

Abstract
The Candida Gene Order Browser (CGOB) was developed as a tool to visualize and analyze synteny relationships in multiple Candida species, and to provide an accurate, manually curated set of orthologous Candida genes for evolutionary analyses. Here, we describe major improvements to CGOB. The underlying structure of the database has been changed significantly. Genomic features are now based directly on genome annotations rather than on protein sequences, which allows non-protein features such as centromere locations in Candida albicans and tRNA genes in all species to be included. The data set has been expanded to 13 species, including genomes of pathogens (C. albicans, C. parapsilosis, C. tropicalis, and C. orthopsilosis), and those of xylose-degrading species with important biotechnological applications (C. tenuis, Scheffersomyces stipitis, and Spathaspora passalidarum). Updated annotations of C. parapsilosis, C. dubliniensis, and Debaryomyces hansenii have been incorporated. We discovered more than 1,500 previously unannotated genes among the 13 genomes, ranging in size from 29 to 3,850 amino acids. Poorly conserved and rapidly evolving genes were also identified. Re-analysis of the mating type loci of the xylose degraders suggests that C. tenuis is heterothallic, whereas both Spa. passalidarum and S. stipitis are homothallic. As well as hosting the browser, the CGOB website (http://cgob.ucd.ie) gives direct access to all the underlying genome annotations, sequences, and curated orthology data.
Keywords: CGOB, comparative genomics, gene order, synteny, yeast, pathogens, Candida, xylose
Introduction
The Candida gene order browser (CGOB) was originally adapted from the yeast gene order browser (YGOB), a tool that facilitates visual comparisons and computational analysis of synteny relationships in yeasts from the Saccharomyces clade (Byrne and Wolfe 2005, 2006). The first version of CGOB (Fitzpatrick et al. 2010) contained 10 genomes from 9 Candida species. Like YGOB, CGOB consists of a database, a browser, and a software engine for whole-genome evolutionary analyses. The database consists of orthologous gene assignments (pillars) that have been extensively manually curated, based on genomic context (local synteny) as well as sequence similarity, providing a “gold-standard” set of orthologs for evolutionary analysis. The browser is an interactive tool for visualizing gene order relationships in any section of the genome. It displays a matrix (fig. 1) where each column shows a set of orthologous genes (a pillar) and each continuous horizontal element (a track) represents a segment of chromosome. The software engine allows the whole database to be searched for particular synteny-related patterns, such as sites where tRNA genes coincide with interspecies rearrangements (Gordon et al. 2009), without users having to manually browse through the whole genome.
Fig. 1.

CGOB display and bioinformatics tools. (A) CGOB screenshot. Each track represents a chromosomal region from one species. Tracks are labeled on the right. Each box presents a feature (with gene name and chromosome) and each color a chromosome (with different color palettes used for each genome). A change in track color indicates a break in synteny. Arrows under boxes denote relative orientation. White boxes represent tRNA features (amino acid and anticodon are displayed) and black boxes centromeres. Solid wide black connectors link adjacent genes and are continued in gray if there is a gap in that genome. Clicking these will output the intergenic sequence between the two features. When these connectors are colored orange it denotes an inversion (visible between the first two pillars in Candida parapsilosis and C. orthopsilosis). Double and single small bars connect nonadjacent genes <5 and <20 genes apart, respectively (not shown). The control console at the bottom of the interface lets users input a gene name, select the window size and the version to display, turn genomes on and off, and turn RNA features on and off. The display is centered on SEC34 from C. albicans, highlighted with a yellow box on the top track. (B) Information from CGD for the C. albicans SEC34 gene (launched by the “i” button on gene box in [A]). The equivalent button on the C. parapsilosis track connects to the same database, and on the Saccharomyces cerevisiae track launches the Yeast Genome Database (SGD). (C) BLASTP results for Sec34 versus all CGOB proteins (launched by “b” button on gene box). Pink shading indicates hits to genes that are in the same pillar as the gene used as a BLAST query. (D) Amino acid sequences for genes in the pillar (launched by “aa” button above the tracks). (E) Nucleotide sequences for genes in the pillar (launched by “nt” button above the tracks). (F) Intergenic sequence between cdub_CGOB_00003 and the adjacent centromere (launched by clicking on connector bar between them). (G) MUSCLE multiple sequence alignment of the proteins in the Sec34 pillar (launched from the “msa” button below the tracks). (H) Pairwise yn00 output for all genes in the SEC34 pillar (launched from “rates” below the tracks). (I) PhyML tree of genes in the SEC34 pillar (launched from the “tree” button below the tracks).
CGOB was designed to facilitate comparative analysis within the “CTG” clade of yeast species that translate the codon CTG as serine instead of the canonical leucine (Santos et al. 1993; Massey et al. 2003; Fitzpatrick et al. 2006). This clade includes important fungal pathogens such as Candida albicans, C. dubliniensis, C. tropicalis, and C. parapsilosis, which are diploid and either asexual or parasexual (Hull et al. 2000; Magee BB and Magee PT 2000; Bennett and Johnson 2003; Butler 2007, 2010; Butler et al. 2009). Related haploid and sexual species are also included, such as Clavispora (previously Candida) lusitaniae, Meyerozyma (previously Pichia) guilliermondii, Scheffersomyces (previously Pichia) stipitis, and Debaryomyces hansenii (also known as C. famata) (Fabre et al. 2005; Jeffries et al. 2007; Reedy et al. 2009). The diploid species Lodderomyces elongisporus is more closely related to C. albicans than to the haploid species, although there are some reports that it may have a sexual cycle (van der Walt 1966; Lockhart et al. 2008). CGOB was previously used to identify clusters of genes associated with metabolic pathways in Candida species (Fitzpatrick et al. 2010). Recently, we used CGOB to help annotate the genome of C. orthopsilosis, a species closely related to C. parapsilosis (Riccombeni et al. 2012), which has now been added to the browser database. We have also included the genomes of the xylose-fermenting yeasts C. tenuis and Spathaspora passalidarum (Wohlbach et al. 2011).
