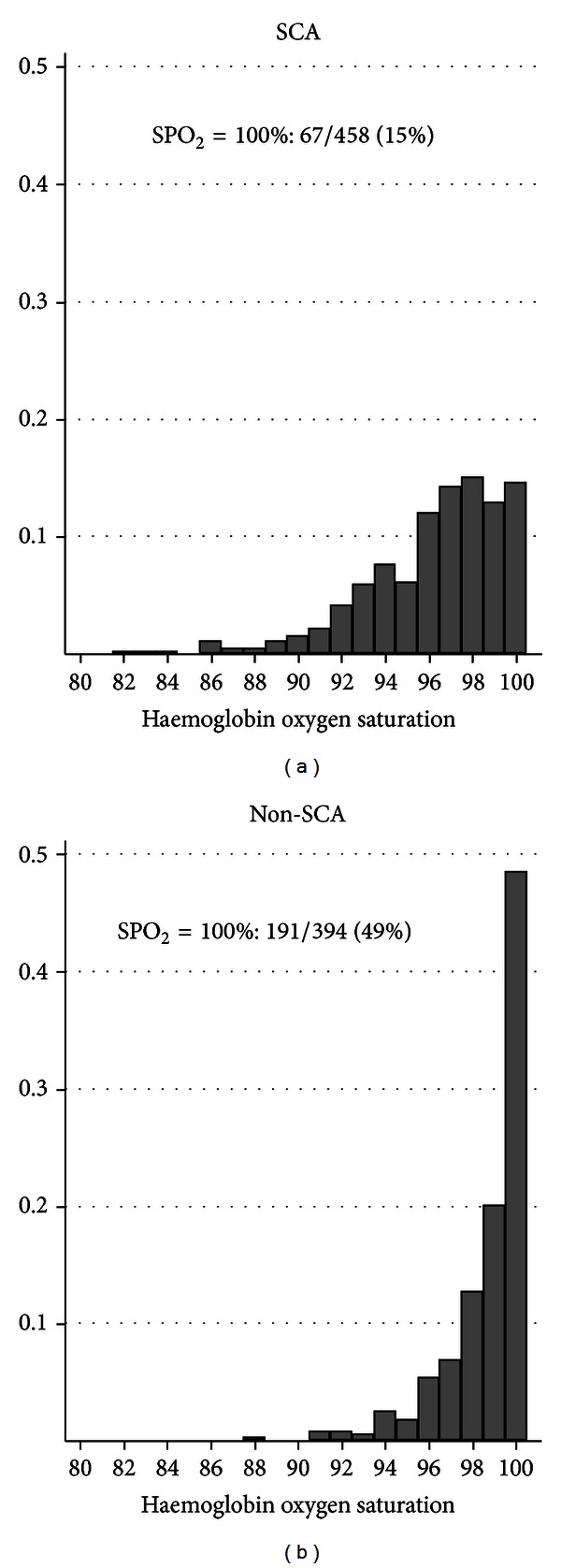
Distribution of daytime hemoglobin oxygen saturation in children with SCA compared to non-SCA.
Predictors of SpO2 in SCA are shown in Table 2. Within the SCA population all of the hematological variables were significantly associated with SpO2. Higher hemoglobin, RBC, and HbF% were associated with greater SpO2, whilst higher MCH, MCHC, and MCV were associated with lower SpO2. MCH was the single biggest contributor to the variation in SpO2 as indicated by the still modest adjusted r 2 value of 0.09. There was weak evidence of a negative association between transferrin saturation and SpO2. In the non-SCA population the same hematological variables were tested for associations with SpO2, as well as Hb phenotype, AS versus AA. However, the only predictor of SpO2 in this group was a positive association for age (beta coefficient 0.05, P = 0.024) with no evidence to suggest even weak effects of any of the hematological indices.
Table 2.
