

Abstract
Background: Platelet microparticles (PM) are the most abundant cell-derived microparticles in the blood, and accumulate in thrombo-inflammatory diseases. Platelets produce PM upon aging via an apoptosis-like process and by activation with strong agonists. We previously showed that long-term treatment of monocytic cells with apoptosis-induced PM (PMap) promotes their differentiation into resident macrophages. Here we investigated shorter term effects of various types of PM on monocyte signalling and function. Methods and results: Flow cytometry and scanning electron microscopy revealed that PM formed upon platelet aging (PMap) or ultra-sonication (PMsonic) expressed activated αIIbβ3 integrins and tended to assemble into aggregates. In contrast, PM formed upon platelet activation with thrombin (PMthr) or Ca2+ ionophore (PMiono) had mostly non-activated αIIbβ3 and little aggregate formation, but had increased CD63 expression. PM from activated and sonicated platelets expressed phosphatidylserine at their surface, while only the latter were enriched in the receptors CD40L and CX3CR1. All PM types expressed P-selectin, interacted with monocytic cells via this receptor, and were internalised into these cells. The various PM types promoted actin cytoskeletal rearrangements and hydrogen peroxide production by monocytic cells. Markedly, both aging- and activation-induced PM types stimulated the phosphoinositide 3-kinase/Akt pathway, suppressing apoptosis induced by several agonists, in a P-selectin-dependent manner. On the other hand, the PM types differentially influenced monocyte signalling in eliciting Ca2+ fluxes (particularly PMap) and in releasing secondary mediators (complement factor C5a with PMap, and pro-inflammatory tumour necrosis factor-α with PMthr). Conclusions: In spite of their common anti-apoptotic potential via Akt activation, aging- and activation-induced PM cause different Ca2+ signalling events and mediator release in monocytic cells. By implication, aging and activated platelets may modulate monocyte function in different way by the shedding of different PM types.
Keywords: Aging, apoptosis, microparticles, monocytes, platelet activation, tumour necrosis factor
Introduction
All blood cells are able to release small membrane vesicles, known as microparticles. Among these platelet microparticles (PM) are the most abundant ones, representing 70-90% of the microparticle population in the circulation in a non-diseased state [1,2]. However, the numbers of PM can further increase under pathological conditions, such as in cardiovascular disease, diabetes or inflammation [3-6]. Given that platelets are increased in activation tendency in all these disorders, it is considered that the higher levels of PM are a consequence of in situ platelet activation. On the other hand, the residual levels of circulating PM in the absence of disease likely originate from aging platelets in the absence of activation [7]. The current insight is that circulating microparticles should not be regarded as inactive cell debris, but as cell fragments that are actively involved in physiological and pathophysiological processes [4,8]. Yet, whether and how the microparticles from platelets influence the functions of blood and vascular cells is hardly understood.
There is limited evidence for the interaction of platelet-derived microparticles with leukocytes and endothelial cells in vitro and in vivo [2,9,10]. Under in vitro conditions, PM with exposed procoagulant membranes are also able to support coagulation and thrombin generation [11]. It is often supposed that PM similarly interact with blood cells as their parental cells, and hence simply propagate the effects of activated platelets. On the other hand, it is known that PM can be shed under different conditions, e.g. from aging platelets and platelets triggered with various agonists [7,12,13]. This raises the question whether the circumstances of PM formation influence their surface characteristics and, thereby, their functional properties. For instance, the shedding of PM from aging platelets occurs in an apoptosis-like process (PMap) that typically differs from agonist-induced platelet activation [14].
In earlier work, we have demonstrated that PMap interact with monocytic cells, and that this interaction, after 2-7 days, promotes the differentiation of these cells to a resident macrophage phenotype [15]. For the present paper, we focused on the shorter-term effects of PM shed by platelets under different conditions, hypothesizing that different types of platelet microparticles produced during aging or activation may have distinct effects on monocyte function. To investigate this, we isolated PM from aging platelets, from platelets activated by strong agonists, and from platelets fragmented by ultrasonication. In these PM types, we characterised the expression of surface glycoproteins. Furthermore, for the most physiological types of PM, we determined functional effects on monocytic cells and primary monocytes.