As well as increasing the number of species included, we also describe significant and fundamental changes that have been made to the structure and function of CGOB. All sequence features are now based on genomic (nucleotide) coordinates, and include tRNA genes as well as protein-coding sequences. We have upgraded the browser interface to add significant new functionality. More importantly, we have substantially improved the annotation of the Candida genomes, by systematic use of homology and synteny searches to identify 1,525 previously unrecognized protein-coding genes. We also removed more than 1,000 predicted introns from S. stipitis gene models. CGOB is a powerful tool for the analysis of gene and genome evolution in the Candida clade, and is the only tool that facilitates comparative genomic analysis of human fungal pathogens and species with species important for biofuel production from wood waste.
Results and Discussion
New Genomes and Interface
The original CGOB and YGOB databases did not contain any DNA sequence information or gene co-ordinates. They consisted of a static file of protein sequences from each species, a static list of the order of the corresponding genes along chromosomes in each species, and an editable (dynamic) orthology database containing information about which proteins were in each pillar. This organizational structure made it difficult for curators to modify the data, even in cases where there were obvious errors in the annotation of a genome. We therefore switched both CGOB and YGOB to a framework where features (genes or other elements) are defined by their genomic coordinates. Each genome sequence is now loaded into the browsers at the DNA level, and all sequence information for a feature is generated dynamically from the corresponding chromosome sequence. Gene order is also calculated dynamically. Genome annotations can now be modified easily—features annotated by the original authors of a genome sequence can be switched OFF by curators if necessary (or turned back ON), new features can be added, and the coordinates of any feature can be modified (fig. 1).
The original version of CGOB contained 10 genomes from species from the Candida (CTG) clade (including two isolates of C. albicans), plus Saccharomyces cerevisiae as a reference. We have now included updated annotations of C. albicans (Bruno et al. 2010; Tuch et al. 2010), C. dubliniensis (Jackson et al. 2009), and D. hansenii (DEHA2 gene models) and made significant changes to the S. stipitis annotation (discussed later). We have also incorporated an annotation of C. parapsilosis based on RNA-seq analysis (Guida et al. 2011) and the recently sequenced genome of C. orthopsilosis (Riccombeni et al. 2012). Finally, we include the genomes of C. tenuis and Spa. passalidarum (Wohlbach et al. 2011). Together with S. stipitis, C. tenuis and Spa. passalidarum are among the few species that can ferment and assimilate the pentose sugar xylose, a major component of plant cell walls (Wohlbach et al. 2011). Xylose fermentation is required for the efficient use of plant material for biofuel production, and CGOB is the only tool that facilitates comparisons between pathogenic species and those that have important applications for biotechnology.
To improve the visualization of larger numbers of genomes, we implemented the more streamlined browser interface shown in figure 1. To save vertical screen space and allow more genomes to be displayed, we moved genome names to the right edge of the screen and compressed the vertical space required for each genome by over a quarter. This flatter display creates space for extra genomes. We flattened the control panel at the bottom of the screen by making a drop-down list of species names that is used to select which genomes are displayed. This change to the control panel also allows us to define a subset of species that will be used as the default group for display; chromosomal tracks from the other species will only be shown if a user chooses to activate them. We use this approach in YGOB, where the default display shows 26 tracks and another 7 are hidden by default, but not in CGOB where all 14 tracks from the current database can fit on most computer screens.
Many of the bioinformatics tools in the original CGOB browser interface have been updated (fig. 1). Information (“i” buttons) in C. albicans and C. parapsilosis launch the Candida genome database (CGD) (Costanzo et al. 2006), and for the Sac. cerevisiae track connects to Saccharomyces genome database (SGD) (Cherry et al. 2012) (fig. 1B). A BLASTP search versus a database of all proteins in CGOB can be launched by clicking the “b” button on any gene’s icon, but the query amino acid sequence is generated dynamically (fig. 1C). Users can now also rerun the BLASTP search with the SEG filter (which removes areas of low compositional complexity) off. Checkboxes in the BLASTP search results page allow users to select multiple genes from the results list, and then to retrieve their sequences (FASTA amino acid or nucleotide sequences), generate a multiple sequence alignment, draw a phylogenetic tree, or calculate levels of synonymous and nonsynonymous sequence divergence (using the same tools that can be launched from the CGOB interface, described later). This feature enables a user to compare or test genes that appear in BLAST results without having to manually extract their sequences, for example, to make a phylogenetic tree of a gene family.
Links at the top of each pillar allow retrieval of the amino acid and nucleotide sequences of all genes in that pillar, extracted dynamically (fig. 1D and E). Users can now also output the intergenic DNA sequence between features by clicking on the black or gray connectors between them (fig. 1F). Links at the bottom of each pillar allow users to generate a multiple sequence alignment using MUSCLE (Edgar 2004) (Fig. 1G), to calculate evolutionary sequence divergence between all pairs of sequences in the pillar using yn00 (Yang and Nielsen 2000) (fig. 1H) or to construct a PhyML phylogenetic tree (Guindon et al. 2009) (fig. 1I). The “+” button in the bottom left hand corner of every CGOB page (fig. 1A) will output the same information seen on screen in a tab delimited text format that is easier to save and manipulate.
CGOB recognizes both systematic names and synonyms for C. albicans genes (taken from CGD; Costanzo et al. 2006). For C. albicans WO-1, C. tropicalis, L. elongisporus, M. guilliermondii, and Cla. lusitaniae the systematic names generated in the sequencing project (Butler et al. 2009) and used in CGD (Inglis et al. 2012) are recognized. The most up-to-date annotations for C. dubliniensis (Jackson et al. 2009), D. hansenii (DEHA2), and C. orthopsilosis (Riccombeni et al. 2012) are also included. For C. parapsilosis, CGOB recognizes gene identifiers from the most recent annotation (cpar2; Guida et al. 2011), as well as from earlier annotations (CPAG; [Jackson et al. 2009] and cpar [Rossignol et al. 2009]).
New Noncoding Features
The original CGOB browser contained only protein-coding genes. We have now annotated transfer RNA genes across all the genomes, using tRNAscan-SE (Lowe and Eddy 1997). tRNA features are displayed on screen as white boxes (e.g., the leucine tRNA pillar in fig. 1A). This enables identification of association of tRNA genes with genomic breakpoints, as was hypothesized to have occurred during the acquisition of a proline racemase gene in the C. parapsilosis lineage by horizontal gene transfer (Fitzpatrick et al. 2008). tRNA gene locations are generally well conserved across species in the CTG clade. For example, we were able to identify probable orthologs in D. hansenii of 48% of the 126 tRNA genes in C. albicans, based on conserved synteny with the nearby protein-coding genes.
