

Abstract
Left ventricular hypertrophy is an independent risk factor for major adverse cardiovascular events. Statins have positive effects on this condition; however, the mechanisms are incompletely understood. In this study, we examined whether the effect of simvastatin on left ventricular hypertrophy can be mediated with the peroxisome proliferator-activated receptor (PPAR)γ-dependent pathway in rabbits with nonischemic heart failure (HF).
We induced aortic insufficiency and constriction in 48 rabbits and divided them equally into control, HF, and HF with simvastatin therapy (HF-SIM) groups. The HF-SIM group was given 10 mg/kg/d of simvastatin. We echocardiographically measured baseline and 8-week cardiac structure and function, and we used Western blot, polymerase chain reaction, and electrophoretic analytic techniques to evaluate messenger RNA expression and protein expression and activity. In comparison with the HF group, the HF-SIM rabbits had an increased ejection fraction and decreased left ventricular mass index, interventricular septal thickness, ventricular posterior-wall thickness, and collagen volume fraction. Moreover, the messenger RNA and protein expression of PPARγ in the HF-SIM rabbits were significantly higher than those in the HF rabbits; and the activity and expression of nuclear factor-κB subunit p65, RhoA, and Rho GTPase were significantly lower. Our results indicate that simvastatin therapy attenuates the PPARγ-dependent pathway in association with the inhibition of RhoA and Rho GTPase signaling to inhibit nuclear factor-κB activation, thus preventing the development of left ventricular hypertrophy and fibrosis in rabbits with nonischemic heart failure.
Key words: Disease models, animal; heart failure/drug therapy; heart ventricles/drug effects; hypertrophy, left ventricular/physiopathology/prevention & control; NF-kappa B/metabolism; PPAR gamma/drug effects/metabolism; rhoA GTP-binding protein; simvastatin; ventricular function, left/drug effects
Left ventricular hypertrophy (LVH) is an adaptational state before heart failure (HF) and an independent risk factor for ischemia, arrhythmia, and sudden death.1 Accordingly, the prevention or regression of LVH is considered to be of primary therapeutic importance.2 Clinically, drugs such as angiotensin II receptor blockers, calcium-channel blockers, angiotensin-converting enzyme inhibitors, and β-blockers are used to reverse or prevent the development of LVH; however, more than half of the patients still develop LVH.3 Therefore, new drugs with antihypertrophic effects are needed.
Hydroxymethylglutaryl coenzyme A reductase inhibitors—statins—have been shown to prevent and reverse LVH.4 The molecular basis of statins' antihypertrophic effect and the benefits of statins in nonischemic HF have been debated.5–7 We reported that simvastatin prevented the progression of cardiac dysfunction associated with alterations of calcium-regulatory proteins in rabbits with nonischemic HF.8 However, the effect of simvastatin on LVH in the same HF model has not been discussed.
The mechanisms through which LVH develops are chiefly neurohormonal, including changes in the catecholaminergic and renin-angiogenesis systems. In addition, the inflammatory signaling pathway has an important role in the development of cardiac hypertrophy. Peroxisome proliferator-activated receptors (PPARs), which are ligand-activated nuclear hormone receptors, inhibit inflammatory response. The PPARs belong to the nuclear-receptor superfamily and consist of 3 isoforms: α, β/δ, and γ. Some studies indicate that PPARγ is crucial in inhibiting cardiac hypertrophy and that the activation of PPARγ negatively regulates nuclear factor-κB (NF-κB), resulting in the attenuation of angiotensin II-induced cardiomyocyte hypertrophy in vitro and in vivo.9,10 Atorvastatin has reportedly prevented cardiac hypertrophy that is partially associated with the PPARγ signaling pathway.11,12 However, the mechanisms of statin-induced PPARγ activation in the heart are incompletely understood.
The pleiotropic effects of statins involve the inhibition of small-molecular-weight guanosine 5′-triphosphate (GTP)-binding proteins (small G proteins) such as Ras and Rho.13 In addition, statins activate PPARγ through the RhoA-dependent signaling pathway in macrophages,14 and statins prevent cardiac hypertrophy partially by inhibiting RhoA signaling.6,13 Therefore, we hypothesized that statins prevent or reverse LVH via the RhoA–PPARγ–NF-κB signaling pathway. In this study, we examined whether the effect of simvastatin on LVH can be mediated via the PPARγ-dependent pathway in rabbits with nonischemic HF induced by combined aortic regurgitation and aortic stenosis.
