

Abstract
Cerebral ischemia initiates a cascade of detrimental events including glutamate-associated excitotoxicity, intracellular calcium accumulation, formation of Reactive oxygen species (ROS), membrane lipid degradation, and DNA damage, which lead to the disruption of cellular homeostasis and structural damage of ischemic brain tissue. Cerebral ischemia also triggers acute inflammation, which exacerbates primary brain damage. Therefore, reducing oxidative stress (OS) and downregulating the inflammatory response are options that merit consideration as potential therapeutic targets for ischemic stroke. Consequently, agents capable of modulating both elements will constitute promising therapeutic solutions because clinically effective neuroprotectants have not yet been discovered and no specific therapy for stroke is available to date. Because of their ability to modulate both oxidative stress and the inflammatory response, much attention has been focused on the role of nitric oxide donors (NOD) as neuroprotective agents in the pathophysiology of cerebral ischemia-reperfusion injury. Given their short therapeutic window, NOD appears to be appropriate for use during neurosurgical procedures involving transient arterial occlusions, or in very early treatment of acute ischemic stroke, and also possibly as complementary treatment for neurodegenerative diseases such as Parkinson or Alzheimer, where oxidative stress is an important promoter of damage. In the present paper, we focus on the role of NOD as possible neuroprotective therapeutic agents for ischemia/reperfusion treatment.
1. Introduction
When the brain blood flow is interrupted, it results in deprivation of oxygen and nutrients to the cells; this situation constitutes an ischemic stroke. Restoration of the flux, or reperfusion, can reduce the damage, but only when this is performed very early after the onset of ischemia, and its efficacy is restricted by secondary injuries, mainly by oxidative stress (OS) and an inflammatory reaction, which lead to cell death by apoptosis [1].
It is noteworthy that ischemic damage not only affects neurons. Thus, in recent years the concept of the neurovascular unit has been highlighted, emphasizing the need to protect not only the neurons, but also all cells in the brain [2–5]. In contrast to the known vulnerability of neurons and astrocytes, it is thought that endothelial cells tend to be more resistant to ischemic or oxidative injury [6]. Hence, to be successful, stroke therapies should be widely effective and must protect all neuronal, glial, and endothelial components in the brain [7].
After focal ischemia, primary neuronal death appears rapidly in the core area and is followed by secondary death in the ischemic penumbra, which evolves from the delayed activation of multiple cellular death pathways. At the core of the ischemic lesion, one of the first events is the rapid decline of adenosine triphosphate (ATP) reserves [8]. Consequently, all energy-dependent processes gradually cease their activity, leading to changes in transmembrane potential. The consequent depolarization (denominated anoxic depolarization) produces massive influx of Na+, Cl−, and Ca2+ inside the cell with K+ efflux [9].
Core anoxic ischemic depolarizations induce release of neurotransmitters such as glutamate. Once released, glutamate generates a phenomenon of peri-infarct depolarization, which increases energy consumption and promotes Ca2+ influx into the cells [10].
The increase in intracellular Ca2+ in neurons and glial cells initiates a set of nuclear and cytoplasmic events that produce deep brain tissue damage that includes the following: Ca2+ mitochondrial overload (which compromises the already affected ATP production and promotes the opening of the mitochondrial transition pore); the increase in OS, and the activation of a number of Ca2+-dependent enzymes. Such enzymes include proteases, kinases, phospholipases, and endonucleases, which destroy biomolecules [10]. Additionally, increased intracellular Ca2+ also promotes the production of NO from constitutive synthases that, together with acidosis and peri-infarct depolarization, contribute to the initiation of damage; later, inflammation and activation of apoptotic phenomena contribute to increased injury [2].
OS is a major mechanism implicated in stroke and in a variety of neurodegenerative diseases, mainly in Alzheimer and Parkinson (reviewed in [11, 12]). The most accepted theory regarding neurodegeneration in Parkinson disease refers to OS as the main cause of damage to neurons in the substantia nigra. In addition, in Alzheimer disease, the OS generated by the action of β-amyloid, which causes massive entry of Ca2+ and caspase activation, leads to neuronal death [13, 14].
During ischemia, reactive oxygen (ROS) and nitrogen species can be generated in the ischemic penumbra but can also be produced during reperfusion injury [15, 16]. Indeed, it is now established that albeit maintenance of partial or complete blood flow is essential for preserving cerebral tissue, it is during reperfusion when it paradoxically induces excessive generation of ROS, such as superoxide anion radical (O2 •−), hydroxyl radical (OH•), hydrogen peroxide (H2O2), and nitric oxide (NO), which contribute to increased neuronal death by oxidizing proteins, damaging DNA, and inducing lipid peroxidation [17].
Reperfusion-induced ROS contribute to a decrease of the NO availability responsible for postischemic endothelial dysfunction [18, 19]. During the ischemic period, reduction in O2 availability reduces the activity of NO synthase, producing O2− instead of NO; later, during reperfusion, the arrival of O2 increases NO synthase activity. These can exert a deleterious effect by promoting nitrosative stress and diminishing the availability of NO for preserving endothelial integrity.
Over the past decade, remarkable advances have been made in understanding the basic molecular mechanisms underlying neuronal death. However, clinically effective neuroprotectants have not yet been discovered and no specific therapy for stroke is available at present. The body of experimental data supports the view that reducing OS should continue to be a potentially viable target for stroke therapy [20]. In addition, the inflammatory response requires consideration as a potential target of therapy for ischemic stroke [21]. Therefore, agents capable of modulating both elements will constitute promising therapeutic solutions [22–25].
2. Ischemic Lesion and Immune Response: Brain Inflammation
It has now been established that the Central nervous system (CNS) is able to raise an immune response to the majority of threatening stimuli, whereby resident cells generate inflammatory mediators including cytokines, prostaglandins, free radicals, complementary chemokines, and adhesion molecules that recruit immune cells and activate glia and microglia (reviewed in [21, 26–28]). The role of microglia and proinflammatory cytokines in the CNS has been characterized in models of brain insults, such as experimental stroke, the most common form of ischemic injury [26]. As mentioned previously, cerebral ischemia triggers acute inflammation, which exacerbates primary brain damage. Although inflammation should be adaptive, the release of proinflammatory cytokines has often been associated with harmful consequences to neurons and myelin [29].
The control of early CNS inflammation is a careful balancing act, as both too much and too little inflammation will lead to decreased or delayed recovery. Whether the inflammation is neurotoxic or protective may depend upon the context and the location of the inflammatory mediator in relation to an injury, and the timing of inflammatory response may determine the outcome (see Table 1 in [27]).
For example, tumor necrosis factor alpha (TNF-α) upregulated in the proximity of an evolving lesion contributes to secondary infarct growth, whereas cytokine induction remote from the ischemic lesion confers neuroprotection [30]. TNF-α could enhance apoptotic processes through its action on its tumor necrosis factor type 1 receptor (TNFR1) in models of acute (ischemia, excitotoxicity) and chronic (Alzheimer disease, multiple sclerosis) neurodegeneration [31]. TNF-α and interleukin 1beta (IL-1β) exert neurotoxicity in cerebral ischemia in the presence of elevated inducible nitric oxide synthase (iNOS), while in the absence of iNOS, both cytokines appear to contribute to neuroprotection and plasticity, highlighting the role of the context [32].
There is important recognition that protection of endothelial function and downregulation of vascular inflammation comprise part of neuroprotection phenomena and may possess added therapeutic benefit against stroke injury [33]. However, research on clinically effective neurovascular protective therapies for brain damage remains at an early phase [34]. Much attention has been focused on the role of NO in vessel protection from OS and inflammation [35]. Because OS coexists with inflammation and endothelial dysfunction, determining antioxidant status may be helpful in monitoring the progress of Nitric oxide donors (NOD) treatment. A variety of structurally different NOD, which release NO either as a free radical (NO•) or as an NO ion (NO+/NO−), have shown to reduce OS/inflammation and to increase cerebral blood flow [35–38]; thus, these can be considered attractive candidates for therapeutic agents in experimental models of stroke.
3. Nitric Oxide Donors (NOD) as Neuroprotective Agents in Ischemic Stroke
3.1. Nitric Oxide in Ischemic Stroke
Nitric oxide (NO) plays a dual role, that is, neuroprotection and neurotoxicity, in the pathophysiology of cerebral ischemia-reperfusion injury [39]. NO is synthesized by NOS, of which there are three known isoforms: nNOS; eNOS, and inducible or immunological NOS (iNOS). The first two are constitutively expressed and their activity is dependent on changes at the intracellular Ca2+ level, while iNOS acts in a Ca2+-independent manner [40]. Baseline concentration of NO in the brain is mainly due to nNOS activity, and secondarily to eNOS. iNOS is not expressed under physiological conditions [41, 42].
In the brain, eNOS is mainly produced by the vascular endothelium and the choroid plexus [43]. Although eNOS-NO production is a minor part of total brain NOS activity, this enzyme is critical for the regulation of cerebrovascular hemodynamics and for the protection of endothelium integrity from inflammatory, oxidative, and procoagulant stimuli. It has been demonstrated that eNOS-derived NO scavenges ROS [44] and inhibits the expression of cellular adhesion molecules [45], platelet aggregation [46], and leukocyte adhesion [47].
During ischemia, NO concentration decreases because of oxygen deficiency [41]. However, immediately after reperfusion, biosynthesis of this molecule is triggered mainly by overactivation of nNOS, as evidenced in nNOS (−/−) mice [48] or with specific inhibitors such as 7-NI [41, 49]. Glutamate-induced Ca2+ overload in ischemic neurons is responsible for the rise of nNOS-derived NO [50]. Concentration of NO returns to physiological levels approximately 1 h after reperfusion [48, 49, 51] and increases again due to iNOS expression between 12 h and up to 8 days later [52, 53]. iNOS sources at this stage comprise microglia [53], astrocytes [54], endothelial cells [55], and infiltrated leukocytes [56]. The amount of iNOS-NO derived is 100–1,000 times than that produced by nNOS [57].
Therefore, NO deriving from different sources (neurons, brain vessels, glia, and neutrophils) may exert an influence on the evolution of brain damage at different time-points after an ischemic insult [42]. Thus, the use of relatively selective inhibitors of NOS isoforms and genetically modified mice has contributed to clarifying the role of NO in cerebral ischemia-reperfusion injury as follows.
Total suppression of eNOS activity in knockout mice (eNOS −/−) renders them hypertensive [48] and more susceptible to ischemia-reperfusion injury, with larger infarcts compared with those of controls and a more severe reduction in cerebral blood flow (CBF) [48, 51, 58]. Conversely, overexpression of eNOS by flavonoids induces a protective effect [59]. In contrast to eNOS, infarct volume and neuronal death are consistently decreased by nNOS gene deficiencies or by nNOS inhibition [48, 60–64]. nNOS abolition also reduces excitotoxicity [65], nitrosative stress [63, 66], and O2− production [67] and downregulates calpain and caspase-3 in ischemic lesion [61, 64]. Additionally, during reperfusion, iNOS-produced NO contributes to brain injury [42, 68]. iNOS expression is transcriptionally regulated by nuclear factor kappa B (NF-κB) secondary to moderate inflammatory stimuli such as TNF-α [69] and IL-1β [70], and also by oxidative radicals [71]. Due to the large amount of NO produced by iNOS, this enzyme is related with high peroxynitrite production and significant nitrosative damage of biological molecules [72].
Consequently, nNOS mediates early neuronal injury, while iNOS contributes to late neuronal injury, whereas eNOS activity might be protective [41, 42, 73]. Whether the effects of this molecule are beneficial or harmful depends on the cellular compartment in which NO is generated, on its concentration, on the environment's redox state, and on the evolutive stage of ischemic brain injury [42, 48, 73].