We also added annotations of ribosomal rRNA genes to the CGOB data set, based either on annotations by the original authors or on BLASTN searches with the C. albicans 18S, 5.8S, 25S, and 5S genes. The location of the rDNA array is conserved among C. albicans, C. dubliniensis, and C. tropicalis, and (at a different site) among C. parapsilosis, C. orthopsilosis, and L. elongisporus (Proux 2012). In other CTG clade species, rDNA arrays are present at species-specific sites (M. guilliermondii, S. stipitis, Spa. passalidarum, and C. tenuis) or near telomeres (Cla. lusitaniae). We cannot find the locus in D. hansenii. It is clear that the rDNA array has moved around the genome during CTG clade evolution, but unlike the situation in the Saccharomyces clade (Proux-Wera et al. 2013) we were unable to identify an ancestral rDNA location in the CTG clade or retrace the history of rDNA movement away from this site.
Unlike species in the Saccharomyces clade that have “point” centromeres (conserved sequences recognized by specific proteins) and are therefore relatively easily identified by sequence analysis, the Candida species have “regional” centromeres, which are longer and more poorly conserved (Ishii 2009). Centromeres have been experimentally verified only in C. albicans and C. dubliniensis (Sanyal et al. 2004; Padmanabhan et al. 2008) and these are now included in CGOB. Centromere locations have been predicted in Cla. lusitaniae and S. stipitis (Lynch et al. 2010) but as these have not yet been experimentally verified they have not been included in the browser. As the non-coding and RNA features are not of interest to all researchers or to all questions they can all be turned off (and back on again) in the control panel.
Gene Discovery
For many of the Candida genomes (apart from C. albicans) their initial gene annotation was performed automatically, with some support from synteny analysis (Butler et al. 2009). We therefore suspected that like the Saccharomyces species (OhEigeartaigh et al. 2011), many protein-coding genes, particularly those with short open reading frames, were likely to have been missed. Many may be important for function. In addition, analysis of gene loss and gain during evolution requires accurate information. We therefore used the SearchDOGS program originally developed for YGOB (OhEigeartaigh et al. 2011) to search for missing orthologs. This program uses BLAST searches combined with synteny information to detect unannotated genes, for example, by re-examining the DNA sequence of a region that is annotated as intergenic in one species, but which is in the same genomic context (the same flanking genes on each side) as an annotated gene in one or more other species (fig. 2). We ran two iterations of SearchDOGS, followed by manual curation of all predicted open reading frames. This analysis identified 1,525 new genes from all 13 genomes (table 1 and supplementary table S1, Supplementary Material online). The new features have been entered into the CGOB database and identified with the prefix “CGOB,” a short species identifier and a number (e.g., lelo_CGOB_00001 from L. elongisporus, or wo-1_CGOB_00025 from C. albicans WO-1).
Fig. 2.

Gene finding in Candida species. Examples of three small genes (orthologs of LSM5, TOM5, and PMP1 from Saccharomyces cerevisiae) identified in multiple Candida species. The upper panels show screen shots from CGOB. The genes highlighted in red were identified by SearchDOGS. The lower panels show multiple alignments of the predicted proteins sequences carried out using SeaView (Gouy et al. 2010). The species are listed in the same order as in the top panels (S.c. = Saccharomyces cerevisiae).
Table 1.
Numbers of Genes Added, Modified, and Removed in Each Species.
| Species | Updated Number of Genes | New Genes Added |
Existing Genes Modified |
Genes Removed | ||
|---|---|---|---|---|---|---|
| Total Genes Added | Intron-Containing Genes Added | Total Genes Modified | Intron Modified | |||
| Candida albicans SC5314 | 6,207 | 13 | 0 | 2 | 0 | 7 |
| C. albicans WO-1 | 6,268 | 114 | 28 | 28 | 26 | 9 |
| C. dubliniensis | 6,070 | 88 | 2 | 13 | 5 | 0 |
| C. tropicalis | 6,445 | 192 | 96 | 32 | 29 | 8 |
| C. parapsilosis | 5,843 | 8 | 2 | 3 | 0 | 0 |
| C. orthopsilosis | 5,707 | 7 | 1 | 14 | 1 | 0 |
| L. elongisporus | 5,931 | 130 | 57 | 17 | 17 | 2 |
| Debaryomyces hansenii | 6,411 | 12 | 3 | 8 | 7 | 1 |
| S. stipitis | 6,026 | 211 | 11 | 31 | 27a | 0 |
| C. tenuis | 5,800 | 267 | 9 | 7 | 5 | 7 |
| Spathaspora passalidarum | 6,071 | 93 | 6 | 31 | 7 | 6 |
| M. guilliermondii | 6,135 | 213 | 91 | 39 | 32 | 3 |
| Clavispora lusitaniae | 6,116 | 177 | 75 | 39 | 27 | 4 |
| Total | 1,525 | 381 | 264 | 183 | 47 | |
aNumber excludes ∼1,200 in-frame “introns” that were removed from the S. stipitis annotation.
The 1,525 newly annotated genes (supplementary table S1, Supplementary Material online) encode proteins ranging in size from 29 amino acids (ctro_CGOB_00073) to 3,850 amino acids (ctro_CGOB_00180). Although most are short (43% of them are <100 amino acids), some are surprisingly long (e.g., 53 ORFs of >1,000 amino acids were identified). We have identified new genes even in manually curated genomes like C. albicans. Thirteen new protein-coding genes were predicted in C. albicans SC5314 and many more (114) in C. albicans WO-1. These include Lsm5 (fig. 2), a component of two heteroheptameric complexes in Sac. cerevisiae that are involved in mRNA degradation and splicing (He and Parker 2000). All the other components of the Lsm complexes were correctly annotated in C. albicans and most of the other species. However, Lsm5 was missed in 7 genomes, probably because it is short (77–101 amino acids) and not called in C. albicans, which was used for annotating most of the other genomes. Tom5, a component required for import of proteins into the mitochondria, was also missed from both C. albicans species and 8 other genomes. It was called correctly only in C. parapsilosis (based on transcriptional data [Guida et al. 2011]), C. orthopsilosis and D. hansenii. Tom5 is very short (47–50 amino acids), but very highly conserved (fig. 2).