Materials and Methods
Establishing the Nonischemic-Heart-Failure Model
All experiments were reviewed and approved by the Animal Ethics Committee of Soochow University. We induced HF through combined aortic insufficiency (AI) and aortic constriction, as previously described.8 Aortic constriction was induced 2 weeks after the induction of AI. The experiments lasted 8 weeks from the induction of AI until the experimental rabbits were euthanized. Forty-eight rabbits were assigned in equal numbers (16 each) to a control group that underwent a sham operation, to a heart-failure group (HF group) treated with 5 mL/d of oral saline solution, or to a heart-failure group treated with simvastatin (HF-SIM group) at a dose of 10 mg/kg/d dissolved in 5 mL of saline solution, beginning one day after the induction of AI. The control-group rabbits were given 5 mL/d of saline solution during the same time period. During the experiment, LVH developed 4 weeks after the induction of AI; 4 rabbits died in the HF group, 1 died in the HF-SIM group, and no rabbits died in the control group.
Measuring Cardiac Structure and Function
Cardiac structure and function were evaluated at baseline and at the end of the experiment, as previously described.8 We obtained echocardiographic values for left atrial diameter, interventricular septal thickness, left ventricular (LV) posterior wall thickness, LV end-diastolic diameter, LV end-systolic diameter, LV ejection fraction, and fractional shortening (FS).
Histology of Left Ventricular Myocardium
At the end of the experiments, the body and LV of all the rabbits were weighed. The LV mass index was equal to the LV-to-body weight ratio. The LV myocardium was fixed in 3.8% perfusion of formaldehyde, embedded in paraffin, sectioned into 4-µm slices, and stained with hematoxylin and eosin (H & E) to detect myocyte size and with Van Gieson stain for the evaluation of fibrosis. Connective-tissue and muscle areas turned gray, collagen fibers red, myocytes yellow, and interstitial space white. The collagen volume fraction (CVF) was determined with use of an Image-Pro Plus® 4.0 automated image analyzer (Media Cybernetics, Inc.; Rockville, Md). The digitized profiles were transferred to a computer that calculated CVF as the sum of all connective-tissue areas in the entire section divided by all connective-tissue and muscle areas, expressed as a percentage. The CVF excluded scars and perivascular collagenous areas. At least 3 sections per heart and 5 fields per section were examined.
Preparation of Membrane, Cytosol, and Nuclear Proteins
Left ventricular tissue samples were cut into pieces and placed in a lysis buffer containing 25 mMol/L tris(hydroxymethyl)aminomethane hydrochloride (Tris-HCl) (pH, 7.4), 0.5 mMol/L EDTA, 0.5 mMol/L ethylene glycol tetraacetic acid (EGTA), 10 mMol/L β-mercaptoethanol, and protease inhibitors including 0.2 µmol/L leupeptin and 15 µmol/L aprotinin on ice. The lysates were homogenized and then centrifuged at 4,000 rpm at 4 °C for 5 min. The supernatants were then centrifuged at 12,000 rpm at 4 °C for 1 hour. At this time, the supernatants contained cytosolic proteins, and the sediments, which consisted of membrane proteins, were resuspended in the same solution. The nuclear proteins were extracted from LV tissue samples with use of a subcellular proteome ProteoExtract® kit (Merck KgaA; Darmstadt, Germany). They were both frozen at −20 °C. The protein concentration was determined by means of a Bradford assay.
Western Blot Analysis of RhoA, PPARγ, and NF-κB p65 Subunit
Membrane proteins were analyzed for RhoA protein, and nuclear proteins were analyzed for PPARγ and the NF-κB p65 subunit. Fifty µg of total proteins was separated by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) on polyacrylamide gradient gels (5%–10% gels; RhoA, 24 kDa; and both PPARγ and p65, 65 kDa) and transferred onto 0.4-µm nitrocellulose membranes with use of a PowerPAC™ Basic 300 (Bio-Rad Laboratories, Inc.; Hercules, Calif). The membranes were probed with anti-RhoA from rabbit at 1:100 dilutions (EMD Millipore Corporation; Billerica, Mass), anti-PPARγ from rabbit at 1:1,000 dilutions (EMD Millipore), and anti-p65 from rabbit at 1:500 dilutions (EMD Millipore). Horseradish peroxidase-conjugated anti-rabbit immunoglobulin G antibodies (Santa Cruz Biotechnology [Shanghai] Co., Ltd.; Shanghai, PRC) were used at 1:5,000 dilution as secondary antibodies. The peroxidase-reaction products were made visible upon the use of a chemiluminescent blotting kit (KPL, Inc.; Gaithersburg, Md). The films were scanned into a computer, and the protein-band densities were quantified by densitometric analysis with use of ImageJ 1.42 freeware. The relative levels of targeted proteins were corrected against the β-actin value in each sample.