In addition to their involvement in ischemia, the expression of iNOS in astrocytes and microglia and the production of large amounts of NO contribute to dopaminergic neuronal death in the neuropathology of experimental Parkinson disease [74, 75]. In the brains of patients with Alzheimer disease, nitrosylation of proteins is a hallmark of tissue damage [76] and is particularly high in hippocampus and cerebral cortex. The presence of beta amyloid is sufficient for triggering iNOS activation in astrocytes and microglia [77]. In addition, nNOS activity in reactive astrocytes surrounds beta-amyloid plaques in entorhinal cortex and is related with DNA fragmentation in CA1 and CA4 fields [78], while there is a correlation between neurofibrillary tangles and senile plaques with a reduction in eNOS capillary levels [79].
According to the dual role of NO in brain ischemia, there is a rationale for the use of NO for promoting treatments shortly after the occurrence of focal cerebral ischemia as neuroprotective strategies [42, 53, 80, 81]. The neurovascular protective mechanism of eNOS-NO suggests that intervention with NO may be most effective when delivered at an optimal amount by a suitable source at the correct time in an appropriate environment [82].
3.2. Nitric Oxide Donors (NOD)
Nitric oxide donors are a heterogeneous group of drugs whose common feature is the ability to release NO or an NO-related species, such as the Nitrosonium ion (NO+) or the Nitroxyl anion (NO−), in vitro or in vivo, independently on its endogenous sources [83]. The following are the NOD most frequently employed in clinical and basic research: organic nitrates (e.g., nitroglycerin, isosorbide-5-mononitrate, nicorandil, pentaerythritol tetranitrate); S-nitrosothiols (e.g., S-nitroso-N-acetylpenicillamine and S-nitroso-glutathione); sydnonimines (e.g., molsidomine, SIN-1); NONOates (JS-K, SPERMINE-NONOate, and PROLI-NONOate), and sodium nitroprusside [84]. Table 1 illustrates NOD effects on experimental cerebral ischemia.
Table 1.
Nitric oxide donors in experimental cerebral ischemia.
| Species | Model | Time of ischemia/reperfusion | Nitric oxide donor | Doses and administration | Effect | Reference |
|---|---|---|---|---|---|---|
| Rat | MCAO | 20′/24 h | GSNO | 1 mg/kg at the onset of reperfusion | Reduction in infarct size Increase in CBF Decrease in cortical expression of TNF-α and IL-1 in penumbral region Attenuation of iNOs expression in microglia, astrocytes and vessels of penumbral region Inhibition of monocyte/macrophage infiltration Downregulates adhesion molecules (ICAM-1, LFA-1) Reduction in TUNEL-positive cells and caspase-3 activity Blocks NF-κB (p65/p50 complex) and is able to bind to DNA in astrocytes and microglial cells in vitro |
[53] |
|
| ||||||
| Rat | MCAO | 20′/24 h | SNP GSNO SNAP MAHMA/NONO-ate PAPA/NONOate SIN-1 |
2 and 3 μmol/kg per 10 min IV at onset of reperfusion | Increase in CBF (except MAHMA and PAPA) Reduction in infarct size (GSNO, SNP, and SNAP) Improvement in neurological score (GSNO, SNP, and SNAP) Reduction in lipid peroxidation in plasma (all of them) Decrease in plasma levels of nitrotyrosine (GSNO, SNP and SNAP) Increase in NO plasma level (except SNAP) Reduction in mRNA expression of ICAM-1 (GSNO, SNAP, SNP) and E-selectin (except SIN-1) |
[38] |
|
| ||||||
| Rat | MCAO | 90′/24 h | ZJM-289 | 0.1 and 0.2 mmol/kg IV 1 h prior to ischemia | Improvement in neurological deficit (motor function) Reduction in infarct size Reduction in brain water content Decrease of neuronal degeneration Inhibition of nNOS expression Increase of NO level ipsilateral to ischemia Increase in cGMP level |
[85] |
|
| ||||||
| Rat | MCAO | 90′/1.5, 3, 4.5, 6 and 12 h | Sodium nitrite | 480 nmol per 1 min at 1.5, 3, 4.5, and 6 h postischemia, IV | Reduction in infarct size (1.5, 3, 4.5 and 6 h) Improvement in motor function (4.5 h) Decrease of microhypoxic areas (12 h) Reduction in free reactive oxygen and nitrogen species (12 h) |
[86] |
|
| ||||||
| Rat | MCAO | 2 h/7 days | SNP Sperm-ine/NONO-ate |
SNP: 0.11 mg/kg per 120 min, trans-ischemia, IV Spermine: 0.36 mg/kg per 120 min, trans-ischemia, IV |
Reduction in infarct size Increase in cerebral perfusion |
[87] |
|
| ||||||
| Rat | MCAO | 2 h/3 days | SIN-1 | 0.1 and 1 mg/kg 30 min before ischemia, IV | Reduction in infarct size in normo- and hyperglycemic rats | [88] |
|
| ||||||
| Rat | Permanent MCAO | 24 h/no reperfusion | SNP SIN-1 |
SNP: 3 mg/kg/h trans-ischemia, IA SIN-1: 1.5, 3, and 6 mg/kg/h trans-ischemia, IA |
Both produced an increase in CBF and a reduction in infarct size SNP decreased platelet aggregation in vitro but not in vivo at the same doses |
[80] |
|
| ||||||
| Rat | Permanent MCAO | 24 h/no reperfusion | SIN-1 | 3 mg/kg/h per 60 min at 3, 15, 30, 60, and 120 min after ischemia, IA | Reduction in infarct size Increase in CBF |
[89] |
|
| ||||||
| Rat | 4-VO | 15′/30′, 6 h, 12 h, 3 and 5 days | SNP | 5 mg/kg, 3 doses: 30 min prior to ischemia, 1 h postischemia, and 2.5 h postischemia, IP | Suppression of JNK3 downstream pathway (30′, 3 h) Increase in Akt and Bad phosphorylation (12h) Inhibition of Cytochrome c release from mitochondria (6 h) Reduction in TUNEL-positive cells and caspase-3 activity (3 h) Augmentation of neuronal survival in CA1 pyramidal layer (3–5 d) |
[61] |
|
| ||||||
| Rat | 4-VO | 15′/6 h, 3 and 5 days | SNP | 5 mg/kg, 3 doses: 30′ prior to ischemia, 1 h postischemia and 2.5 h postischemia, IP | Decreased hippocampal activation of nNOS by nitrosylation and phosphorylation (6 h) Suppression of JNK3 downstream pathway (6 h) Inhibition of release of Cytochrome C into cytoplasm (6 h) Attenuation of caspase-3 activity (6 h) Reduction in neuronal degeneration (5 d) and TUNEL-positive cells (3 d) in CA1 pyramidal layer |
[65] |
|
| ||||||
| Rabbit and rat | MCAO | 60′/2, 4 h respectively | ProliNO/ NONO-ate |
Rabbit: 10−6 mol/L Rat: 10−5 mol/L At the onset of reperfusion per 60 min, IA |
Reduction in free reactive oxygen species Reduction in infarct size |
[90] |
|
| ||||||
| Goat | MCAO | 20′/7 days | SNP DEA/NONOate DETA/NONOate |
SNP: 10−9–3 × 10−4 mol/L, IV DEA: 10−9–3 × 10−4 mol/L, IV DETA: 10−7–3 × 10−4 mol/L, IV |
MCA relaxation | [91] |
Lines of evidence are ordered first by animal model and then by surgical procedure and severity of the ischemia. In cases in which the effects were different at different reperfusion times, this is indicated after each effect by the corresponding time as a superscript between parentheses. MAP: Mean arterial pressure; CA1: Cornu Ammonis; CBF: Cerebral blood flow; GSNO: S-nitrosoGlutathione; IA: IntraArterial; ICAM-1: Intercellular adhesion molecule-1; IL1: Interleukin 1; iNOS: inducible Nitric oxide synthase; IP: Intraperitoneal; IV: Intravenous; JNK3: c-Jun N-terminal kinase-3; LFA: Lymphocyte function-associated antigen-1; MAHMA: Methylamine hexamethylene methylamine NONOate; MCA: Middle cerebral artery; MCAO: Middle cerebral artery occlusion; nNOS: neuronal Nitric oxide synthase; NO: Nitric oxide; PAPA: Propylamine propylamine NONOate; SAP: Systolic arterial pressure; SIN-1: 3-morpholinoSydnonimine; SNAP: S-nitroso-N-acetyl-penicillamine; SNP: Sodium nitroprusside; TNF: Tumor necrosis factor; TUNEL: Terminal dUTP nick end labeling; 4-VO: four Vessel occlusion model.
Despite the fact that all of these are considered NOD, they have different pharmacokinetic and dynamic profiles that determine the type and extent of their biological effects. The main determinant of these effects is the manner in which NO is released, the amount of NO generated, and the time during which it is released. Moreover, some of these generate alternative products that may arise during their metabolism or decomposition. These products may even be present in quantities exceeding NO with independent or side effects [92].
For example, organic nitrates, the most common NOD utilized in coronary artery disease, require enzymatic bioactivation in order to deliver NO [93]. Their principal effect is at the vascular level, by increasing venous capacitance and coronary vasodilation [84]. S-nitrosothiols are a heterogeneous group characterized by a nitroso group attached by a single chemical bond to the sulfur atom of a thiol [83]. S-nitrosoGlutathione (GSNO) is found in vivo and is an important intermediary in organic nitrate metabolism. The remaining nitrosothiols are synthetic. These compounds act as NO-carriers, NO-reservoirs, and intermediates in protein nitrosylation. They also possess the ability to transfer the different NO species through chains of thiols, without releasing the NO molecule itself. This feature decreases the possibility of NO reacting with O2− to form ONOO−, or that of reacting with other molecules to nitrosylate these [94, 95]. Sydnonimines release NO spontaneously, without enzymatic activity. Superoxide is generated concomitantly and may combine with NO to generate ONOO−. This process also releases significant quantities of hydroxyl radical, which increases its prooxidant potential [96]. Therefore, these compounds are considered peroxynitrite donors more than NOD and are utilized as nitrosative stress inducers in experimental models. NONOates decompose spontaneously in solution, at physiological pH and temperature, without interaction with biological molecules and in a concentration-dependent manner; thus, they usually are employed as NO-release models [90].
Sodium nitroprusside (SNP) is a compound consisting of an iron core surrounded by five Cyanide ion (CN−) molecules and one molecule of the Nitrosonium ion (NO+). SNP does not liberate NO spontaneously in vitro, but does require partial reduction (one-electron transfer) by a variety of reducing agents present in membrane cells. It is also possible to release NO from SNP by photolysis. In addition to NO, SNP can release, in aqueous solution, a range of oxidant and free radical species, such as iron, cyanide, superoxide, H2O2, and hydroxyl radical in direct proportion to its concentration [97–99]. Because of the nitrosative and prooxidant potential inherent in the different NOD, these have been widely used as models of neuronal damage (for a more detailed review, see [83, 84, 94]).
3.3. Sodium Nitroprusside-Induced Neurotoxicity
The potentially adverse effects of SNP on cells and tissues have been widely used in vivo and in vitro to study the mechanisms involved in nitrosative and OS injury. While many of the pharmacological effects elicited by SNP are attributed to the NO molecule, several in vitro studies [100, 101] revealed other biological SNP properties that are independent of the NO moiety, due to the huge number of by-products released during its decomposition (e.g., cyanide, iron, and ROS).
In vitro, SNP is usually neurotoxic. This compound is able to cause cytotoxicity in the human neuroblastoma cell line SH-SY5Y by means of OS. In addition, SNP treatment activates the (ERK1/2) pathway and inactivates the Akt pathway, leading to cell death [102]. Additionally, inhibition of ERK activation or exogenous Superoxide dismutase (SOD) treatment protects human melanoma from SNP toxicity [103]. Furthermore, in hippocampal neurons, SNP and SIN-1 are capable of decreasing Bcl-2 and increasing Bax expression along with caspase-3 activation, leading to neuronal apoptosis [104]. Concentration-dependent neuronal death was induced in cerebellar granular cells after exposure to SNP by hydroxyl radical generation, as well as by increasing the level of iron and lipid peroxidation [105]. In cultured cholinergic cells, SNP impairs oxidative metabolism of Acetyl Co-A by suppression of choline acetyltransferase and pyruvate dehydrogenase activities in mitochondria and cytoplasm. This effect triggers OS and a reduction in neuronal viability [106].