Predictors of daytime SpO2 in SCA.
| Explanatory variable | β-coefficient | P value | Adj r 2 |
|---|---|---|---|
| Hematological indices | |||
| Hemoglobin (g/dL) | 0.35 | 0.007 | 0.014 |
| RBC × 10−9 | 1.10 | <0.001 | 0.050 |
| MCHC (g/dL) | −0.57 | <0.001 | 0.073 |
| MCH | −0.29 | <0.001 | 0.090 |
| MCV (fl) | −0.02 | 0.041 | 0.007 |
| HbF% (N = 315)† | 0.12 | <0.001 | 0.026 |
| Iron status and nutritional status | |||
| Transferrin saturation % | −0.03 | 0.03 | 0.008 |
| BMI Z-score (N = 451) | 0.18 | 0.049 | 0.006 |
| Hemolytic markers | |||
| Lactate dehydrogenase (IU) {natural log} (N = 427) | −0.72 | 0.011 | 0.013 |
| Unconjugated bilirubin (μmol/L) {natural log} (N = 415) | −0.34 | 0.029 | 0.009 |
| α-thalassemia genotype | |||
| 3.7 heterozygote versus Wild Type | 1.29 | <0.001 | |
| 3.7 homozygote versus Wild Type | 2.04 | <0.001 | 0.056* |
| G6PD genotype | |||
| Heterozygote females versus Wild Type | −0.52 | 0.276 | |
| Homozygote females and affected males versus Wild Type | −1.30 | 0.005 | 0.014* |
N = 458 unless otherwise stated. *Adj r 2 for whole model. †Analysis of SpO2 limited to those with HbF and SpO2 measured when patients were 5 years or older.
In the SCA population for whom genotypes were available, coinheritance of the alpha-thalassemia 3.7 genotype was associated with increased SpO2, whilst co-inheritance of G6PD deficiency was associated with decreased SpO2.
Pair-wise associations between the different explanatory variables for daytime SpO2 within the SCA group were investigated and are presented in full in Supplementary Table 1 available online at http://dx.doi.org/10.1155/2013/472909. Alpha-thalassemia genotype was highly associated with all of the hematological variables, with the number of 3.7 deletions being associated with higher hemoglobin (Pearson correlation coefficient 0.23); RBC count (0.55); lower MCV (−0.24), MCHC (−0.32), and MCH (−0.60); and also lower hemolytic markers, particularly unconjugated bilirubin (−0.26), all with P values <0.0001. Interestingly, presence of the 3.7 alpha-thalassemia deletion was also associated with improved nutritional status as determined by body-mass-index z-score. Higher MCH and MCV were associated with lower hemoglobin in the SCA population whilst HbF% was associated with higher hemoglobin. Transferrin saturation was positively associated with hemoglobin (0.17) and also higher MCH (0.33), both P values <0.0005, but was not associated with MCV. Interestingly, G6PD genotype was not significantly associated with any of the other variables.
Correlations between the hematological variables were similar in the non-SCA population compared to SCA (data not shown) except that higher MCV was correlated with higher hemoglobin (0.64, P < 0.001) but was not associated with RBC count and no association was apparent between hemoglobin and MCHC.
In multivariable models predicting SpO2, the inclusion of MCH with alpha-thalassemia genotype resulted in a diminished and nonsignificant association for alpha-thalassemia. However, in models including either MCH or alpha-thalassemia there were an independent effect of HbF% and weak evidence of an independent effect of G6PD limited to affected males and homozygote females (Supplementary Table 2), but with the greatest variation explained by a model including MCH and HbF% (adjusted r 2 = 0.15, P < 0.001, N = 315).
4. Discussion
Few studies have attempted to determine the hematological or genetic correlates of daytime SpO2. In this study we report that higher daytime SpO2 in children with SCA is associated with higher red cell count and hemoglobin, but with lower MCH or MCHC. Coinheritance of the alpha-thalassemia 3.7 deletion in our SCA population is strongly associated with increased hemoglobin, red cell count and decreased MCH and MCHC and also with increased SpO2, but not independently of these factors. Thus the basis of the association between alpha-thalassemia genotype and SpO2 would appear to be via its effects on red cell indices. In Kenyan nonsickle populations, alpha-thalassemia 3.7 deletion copy number is also associated with slightly increased hemoglobin levels and increased red cell counts, but effects on anaemia appear to be modulated by sickle trait status [20]. This suggests that the decreased anemia and increased red cells observed in SCA plus alpha-thalassemia results from decreased rate of red cell destruction, proposed to be due to lower HbS concentration in red cells, and thus reduced HbS polymerization which is concentration dependent as well as being increased by desaturation [11]. This is supported by the observation that alpha thalassemia was associated with lower levels of LDH and unconjugated bilirubin as markers of hemolysis. Red cell count and hemoglobin were not associated with SpO2 when adjusting for MCH and thus anemia does not appear to be a direct cause of low SpO2 in our population. A beneficial effect of alpha thalassemia on daytime SpO2 has also been observed in Jamaican children with SCA, [21] but in contrast to our study, there were also independent effects of hemoglobin, whilst neither MCH, or MCHC was associated with SpO2 [21].