Real-Time Polymerase Chain Reaction Analysis of PPARγ
Total RNA was extracted from LV tissue with use of Invitrogen™ TRIzol® reagent (Life Technologies; Grand Island, NY). The RNA concentrations were evaluated with use of a Shimadzu UV-1700 spectrophotometer (Shimadzu Corporation; Kyoto, Japan) at A260 nm, and 5 µg of total RNA from each sample was reverse-transcribed into complementary DNA (cDNA) with use of a Fermentas reverse transcriptase kit (Thermo Fisher Scientific Inc.; Waltham, Mass). The reverse transcriptase polymerase chain reaction (PCR) mixtures included 1.25 µL of cDNA, 0.2 µmol/L each of forward and reverse primer, 200 µmol/L 4× deoxyribonucleotide triphosphates (dNTPs), 1 U of Taq DNA polymerase, 2.5 mMol/L magnesium chloride, and 2.5 µL of 10× buffer. The amplifications were performed under the following conditions: denaturing at 94 °C for 4 min, 35 cycles of denaturing at 94 °C for 1 min, annealing at 55 °C for 1 min, and extension at 72 °C for 1 min and then for 10 min. The PPARγ and β-actin PCR primers were designed by Shanghai Sangon Biological Engineering Technology & Services Co. Ltd. (Shanghai, PRC) as follows:
PPARγ forward primer 5′- TTCCTGTCAAGATCGCCCTCG-3′ and reverse primer 5′- TGGGGATG TCTCATAATGCCA-3′; and
β-actin forward primer 5′-CTTCTCCTTGATGTCCCGCACGAT-3′ and reverse primer 5′-GTGCTGTCCCTG TACGCCTCTGG-3′.
The lengths of the amplified fragments of PPARγ and β-actin were 193 and 231 base pairs, respectively. The PCR products were analyzed by means of electrophoresis, and messenger RNA (mRNA) bands were quantified on a densitometer that used a gel-imaging system (Bio-Rad). The relative levels of PPARγ mRNA were normalized to the β-actin mRNA level in each sample.
Analysis of GTP-Binding Activity
The GTP-binding activity of Rho GTPase was analyzed after the separation of cytosolic proteins by means of SDS-PAGE (8% gel). After being blotted to nitrocellulose, the proteins were renatured by overnight incubation in buffer containing 25 mM Tris and 192 mM glycine. After 20 min of preincubation in binding buffer (50 mM Tris [pH, 7.5], 0.3% Tween 20, 5 mM magnesium chloride, and 1 mM EGTA), [γ-32P]GTP (1 mCi/mL) was added. After a further incubation period of 90 min at room temperature, the filters were washed 3 times for 30 min with binding buffer. Subsequently, the gels were dried and the level of [γ-32P]GTP binding was displayed on a phosphor screen by means of autoradiography; the data were acquired on a Storm® PhosphoImager® (Molecular Dynamics Inc.; Sunnyvale, Calif) and analyzed with use of that system's associated ImageQuant® software.