In addition to SNP, other NOD are able to elicit cytotoxicity in vitro. In cortical neuronal cultures, SIN-1 induced neurotoxicity by ATP depletion and protein nitration, which was counteracted by the addition of hemoglobin (a NO scavenger), SOD, and an ONOO scavenger, demonstrating that the main mediator of damage in this case is ONOO− [107]. In the same cell culture type, neuronal viability was significantly reduced when compared with that of controls after DETANONOate exposure. This effect was associated with a decrease in catalase activity and expression [108].
Likewise, SNP has been used to induce neurotoxicity in vivo. While not mimicking a specific neuropathology, the rapid and localized neurodegeneration and demyelination caused by the SNP, when injected into the brain, provides a very practical tool for studying the role of the individual molecular players that can be involved in the immediate and consequent damage implicit in neurodegenerative processes. In animal models, SNP causes acute and localized excitotoxic cell death when infused within the brain parenchyma. This damage is also associated with a transient inflammatory response [109]. The neurodegeneration caused by SNP is accompanied by microglial activation and the induction of the proinflammatory cytokines TNF-α and IL-1β. Injection of exogenous TNF-α was shown to exacerbate the damage and inflammation caused by SNP through specific and transient activation of resident microglia [109]; in contrast, the abolition of the endogenous production of TNF-α genetically is also detrimental, because it delays microglial activation, which is later expressed in an excessive manner. However, these effects do not extend to the IL-1B. Thus, this suggests that the source, timing, and dose of TNF-α are preponderant in determining the fate of neurons and myelin during SNP-induced neurotoxicity [29].
Additionally, when infused into the substantia nigra, SNP induces an acute increase in lipid peroxidation, which is blocked by NO, oxyhemoglobin, and deferoxamine (an iron chelator), suggesting that OS is elicited, at least in part, by the iron moiety of SNP [110, 111]. Thus, the addition of SNP and other NOD to neuronal cultures or into brain parenchyma causes damage through the establishment of OS, nitrosative stress, and the disturbance of cellular oxidative metabolism. The death pathway activated through these mechanisms is mainly apoptotic.
However, it should be considered that the cytotoxic effects of NOD are not necessarily due to the presence of NO, because its addition to culture media alone does not cause neurotoxicity [112]; therefore, other compounds that are also part of the NOD molecules can be delivered with different effects. Such is the case of SNP, in which its toxicity lies more in its content of iron [110, 112, 113] and cyanide ions [114] rather than in its NO content or, as the case of SIN-1, whose decomposition releases superoxide anion and hydroxyl radicals along with NO, leading to the production of large amounts of ONOO− [96].
Finally, it should be taken into account that the cytoprotective and physiological effects of NO described (e.g., vasodilatation, neurotransmission, endothelial protection) require extremely small concentrations (pico- to nanomolar), while harmful effects take place at higher concentrations, particularly under OS [93]. In culture and in intracerebral application, the NOD concentrations usually administered fall within the micro- to millimolar range. Thus, direct contact of NOD with neurons in culture or intraparenchymally coupled with the high concentration of these could be mimicking overactivation of nNOS or iNOS during the postischemic reperfusion period or in other neurodegenerative disorders [83]. Figure 1 depicts the signaling pathways involved in the neurotoxic effects of NO.
Figure 1.
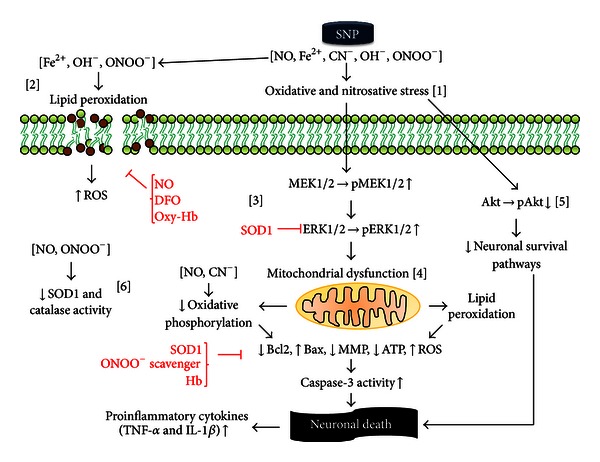
In vitro and intracerebral effects of sodium nitroprusside and other nitric oxide donors (NOD) on neuronal survival. SNP is capable of releasing or producing diverse byproducts, such as nitric oxide (NO), iron, cyanide anions, hydroxyl radicals, and peroxynitrite. Collectively, these are all capable of inducing oxidative and nitrosative stress [1], with the possibility of modifying the structure and function of proteins, nucleic acids, and lipids by means of oxidation and nitrosylation. Iron, via the Fenton reaction, generates OH− that, together with ONOO− and other reactive species, damage membranes by lipid peroxidation [2] with decreased cellular viability. This effect is blocked by the addition of NO, oxyhemoglobin, and deferoxamine, which suggests the important role of iron and NO in this reaction. The oxidative stress (OS) produced by SNP increases the activation of MEK1/2 and its substrate ERK1/2 by phosphorylation [3]. Both effects are blocked by SOD, suggesting the participation of (O2−) in this reaction, probably in the form of ONOO−. Activation of ERK1/2 is associated with a reduction of Bcl2 and an increase in (Bax), and both conditions are associated with an activation of mitochondrial apoptotic pathways. Mitochondria are a target of SNP at different levels: SNP induces lipid peroxidation of its membrane with the subsequent activation of proapoptotic pathways via caspases. In addition, NO and CN− affect the functioning of the mitochondrial respiratory chain, thereby altering mitochondrial membrane potential, reducing ATP production and the generation of large amounts of reactive oxygen species [4]. The addition of ONOO− scavengers and SOD1 counteracts this effect. Also, SNP decreases Akt phosphorylation [5] and reduces the expression and function of SOD1 and catalase [6]. These actions decrease antioxidant responsiveness and the activation of neuronal survival pathways. OH−, hydroxyl radical; ONOO−, peroxynitrite; Akt, protein kinase B (PKB); Bax, Bcl-2-associated X protein; Bcl2, B-cell lymphoma 2; CN−, cyanide anion; ERK1/2, extracellular signal-regulated kinase 1/2; IL-1β, interleukin 1 beta; MEK1/2, mitogen-activated protein kinase kinase 1/2; MMP, mitochondrial membrane potential; NO, nitric oxide; ROS, reactive oxygen species; SNP, sodium nitroprusside; SOD1, superoxide dismutase (Cu-Zn); TNF-α, tumor necrosis factor alpha.
3.4. Nitric Oxide as an Anti-Inflammatory and Neuroprotective Agent
In contrast to the evidence presented in the previous section based on the reactivity of NO with iron and ROS, in 1994 Chiueh proposed that NO and related donor compounds may protect against the OS induced by small-molecular-weight iron complexes in the dopaminergic nigrostriatal system [115]. Since then, a growing number of reports have confirmed the potent neuroprotective and antioxidant actions of NO in the brain in experimental models of Parkinson disease [116–120]. In addition, NO has shown to inhibit lipid peroxidation of low-density lipoprotein oxidation [121, 122] in order to protect against neurotoxin-induced dopaminergic neurotoxicity [112, 118, 123, 124] and to shield cells from OS [125–127], protecting these in vivo through both antioxidative and -apoptotic mechanisms [118].
In the hippocampus, NO mediates cellular transduction mechanisms, regulates neuronal plasticity [128], and suppresses neuronal apoptotic cell death [129]. Thus, NO may be neuroprotective or restorative after a stroke [130–132], after traumatic brain injury [133, 134], and during Alzheimer disease [135] and depression [136].
3.5. NO Donors Exerted a Neuroprotective Effect against Cerebral Ischemia-Reperfusion Injury at Different Levels by Influencing Cellular Oxidative Status
SNP and SPERMINE-NONOate are able to reduce infarct size after transient focal cerebral ischemia when administered early [87]. Likewise, S-nitrosothiols (GSNO and SNAP) and SIN-1 additionally reduced infarct volume and improved neurological performance [38, 88, 89]. Hemodynamically, SNP, GSNO, and SNAP increase CBF in the penumbral region when administered at the onset of reperfusion [38, 80, 89].
In addition, pre- and postischemic administration of SNP attenuates the ischemia-induced increase of caspase-3 at 6 h of reperfusion and downregulates neuronal apoptosis by inhibiting increased phosphorylation of JNK, c-Jun, and Bcl-2 [61, 65]. This effect is achieved through nitrosylation of nNOS, which decreases its NO production. This means that SNP can regulate NO metabolism in the target cells. In focal ischemia, SNP and S-nitrosothiols decrease lipid peroxidation and nitrotyrosine formation in plasma, which is associated with less oxidative and nitrosative stress, neuroprotection, and fewer anti-inflammatory effects [38].
The effects of NOD on ischemia-reperfusion injury are also related with modulation of the inflammatory response, and these effects are probably the neuroprotective effects with the greatest impact after cerebral ischemia and reperfusion of this drug type. To shield nerve tissue from ischemia-reperfusion injury is not sufficient to protect the brain parenchyma, but also must preserve the integrity of the Blood brain barrier (BBB). Thus, the cerebral vascular endothelium is essential in the control of vascular inflammatory and oxidative responses, leukocyte migration, and the production of inflammatory mediators capable of spreading to nerve tissue [26]. Under physiological conditions, eNOS-NO is responsible for maintaining the integrity of the vascular endothelium. But under ischemic conditions, endothelial dysfunction could be offset by mimicking eNOS-derived NO neuroprotective functions by intravascular administration of a NOD [137]. Control of endothelial inflammatory and oxidative responses in turn allows restriction of their impact on resident brain cells, particularly on those with an inflammatory phenotype, such as microglia and astrocytes. Therefore, effective neuroprotection should include protection of the BBB and of the elements within it [138, 139].
In vivo, high expression of TNF-α, IL-1β, and iNOS in microglia and astrocytes after focal cerebral ischemia is reduced by GSNO. Likewise, GSNO induces a reduction in microglial and macrophage cells in the penumbral region, which is associated with less expression of cellular adhesion molecules such as ICAM-1 in endothelial cells [56]. Decreased expression of adhesion molecules (ICAM-1 and E-selectin) was also demonstrated with SNP and SNAP in the same model [38].
Anti-inflammatory effects are also demonstrated in other neuronal-damaged models, such as experimental autoimmune encephalomyelitis [140] and traumatic brain injury [134]. Under both conditions, NOD inhibited the expression of cell adhesion molecules and infiltration of vascular immune cells into the CNS, which subsequently led to reduction in the expression of proinflammatory cytokines at the site of injury. This suggests less damage to BBB integrity, which is an indicator of neuroprotection.
Beyond the CNS, SNP protects other organs from inflammatory damage. In cardiac surgery, SNP decreases cardiac cytokine release [141–143] and improves postischemic cardiac function [143]. In experimental models of ischemia-reperfusion injury, such as those in kidney [144] and lung [145], SNP attenuates the expression of proinflammatory cytokines and reduces leukocyte-endothelium adhesion, respectively.
3.6. Anti-Inflammatory Mechanisms of Nitric Oxide Donors
In vitro studies have elucidated some of the mechanisms involved in the anti-inflammatory effect of NOD. It is well documented that cerebral ischemia, and particularly reperfusion, leads to nuclear translocation of NF-κB into the core and ischemic penumbra [146, 147], as well as into the microvessels of the affected region [148, 149]. NF-κB is a key regulator of innate immunity, inflammation, and of cell survival and proliferation [150]. This inducible transcription factor is comprised of two subunits. There are five subunits that can be combined to yield homo- or heterodimers of NF-κB as follows: p50, p52, c-Rel, p65 (RelA), and RelB [151]. C-Rel-containing dimer activation increases neuron resistance to ischemia [152]. Moreover, the prevalent heterodimer during cerebral ischemia and reperfusion is formed by p50- and p65-inducible subunits, and its activation contributes to the pathogenesis of postischemic injury [146, 152, 153]. NF-κB is maintained in latent form in the cytoplasm of cells bound to inhibitory IκB proteins. Phosphorylation of IκB releases NF-κB by permitting its translocation into the nucleus, its binding with NF-κB motifs, and the subsequent activation of its target genes. There is, in turn, an enzymatic complex responsible for IκB phosphorylation in specific serine residues, the so-called IκB kinases (IKK). Activation of IKK is essential to induce NF-κB activity [154].