Figure 2 shows a further example of a very small gene, orthologous to CPAR2_602480 in C. parapsilosis, which was added to 11 genomes including C. albicans. The protein is 38 amino acids long, highly conserved, and homologous to Pmp1 from Sac. cerevisiae, where it functions as a regulatory subunit of the yeast plasma membrane H(+)-ATPase (Mousson et al. 2002). CPAR2_602480 was annotated in C. parapsilosis using transcriptional data (Guida et al. 2011) and extended to other species using synteny information from CGOB.
Many genes were not originally annotated because introns were not correctly assigned. We identified 381 novel genes containing introns (by bioinformatic analysis) and modified or added introns to 183 additional genes (table 1). A small number of unconserved open reading frames that overlapped with alternative conserved translations were removed.
Before the SearchDOGS iterations, the best-annotated genomes were those of D. hansenii (12 additional genes predicted), and C. parapsilosis and C. orthopsilosis (8 and 7 genes predicted respectively). The D. hansenii genome was sequenced and annotated by the Génolevures consortium (Dujon et al. 2004; original gene identifiers beginning with DEHA0). Significant improvements in the annotation were later reported (gene identifiers beginning with DEHA2). The high quality of the current D. hansenii annotation is a reflection of the substantial manual curation and the expertise of the consortium in annotating genomes of Saccharomycotina species, through the application of a web-based collaborative system (Magus; Martin, Sherman, et al. 2011). The current C. parapsilosis genome annotation is based on transcriptional data, which was used to correct several hundred gene models and to identify 300 novel protein-coding genes with respect to the original automated gene calling (Guida et al. 2011). The C. orthopsilosis genome annotation is based on similarity and synteny data from CGOB (Riccombeni et al. 2012) and is an excellent illustration of the power of this approach.
The annotations of the xylose-degrading yeasts, S. stipitis, Spa. passalidarum, and C. tenuis, all sequenced and annotated by the Joint Genome Institute, presented the most problems. We identified between 93 and 267 new protein-coding genes in each species. In addition, while loading the genome data for S. stipitis (Jeffries et al. 2007), we noticed that a large number of gene models (1,611) included introns, with multiple introns (totaling 2,567) in single genes in several cases. This number of predicted introns is unusual, as species in the Saccharomycotina have very few (e.g., 415 in 381 genes in C. albicans [Mitrovich et al. 2007] and 422 in 387 genes in C. parapsilosis [Guida et al. 2011]). None of the predicted introns in S. stipitis have experimental support. On closer investigation, we noticed that the many of the predicted introns were in-frame, and when included in the translation often generated a predicted protein with greater similarity to orthologs in other species than the translation of the spliced gene model (fig. 3). We therefore carried out a systematic search for annotated in-frame introns in S. stipitis that are not conserved in other species. We removed 1,231 introns (not listed in table 1) that we believe are incorrectly annotated. Because this resulted in a major change in the genome annotation, we retained the original S. stipitis gene identifiers rather than introducing new ones for every modified gene. The genomes of C. tenuis and Spa. passalidarum also contain high numbers of genes with predicted introns (974 and 994 respectively), which we suspect result from applying a gene finding model developed for filamentous fungi rather than from species in the Saccharomycotina (Jeffries et al. 2007; Wohlbach et al. 2011). However, we have not systematically investigated nor removed introns in these two species.
Fig. 3.

Invalid intron annotation in Scheffersomyces stipitis. Alignment of a region of Sla1 from Candida albicans, Debaryomyces hansenii, and two alternative gene models for S. stipitis. The red box highlights a region that was originally annotated as an in-frame intron (A), but where a revised model that ignores the intron increases sequence similarity (B). We also removed four other predicted in-frame introns in PICST_82711.
Finding Poorly Conserved Orthologs and “Hidden Homology”
In assigning genes to pillars, we first use BLASTP searches and reciprocal best hits, with a conservative cutoff. However, poorly conserved orthologs can be difficult to identify based solely on protein similarity, and remain as “singleton” pillars instead of being incorporated into other pillars. To tackle this problem, we developed an algorithm called Synteno-BLAST, which interprets weak BLAST scores in combination with synteny information. Synteno-BLAST systematically searches for putative orthologs by looking for singleton pillars that can be merged with another pillar on the basis of a BLASTP (E < 1e−5) hit to at least one gene in the other pillar, provided that the assignment is also supported by the syntenic context.
CGOB’s Synteno-BLAST based approach reveals “hidden homology” between genes that cannot be found by BLASTP alone, and shows the importance of establishing orthology in the context of synteny. As much editing as possible is automated. However, singleton genes with only very weak BLASTP hits (up to E = 10) to a nearby pillar (and not necessarily having hits to all the genes in that pillar), that were not assigned to that pillar automatically, can be placed in it manually with sufficient syntenic evidence. This manual curation of orthology data provides one of the main strengths of the CGOB database. For example, 604 genes from D. hansenii, 580 from C. tenuis, and 334 from Spa. passalidarum were added to pillars following analysis of synteny. One example is shown in figure 4. The gene EED1, which is required for filamentation in C. albicans, was originally described as unique to this species (Martin, Moran, et al. 2011). Comparing C. albicans EED1 with the C. dubliniensis ORF (CD36_34980) shows the two proteins have significant regions of similarity, with some apparent deletions in EED1 (fig. 4B). One C-terminal region missing from the C. albicans protein is predicted to encode a SANT domain, a DNA binding domain shared by several chromatin remodeling machines (Aasland et al. 1996). The SANT domain is present in genes at the equivalent syntenic position in most of the other CTG species (fig. 4A, C). The C. albicans and C. dubliniensis proteins are significantly longer than the predicted proteins from the other species. It is therefore likely that EED1 was present in the common ancestor of the CTG clade, but is rapidly evolving, and has undergone some particularly significant changes in the C. albicans lineage (e.g., loss of the SANT domain). As Eed1 is a repressor of the hyphal-to-yeast transition in C. albicans, the divergence in gene sequence may be associated with the ability of this species to undergo true hyphal growth, a phenotype that is almost unique in the CTG clade.