Electrophoretic Mobility Shift Assay of p65 Activity
An electrophoretic mobility shift assay was performed in nuclear proteins with use of double-stranded oligonucleotides (Promega Corporation; Madison, Wisc) labeled with [γ-32P] adenosine 5′-triphosphate (ATP) (3,000 Ci/mMol). The 5′AGTTGAGGGGACTTTCCCAGGC-3′ oligonucleotide of the NF-κB nucleotide was used as the consensus binding site. Oligonucleotides were labeled in the following reaction: 2 µL of oligonucleotide (1.75 pMol/µL), 2 µL of T4 polynucleotide kinase buffer, 1 µL of T4 polynucleotide kinase (10 U/µL), and 2.5 µL of [γ-32P]ATP (3,000 Ci/mMol at 10 mCi/mL) incubated at 37 °C for 1 hour. The reaction was stopped by adding 90 µL of Tris-EDTA (TE) buffer (10 mM Tris-HCl [pH, 7.4] and 1 mM EDTA). To separate the labeled probe from the unbound ATP, the reaction mixture was eluted in a column for nick translation (Pharmacia Spain S.A.; Sant Cugat del Valles, Spain) in accordance with the manufacturer's instructions. Ten µg of crude nuclear proteins was incubated for 10 min on ice in binding buffer (10 mM Tris-HCl [pH, 8.0], 25 mM potassium chloride, 0.5 mM dithiotreitol [DTT], 0.1 mM EDTA [pH, 8.0], 5% glycerol, 5 mg/mL bovine serum albumin [BSA], 100 µg/mL transfer RNA, and 50 µg/mL poly[deoxyinosinic-deoxycytidylic] acid sodium salt [poly dI–dC]). In supershift experiments, extracts were preincubated with p65 antibody (Santa Cruz Biotechnology) for 30 min at room temperature before the labeled oligonucleotides were added. For oligonucleotide competition experiments, a 100-fold molar excess of unlabeled oligonucleotide was included in the reaction mix and incubated for 30 min at room temperature before the labeled oligonucleotides were added. Protein–DNA complexes were separated by means of electrophoresis at 4 °C on a 5% acrylamide gel. The gels were dried and exposed for autoradiography on a phosphor screen; data were acquired on a Storm PhosphoImager (Molecular Dynamics) and quantified with use of ImageQuant software.
Statistical Analysis
Data for each group are expressed as mean ± SE. Differences among groups were evaluated by means of one-way analysis of variance followed by a post hoc Bonferroni multiple-comparison test. All statistical analyses were performed with use of SPSS 13.0 software (IBM Corporation; Armonk, NY). Values of P <0.05 were considered to be statistically significant.
Results
Effects of Simvastatin on Left Ventricular Hypertrophy and Function
Table I shows that the HF group exhibited significantly increased LV mass index, thicker interventricular septa and LV posterior walls, and lower LV ejection fractions and FS than did the control group, indicating LVH with decreased LV systolic function. However, simvastatin inhibited these changes in the HF-SIM group (Fig. 1).
TABLE I. Left Ventricular Mass Index and Echocardiographic Measurements after the Study
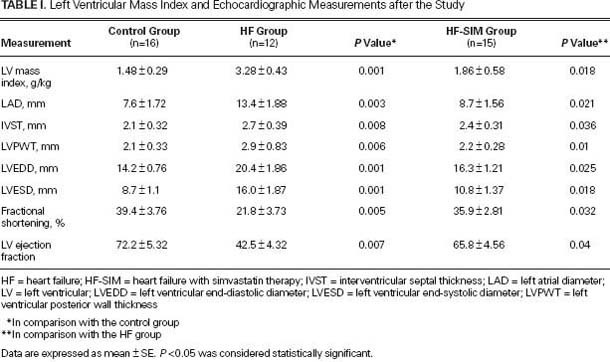
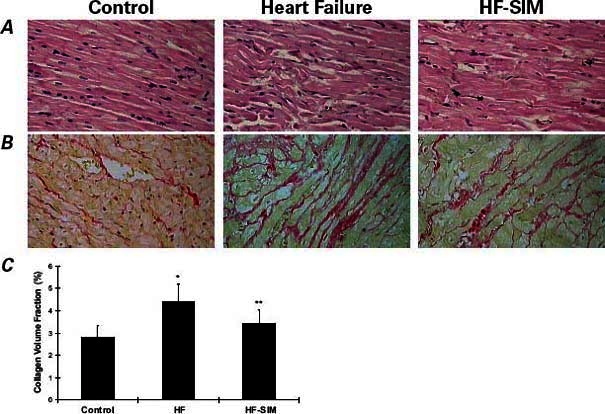
Fig. 1 Representative samples from one rabbit in each group. Left ventricular sections stained with A) H & E (orig. ×400) and B) Van Gieson stain (orig. ×400) show the effects of simvastatin on left ventricular fibrosis and hypertrophy in comparisons of the control group (first column), the heart-failure (HF) group (2nd column), and the heart failure-with-simvastatin therapy (HF-SIM) group (3rd column). The sections from the HF group show cardiomyocyte hypertrophy, myocyte disarray, and increased interstitial fibrosis; in contrast, sections from the HF-SIM group display only mild changes. C) Collagen volume fraction was decreased in the HF-SIM group in comparison with the HF group.