It is interesting to note that within the non-SCA population none of the tested variables were associated with SpO2. This may have been the result of the reduced variability in SpO2 in the non-SCA population, or reflect sickle-specific effects of red cell indices on SpO2. Again, this contrasts with observations in older Jamaican children who were HbAA (15–18 years) in whom hemoglobin and MCV (but not MCH or MCHC) were associated with SpO2.
Our observation of a beneficial effect of co-inheritance of alpha-thalassemia on daytime SpO2 in children with SCA appears to be in direct contrast to our previous observation of a negative effect on mean overnight SpO2, measured in a small group (N = 30) of similar aged Tanzanian children with SCA [22]. However, this effect of alpha-thalassemia appeared to be confounded by the strong negative association between transferrin saturation and mean overnight SpO2. We concluded that this association was most likely the result of reverse causality from nocturnal chronic and intermittent hemoglobin desaturation causing increased transferrin saturation due to upregulation of hypoxia-inducible factor, which has downstream effects of increasing iron absorption, which may also be increased by alpha-thalassemia [20]. Thus we suggest that in a larger dataset, alpha-thalassemia may also have a beneficial effect on mean overnight SpO2.
Similar to Jamaican [21] and American SCA children [6], HbF% was positively and independently associated with SpO2. It is not possible in the current study to determine if an effect of HbF% is via direct effects on increased hemoglobin oxygen affinity or through indirect effects on rates of sickling or red cell rheology. Interestingly alpha-thalassemia was also associated with increased HbF%, perhaps through increased rate of release of immature red cells from ineffective erythropoiesis. The co-inheritance of G6PD deficiency was negatively associated with SpO2. As there was no evidence of an association between G6PD and levels of hemolytic markers, as has been observed previously in SCA [9], the effect on SpO2 is likely through another mechanism.
Lower daytime and nocturnal SpO2 in SCA are associated with poor clinical outcomes. This includes elevated cerebral blood flow velocities [1, 2] and an increased risk of stroke [3, 23]. Co-inheritance of alpha-thalassemia is protective against elevated cerebral blood flow velocities in children with SCA [24, 25] and is associated with reduced stroke risk [26], whilst G6PD increases the risk [24]. Thus the mechanisms behind these associations may, at least in part, be through effects on hemoglobin oxygen saturation and subsequent endothelial dysfunction [27, 28] as well as other potential effects of red cell deformability and endothelial adhesion [24].
In conclusion, in our population of Tanzanian children with SCA, the strongest predictor of lower SpO2 is increased mean cell hemoglobin, which is strongly correlated with a decreased copy number of the 3.7 alpha-thalassemia deletion. The degree of anemia, red cell count, and hemolytic markers do not independently correlate with SpO2, thus suggesting that effects of mean cell hemoglobin and the alpha-thalassemia genotype on SpO2 are mediated through effects on red cell rheology.
Supplementary Material
Table 1 of the supplementary material contains the results of investigating the associations between the exposure variables in order to assess for potential confounding or co-linearity before including variables in multivariable models or to consider possible mechanisms of action. Table 2 reports four different possible multivariable models of independent predictors of daytime SpO2 in SCA with either the alpha-thalassemia 3.7 deletion or mean cell haemoglobin plus G6PD genotype or HbF%.
Ethics Approval
Ethical permission was granted by the Muhimbili University of Health and Allied Sciences Ethics Committee (Ref: MU/RP/AECNoI.XII/77).
Authors' Contributions
S. E. Cox and F. J. Kirkham conceived and designed the study; S. E. Cox conducted the statistical analyses and wrote the paper. All authors contributed to critical evaluation and final drafting of the paper.
Conflicts of Interests
The authors declare no conflicts of interests.