In the ischemic brain, a wide range of stimuli may trigger activation of NF-κB including, among others, the following: hypoxia [155]; IL-1, and TNF-α [156]; OS [157]; glutamate [158], and NOS activity, such as nNOS and iNOS [159]. Overactivation of NF-κB after ischemia has been documented in neurons [147], astrocytes [53], microglia [160], and in endothelial cells [149]. Although in some hippocampal neurons NF-κB have a constitutive action related with neuronal survival [150], overactivation of the p50/p65 heterodimer in neurons, glial, and endothelial cells due to ischemia, appears to contribute to acute neurodegeneration. In neurons, NF-κB translocation has been associated with apoptosis [146, 147], while in glia and in vascular endothelium, NF-κB activates a proinflammatory phenotype [53, 149, 160].
Therefore, blocking inflammatory phenotype activation of NF-κB could disrupt the cascade of events that culminate in proinflammatory brain tissue destruction. In human endothelial cells, the addition of exogenous NO through GSNO limits TNF-α activation of NF-κB in a time- and concentration-dependent manner [161]. This is also achieved by SNP upon stimulation with IL-1α, IL-1β, IL-4, and LPS [162]. NF-κB inhibition is in fact sustained by the constitutive activity of eNOS, because its inhibition, without an inflammatory stimulus, triggers nuclear translocation of NF-κB [156, 161, 162]. By inhibiting activation of the transcription factor, NO effectively blocks monocyte adhesion, as well as the expression of the proinflammatory target genes of NF-κB, such as TNF-α, IL-6, iNOS, V-CAM, ICAM-1, E-selectin, and COX-2 [70, 156, 162–164].
In astrocytes and microglial cells, NOD also exhibits an anti-inflammatory profile through downregulation of NF-κB. In primary rat astrocytes and in a BV2 microglial cell line, GSNO mitigates iNOS production by inhibiting the ability of NF-κB to bind to DNA [53]. Therefore, NOD are capable not only of regulating NF-κB at the vascular level, but also they possess the capability of influencing glial cell reactivity and limiting their production of iNOS, proinflammatory cytokines, and other molecules and prooxidant enzymes that are potentially harmful to neuronal cells.
Inhibition of NF-κB by exogenous NO has been documented at different levels. In the brain ischemic environment, activation of NF-κB occurs, at least in part, via ROS [165]. Former researches have shown that one of the most significant sources of ROS in the ischemic brain is through the metabolism of arachidonic acid by Cyclooxygenase (COX) [166, 167]. COX-2 expression is increased in brain tissue after global [168] and focal [167, 169] cerebral ischemia. ROS are produced by the peroxidase step of the COX reaction in which prostaglandin G2 is converted into prostaglandin H2 [170, 171]. Hence, reducing COX-2 activity reduces oxidative damage of the ischemic brain [167, 169]. The NO donors GSNO and SNP are able to downregulate LPS-induced COX-2 protein expression via inhibition of NF-κB DNA binding activity in murine monocytes [172]. Therefore, both drugs may be candidates for neuroprotective antioxidants in cerebral ischemia. In addition, NO is a superoxide scavenger; hence, NO may inhibit NF-κB by scavenging superoxide anion [162].
Furthermore, SNP is capable of interfering directly with the ability of NF-κB to translocate into the nucleus. Specifically, SNP inactivates NF-κB by nitration of the p65 subunit at Tyr-66 and Tyr-152. This protein modification suppresses iNOS mRNA expression and prevents the activation of NF-κB target genes by TNF-α stimulation [164]. S-nitrosylation of the p50 subunit at Cys-62 has also been demonstrated as a major mechanism of NO regulation by inhibition of the p50 binding to its consensus DNA target sequence [173].
With respect to IκB-α, exogenous NO increases mRNA IκB levels and stabilizes the complex formed with NF-κB [161, 172]. This stabilization is related with S-nitrosylation of the Cys-179 of IKKß, which decreases its ability to phosphorylate IκB [174]. Additionally, NO interferes with the transient degradation of IκB-α induced by cytokines [70]. These three actions induce negative regulation of NF-κB DNA-binding activity by NOD. Figure 2 sums up the neuroprotective actions of NOD.
Figure 2.
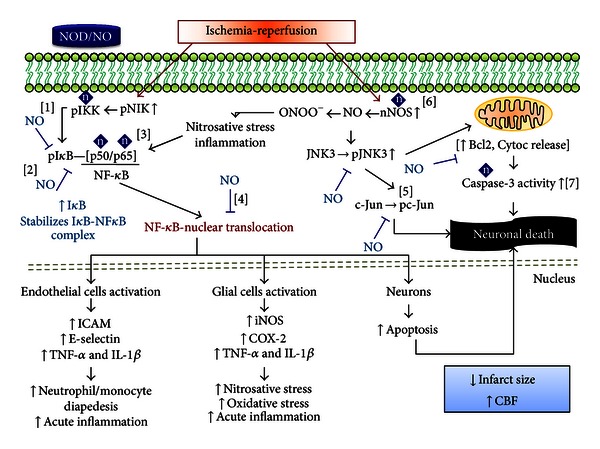
Cerebral ischemia-reperfusion activates two major signaling pathways which exert an effect on NOD. (A) NF-κB pathway. Oxidative stress (OS) and inflammatory stimuli phosphorylate NIK. Subsequently, IKK phosphorylates NIK, which in turn phosphorylates IkB, resulting in Iκb degradation and NF-κB translocation and activation. This action exerts different effects depending on the cell line. In endothelial cells, NF-κB promotes a proinflammatory phenotype, with the expression of cellular adhesion molecules and proinflammatory cytokines that induce leukocyte migration to the ischemic territory and promote acute inflammation. In glial cells, NF-κB leads to the expression of iNOS, COX-2, and proinflammatory cytokines. These effects contribute to nitrosative, oxidative, and inflammatory damage. Finally, in neurons, NF-κB induces the expression of apoptosis pathways. NOD can act at different levels in this pathway: NOD-derived NO diffuses across target cell membranes, where it is able to nitrosylate kinases located upstream of NF-κB, such as IKK, thereby suppressing their ability to phosphorylate [1]. This inhibition prevents Iκb phosphorylation and its degradation; thus the release of NF-κB. NO can also increase Iκb gene transcription and stabilize the complex formed by Iκb and NF-κB [2]. Furthermore, NOD-derived NO is capable of nitrosylating directly into the p50 and p65 subunits of NF-κB, which blocks their ability to migrate to the nucleus [3]. All of these actions prevent the nuclear translocation of NF-κB, therefore the expression of their target genes [4]. (B) Cerebral ischemia-reperfusion increases nNOS activity, which enhances its NO production. This NO can react with free radicals to produce ONOO− and also activates the JNK3 pathway. The result is c-Jun phosphorylation and mitochondrial dysfunction, with an increase in Bcl2 phosphorylation and cytochrome C release into the cytoplasm. In addition, this activates caspase-3 and leads to neuronal apoptosis. NOD-derived NO downregulates neuronal apoptosis by inhibiting increased phosphorylation of JNK, c-Jun, and Bcl-2 [5]. This is achieved by S-nitrosylation of nNOS, which interferes with its NO production [6]. NO is also capable of nitrosylating caspase-3 directly [7]. All of these effects, along with an increase in CBF, reduce brain damage after the ischemia-reperfusion event. Bcl2, B-cell lymphoma 2; CBF, cerebral blood flow; COX-2, cyclooxygenase 2; Cytoc, cytochrome; ICAM, intercellular adhesion molecule; IkB, inhibitors of kB; IKK, Iκb kinase; IL-1β, interleukin 1 beta; iNOS, inducible nitric oxide synthase; JNK3, c-Jun N-terminal kinases 3; NF-κB, nuclear factor kappa B; NIK, NF-κB-inducing kinase; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; NOD, nitric oxide donors; ONOO−, peroxynitrite; TNF-α, tumor necrosis factor alpha; [n] S-nitrosylation.
4. Concluding Remarks
Understanding the interaction between the CNS and the immune system will provide greater insight into several different pathologies that involve CNS inflammation and the increase in the number of potential pharmacological targets.
The great variability in the observed effects elicited by NOD, from neuroprotection to toxicity, could be due to the great diversity in doses used in the experiments, which in fact are mainly distant from the existing physiological concentrations. Clarity about the NO concentrations that exists physiologically is essential for developing a quantitative understanding of NO signaling, for performing experiments with NO that emulate reality, and for knowing whether or not NO concentrations become abnormal in disease states [175]. Several independent lines of evidence suggest that NO operates physiologically at concentrations that are orders of magnitude lower than the near-micromolar order once considered correct.
Accordingly, physiological NO concentrations range from 100 pM (or below) up to 5 nM (reviewed in [175]). Therefore, the establishment of reliable methods for directing microelectrode measurement of NO concentrations and the (most foreseeable) progression of newly developed NO biosensors for quantitative imaging of NO signaling in subcellular dimensions and in real time in tissues in vivo will facilitate advances in this fundamental, but yet unsettled, area.
In addition to NOD concentration, it is relevant to consider the administration pathway (intravascular, intraperitoneal, or directly into the culture media in vitro), as well as the cell type in which the donor exerts its action, together with the cell redox state (reduction-oxidation), because these factors are determining ones in selecting the signaling pathway that will be affected or modified by the action of these.
Therefore, therapeutic use of these molecules must be performed carefully, because they can be beneficial for one tissue or cell type and harmful for others. Given their short therapeutic window, NOD appear appropriate for use during neurosurgical procedures involving transient arterial occlusions or in very early treatment of acute ischemic stroke [87].
At present, translation from in vitro to in vivo preclinical stroke models requires further research, as clearly as that required for the case for translation from in vivo animal models to the clinical condition of drugs for treatment of acute ischemic stroke, which requires overcoming phase III trials in patients.
Acknowledgments
This work was partially supported by Universidad de Guadalajara (UdeG) under Grant P3E/2012/137505 to D. Ortuño-Sahagún, and Grants CONACyT-México grant 2012-180268 and PROMEP/103.5/12/8143 to A. E. Rojas-Mayorquín. The authors apologize to authors whose works have not been reviewed and to those whose papers have not received the emphasis that they merit. They also apologize to authors whose work has not been appropriately cited due to space limitations and/or to limitations of their knowledge.