*P <0.01 in comparison with the control group
**P <0.05 in comparison with the HF group
Effects of Simvastatin on Histologic Phenotypes of Left Ventricular Myocardium
As Figure 1 shows, histologic analysis of H & E- and Van Gieson-stained cardiac sections revealed cardiomyocyte hypertrophy, myocyte disarray, and increased interstitial fibrosis in the HF group when compared with the control group; in contrast, the HF-SIM group displayed only mild changes. The CVF in both the HF (4.39% ± 0.8%) and HF-SIM groups (3.43% ± 0.59%) was increased in comparison with the control group (2.81% ± 0.51%; P <0.01), and the HF-SIM values were significantly better than the HF values (P <0.05) because simvastatin prevented an increase in CVF (Fig. 1C).
Effects of Simvastatin on RhoA Protein Abundance and Rho GTPase Activity
As Figure 2 shows, RhoA protein abundances (0.93 ± 0.025 vs 0.37 ± 0.015; P=0.007) and Rho GTPase activities ([γ-32P]GTP, 5,963.9 ± 243.47 vs 2,479.7 ± 311.36; P=0.008) in the HF group were significantly increased when compared with the control group; however, these increases were largely prevented in the HF-SIM group (RhoA protein, 0.54 ± 0.042; P=0.034; and Rho GTPase activity, [[γ-32P]GTP, 2,618.7 ± 290.11; P=0.005]) when compared with the HF group.
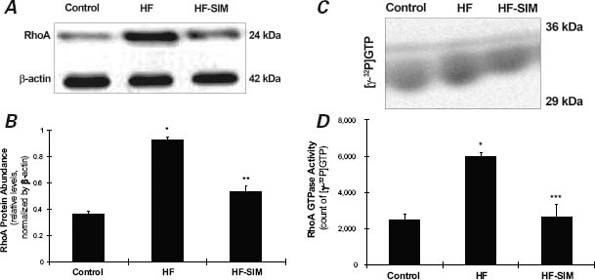
Fig. 2 Representative sample results from one rabbit in each group show the effects of simvastatin on RhoA protein abundance and Rho GTPase activity. A) RhoA protein abundance measured by Western blot assay. B) RhoA protein abundance in the heart failure-with-simvastatin therapy (HF-SIM) group was significantly decreased in comparison with the heart-failure (HF) group. C) Representative sample results from one rabbit in each group show Rho GTPase activity measured via a guanosine 5′-triphosphate (GTP)-binding assay. D) Data from 10 rabbits show significantly decreased Rho GTPase activity in the HF-SIM group in comparison with the HF group.
*P <0.01 in comparison with the control group
**P <0.05 in comparison with the HF group
***P <0.01 in comparison with the HF group
Effects of Simvastatin on PPARγ mRNA and Protein Abundance
As Figure 3 shows, PPARγ mRNA (0.31 ± 0.01 vs 0.72 ± 0.018; P=0.003) and protein abundance (0.4 ± 0.016 vs 0.82 ± 0.038; P=0.006) were significantly decreased in the HF group when compared with the control group. In the HF-SIM group, levels of PPARγ mRNA (0.55 ± 0.009; P=0.048) and protein abundance (0.64 ± 0.017; P=0.041) were restored to normal.
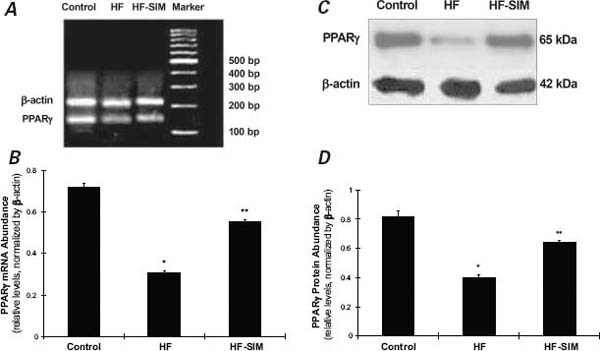
Fig. 3 Effects of simvastatin on peroxisome proliferator-activated receptor (PPAR)γ expressions. A) PPARγ messenger RNA (mRNA) abundance measured by reverse transcriptase polymerase chain reaction (base pairs). B) PPARγ mRNA in the heart failure-with-simvastatin therapy (HF-SIM) group was significantly increased in comparison with the heart-failure (HF) group. C) Representative samples from one rabbit in each group show PPARγ protein abbundance measured by Western blot assay. D) PPARγ protein abundance in the HF-SIM group was significantly increased in comparison with the HF group.