Acknowledgments
This research was supported by Wellcome Trust, UK, project Grant 080025 (S. E. Cox), personal Fellowship 072064; (J. Makani), and Wellcome Trust Strategic Award 085438 (C. R. Newton, J. Makani, and S. E. Cox). The authors warmly thank the patients and staff of MNH and MUHAS, Dar-es-Salaam, Tanzania, who made this work possible. The authors also thank the staff in the Clinical Chemistry Unit at MNH Central Pathology Laboratory for clinical chemistry and transferrin saturation analyses and Josephine Mgaya and Harvest Mariki for the hematology analyses and sample archiving.
References
- 1.Makani J, Kirkham FJ, Komba A, et al. Risk factors for high cerebral blood flow velocity and death in Kenyan children with Sickle Cell Anaemia: role of haemoglobin oxygen saturation and febrile illness. British Journal of Haematology. 2009;145(4):529–532. doi: 10.1111/j.1365-2141.2009.07660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quinn CT, Variste J, Dowling MM. Haemoglobin oxygen saturation is a determinant of cerebral artery blood flow velocity in children with sickle cell anaemia. British Journal of Haematology. 2009;145(4):500–505. doi: 10.1111/j.1365-2141.2009.07652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quinn CT, Sargent JW. Daytime steady-state haemoglobin desaturation is a risk factor for overt stroke in children with sickle cell anaemia. British Journal of Haematology. 2008;140(3):336–339. doi: 10.1111/j.1365-2141.2007.06927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quinn CT, Ahmad N. Clinical correlates of steady-state oxyhaemoglobin desaturation in children who have sickle cell disease. British Journal of Haematology. 2005;131(1):129–134. doi: 10.1111/j.1365-2141.2005.05738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beutler E, Paniker NV, West C. The effect of 2,3-DPG on the sickling phenomenon. Blood. 1971;37(2):184–186. [PubMed] [Google Scholar]
- 6.Rackoff WR, Kunkel N, Silber JH, Asakura T, Ohene-Frempong K. Pulse oximetry and factors associated with hemoglobin oxygen desaturation in children with sickle cell disease. Blood. 1993;81(12):3422–3427. [PubMed] [Google Scholar]
- 7.Johnson MC, Kirkham FJ, Redline S, et al. Left ventricular hypertrophy and diastolic dysfunction in children with sickle cell disease are related to asleep and waking oxygen desaturation. Blood. 2010;116(1):16–21. doi: 10.1182/blood-2009-06-227447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Embury SH, Dozy AM, Miller J. Concurrent sickle-cell anemia and α-thalassemia. Effect on severity of anemia. The New England Journal of Medicine. 1982;306(5):270–274. doi: 10.1056/NEJM198202043060504. [DOI] [PubMed] [Google Scholar]
- 9.Nouraie M, Reading NS, Campbell A, et al. Association of G6PD202A,376G with lower haemoglobin concentration but not increased haemolysis in patients with sickle cell anaemia. British Journal of Haematology. 2010;150(2):218–225. doi: 10.1111/j.1365-2141.2010.08215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stevens MCG, Maude GH, Beckford M. α thalassemia and the hematology of homozygous sickle cell disease in childhood. Blood. 1986;67(2):411–414. [PubMed] [Google Scholar]
- 11.Embury SH, Clark MR, Monroy G, Mohandas N. Concurrent sickle cell anemia and α-thalassemia. Effect of pathological properties of sickle erythrocytes. Journal of Clinical Investigation. 1984;73(1):116–123. doi: 10.1172/JCI111181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kulozik AE, Kar BC, Serjeant GR, Serjeant BE, Weatherall DJ. The molecular basis of α thalassemia in India. Its interaction with the sickle cell gene. Blood. 1988;71(2):467–472. [PubMed] [Google Scholar]
- 13.Serjeant BE, Mason KP, Kenny MW. Effect of alpha thalassaemia on the rheology of homozygous sickle cell disease. British Journal of Haematology. 1983;55(3):479–486. doi: 10.1111/j.1365-2141.1983.tb02163.x. [DOI] [PubMed] [Google Scholar]
- 14.Castro O, Poillon WN, Finke H, Massac L. Improvement of sickle cell anemia by iron-limited erythropoiesis. American Journal of Hematology. 1994;47(2):74–81. doi: 10.1002/ajh.2830470203. [DOI] [PubMed] [Google Scholar]