References
- 1.Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. Journal of Translational Medicine. 2009;7, article 97 doi: 10.1186/1479-5876-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nature Reviews Neuroscience. 2003;4(5):399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 3.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nature Reviews Neuroscience. 2004;5(5):347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 4.del Zoppo GJ. Stroke and neurovascular protection. The New England Journal of Medicine. 2006;354(6):553–555. doi: 10.1056/NEJMp058312. [DOI] [PubMed] [Google Scholar]
- 5.Lok J, Gupta P, Guo S, et al. Cell-cell signaling in the neurovascular unit. Neurochemical Research. 2007;32(12):2032–2045. doi: 10.1007/s11064-007-9342-9. [DOI] [PubMed] [Google Scholar]
- 6.Lu D, Maulik N, Moraru II, Kreutzer DL, Das DK. Molecular adaptation of vascular endothelial cells to oxidative stress. The American Journal of Physiology. 1993;264(3, part 1):C715–C722. doi: 10.1152/ajpcell.1993.264.3.C715. [DOI] [PubMed] [Google Scholar]
- 7.Lee BJ, Egi Y, van Leyen K, Lo EH, Arai K. Edaravone, a free radical scavenger, protects components of the neurovascular unit against oxidative stress in vitro. Brain Research. 2010;1307:22–27. doi: 10.1016/j.brainres.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warner DS, Sheng H, Batinić-Haberle I. Oxidants, antioxidants and the ischemic brain. The Journal of Experimental Biology. 2004;207(18):3221–3231. doi: 10.1242/jeb.01022. [DOI] [PubMed] [Google Scholar]
- 9.Syntichaki P, Tavernarakis N. The biochemistry of neuronal necrosis: rogue biology? Nature Reviews Neuroscience. 2003;4(8):672–684. doi: 10.1038/nrn1174. [DOI] [PubMed] [Google Scholar]
- 10.Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Research Reviews. 2007;54(1):34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 12.Gandhi S, Abramov AY. Mechanism of oxidative stress in neurodegeneration. Oxidative Medicine and Cellular Longevity. 2012;2012:11 pages. doi: 10.1155/2012/428010.428010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies KJ. Oxidative stress: the paradox of aerobic life. Biochemical Society Symposium. 1995;61:1–31. doi: 10.1042/bss0610001. [DOI] [PubMed] [Google Scholar]
- 14.Mastaloudis A, Leonard SW, Traber MG. Oxidative stress in athletes during extreme endurance exercise. Free Radical Biology & Medicine. 2001;31(7):911–922. doi: 10.1016/s0891-5849(01)00667-0. [DOI] [PubMed] [Google Scholar]
- 15.Shi H, Liu KJ. Cerebral tissue oxygenation and oxidative brain injury during ischemia and reperfusion. Frontiers in Bioscience. 2007;12(4):1318–1328. doi: 10.2741/2150. [DOI] [PubMed] [Google Scholar]
- 16.Broughton BRS, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40(5):e331–e339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- 17.Barber PA, Demchuk AM, Hirt L, Buchan AM. Biochemistry of ischemic stroke. Advances in Neurology. 2003;92:151–164. [PubMed] [Google Scholar]
- 18.Drexler H, Hornig B. Endothelial dysfunction in human disease. Journal of Molecular and Cellular Cardiology. 1999;31(1):51–60. doi: 10.1006/jmcc.1998.0843. [DOI] [PubMed] [Google Scholar]
- 19.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circulation Research. 2000;87(10):840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 20.Niizuma K, Endo H, Chan PH. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. Journal of Neurochemistry. 2009;109(supplement 1):133–138. doi: 10.1111/j.1471-4159.2009.05897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmad M, Graham SH. Inflammation after stroke: mechanisms and therapeutic approaches. Translational Stroke Research. 2010;1(2):74–84. doi: 10.1007/s12975-010-0023-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong CHY, Crack PJ. Modulation of neuro-inflammation and vascular response by oxidative stress following cerebral ischemia-reperfusion injury. Current Medicinal Chemistry. 2008;15(1):1–14. doi: 10.2174/092986708783330665. [DOI] [PubMed] [Google Scholar]
- 23.Beer C, Blacker D, Hankey GJ, Puddey IB. Association of clinical and aetiologic subtype of acute ischaemic stroke with inflammation, oxidative stress and vascular function: a cross-sectional observational study. Medical Science Monitor. 2011;17(9):467–473. doi: 10.12659/MSM.881931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pandya RS, Mao L, Zhou H, et al. Central nervous system agents for ischemic stroke: neuroprotection mechanisms. Central Nervous System Agents in Medicinal Chemistry. 2011;11(2):81–97. doi: 10.2174/187152411796011321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schnegg CI, Robbins ME. Neuroprotective mechanisms of PPARδ: modulation of oxidative stress and inflammatory processes. PPAR Research. 2011;2011:10 pages. doi: 10.1155/2011/373560.373560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ceulemans AG, Zgavc T, Kooijman R, Hachimi-Idrissi S, Sarre S, Michotte Y. The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia. Journal of Neuroinflammation. 2010;7, article 74 doi: 10.1186/1742-2094-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Downes CE, Crack PJ. Neural injury following stroke: are Toll-like receptors the link between the immune system and the CNS? British Journal of Pharmacology. 2010;160(8):1872–1888. doi: 10.1111/j.1476-5381.2010.00864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zierath D, Thullbery M, Hadwin J, et al. CNS immune responses following experimental stroke. Neurocritical Care. 2010;12(2):274–284. doi: 10.1007/s12028-009-9270-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turrin NP, Rivest S. Tumor necrosis factor α but not interleukin 1β mediates neuroprotection in response to acute nitric oxide excitotoxicity. The Journal of Neuroscience. 2006;26(1):143–151. doi: 10.1523/JNEUROSCI.4032-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stoll G, Jander S, Schroeter M. Detrimental and beneficial effects of injury-induced inflammation and cytokine expression in the nervous system. Advances in Experimental Medicine and Biology. 2002;513:87–113. doi: 10.1007/978-1-4615-0123-7_3. [DOI] [PubMed] [Google Scholar]
- 31.Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. The Journal of Neuroscience. 2002;22(7):p. RC216. doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stoll G, Jander S, Schroeter M. Cytokines in CNS disorders: neurotoxicity versus neuroprotection. Journal of Neural Transmission, Supplement. 2000;(59):81–89. doi: 10.1007/978-3-7091-6781-6_11. [DOI] [PubMed] [Google Scholar]
- 33.Lo EH. Combination stroke therapy: easy as APC? Nature Medicine. 2004;10(12):1295–1296. doi: 10.1038/nm1204-1295. [DOI] [PubMed] [Google Scholar]
- 34.Fagan SC, Hess DC, Machado LS, Hohnadel EJ, Pollock DM, Ergul A. Tactics for vascular protection after acute ischemic stroke. Pharmacotherapy. 2005;25(3):387–395. doi: 10.1592/phco.25.3.387.61592. [DOI] [PubMed] [Google Scholar]
- 35.Cirino G, Fiorucci S, Sessa WC. Endothelial nitric oxide synthase: the cinderella of inflammation? Trends in Pharmacological Sciences. 2003;24(2):91–95. doi: 10.1016/S0165-6147(02)00049-4. [DOI] [PubMed] [Google Scholar]
- 36.Cirino G, Distrutti E, Wallace JL. Nitric oxide and inflammation. Inflammation & Allergy-Drug Targets. 2006;5(2):115–119. doi: 10.2174/187152806776383143. [DOI] [PubMed] [Google Scholar]
- 37.Rodrigo J, Fernández AP, Serrano J, Peinado MA, Martínez A. The role of free radicals in cerebral hypoxia and ischemia. Free Radical Biology & Medicine. 2005;39(1):26–50. doi: 10.1016/j.freeradbiomed.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 38.Khan M, Jatana M, Elango C, Paintlia AS, Singh AK, Singh I. Cerebrovascular protection by various nitric oxide donors in rats after experimental stroke. Nitric Oxide. 2006;15(2):114–124. doi: 10.1016/j.niox.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 39.Iadecola C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends in Neurosciences. 1997;20(3):132–139. doi: 10.1016/s0166-2236(96)10074-6. [DOI] [PubMed] [Google Scholar]
- 40.Guix FX, Uribesalgo I, Coma M, Muñoz FJ. The physiology and pathophysiology of nitric oxide in the brain. Progress in Neurobiology. 2005;76(2):126–152. doi: 10.1016/j.pneurobio.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 41.Adachi N, Lei B, Soutani M, Arai T. Different roles of neuronal and endothelial nitric oxide synthases on ischemic nitric oxide production in gerbil striatum. Neuroscience Letters. 2000;288(2):151–154. doi: 10.1016/s0304-3940(00)01222-2. [DOI] [PubMed] [Google Scholar]
- 42.Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. The Journal of Neuroscience. 1997;17(23):9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stanarius A, Töpel I, Schulz S, Noack H, Wolf G. Immunocytochemistry of endothelial nitric oxide synthase in the rat brain: a light and electron microscopical study using the tyramide signal amplification technique. Acta Histochemica. 1997;99(4):411–429. doi: 10.1016/S0065-1281(97)80034-7. [DOI] [PubMed] [Google Scholar]
- 44.Kuhlencordt PJ, Rosel E, Gerszten RE, et al. Role of endothelial nitric oxide synthase in endothelial activation: insights from eNOS knockout endothelial cells. The American Journal of Physiology. 2004;286(5):C1195–C1202. doi: 10.1152/ajpcell.00546.2002. [DOI] [PubMed] [Google Scholar]
- 45.Abu Nabah YN, Mateo T, Cerdá-Nicolás M, et al. L-NAME induces direct arteriolar leukocytes adhesion, which is mainly mediated by angiotensin-II. Microcirculation. 2005;12(5):443–453. doi: 10.1080/10739680590960962. [DOI] [PubMed] [Google Scholar]
- 46.Moore C, Sanz-Rosa D, Emerson M. Distinct role and location of the endothelial isoform of nitric oxide synthase in regulating platelet aggregation in males and females in vivo. European Journal of Pharmacology. 2011;651(1–3):152–158. doi: 10.1016/j.ejphar.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 47.Hossain M, Qadri SM, Liu L. Inhibition of nitric oxide synthesis enhances leukocyte rolling and adhesion in human microvasculature. Journal of Inflammation. 2012;9, article 28 doi: 10.1186/1476-9255-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ito Y, Ohkubo T, Asano Y, et al. Nitric oxide production during cerebral ischemia and reperfusion in eNOS- and nNOS-knockout mice. Current Neurovascular Research. 2010;7(1):23–31. doi: 10.2174/156720210790820190. [DOI] [PubMed] [Google Scholar]
- 49.Liu K, Li Q, Zhang L, Zheng X. The dynamic detection of NO during stroke and reperfusion in vivo. Brain Injury. 2009;23(5):450–458. doi: 10.1080/02699050902838173. [DOI] [PubMed] [Google Scholar]
- 50.Zhou L, Zhu DY. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide. 2009;20(4):223–230. doi: 10.1016/j.niox.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 51.Wei G, Dawson VL, Zweier JL. Role of neuronal and endothelial nitric oxide synthase in nitric oxide generation in the brain following cerebral ischemia. Biochimica et Biophysica Acta. 1999;1455(1):23–34. doi: 10.1016/s0925-4439(99)00051-4. [DOI] [PubMed] [Google Scholar]