Fig. 4.

EED1 orthologs in Candida species. (A) Screen shot from CGOB, showing synteny around the EED1 gene from C. albicans strain SC5314. The species are in the same order as shown in figure 1. In C. albicans strain WO-1, there is a short gap in the genome sequence internal to EED1 with two frameshifting indels, making it a possible pseudogene (indicated with a “p”). (B) Alignment of putative EED1 orthologs from C. albicans SC5314 and C. dubliniensis, generated using T-coffee (Notredame et al. 2000). The boxed region highlights the SANT domain which is missing from the C. albicans protein. (C) Alignment of SANT domains from Eed1 proteins excluding C. albicans.
Identification of MTL Locus
The term “Candida” means imperfect or asexual species. Although a parasexual cycle has been identified in C. albicans, C. dubliniensis, and C. tropicalis, resulting from mating of haploid and diploid cells, these species have never been shown to undergo meiosis (Pujol et al. 2004; Bennett and Johnson 2005; Porman et al. 2011; Hickman et al. 2013). No sexual cycle has been identified in some of the other diploid pathogens, such as C. parapsilosis and C. orthopsilosis (Logue et al. 2005; Sai et al. 2011). However, not all CTG clade species are asexual. Some, such as D. hansenii and S. stipitis, have haplontic life cycles—they undergo conjugation and almost immediately go through meiosis (van del Walt et al. 1977; Melake et al. 1996). Mating and meiosis of haploid isolates of Cla. lusitaniae and M. guilliermondii has also been observed (Wickerham and Burton 1954; Reedy et al. 2009).
Mating type is determined by the genes at the MTL, or mating type-like locus. Many Candida species (including diploid asexual species) have heterothallic MTL idiomorphs, and mating occurs between cells of opposite mating type, MTLa and MTLα (Butler 2007, 2010). Cell type is determined by the mating genes, a1 and a2 at the MTLa locus, and α1 and α2 at the MTLα locus. Alleles of other genes (PIK, PAP, and OBP) within the idiomorphs have no apparent roles in mating, but may be involved in biofilm development (Srikantha et al. 2012). Some CTG clade species (such as D. hansenii and S. stipitis) are homothallic, with mating occurring between genetically identical cells. There are genes from both MTLa and MTLα (a1, a2, and α1) at a single locus in these species (Fabre et al. 2005; Butler 2010).
Examination of the MTL loci of C. tenuis and Spa. passalidarum suggests that the former is heterothallic (with MTLα in the sequenced isolate) and the latter is homothallic (fig. 5A). The designation was not immediately obvious because the genes MTLα1 and MTLα2 in C. tenuis and MTLa1 in Spa. passalidarum were not present in the original genome annotations. We also identified MTLα1 in S. stipitis. The structure of the MTLα locus in C. tenuis is similar to the equivalent idiomorph in C. albicans (fig. 5A). As previously reported, the S. stipitis idiomorph contains a1, a2, and α1 genes at the same location, similar to D. hansenii (Butler 2010). Spathaspora passalidarum also has a homothallic structure, but has only one MTLa gene (a1) and both MTLα genes (α1 and α2). Like S. stipitis, the homothallic Spa. passalidarum MTL appears to have arisen from acquisition of information from an MTLa idiomorph integrated at an MTLα locus (Butler 2010).
Fig. 5.
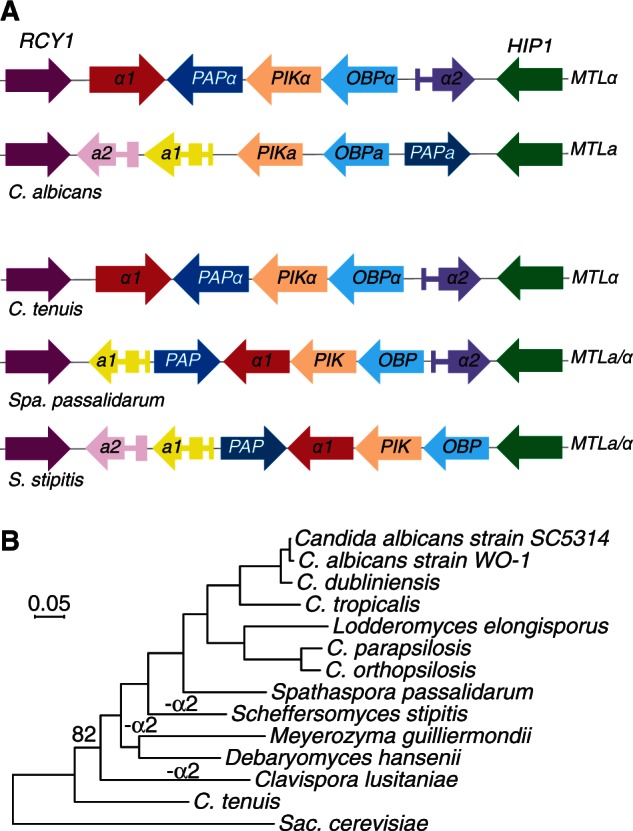
Identification of MTL loci. (A) The putative MTL loci from Candida tenuis, Scheffersomyces stipitis, and Spa. passalidarum are shown in comparison with the MTLa and MTLα idiomorphs from C. albicans. Introns are indicated with narrow rectangles. The structure of the C. tenuis idiomorph closely resembles MTLα from C. albicans. Both S. stipitis and Spa. passalidarum have homothallic-like structures, with mating genes from both MTLa and MTLα (B) Phylogenetic relationship of species in the CGOB database, rooted using Saccharomyces cerevisiae. The tree was constructed using PhyML. A bootstrap value of 82% is shown for one branch; all other branches had 100% support from 100 replicates. Various supertree methods gave either this topology, or an alternative one in which the positions of C. tenuis and Clavispora lusitaniae were swapped. Species that have lost MTLα2 are marked.
Earlier genomic analysis suggested that α2 was lost from the sexual species (D. hansenii, S. stipitis, M. guilliermondii, and Cla. lusitaniae) in the CTG clade, and this was correlated with differences in sporulation between Candida and Saccharomyces species (Butler et al. 2009; Butler 2010). However, both C. tenuis and Spa. passalidarum have retained α2. Examination of the evolutionary relationship of the species (fig. 5B) shows that it is no longer clear that the loss of α2 occurred on a single ancestral branch, and may indeed have occurred independently in several lineages.