- 15.Makani J, Cox SE, Soka D, et al. Mortality in sickle cell anemia in africa: a prospective cohort study in Tanzania. PLoS ONE. 2011;6(2) doi: 10.1371/journal.pone.0014699.e14699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams TN, Mwangi TW, Wambua S, et al. Negative epistasis between the malaria-protective effects of α+-thalassemia and the sickle cell trait. Nature Genetics. 2005;37(11):1253–1257. doi: 10.1038/ng1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox SE, Doherty CP, Atkinson SH, et al. Haptoglobin genotype, anaemia and malaria in Gambian children. Tropical Medicine & International Health. 2007;13(1):76–82. doi: 10.1111/j.1365-3156.2007.01976.x. [DOI] [PubMed] [Google Scholar]
- 18.Vuong QH. Likelihood ratio tests for model selection and non-nested hypotheses. Econometrica. 1989;57(2):307–333. [Google Scholar]
- 19.Town M, Bautista JM, Mason PJ, Luzzatto L. Both mutations in G6PD A- are necessary to produce the G6PD deficient phenotype. Human Molecular Genetics. 1992;1(3):171–174. doi: 10.1093/hmg/1.3.171. [DOI] [PubMed] [Google Scholar]
- 20.Zimmermann MB, Fucharoen S, Winichagoon P, et al. Iron metabolism in heterozygotes for hemoglobin E (HbE), α-thalassemia 1, or β-thalassemia and in compound heterozygotes for HbE/β-thalassemia. American Journal of Clinical Nutrition. 2008;88(4):1026–1031. doi: 10.1093/ajcn/88.4.1026. [DOI] [PubMed] [Google Scholar]
- 21.Homi J, Levee L, Higgs D, Thomas P, Serjeant G. Pulse oximetry in a cohort study of sickle cell disease. Clinical and Laboratory Haematology. 1997;19(1):17–22. doi: 10.1046/j.1365-2257.1997.00215.x. [DOI] [PubMed] [Google Scholar]
- 22.Cox SE, L'Esperance V, Makani J, et al. Sickle cell anemia: iron availability and nocturnal oximetry. Journal of Clinical Sleep Medicine. 2012;8(5):541–545. doi: 10.5664/jcsm.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirkham FJ, Hewes DKM, Prengler M, Wade A, Lane R, Evans JPM. Nocturnal hypoxaemia and central-nervous-system events in sickle-cell disease. The Lancet. 2001;357(9269):1656–1659. doi: 10.1016/s0140-6736(00)04821-2. [DOI] [PubMed] [Google Scholar]
- 24.Bernaudin F, Verlhac S, Chevret S, et al. G6PD deficiency, absence of α-thalassemia, and hemolytic rate at baseline are significant independent risk factors for abnormally high cerebral velocities in patients with sickle cell anemia. Blood. 2008;112(10):4314–4317. doi: 10.1182/blood-2008-03-143891. [DOI] [PubMed] [Google Scholar]
- 25.Belisário AR, Rodrigues CV, Martins ML, Silva CM, Viana MB. Coinheritance of α-thalassemia decreases the risk of cerebrovascular disease in a cohort of children with sickle cell anemia. Hemoglobin. 2010;34(6):516–529. doi: 10.3109/03630269.2010.526003. [DOI] [PubMed] [Google Scholar]
- 26.Flanagan JM, Frohlich DM, Howard TA, et al. Genetic predictors for stroke in children with sickle cell anemia. Blood. 2011;117(24):6681–6684. doi: 10.1182/blood-2011-01-332205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Setty BNY, Stuart MJ, Dampier C, Brodecki D, Allen JL. Hypoxaemia in sickle cell disease: biomarker modulation and relevance to pathophysiology. The Lancet. 2003;362(9394):1450–1455. doi: 10.1016/S0140-6736(03)14689-2. [DOI] [PubMed] [Google Scholar]
- 28.Niu X, Nouraie M, Campbell A, et al. Angiogenic and inflammatory markers of cardiopulmonary changes in children and adolescents with sickle cell disease. PLoS ONE. 2009;4(11) doi: 10.1371/journal.pone.0007956.e7956 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table 1 of the supplementary material contains the results of investigating the associations between the exposure variables in order to assess for potential confounding or co-linearity before including variables in multivariable models or to consider possible mechanisms of action. Table 2 reports four different possible multivariable models of independent predictors of daytime SpO2 in SCA with either the alpha-thalassemia 3.7 deletion or mean cell haemoglobin plus G6PD genotype or HbF%.