- 52.Iadecola C, Zhang F, Xu X. Inhibition of inducible nitric oxide synthase ameliorates cerebral ischemic damage. The American Journal of Physiology. 1995;268(1):R286–R292. doi: 10.1152/ajpregu.1995.268.1.R286. [DOI] [PubMed] [Google Scholar]
- 53.Khan M, Sekhon B, Giri S, et al. S-Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. Journal of Cerebral Blood Flow and Metabolism. 2005;25(2):177–192. doi: 10.1038/sj.jcbfm.9600012. [DOI] [PubMed] [Google Scholar]
- 54.Gibson CL, Coughlan TC, Murphy SP. Glial nitric oxide and ischemia. GLIA. 2005;50(4):417–426. doi: 10.1002/glia.20143. [DOI] [PubMed] [Google Scholar]
- 55.Niwa M, Inao S, Takayasu M, et al. Time course of expression of three nitric oxide synthase isoforms after transient middle cerebral artery occlusion in rats. Neurologia Medico-Chirurgica. 2001;41(2):63–73. doi: 10.2176/nmc.41.63. [DOI] [PubMed] [Google Scholar]
- 56.Suzuki M, Tabuchi M, Ikeda M, Tomita T. Concurrent formation of peroxynitrite with the expression of inducible nitric oxide synthase in the brain during middle cerebral artery occlusion and reperfusion in rats. Brain Research. 2002;951(1):113–120. doi: 10.1016/s0006-8993(02)03145-1. [DOI] [PubMed] [Google Scholar]
- 57.Pannu R, Singh I. Pharmacological strategies for the regulation of inducible nitric oxide synthase: neurodegenerative versus neuroprotective mechanisms. Neurochemistry International. 2006;49(2):170–182. doi: 10.1016/j.neuint.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 58.Huang Z, Huang PL, Ma J, et al. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. Journal of Cerebral Blood Flow and Metabolism. 1996;16(5):981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- 59.Li R, Guo M, Zhang G, Xu X, Li Q. Nicotiflorin reduces cerebral ischemic damage and upregulates endothelial nitric oxide synthase in primarily cultured rat cerebral blood vessel endothelial cells. Journal of Ethnopharmacology. 2006;107(1):143–150. doi: 10.1016/j.jep.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 60.Hara H, Huang PL, Panahian N, Fishman MC, Moskowitz MA. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. Journal of Cerebral Blood Flow and Metabolism. 1996;16(4):605–611. doi: 10.1097/00004647-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 61.Pei D-S, Song Y-J, Yu H-M, Hu W-W, Du Y, Zhang G-Y. Exogenous nitric oxide negatively regulates c-Jun N-terminal kinase activation via inhibiting endogenous NO-induced S-nitrosylation during cerebral ischemia and reperfusion in rat hippocampus. Journal of Neurochemistry. 2008;106(4):1952–1963. doi: 10.1111/j.1471-4159.2008.05531.x. [DOI] [PubMed] [Google Scholar]
- 62.Nanri K, Montécot C, Springhetti V, Seylaz J, Pinard E. The selective inhibitor of neuronal nitric oxide synthase, 7- nitroindazole, reduces the delayed neuronal damage due to forebrain ischemia in rats. Stroke. 1998;29(6):1248–1254. doi: 10.1161/01.str.29.6.1248. [DOI] [PubMed] [Google Scholar]
- 63.Gürsoy-Özdemir Y, Bolay H, Saribaş O, Dalkara T. Role of endothelial nitric oxide generation and peroxynitrite formation in reperfusion injury after focal cerebral ischemia. Stroke. 2000;31(8):1974–1981. doi: 10.1161/01.str.31.8.1974. [DOI] [PubMed] [Google Scholar]
- 64.Sun M, Zhao Y, Gu Y, Xu C. Inhibition of nNOS reduces ischemic cell death through down-regulating calpain and caspase-3 after experimental stroke. Neurochemistry International. 2009;54(5-6):339–346. doi: 10.1016/j.neuint.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 65.Yu HM, Xu J, Li C, et al. Coupling between neuronal nitric oxide synthase and glutamate receptor 6-mediated c-Jun N-terminal kinase signaling pathway via S-nitrosylation contributes to ischemia neuronal death. Neuroscience. 2008;155(4):1120–1132. doi: 10.1016/j.neuroscience.2008.03.061. [DOI] [PubMed] [Google Scholar]
- 66.Eliasson MJL, Huang Z, Ferrante RJ, et al. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. The Journal of Neuroscience. 1999;19(14):5910–5918. doi: 10.1523/JNEUROSCI.19-14-05910.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gürsoy-Ozdemir Y, Can A, Dalkara T. Reperfusion-induced oxidative/nitrative injury to neurovascular unit after focal cerebral ischemia. Stroke. 2004;35(6):1449–1453. doi: 10.1161/01.STR.0000126044.83777.f4. [DOI] [PubMed] [Google Scholar]
- 68.Danielisova V, Burda J, Nemethova M, Gottlieb M. Aminoguanidine administration ameliorates hippocampal damage after middle cerebral artery occlusion in rat. Neurochemical Research. 2011;36(3):476–486. doi: 10.1007/s11064-010-0366-1. [DOI] [PubMed] [Google Scholar]
- 69.Trickler WJ, Mayhan WG, Miller DW. Brain microvessel endothelial cell responses to tumor necrosis factor-alpha involve a nuclear factor kappa B (NF-κB) signal transduction pathway. Brain Research. 2005;1048(1-2):24–31. doi: 10.1016/j.brainres.2005.04.028. [DOI] [PubMed] [Google Scholar]
- 70.Katsuyama K, Shichiri M, Marumo F, Hirata Y. NO inhibits cytokine-induced iNOS expression and NF-κB activation by interfering with phosphorylation and degradation of IκB-α . Arteriosclerosis, Thrombosis, and Vascular Biology. 1998;18(11):1796–1802. doi: 10.1161/01.atv.18.11.1796. [DOI] [PubMed] [Google Scholar]
- 71.Foncea R, Carvajal C, Almarza C, Leighton F. Endothelial cell oxidative stress and signal transduction. Biological Research. 2000;33(2):89–96. doi: 10.4067/s0716-97602000000200008. [DOI] [PubMed] [Google Scholar]
- 72.ArunaDevi R, Ramteke VD, Kumar S, et al. Neuroprotective effect of s-methylisothiourea in transient focal cerebral ischemia in rat. Nitric Oxide. 2010;22(1):1–10. doi: 10.1016/j.niox.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 73.Samdani AF, Dawson TM, Dawson VL. Nitric oxide synthase in models of focal ischemia. Stroke. 1997;28(6):1283–1288. doi: 10.1161/01.str.28.6.1283. [DOI] [PubMed] [Google Scholar]
- 74.Jackson-Lewis V, Smeyne RJ. MPTP and SNpc DA neuronal vulnerability: role of dopamine, superoxide and nitric oxide in neurotoxicity. Neurotoxicity Research. 2005;7(3):193–201. doi: 10.1007/BF03036449. [DOI] [PubMed] [Google Scholar]
- 75.Marchetti B, Serra PA, Tirolo C, et al. Glucocorticoid receptor-nitric oxide crosstalk and vulnerability to experimental parkinsonism: pivotal role for glia-neuron interactions. Brain Research Reviews. 2005;48(2):302–321. doi: 10.1016/j.brainresrev.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 76.Aliev G, Palacios HH, Lipsitt AE, et al. Nitric oxide as an initiator of brain lesions during the development of Alzheimer disease. Neurotoxicity Research. 2009;16(3):293–305. doi: 10.1007/s12640-009-9066-5. [DOI] [PubMed] [Google Scholar]
- 77.Malinski T. Nitric oxide and nitroxidative stress in Alzheimer’s disease. Journal of Alzheimer’s Disease. 2007;11(2):207–218. doi: 10.3233/jad-2007-11208. [DOI] [PubMed] [Google Scholar]
- 78.Simic G, Lucassen PJ, Krsnik Ž, et al. nNOS expression in reactive astrocytes correlates with increased cell death related DNA damage in the hippocampus and entorhinal cortex in Alzheimer’s disease. Experimental Neurology. 2000;165(1):12–26. doi: 10.1006/exnr.2000.7448. [DOI] [PubMed] [Google Scholar]
- 79.Jeynes B, Provias J. Significant negative correlations between capillary expressed eNOS and Alzheimer lesion burden. Neuroscience Letters. 2009;463(3):244–248. doi: 10.1016/j.neulet.2009.07.091. [DOI] [PubMed] [Google Scholar]
- 80.Zhang F, White JG, Iadecola C. Nitric oxide donors increase blood flow and reduce brain damage in focal ischemia: evidence that nitric oxide is beneficial in the early stages of cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism. 1994;14(2):217–226. doi: 10.1038/jcbfm.1994.28. [DOI] [PubMed] [Google Scholar]
- 81.Zhang F, Xu S, Iadecola C. Time dependence of effect of nitric oxide synthase inhibition on cerebral ischemic damage. Journal of Cerebral Blood Flow and Metabolism. 1995;15(4):595–601. doi: 10.1038/jcbfm.1995.73. [DOI] [PubMed] [Google Scholar]
- 82.Willmot MR, Bath PMW. The potential of nitric oxide therapeutics in stroke. Expert Opinion on Investigational Drugs. 2003;12(3):455–470. doi: 10.1517/13543784.12.3.455. [DOI] [PubMed] [Google Scholar]
- 83.Miller MR, Megson IL. Recent developments in nitric oxide donor drugs. British Journal of Pharmacology. 2007;151(3):305–321. doi: 10.1038/sj.bjp.0707224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scatena R, Bottoni P, Pontoglio A, Giardina B. Pharmacological modulation of nitric oxide release: new pharmacological perspectives, potential benefits and risks. Current Medicinal Chemistry. 2010;17(1):61–73. doi: 10.2174/092986710789957841. [DOI] [PubMed] [Google Scholar]
- 85.Zhuang P, Ji H, Zhang YH, Min ZL, Ni QG, You R. ZJM-289, a novel nitric oxide donor, alleviates the cerebral ischaemic-reperfusion injury in rats. Clinical and Experimental Pharmacology and Physiology. 2010;37(3):e121–e127. doi: 10.1111/j.1440-1681.2010.05353.x. [DOI] [PubMed] [Google Scholar]
- 86.Jung KH, Chu K, Lee ST, et al. Augmentation of nitrite therapy in cerebral ischemia by NMDA receptor inhibition. Biochemical and Biophysical Research Communications. 2009;378(3):507–512. doi: 10.1016/j.bbrc.2008.11.081. [DOI] [PubMed] [Google Scholar]
- 87.Salom JB, Ortí M, Centeno JM, Torregrosa G, Alborch E. Reduction of infarct size by the NO donors sodium nitroprusside and spermine/NO after transient focal cerebral ischemia in rats. Brain Research. 2000;865(2):149–156. doi: 10.1016/s0006-8993(00)02095-3. [DOI] [PubMed] [Google Scholar]
- 88.Coert BA, Anderson RE, Meyer FB. Effects of the nitric oxide donor 3-morpholinosydnonimine (SIN-1) in focal cerebral ischemia dependent on intracellular brain pH. Journal of Neurosurgery. 2002;97(4):914–921. doi: 10.3171/jns.2002.97.4.0914. [DOI] [PubMed] [Google Scholar]
- 89.Zhang F, Iadecola C. Reduction of focal cerebral ischemic damage by delayed treatment with nitric oxide donors. Journal of Cerebral Blood Flow and Metabolism. 1994;14(4):574–580. doi: 10.1038/jcbfm.1994.71. [DOI] [PubMed] [Google Scholar]
- 90.Pluta RM, Rak R, Wink DA, et al. Effects of nitric oxide on reactive oxygen species production and infarction size after brain reperfusion injury. Neurosurgery. 2001;48(4):884–893. doi: 10.1097/00006123-200104000-00039. [DOI] [PubMed] [Google Scholar]
- 91.Salom JB, Barberá MD, Centeno JM, Ortí M, Torregrosa G, Alborch E. Relaxant effects of sodium nitroprusside and NONOates in goat middle cerebral artery: delayed impairment by global ischemia-reperfusion. Nitric Oxide. 1999;3(1):85–93. doi: 10.1006/niox.1999.0212. [DOI] [PubMed] [Google Scholar]
- 92.Yamamoto T, Bing RJ. Nitric oxide donors. Proceedings of the Society for Experimental Biology and Medicine. 2000;225(3):200–206. doi: 10.1046/j.1525-1373.2000.22525.x. [DOI] [PubMed] [Google Scholar]
- 93.Thatcher GRJ, Nicolescu AC, Bennett BM, Toader V. Nitrates and no release: contemporary aspects in biological and medicinal chemistry. Free Radical Biology & Medicine. 2004;37(8):1122–1143. doi: 10.1016/j.freeradbiomed.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 94.Granik VG, Grigor’ev NB. Exogenous donors of nitric oxide (a chemical aspect) Russian Chemical Bulletin. 2002;51(8):1375–1422. [Google Scholar]