Conclusion
We report here the development of new nucleotide-based data structures for CGOB and YGOB, and illustrate the application of syntenic information for gene discovery. We have greatly improved the annotation of most of the Candida species, though some errors still remain. In the entire CGOB database (78,505 protein-coding genes excluding Sac. cerevisiae), 824 genes do not begin with an ATG, and 1,281 do not have an annotated stop codon. These are indicated by warning messages in the CGOB pillars. The majority (>60%) of the problematic genes are from the genomes of S. stipitis, C. tenuis, and Spa. passalidarum. However, the new annotations provide an important tool for the research community; for example, browser pillar information has been used recently to help characterize Gene Ontology annotation in Candida species (Inglis et al. 2013) and to study xylose pathway evolution (Riccombeni et al. 2012).
Materials and Methods
Construction of Nucleotide-Based Databases
Both YGOB (Byrne and Wolfe 2005, 2006) and CGOB were converted to nucleotide-based frameworks; only CGOB is described here, but similar changes have been implemented in both browsers. This was accomplished by replacing the static gene order lists and protein sequence files for each species with information calculated dynamically from genome sequences. For each species, we store a local version of the genome annotation and sequence, derived initially from NCBI, EMBL, SGD (Cherry et al. 2012), or CGD (Costanzo et al. 2006) annotations. Each genome annotation file stores the following information for each gene or other genomic feature:
– Name: the unique name used to identify the feature.
– Orientation: 0 or 1 for Crick or Watson strand, respectively.
– Start co-ordinate: the lowest-numbered coordinate in the range of the feature.
– Stop coordinate: the highest-numbered coordinate in the range of the feature.
– On/Off: determines whether the feature is displayed in CGOB/YGOB.
– Chromosome/Contig/Scaffold number: identifying number of source sequence.
– Short Name: the shorter name that will appear in the feature’s on-screen box.
– Coordinates: complete coordinates of the feature with intron/exon annotation and complement tag if appropriate.
– Notes: tags imported from GenBank, CGD/SGD descriptions, or added by CGOB/YGOB curators.
Nonprotein-coding features such as tRNA and rRNA genes and centromeres are annotated in an identical but parallel way to protein-coding features, with each type of feature having its own annotation file (with the above format). This parallel approach facilitates turning nonprotein-coding features on/off and the different on-screen and backend treatment of them, but most importantly stops the mixing of different feature types in the same pillar. It also allows for the easy later addition of new feature types to a genome that was initially loaded without them.
The genome annotation files for each species are associated with a particular FASTA DNA sequence file containing the corresponding genome sequence. The On/Off function allows us to choose to ignore certain features, such as “dubious” genes that were present in the annotation we imported but which we do not wish to display in CGOB/YGOB, without losing trace of them (e.g., they are listed if the intergenic region they are in is examined).
The annotation files and sequence file for each genome are then used as the source from which all other sequence information in the browsers is generated, including the amino acid and nucleotide sequences of individual genes, the DNA sequences of intergenic regions, and the internal BLAST databases. The order in which the features are displayed on-screen is determined by the order of their Start coordinates.
An editor feature allows CGOB/YGOB curators to modify the coordinates of features, or to create new features such as previously unannotated genes. Thus, we have the ability to modify the annotations we imported from other databases, but not to edit the genome sequence itself.
The structure of the database of homologous gene assignments across species (pillars) is unchanged from the original versions of CGOB and YGOB. We migrated pillar assignments from the protein-based versions to the nucleotide-based versions of the databases to the greatest extent possible.
Novel genes were identified by two iterations of SearchDOGS (OhEigeartaigh et al. 2011) and by manual investigation.
Phylogenetic Analysis
The species tree was constructed using PhyML (BLOSUM + I + Γ with 8 rate classes [Guindon and Gascuel 2003]) using as input 100,000 informative amino acid sites from proteins that are present in all 14 species, randomly chosen from Muscle alignments filtered by Gblocks (Castresana 2000; Edgar 2004). Other alignments were generated using T-coffee (Notredame et al. 2000) and ClustalW implemented through SeaView (Gouy et al. 2010).
Supplementary Material
Supplementary table S1 is available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank the many present and former colleagues in the Wolfe lab who helped with the development of features in YGOB that were carried over into CGOB. They thank Gaël Jalowicki for constructing the phylogenetic tree. This work was supported by Science Foundation Ireland (08/N1B1865) to G.B., the Wellcome Trust Computational Infection Biology PhD programme (0997419/Z/11/Z) to G.B., and the European Research Council (Advanced Grant 268893) to K.H.W.