*P <0.01 in comparison with the control group
**P <0.05 in comparison with the HF group
Effects of Simvastatin on NF-κB p65 Protein Abundance and Activity
As Figures 4A and 4B show, the protein abundance of NF-κB p65 was significantly increased in the HF group when compared with the control group (0.72 ± 0.038 vs 0.42 ± 0.013; P=0.008), and the increases were smaller in the HF-SIM group (0.49 ± 0.015; P=0.039) than in the HF group. As Figures 4C and 4D show, NF-κB p65 activity was significantly increased in the HF group when compared with the control group ([γ-32P]ATP, 3,922.8 ± 358.44 vs 1,464.9 ± 130.09; P=0.004). In the HF-SIM group, levels of NF-κB p65 activity ([γ-32P]ATP, 1,897.2 ± 263.71; P=0.006) were reduced toward normal.
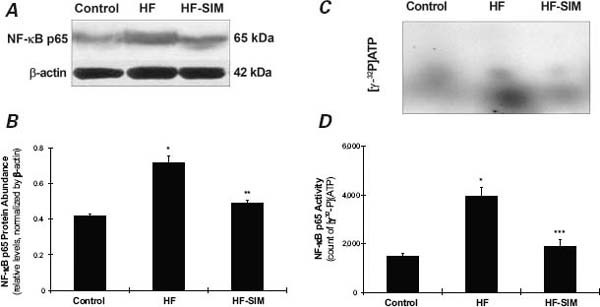
Fig. 4 Effects of simvastatin therapy on nuclear factor (NF)-κB subunit p65 protein abundance and activity. A) NF-κB p65 protein abundance measured by Western blot assay (sample results from one rabbit). B) Data from 10 rabbits show significantly decreased NF-κB p65 protein abundance in the heart failure-with-simvastatin therapy (HF-SIM) group in comparison with the heart-failure (HF) group. C) [γ-32P]ATP-labeled NF-κB p65 DNA in one rabbit, measured by electrophoretic mobility shift assay. D) Average data from all rabbits show that NF-κB p65 protein activity in the HF-SIM group was significantly decreased in comparison with the HF group.
ATP = adenosine 5′-triphosphate
*P <0.01 in comparison with the control group
**P <0.05 in comparison with the HF group
***P <0.01 in comparison with the HF group
Discussion
Our findings suggest that simvastatin therapy prevents the development of LVH and fibrosis in an experimental model of nonischemic HF and could be associated with the activation of the PPARγ-dependent pathway. Some studies have indicated that PPARγ activation negatively regulates NF-κB, resulting in the attenuation of angiotensin II-induced cardiomyocyte hypertrophy in vitro and in vivo.9,10 Statins have had positive effects on the PPARγ-dependent pathway, with resultant improvement in cardiac hypertrophy.11,12 To our knowledge, our study is the first to show that simvastatin inhibits RhoA signaling that contributes to activation of the PPARγ-dependent pathway in rabbits with nonischemic HF and LVH.
Effects of Simvastatin on the PPARγ–NF-κB Pathway
The transcription factor PPARγ belongs to the nuclear-receptor superfamily.15 It is highly expressed in adipose tissue, where it plays a major regulatory role in adipocyte differentiation and the expression of adipocyte-specific genes involved in lipid metabolism.16 In addition, PPARγ has been identified in the heart, where it plays a crucial role in inhibiting cardiac hypertrophy; and PPARγ activators inhibit cardiac hypertrophy in cardiac myocytes at least partially through the NF-κB pathway. There is negative crosstalk between PPARγ and NF-κB.17,18 Some studies have shown that atorvastatin attenuates the down-regulation of PPARγ and improves PPARγ signaling to inhibit NF-κB activation, thus preventing cardiac hypertrophy.11,12
In our experiments, both mRNA and the protein expression of PPARγ significantly decreased in the HF groups with LVH, and the protein expression and activity of the p65 subunit of NF-κB increased. In addition, not only were LVH and dysfunction prevented by simvastatin therapy, but the positive changes from PPARγ and the NF-κB subunit p65 were restored in the HF-SIM group. These results indicate that the PPARγ–NF-κB signaling pathway participates in the development of LVH and that simvastatin prevents LVH associated with the PPARγ-dependent pathway. However, the molecular mechanism behind simvastatin's activation of PPARγ remains to be determined.