- 95.Hogg N. The biochemistry and physiology of S-nitrosothiols. Annual Review of Pharmacology and Toxicology. 2002;42:585–600. doi: 10.1146/annurev.pharmtox.42.092501.104328. [DOI] [PubMed] [Google Scholar]
- 96.Hogg N, Darley-Usmar VM, Wilson MT, Moncada S. Production of hydroxyl radicals from the simultaneous generation of superoxide and nitric oxide. Biochemical Journal. 1992;281(2):419–424. doi: 10.1042/bj2810419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(14):6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yamada M, Momose K, Richelson E, Yamada M. Sodium nitroprusside-induced apoptotic cellular death via production of hydrogen peroxide in murine neuroblastoma N1E-115 cells. Journal of Pharmacological and Toxicological Methods. 1996;35(1):11–17. doi: 10.1016/1056-8719(95)00111-5. [DOI] [PubMed] [Google Scholar]
- 99.Terwel D, Nieland LJM, Schutte B, Reutelingsperger CPM, Ramaekers FCS, Steinbusch HWM. S-nitroso-N-acetylpenicillamine and nitroprusside induce apoptosis in a neuronal cell line by the production of different reactive molecules. European Journal of Pharmacology. 2000;400(1):19–33. doi: 10.1016/s0014-2999(00)00379-4. [DOI] [PubMed] [Google Scholar]
- 100.Cardaci S, Filomeni G, Rotilio G, Ciriolo MR. Reactive oxygen species mediate p53 activation and apoptosis induced by sodium nitroprusside in SH-SY5Y cells. Molecular Pharmacology. 2008;74(5):1234–1245. doi: 10.1124/mol.108.048975. [DOI] [PubMed] [Google Scholar]
- 101.Kim HJ, Tsoy I, Park MK, et al. Iron released by sodium nitroprusside contributes to heme oxygenase-1 induction via the cAMP-protein kinase A-mitogen-activated protein kinase pathway in RAW 264.7 cells. Molecular Pharmacology. 2006;69(5):1633–1640. doi: 10.1124/mol.105.020487. [DOI] [PubMed] [Google Scholar]
- 102.Ignjatović D, Vojnović Milutinović D, Nikolić-Kokić A, et al. The mechanisms responsible for neuroprotective capacity of arylpiperazine dopaminergic ligands against cell death induced by sodium nitroprusside. European Journal of Pharmacology. 2012;683(1–3):93–100. doi: 10.1016/j.ejphar.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 103.Gomez-Sarosi LA, Strasberg-Rieber M, Rieber M. ERK activation increases nitroprusside induced apoptosis in human melanoma cells irrespective of p53 status: role of superoxide dismutases. Cancer Biology and Therapy. 2009;8(12):1173–1182. doi: 10.4161/cbt.8.12.8561. [DOI] [PubMed] [Google Scholar]
- 104.Matsuzaki H, Tamatani M, Mitsuda N, et al. Activation of Akt kinase inhibits apoptosis and changes in Bcl-2 and Bax expression induced by nitric oxide in primary hippocampal neurons. Journal of Neurochemistry. 1999;73(5):2037–2046. [PubMed] [Google Scholar]
- 105.Gepdiremen A, Hacimüftüoğlu A, Büyükokuroğlu ME, Süleyman H. Nitric oxide donor sodium nitroprusside induces neurotoxicity in cerebellar granular cell culture in rats by an independent mechanism fromL-type or dantrolene-sensitive calcium channels. Biological & Pharmaceutical Bulletin. 2002;25(10):1295–1297. doi: 10.1248/bpb.25.1295. [DOI] [PubMed] [Google Scholar]
- 106.Bielarczyk H, Gul S, Ronowska A, Bizon-Zygmanska D, Pawelczyk T, Szutowicz A. RS-α-lipoic acid protects cholinergic cells against sodium nitroprusside and amyloid-β neurotoxicity through restoration of acetyl-CoA level. Journal of Neurochemistry. 2006;98(4):1242–1251. doi: 10.1111/j.1471-4159.2006.03966.x. [DOI] [PubMed] [Google Scholar]
- 107.Zhang Y, Rosenberg PA. Caspase-1 and poly (ADP-ribose) polymerase inhibitors may protect against peroxynitrite-induced neurotoxicity independent of their enzyme inhibitor activity. The European Journal of Neuroscience. 2004;20(7):1727–1736. doi: 10.1111/j.1460-9568.2004.03651.x. [DOI] [PubMed] [Google Scholar]
- 108.Gray E, Ginty M, Kemp K, Scolding N, Wilkins A. Peroxisome proliferator-activated receptor-α agonists protect cortical neurons from inflammatory mediators and improve peroxisomal function. The European Journal of Neuroscience. 2011;33(8):1421–1432. doi: 10.1111/j.1460-9568.2011.07637.x. [DOI] [PubMed] [Google Scholar]
- 109.Blais V, Rivest S. Effects of TNF-α and IFN-γ on nitric oxide-induced neurotoxicity in the mouse brain. Journal of Immunology. 2004;172(11):7043–7052. doi: 10.4049/jimmunol.172.11.7043. [DOI] [PubMed] [Google Scholar]
- 110.Rauhala P, Khaldi A, Mohanakumar KP, Chiueh CC. Apparent role of hydroxyl radicals in oxidative brain injury induced by sodium nitroprusside. Free Radical Biology & Medicine. 1998;24(7-8):1065–1073. doi: 10.1016/s0891-5849(97)00386-9. [DOI] [PubMed] [Google Scholar]
- 111.Rabkin SW, Kong JY. Nitroprusside induces cardiomyocyte death: interaction with hydrogen peroxide. The American Journal of Physiology. 2000;279(6):H3089–H3100. doi: 10.1152/ajpheart.2000.279.6.H3089. [DOI] [PubMed] [Google Scholar]
- 112.Mohanakumar KP, Hanbauer I, Chiueh CC. Neuroprotection by nitric oxide against hydroxyl radical-induced nigral neurotoxicity. Journal of Chemical Neuroanatomy. 1998;14(3-4):195–205. doi: 10.1016/s0891-0618(98)00032-5. [DOI] [PubMed] [Google Scholar]
- 113.Rao DNR, Cederbaum AI. Generation of reactive oxygen species by the redox cycling of nitroprusside. Biochimica et Biophysica Acta. 1996;1289(2):195–202. doi: 10.1016/0304-4165(95)00158-1. [DOI] [PubMed] [Google Scholar]
- 114.Belani KG, Singh H, Beebe DS, et al. Cyanide toxicity in juvenile pigs and its reversal by a new prodrug, sulfanegen sodium. Anesthesia and Analgesia. 2012;114(5):956–961. doi: 10.1213/ANE.0b013e31824c4eb5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chiueh CC. Neurobiology of NO. and .OH: basic research and clinical relevance. Annals of the New York Academy of Sciences. 1994;738:279–281. [PubMed] [Google Scholar]
- 116.Chiueh CC, Andoh T, Lai AR, Lai E, Krishna G. Neuroprotective strategies in Parkinson's disease: protection against progressive nigral damage induced by free radicals. Neurotoxicity Research. 2000;2:293–310. doi: 10.1007/BF03033799. [DOI] [PubMed] [Google Scholar]
- 117.Mohanakumar KP, Thomas B, Sharma SM, Muralikrishnan D, Chowdhury R, Chiueh CC. Nitric oxide: an antioxidant and neuroprotector. Annals of the New York Academy of Sciences. 2002;962:389–401. doi: 10.1111/j.1749-6632.2002.tb04083.x. [DOI] [PubMed] [Google Scholar]
- 118.Rauhala P, Andoh T, Yeh K, Chiueh CC. Contradictory effects of sodium nitroprusside and S-nitroso-N-acetylpenicillamine on oxidative stress in brain dopamine neurons in vivo. Annals of the New York Academy of Sciences. 2002;962:60–72. doi: 10.1111/j.1749-6632.2002.tb04056.x. [DOI] [PubMed] [Google Scholar]
- 119.Heales SJR, Lam AAJ, Duncan AJ, Land JM. Neurodegeneration or neuroprotection: the pivotal role of astrocytes. Neurochemical Research. 2004;29(3):513–519. doi: 10.1023/b:nere.0000014822.69384.0f. [DOI] [PubMed] [Google Scholar]
- 120.Banerjee R, Saravanan KS, Thomas B, Sindhu KM, Mohanakumar KP. Evidence for hydroxyl radical scavenging action of nitric oxide donors in the protection against 1-methyl-4-phenylpyridinium-induced neurotoxicity in rats. Neurochemical Research. 2008;33(6):985–995. doi: 10.1007/s11064-007-9473-z. [DOI] [PubMed] [Google Scholar]
- 121.Hogg N, Kalyanaraman B, Joseph J, Struck A, Parthasarathy S. Inhibition of low-density lipoprotein oxidation by nitric oxide. Potential role in atherogenesis. FEBS Letters. 1993;334(2):170–174. doi: 10.1016/0014-5793(93)81706-6. [DOI] [PubMed] [Google Scholar]
- 122.Rauhala P, Mohanakumar KP, Sziraki I, Lin AM, Chiueh CC. S-Nitrosothiols and nitric oxide, but not sodium nitroprusside, protect nigrostriatal dopamine neurons against iron-induced oxidative stress in vivo. Synapse. 1996;23(1):58–60. doi: 10.1002/(SICI)1098-2396(199605)23:1<58::AID-SYN7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 123.Tsai MJ, Lee EHY. Nitric oxide donors protect cultured rat astrocytes from 1-methyl-4- phenylpyridinium-induced toxicity. Free Radical Biology & Medicine. 1998;24(5):705–713. doi: 10.1016/s0891-5849(97)00329-8. [DOI] [PubMed] [Google Scholar]
- 124.Genc S, Kuralay F, Genc K, et al. Erythropoietin exerts neuroprotection in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated C57/BL mice via increasing nitric oxide production. Neuroscience Letters. 2001;298(2):139–141. doi: 10.1016/s0304-3940(00)01716-x. [DOI] [PubMed] [Google Scholar]
- 125.Wink DA, Hanbauer I, Krishna MC, DeGraff W, Gamson J, Mitchell JB. Nitric oxide protects against cellular damage and cytotoxicity from reactive oxygen species. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(21):9813–9817. doi: 10.1073/pnas.90.21.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Suschek CV, Briviba K, Bruch-Gerharz D, Sies H, Kröncke KD, Kolb-Bachofen V. Even after UVA-exposure will nitric oxide protect cells from reactive oxygen intermediate-mediated apoptosis and necrosis. Cell Death and Differentiation. 2001;8(5):515–527. doi: 10.1038/sj.cdd.4400839. [DOI] [PubMed] [Google Scholar]
- 127.Wenk GL, McGann-Gramling K, Hauss-Wegrzyniak B, et al. Attenuation of chronic neuroinflammation by a nitric oxide-releasing derivative of the antioxidant ferulic acid. Journal of Neurochemistry. 2004;89(2):484–493. doi: 10.1111/j.1471-4159.2004.02359.x. [DOI] [PubMed] [Google Scholar]
- 128.Palumbo ML, Fosser NS, Rios H, et al. Loss of hippocampal neuronal nitric oxide synthase contributes to the stress-related deficit in learning and memory. Journal of Neurochemistry. 2007;102(1):261–274. doi: 10.1111/j.1471-4159.2007.04528.x. [DOI] [PubMed] [Google Scholar]
- 129.Contestabile A, Ciani E. Role of nitric oxide in the regulation of neuronal proliferation, survival and differentiation. Neurochemistry International. 2004;45(6):903–914. doi: 10.1016/j.neuint.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 130.Zhang R, Zhang L, Zhang Z, et al. A nitric oxide donor induces neurogenesis and reduces functional deficits after stroke in rats. Annals of Neurology. 2001;50(5):602–611. doi: 10.1002/ana.1249. [DOI] [PubMed] [Google Scholar]
- 131.Endres M, Gertz K, Lindauer U, et al. Mechanisms of stroke protection by physical activity. Annals of Neurology. 2003;54(5):582–590. doi: 10.1002/ana.10722. [DOI] [PubMed] [Google Scholar]
- 132.Hashiguchi A, Yano S, Morioka M, et al. Up-regulation of endothelia nitric oxide synthase via phosphatidylinositol 3-kinase pathway contributes to ischemic tolerance in the CA1 subfield of the gerbil hippocampus. Journal of Cerebral Blood Flow and Metabolism. 2004;24(3):271–279. doi: 10.1097/01.WCB.0000110539.96047.FC. [DOI] [PubMed] [Google Scholar]
- 133.Lu D, Mahmood A, Zhang R, Copp M. Upregulation of neurogensis and reduction in functional deficits following administration of DEtA/NONOate, a nitric oxide donor, after traumatic brain injury in rats. Journal of Neurosurgery. 2003;99(2):351–361. doi: 10.3171/jns.2003.99.2.0351. [DOI] [PubMed] [Google Scholar]