References
- Aasland R, Stewart AF, Gibson T. The SANT domain: a putative DNA-binding domain in the SWI-SNF and ADA complexes, the transcriptional co-repressor N-CoR and TFIIIB. Trends Biochem Sci. 1996;21:87–88. [PubMed] [Google Scholar]
- Bennett RJ, Johnson AD. Completion of a parasexual cycle in Candida albicans by induced chromosome loss in tetraploid strains. EMBO J. 2003;22:2505–2515. doi: 10.1093/emboj/cdg235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RJ, Johnson AD. Mating in Candida albicans and the search for a sexual cycle. Annu Rev Microbiol. 2005;59:233–255. doi: 10.1146/annurev.micro.59.030804.121310. [DOI] [PubMed] [Google Scholar]
- Bruno VM, Wang Z, Marjani SL, Euskirchen GM, Martin J, Sherlock G, Snyder M. Comprehensive annotation of the transcriptome of the human fungal pathogen Candida albicans using RNA-seq. Genome Res. 2010;20:1451–1458. doi: 10.1101/gr.109553.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler G. The evolution of MAT: the ascomycetes. In: Heitman J, Kronstad JW, Taylor JW, Casselton LA, editors. Sex in fungi. Washington: ASM Press; 2007. pp. 3–18. [Google Scholar]
- Butler G. Fungal sex and pathogenesis. Clin Microbiol Rev. 2010;23:140–159. doi: 10.1128/CMR.00053-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler G, Rasmussen MD, Lin MF, et al. (51 co-authors) Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature. 2009;459:657–662. doi: 10.1038/nature08064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne KP, Wolfe KH. The yeast gene order browser: combining curated homology and syntenic context reveals gene fate in polyploid species. Genome Res. 2005;15:1456–1461. doi: 10.1101/gr.3672305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne KP, Wolfe KH. Visualizing syntenic relationships among the hemiascomycetes with the yeast gene order browser. Nucleic Acids Res. 2006;34:D452–D455. doi: 10.1093/nar/gkj041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Cherry JM, Hong EL, Amundsen C, et al. (21 co-authors) Saccharomyces genome database: the genomics resource of budding yeast. Nucleic Acids Res. 2012;40:D700–D705. doi: 10.1093/nar/gkr1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo MC, Arnaud MB, Skrzypek MS, Binkley G, Lane C, Miyasato SR, Sherlock G. The Candida genome database: facilitating research on Candida albicans molecular biology. FEMS Yeast Res. 2006;6:671–684. doi: 10.1111/j.1567-1364.2006.00074.x. [DOI] [PubMed] [Google Scholar]
- Dujon B, Sherman D, Fischer G, et al. (71 co-authors) Genome evolution in yeasts. Nature. 2004;430:35–44. doi: 10.1038/nature02579. [DOI] [PubMed] [Google Scholar]
- Edgar RC. Muscle: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre E, Muller H, Therizols P, Lafontaine I, Dujon B, Fairhead C. Comparative genomics in hemiascomycete yeasts: evolution of sex, silencing, and subtelomeres. Mol Biol Evol. 2005;22:856–873. doi: 10.1093/molbev/msi070. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick DA, Logue ME, Butler G. Evidence of recent interkingdom horizontal gene transfer between bacteria and Candida parapsilosis. BMC Evol Biol. 2008;8:181. doi: 10.1186/1471-2148-8-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick DA, Logue ME, Stajich JE, Butler G. A fungal phylogeny based on 42 complete genomes derived from supertree and combined gene analysis. BMC Evol Biol. 2006;6:99. doi: 10.1186/1471-2148-6-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick DA, O’Gaora P, Byrne KP, Butler G. Analysis of gene evolution and metabolic pathways using the Candida gene order browser. BMC Genomics. 2010;11:290. doi: 10.1186/1471-2164-11-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JL, Byrne KP, Wolfe KH. Additions, losses, and rearrangements on the evolutionary route from a reconstructed ancestor to the modern Saccharomyces cerevisiae genome. PLoS Genet. 2009;5:e1000485. doi: 10.1371/journal.pgen.1000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O. Seaview version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- Guida A, Lindstadt C, Maguire SL, Ding C, Higgins DG, Corton NJ, Berriman M, Butler G. Using RNA-seq to determine the transcriptional landscape and the hypoxic response of the pathogenic yeast Candida parapsilosis. BMC Genomics. 2011;12:628. doi: 10.1186/1471-2164-12-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Delsuc F, Dufayard JF, Gascuel O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol. 2009;537:113–137. doi: 10.1007/978-1-59745-251-9_6. [DOI] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- He W, Parker R. Functions of Lsm proteins in mRNA degradation and splicing. Curr Opin Cell Biol. 2000;12:346–350. doi: 10.1016/s0955-0674(00)00098-3. [DOI] [PubMed] [Google Scholar]
- Hickman MA, Zeng G, Forche A, et al. (11 co-authors) The “obligate diploid” Candida albicans forms mating-competent haploids. Nature. 2013;494:55–59. doi: 10.1038/nature11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull CM, Raisner RM, Johnson AD. Evidence for mating of the “asexual” yeast Candida albicans in a mammalian host. Science. 2000;289:307–310. doi: 10.1126/science.289.5477.307. [DOI] [PubMed] [Google Scholar]
- Inglis DO, Arnaud MB, Binkley J, Shah P, Skrzypek MS, Wymore F, Binkley G, Miyasato SR, Simison M, Sherlock G. The Candida genome database incorporates multiple Candida species: multispecies search and analysis tools with curated gene and protein information for Candida albicans and Candida glabrata. Nucleic Acids Res. 2012;40:D667–D674. doi: 10.1093/nar/gkr945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis DO, Skrzypek MS, Arnaud MB, Binkley J, Shah P, Wymore F, Sherlock G. Improved gene ontology annotation for biofilm formation, filamentous growth and phenotypic switching in Candida albicans. Eukaryot Cell. 2013;12:101–108. doi: 10.1128/EC.00238-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii K. Conservation and divergence of centromere specification in yeast. Curr Opin Microbiol. 2009;12:616–622. doi: 10.1016/j.mib.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Jackson AP, Gamble JA, Yeomans T, et al. (26 co-authors) Comparative genomics of the fungal pathogens Candida dubliniensis and C. albicans. Genome Res. 2009;19:2231–2244. doi: 10.1101/gr.097501.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffries TW, Grigoriev IV, Grimwood J, et al. (13 co-authors) Genome sequence of the lignocellulose-bioconverting and xylose-fermenting yeast Pichia stipitis. Nat Biotechnol. 2007;25:319–326. doi: 10.1038/nbt1290. [DOI] [PubMed] [Google Scholar]