Effects of Simvastatin on RhoA Signaling
The effects of another statin, pitavastatin, on load-induced cardiac hypertrophy and fibrosis are mediated (at least in part) through inhibition of the RhoA signaling pathway.6 In our study, both RhoA protein expression and Rho GTPase activity significantly increased in the groups with HF and LVH, whereas these changes were prevented by simvastatin therapy. Our results are in accord with those of previous studies.6,19,20
It is well known that statins inhibit the synthesis of the isoprenoid intermediates of the cholesterol pathway, including farnesylpyrophosphate and geranylgeranyl–pyrophosphate.21 The Rho family is activated by the attachment of geranylgeraniol. This post-translational lipid modification is necessary for the translocation of inactive GTPase from the cytosol to the membrane.22 Statins that block geranylgeraniol synthesis, or isoprenoid-transferase inhibitors that prevent the attachment of specific isoprenoids, might inhibit the membrane translocation and activity of small G proteins in cardiomyocytes.23
In our study, simvastatin markedly attenuated the development of cardiac hypertrophy and fibrosis. These effects were accompanied by inhibited RhoA expression and Rho GTPase activity and by activated PPARγ–NF-κB signaling in rabbits with nonischemic HF induced by combined AI and aortic constriction. It has been reported that statins activate PPARγ through the RhoA-dependent signaling pathway in macrophages.14 It is therefore possible that all these effects of simvastatin are ultimately attributable, at least in part, to inhibition of the isoprenylation of Rho family members in the heart.
In our study, we found that simvastatin prevents the development of LVH and left-sided cardiac dysfunction. In previous studies,11,12,23 only the effect of statins on LVH was observed. In addition, the indicators of LVH in our study were different from those in the previous studies. We used echocardiography to detect the interventricular septal thickness and LV posterior wall thickness, and we stained LV myocardium with H & E stain to detect myocyte size and with Van Gieson stain to evaluate fibrosis. The heart–body weight ratio, some fetal-type genes (such as atrial natriuretic factor), and contractile elements (such as myosin light chain-2) were used as indicators of LVH in previous studies. We think that the indicators of LVH in our study might be more useful than those in previous studies.
Some studies have shown that statins prevent cardiac hypertrophy partially associated with the PPARγ and the RhoA signaling pathway.11,12,23 However, in these studies, neither the upstream pathway of activation of PPARγ nor the downstream effect of inhibition of RhoA was discussed. Yano and colleagues14 indicated that statins activate PPARγ through the RhoA-dependent signaling pathway in macrophages. In our study, the results also suggest that simvastatin activates the PPARγ–NF-κB signaling pathway that is associated with the inhibition of RhoA and Rho GTPase activity in the heart. To our knowledge, our results are the first indication that simvastatin prevents the progression of LVH in a nonischemic-HF model because of inhibition of the RhoA–PPARγ–NF-κB signaling axis. Our findings perhaps yield a new insight into the prevention of LVH in nonischemic heart failure.
Footnotes
Address for reprints: Cao Zou, MD, Department of Cardiology, First Affiliated Hospital of Soochow University, No. 188, Shizi Rd., Suzhou City 215006, PRC
E-mail: nkzc75@163.com
References
- 1.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 1990;322(22):1561–6. [DOI] [PubMed]
- 2.Simko F. Physiologic and pathologic myocardial hypertrophy–physiologic and pathologic regression of hypertrophy? Med Hypotheses 2002;58(1):11–4. [DOI] [PubMed]
- 3.Klingbeil AU, Schneider M, Martus P, Messerli FH, Schmieder RE. A meta-analysis of the effects of treatment on left ventricular mass in essential hypertension. Am J Med 2003;115 (1):41–6. [DOI] [PubMed]
- 4.Simko F. Statins: a perspective for left ventricular hypertrophy treatment. Eur J Clin Invest 2007;37(9):681–91. [DOI] [PubMed]