- 134.Khan M, Im YB, Shunmugavel A, et al. Administration of S-nitrosoglutathione after traumatic brain injury protects the neurovascular unit and reduces secondary injury in a rat model of controlled cortical impact. Journal of Neuroinflammation. 2009;6, article 32 doi: 10.1186/1742-2094-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Puzzo D, Vitolo O, Trinchese F, Jacob JP, Palmeri A, Arancio O. Amyloid-β peptide inhibits activation of the nitric oxide/cGMP/cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity. The Journal of Neuroscience. 2005;25(29):6887–6897. doi: 10.1523/JNEUROSCI.5291-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hua Y, Huang XY, Zhou L, et al. DETA/NONOate, a nitric oxide donor, produces antidepressant effects by promoting hippocampal neurogenesis. Psychopharmacology. 2008;200(2):231–242. doi: 10.1007/s00213-008-1200-1. [DOI] [PubMed] [Google Scholar]
- 137.Gertz K, Endres M. eNOS and stroke: prevention, treatment and recovery. Future Neurology. 2008;3(5):537–550. [Google Scholar]
- 138.Yilmaz G, Granger DN. Cell adhesion molecules and ischemic stroke. Neurological Research. 2008;30(8):783–793. doi: 10.1179/174313208X341085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yasuda Y, Shimoda T, Uno K, et al. Temporal and sequential changes of glial cells and cytokine expression during neuronal degeneration after transient global ischemia in rats. Journal of Neuroinflammation. 2011;8, article 70 doi: 10.1186/1742-2094-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Prasad R, Giri S, Nath N, Singh I, Singh AK. GSNO attenuates EAE disease by S-nitrosylation-mediated modulation of endothelial-monocyte interactions. GLIA. 2007;55(1):65–77. doi: 10.1002/glia.20436. [DOI] [PubMed] [Google Scholar]
- 141.Massoudy P, Zahler S, Freyholdt T, et al. Sodium nitroprusside in patients with compromised left ventricular function undergoing coronary bypass: reduction of cardiac proinflammatory substances. Journal of Thoracic and Cardiovascular Surgery. 2000;119(3):566–574. doi: 10.1016/s0022-5223(00)70138-3. [DOI] [PubMed] [Google Scholar]
- 142.Göl MK, Nisanoğlu V, Işcan Z, Balci M, Kandemir O, Taşdemir O. Inhibition of systemic inflammatory response with sodium nitroprusside in open heart surgery. Journal of Cardiovascular Surgery. 2002;43(6):803–809. [PubMed] [Google Scholar]
- 143.Freyholdt T, Massoudy P, Zahler S, et al. Beneficial effect of sodium nitroprusside after coronary artery bypass surgery: pump function correlates inversely with cardiac release of proinflammatory cytokines. Journal of Cardiovascular Pharmacology. 2003;42(3):372–378. doi: 10.1097/00005344-200309000-00008. [DOI] [PubMed] [Google Scholar]
- 144.Sánchez-Pérez-Verdía E, López-Neblina F, Portilla E, Ortíz GG, González-Ojeda A, Álvares R. Exogenous nitric oxide protects kidney from ischemia/reperfusion. Journal of Investigative Surgery. 2001;14(6):313–320. doi: 10.1080/089419301753435684. [DOI] [PubMed] [Google Scholar]
- 145.Anaya-Prado R, Toledo-Pereyra LH, Walsh J, Guo RF, Reuben J, Ward PA. Exogenous nitric oxide donor and related compounds protect against lung inflammatory response after hemorrhagic shock and resuscitation. Journal of Trauma. 2004;57(5):980–988. doi: 10.1097/01.ta.0000135354.72494.8d. [DOI] [PubMed] [Google Scholar]
- 146.Clemens JA, Stephenson DT, Dixon EP, et al. Global cerebral ischemia activates nuclear factor-κKB prior to evidence of DNA fragmentation. Molecular Brain Research. 1997;48(2):187–196. doi: 10.1016/s0169-328x(97)00092-2. [DOI] [PubMed] [Google Scholar]
- 147.Zhang W, Potrovita I, Tarabin V, et al. Neuronal activation of NF-κB contributes to cell death in cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism. 2005;25(1):30–40. doi: 10.1038/sj.jcbfm.9600004. [DOI] [PubMed] [Google Scholar]
- 148.Howard EF, Chen Q, Cheng C, Carroll JE, Hess D. NF-κB is activated and ICAM-1 gene expression is upregulated during reoxygenation of human brain endothelial cells. Neuroscience Letters. 1998;248(3):199–203. doi: 10.1016/s0304-3940(98)00239-0. [DOI] [PubMed] [Google Scholar]
- 149.Kolev K, Skopál J, Simon L, Csonka É, Machovich R, Nagy Z. Matrix metalloproteinase-9 expression in post-hypoxic human brain capillary endothelial cells: H2O2 as a trigger and NF-κB as a signal transducer. Thrombosis and Haemostasis. 2003;90(3):528–537. doi: 10.1160/TH03-02-0070. [DOI] [PubMed] [Google Scholar]
- 150.Bhakar AL, Tannis LL, Zeindler C, et al. Constitutive nuclear factor-κB activity is required for central neuron survival. The Journal of Neuroscience. 2002;22(19):8466–8475. doi: 10.1523/JNEUROSCI.22-19-08466.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sarkar FH, Li Y, Wang Z, Kong D. NF-κB signaling pathway and its therapeutic implications in human diseases. International Reviews of Immunology. 2008;27(5):293–319. doi: 10.1080/08830180802276179. [DOI] [PubMed] [Google Scholar]
- 152.Sarnico I, Lanzillotta A, Boroni F, et al. NF-κB p50/RelA and c-Rel-containing dimers: opposite regulators of neuron vulnerability to ischaemia. Journal of Neurochemistry. 2009;108(2):475–485. doi: 10.1111/j.1471-4159.2008.05783.x. [DOI] [PubMed] [Google Scholar]
- 153.Stephenson D, Yin T, Smalstig EB, et al. Transcription factor nuclear factor-kappa B is activated in neurons after focal cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism. 2000;20(3):592–603. doi: 10.1097/00004647-200003000-00017. [DOI] [PubMed] [Google Scholar]
- 154.Ridder DA, Schwaninger M. NF-κB signaling in cerebral ischemia. Neuroscience. 2009;158(3):995–1006. doi: 10.1016/j.neuroscience.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 155.Cummins EP, Berra E, Comerford KM, et al. Prolyl hydroxylase-1 negatively regulates IκB kinase-β, giving insight into hypoxia-induced NFκB activity. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(48):18154–18159. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Blais V, Rivest S. Inhibitory action of nitric oxide on circulating tumor necrosis factor-induced NF-κB activity and COX-2 transcription in the endothelium of the brain capillaries. Journal of Neuropathology and Experimental Neurology. 2001;60(9):893–905. doi: 10.1093/jnen/60.9.893. [DOI] [PubMed] [Google Scholar]
- 157.Huang CY, Fujimura M, Noshita N, Chang YY, Chan PH. SOD1 down-regulates NF-κB and c-Myc expression in mice after transient focal cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism. 2001;21(2):163–173. doi: 10.1097/00004647-200102000-00008. [DOI] [PubMed] [Google Scholar]
- 158.Guerrini L, Blasi F, Denis-Donini S. Synaptic activation of NF-κB by glutamate in cerebellar granule neurons in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(20):9077–9081. doi: 10.1073/pnas.92.20.9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Greco R, Mangione AS, Amantea D, Bagetta G, Nappi G, Tassorelli C. IkappaB-alpha expression following transient focal cerebral ischemia is modulated by nitric oxide. Brain Research. 2011;1372:145–151. doi: 10.1016/j.brainres.2010.11.071. [DOI] [PubMed] [Google Scholar]
- 160.Gabriel C, Justicia C, Camins A, Planas AM. Activation of nuclear factor-κ-B in the rat brain after transient focal ischemia. Molecular Brain Research. 1999;65(1):61–69. doi: 10.1016/s0169-328x(98)00330-1. [DOI] [PubMed] [Google Scholar]
- 161.Peng HB, Libby P, Liao JK. Induction and stabilization of IκBα by nitric oxide mediates inhibition of NF-κB. The Journal of Biological Chemistry. 1995;270(23):14214–14219. doi: 10.1074/jbc.270.23.14214. [DOI] [PubMed] [Google Scholar]
- 162.de Caterina R, Libby P, Peng HB, et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. The Journal of Clinical Investigation. 1995;96(1):60–68. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Walley KR, McDonald TE, Higashimoto Y, Hayashi S. Modulation of proinflammatory cytokines by nitric oxide in murine acute lung injury. American Journal of Respiratory and Critical Care Medicine. 1999;160(2):698–704. doi: 10.1164/ajrccm.160.2.9809081. [DOI] [PubMed] [Google Scholar]
- 164.Park SW, Huq MDM, Hu X, Wei LN. Tyrosine nitration on p65: a novel mechanism to rapidly inactive nuclear factor-κB. Molecular and Cellular Proteomics. 2005;4(3):300–309. doi: 10.1074/mcp.M400195-MCP200. [DOI] [PubMed] [Google Scholar]
- 165.Song YS, Kim MS, Kim HA, et al. Oxidative stress increases phosphorylation of IB kinase-α by enhancing NF-κB-inducing kinase after transient focal cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism. 2010;30(7):1265–1274. doi: 10.1038/jcbfm.2010.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Candelario-Jalil E, González-Falcón A, García-Cabrera M, et al. Assessment of the relative contribution of COX-1 and COX-2 isoforms to ischemia-induced oxidative damage and neurodegeneration following transient global cerebral ischemia. Journal of Neurochemistry. 2003;86(3):545–555. doi: 10.1046/j.1471-4159.2003.01812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Candelario-Jalil E, Alvarez D, Merino N, León OS. Delayed treatment with nimesulide reduces measures of oxidative stress following global ischemic brain injury in gerbils. Neuroscience Research. 2003;47(2):245–253. doi: 10.1016/s0168-0102(03)00184-6. [DOI] [PubMed] [Google Scholar]
- 168.Nakayama M, Uchimura K, Zhu RL, et al. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(18):10954–10959. doi: 10.1073/pnas.95.18.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ahmad M, Zhang Y, Liu H, Rose ME, Graham SH. Prolonged opportunity for neuroprotection in experimental stroke with selective blockade of cyclooxygenase-2 activity. Brain Research. 2009;1279:168–173. doi: 10.1016/j.brainres.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Im JY, Kim D, Paik SG, Han PL. Cyclooxygenase-2-dependent neuronal death proceeds via superoxide anion generation. Free Radical Biology & Medicine. 2006;41(6):960–972. doi: 10.1016/j.freeradbiomed.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 171.Speed N, Blair AI. Cyclooxygenase- and lipoxygenase-mediated DNA damage. Cancer Metastasis Reviews. 2011;30(3-4):437–447. doi: 10.1007/s10555-011-9298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.D’Acquisto F, Maiuri MC, de Cristofaro F, Carnuccio R. Nitric oxide prevents inducible cyclooxygenase expression by inhibiting nuclear factor-κB and nuclear factor-interleukin-6 activation. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2001;364(2):157–165. doi: 10.1007/s002100100435. [DOI] [PubMed] [Google Scholar]
- 173.Marshall HE, Stamler JS. Inhibition of NF-κB by S-nitrosylation. Biochemistry. 2001;40(6):1688–1693. doi: 10.1021/bi002239y. [DOI] [PubMed] [Google Scholar]
- 174.Reynaert NL, Ckless K, Korn SH, et al. Nitric oxide represses inhibitory κB kinase through S-nitrosylation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(24):8945–8950. doi: 10.1073/pnas.0400588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hall CN, Garthwaite J. What is the real physiological NO concentration in vivo? Nitric Oxide. 2009;21(2):92–103. doi: 10.1016/j.niox.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]