- Lockhart SR, Messer SA, Pfaller MA, Diekema DJ. Lodderomyces elongisporus masquerading as Candida parapsilosis as a cause of bloodstream infections. J Clin Microbiol. 2008;46:374–376. doi: 10.1128/JCM.01790-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue ME, Wong S, Wolfe KH, Butler G. A genome sequence survey shows that the pathogenic yeast Candida parapsilosis has a defective MTLa1 allele at its mating type locus. Eukaryot Cell. 2005;4:1009–1017. doi: 10.1128/EC.4.6.1009-1017.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-se: a program for improved detection of transferRNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch DB, Logue ME, Butler G, Wolfe KH. Chromosomal G + C content evolution in yeasts: systematic interspecies differences, and GC-poor troughs at centromeres. Genome Biol Evol. 2010;2:572–583. doi: 10.1093/gbe/evq042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee BB, Magee PT. Induction of mating in Candida albicans by construction of MTLa and MTLalpha strains. Science. 2000;289:310–313. doi: 10.1126/science.289.5477.310. [DOI] [PubMed] [Google Scholar]
- Martin R, Moran GP, Jacobsen ID, Heyken A, Domey J, Sullivan DJ, Kurzai O, Hube B. The Candida albicans-specific gene EED1 encodes a key regulator of hyphal extension. PLoS One. 2011;6:e18394. doi: 10.1371/journal.pone.0018394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin T, Sherman DJ, Durrens P. The Genolevures database. C R Biol. 2011;334:585–589. doi: 10.1016/j.crvi.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Massey SE, Moura G, Beltrao P, Almeida R, Garey JR, Tuite MF, Santos MA. Comparative evolutionary genomics unveils the molecular mechanism of reassignment of the CTG codon in Candida spp. Genome Res. 2003;13:544–557. doi: 10.1101/gr.811003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melake T, Passoth VV, Klinner U. Characterization of the genetic system of the xylose-fermenting yeast Pichia stipitis. Curr Microbiol. 1996;33:237–242. doi: 10.1007/s002849900106. [DOI] [PubMed] [Google Scholar]
- Mitrovich QM, Tuch BB, Guthrie C, Johnson AD. Computational and experimental approaches double the number of known introns in the pathogenic yeast Candida albicans. Genome Res. 2007;17:492–502. doi: 10.1101/gr.6111907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousson F, Coic YM, Baleux F, Beswick V, Sanson A, Neumann JM. Deciphering the role of individual acyl chains in the interaction network between phosphatidylserines and a single-spanning membrane protein. Biochemistry. 2002;41:13611–13616. doi: 10.1021/bi026274b. [DOI] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- OhEigeartaigh SS, Armisen D, Byrne KP, Wolfe KH. Systematic discovery of unannotated genes in 11 yeast species using a database of orthologous genomic segments. BMC Genomics. 2011;12:377. doi: 10.1186/1471-2164-12-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan S, Thakur J, Siddharthan R, Sanyal K. Rapid evolution of Cse4p-rich centromeric DNA sequences in closely related pathogenic yeasts, Candida albicans and Candida dubliniensis. Proc Natl Acad Sci U S A. 2008;105:19797–19802. doi: 10.1073/pnas.0809770105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porman AM, Alby K, Hirakawa MP, Bennett RJ. Discovery of a phenotypic switch regulating sexual mating in the opportunistic fungal pathogen Candida tropicalis. Proc Natl Acad Sci U S A. 2011;108:21158–21163. doi: 10.1073/pnas.1112076109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proux E. Dublin (Ireland): Trinity College, University of Dublin; 2012. Automated annotation of yeast genomes [PhD thesis] [Google Scholar]
- Proux-Wéra E, Byrne KP, Wolfe KH. Evolutionary mobility of the ribosomal DNA array in yeasts. Genome Biol Evol. 2013;5:525–531. doi: 10.1093/gbe/evt022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol C, Daniels KJ, Lockhart SR, Srikantha T, Radke JB, Geiger J, Soll DR. The closely related species Candida albicans and Candida dubliniensis can mate. Eukaryot Cell. 2004;3:1015–1027. doi: 10.1128/EC.3.4.1015-1027.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reedy JL, Floyd AM, Heitman J. Mechanistic plasticity of sexual reproduction and meiosis in the Candida pathogenic species complex. Curr Biol. 2009;19:891–899. doi: 10.1016/j.cub.2009.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccombeni A, Vidanes G, Proux-Wera E, Wolfe KH, Butler G. Sequence and analysis of the genome of the pathogenic yeast Candida orthopsilosis. PLoS One. 2012;7:e35750. doi: 10.1371/journal.pone.0035750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol T, Ding C, Guida A, d’Enfert C, Higgins DG, Butler G. Correlation between biofilm formation and the hypoxic response in Candida parapsilosis. Eukaryot Cell. 2009;8:550–559. doi: 10.1128/EC.00350-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sai S, Holland L, McGee CF, Lynch DB, Butler G. Evolution of mating within the Candida parapsilosis species group. Eukaryot Cell. 2011;10:578–587. doi: 10.1128/EC.00276-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos MA, Keith G, Tuite MF. Non-standard translational events in Candida albicans mediated by an unusual seryl-tRNA with a 5′-CAG-3′ (leucine) anticodon. EMBO J. 1993;12:607–616. doi: 10.1002/j.1460-2075.1993.tb05693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal K, Baum M, Carbon J. Centromeric DNA sequences in the pathogenic yeast Candida albicans are all different and unique. Proc Natl Acad Sci U S A. 2004;101:11374–11379. doi: 10.1073/pnas.0404318101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikantha T, Daniels KJ, Pujol C, Sahni N, Yi S, Soll DR. Nonsex genes in the mating type locus of Candida albicans play roles in a/alpha biofilm formation, including impermeability and fluconazole resistance. PLoS Pathog. 2012;8:e1002476. doi: 10.1371/journal.ppat.1002476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuch BB, Mitrovich QM, Homann OR, Hernday AD, Monighetti CK, De La Vega FM, Johnson AD. The transcriptomes of two heritable cell types illuminate the circuit governing their differentiation. PLoS Genet. 2010;6:e1001070. doi: 10.1371/journal.pgen.1001070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt JP. Lodderomyces, a new genus of the Saccharomycetacea. Antonie Van Leeuwenhoek. 1966;32:1–5. doi: 10.1007/BF02097439. [DOI] [PubMed] [Google Scholar]
- van del Walt JP, Taylor MB, Liebenberg MV. Ploidy, ascus formation and recombination in Torulaspora (Debaryomyces) hansenii. Antonie Van Leeuwenhoek. 1977;43:205–218. doi: 10.1007/BF00395675. [DOI] [PubMed] [Google Scholar]
- Wickerham LJ, Burton KA. A clarification of the relationship of Candida guilliermondii to other yeasts by a study of their mating types. J Bacteriol. 1954;68:594–597. doi: 10.1128/jb.68.5.594-597.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlbach DJ, Kuo A, Sato TK, et al. (21 co-authors) Comparative genomics of xylose-fermenting fungi for enhanced biofuel production. Proc Natl Acad Sci U S A. 2011;108:13212–13217. doi: 10.1073/pnas.1103039108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000;17:32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.