- 5.Sola S, Mir MQ, Lerakis S, Tandon N, Khan BV. Atorvastatin improves left ventricular systolic function and serum markers of inflammation in nonischemic heart failure. J Am Coll Cardiol 2006;47(2):332–7. [DOI] [PubMed]
- 6.Saka M, Obata K, Ichihara S, Cheng XW, Kimata H, Noda A, et al. Attenuation of ventricular hypertrophy and fibrosis in rats by pitavastatin: potential role of the RhoA-extracellular signal-regulated kinase-serum response factor signalling pathway. Clin Exp Pharmacol Physiol 2006;33(12):1164–71. [DOI] [PubMed]
- 7.Chang SA, Kim YJ, Lee HW, Kim DH, Kim HK, Chang HJ, et al. Effect of rosuvastatin on cardiac remodeling, function, and progression to heart failure in hypertensive heart with established left ventricular hypertrophy. Hypertension 2009;54(3):591–7. [DOI] [PubMed]
- 8.Zou C, Liu Z, Qu F, Lu W, Han L, Song J, et al. Simvastatin prevents decreased SERCA2a activity in non-ischemic heart failure in rabbits via inhibition of beta-adrenergic signaling. Biomedicine & Aging Pathology 2011;1(1):22–7. [DOI] [PubMed]
- 9.Asakawa M, Takano H, Nagai T, Uozumi H, Hasegawa H, Kubota N, et al. Peroxisome proliferator-activated receptor gamma plays a critical role in inhibition of cardiac hypertrophy in vitro and in vivo. Circulation 2002;105(10):1240–6. [DOI] [PubMed]
- 10.Yamamoto K, Ohki R, Lee RT, Ikeda U, Shimada K. Peroxisome proliferator-activated receptor gamma activators inhibit cardiac hypertrophy in cardiac myocytes. Circulation 2001; 104(14):1670–5. [DOI] [PubMed]
- 11.Planavila A, Laguna JC, Vazquez-Carrera M. Atorvastatin improves peroxisome proliferator-activated receptor signaling in cardiac hypertrophy by preventing nuclear factor-kappa B activation. Biochim Biophys Acta 2005;1687(1–3):76–83. [DOI] [PubMed]
- 12.Ye P, Sheng L, Zhang C, Liu Y. Atorvastatin attenuating down-regulation of peroxisome proliferator-activated receptor gamma in preventing cardiac hypertrophy of rats in vitro and in vivo. J Pharm Pharm Sci 2006;9(3):365–75. [PubMed]
- 13.Morikawa-Futamatsu K, Adachi S, Maejima Y, Tamamori-Adachi M, Suzuki J, Kitajima S, et al. HMG-CoA reductase inhibitor fluvastatin prevents angiotensin II-induced cardiac hypertrophy via Rho kinase and inhibition of cyclin D1. Life Sci 2006;79(14):1380–90. [DOI] [PubMed]
- 14.Yano M, Matsumura T, Senokuchi T, Ishii N, Murata Y, Taketa K, et al. Statins activate peroxisome proliferator-activated receptor gamma through extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase-dependent cyclooxygenase-2 expression in macrophages. Circ Res 2007; 100(10):1442–51. [DOI] [PubMed]
- 15.Schoonjans K, Martin G, Staels B, Auwerx J. Peroxisome proliferator-activated receptors, orphans with ligands and functions. Curr Opin Lipidol 1997;8(3):159–66. [DOI] [PubMed]
- 16.Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science 2001;294(5548):1866–70. [DOI] [PubMed]
- 17.Yuan Z, Liu Y, Liu Y, Zhang J, Kishimoto C, Wang Y, et al. Cardioprotective effects of peroxisome proliferator activated receptor gamma activators on acute myocarditis: anti-inflammatory actions associated with nuclear factor kappaB blockade. Heart 2005;91(9):1203–8. [DOI] [PMC free article] [PubMed]
- 18.Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, et al. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem 1999;274(45):32048–54. [DOI] [PubMed]
- 19.Kansui Y, Fujii K, Goto K, Oniki H, Iida M. Chronic fluvastatin treatment alters vascular contraction by inhibiting the Rho/Rho-kinase pathway. Clin Exp Pharmacol Physiol 2006;33(8):673–8. [DOI] [PubMed]
- 20.Wang YX, da Cunha V, Martin-McNulty B, Vincelette J, Li W, Choy DF, et al. Inhibition of Rho-kinase by fasudil attenuated angiotensin II-induced cardiac hypertrophy in apolipoprotein E deficient mice. Eur J Pharmacol 2005;512(2–3): 215–22. [DOI] [PubMed]
- 21.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature 1990;343(6257):425–30. [DOI] [PubMed]
- 22.Van Aelst L, D'Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev 1997;11(18):2295–322. [DOI] [PubMed]
- 23.Laufs U, Kilter H, Konkol C, Wassmann S, Bohm M, Nickenig G. Impact of HMG CoA reductase inhibition on small GTPases in the heart. Cardiovasc Res 2002;53(4):911–20. [DOI] [PubMed]