Materials and methods
Monocyte isolation and THP-1 cell culturing
Peripheral blood was obtained from healthy donors, who had given full informed consent. Leukocytes were isolated from blood buffy coats, as described before [15]. Monocytes were separated from neutrophils by Ficoll density gradient centrifugation, and further purified by negative selection using a Macs monocyte isolation kit II (Miltenyi Biotech). Purity of the monocyte preparations was determined and amounted to > 97%, based on flow cytometry. Human acute monocytic leukaemia THP-1 cells were cultured in RPMI-1640 medium with L-glutamine and 10% foetal calf serum, as described [15].
Generation of different PM types
Apoptosis-induced platelet microparticles (PMap) were isolated from platelet concentrates stored in plasma for 5 days at standard blood bank conditions (Uniklinikum Aachen, Germany) [7]. Platelet-poor plasma (PPP) was prepared by centrifugation at 4,000 g for 5 minutes. From the PPP, PMap were pelleted at 20,000 g for 60 minutes. Pellets were resuspended in Hepes buffer pH 7.45 (136 mM NaCl, 10 mM Hepes, 2.7 mM KCl, 2 mM MgCl2) containing 0.1% bovine serum albumin (BSA) and 0.1% glucose. Suspensions were filtered through a Minisart filter with a 0.8 μm pore size (Sartorius), pelleted again at 20,000 g for 40 minutes, and resuspended in Hepes buffer pH 7.45. The PM-free supernatants were used as a control.
Washed human platelets were isolated from peripheral blood, as described [16], and used at a concentration of 2 × 108 platelets/ml in Hepes buffer pH 7.45 with 2 mM CaCl2, under sterile conditions. To obtain thrombin-activated PM (PMthr), the platelets were activated with 11 nM (1 U/ml) thrombin (Enzyme Research Laboratories). After 30 minutes, residual thrombin was inactivated with 25 nM PPACK (Calbiochem), and the activated platelets were removed by centrifugation at 300 g for 5 minutes. Following filtration through a 0.8 μm filter, PMthr were pelleted at 20,000 g for 40 minutes, and resuspended in Hepes buffer pH 7.45. Calcium ionophore-activated microparticles (PMiono) were obtained similarly, except that the platelets were stimulated with 15 μM A23187 (Calbiochem). Sonicated microparticles (PMsonic) were prepared by ultrasound treatment of washed platelets for 3 × 30 seconds [17]. The microparticles were pelleted by centrifugation twice at 20,000 g for 20 minutes, and resuspended in Hepes buffer pH 7.45. The various microparticle suspensions were snap-frozen in liquid nitrogen, and stored at -20°C until use.
Quantification of PM types
Suspensions of PM were counted as flow-cytometric events at minimal forward/side scatter gating and staining with anti-CD61 mAb. Numbers of PM were obtained by adding fixed amounts of fluorescent beads to the PM suspensions [15]. Furthermore, the PM were quantified by measuring procoagulant membranes in the suspensions, using an in-house developed thrombin generation test [18]. Briefly, preparations of PM were diluted to 3330/μl (based on flow-cytometric counts) in BSA-free Hepes buffer pH 7.45. Samples were then sonicated for 3 minutes on ice to cause complete scrambling of the membrane phospholipids. Subsamples of 7.5 μl were added to microwells, containing 60 μl platelet-free plasma and 5 pM tissue factor (final concentration). Thrombin generation (37°C) was started by addition of 15 μl substrate solution (0.41 mM Z-GGR AMC and 16.6 mM CaCl2 in saline). Nanomolar concentrations of phospholipids were obtained by addition to the plasma of fixed amounts of procoagulant phospholipids (phosphatidylserine : phosphatidylethanolamine : phosphatidylcholine, 1 : 1 : 3, mol/mol) at a concentration range of 1.4-1000 nM.
Scanning electron microscopy
Platelets or PM were immobilised on polylysine-coated surfaces, and fixed overnight with 4% paraformaldehyde in phosphate-buffered saline (PBS). Samples were dehydrated, and subjected to scanning electron microscopy, as described elsewhere [19].
Flow cytometry determination of PM surface markers
Surface characterisation of PM types and platelets was performed with a FACS Canto II flow cytometer (BD Biosciences) [15]. The microparticles were labelled for 30 minutes with fluorescent monoclonal antibodies (mAbs) against CD61, CD62P or activated integrin αIIbβ3 (PAC-1, BD Biosciences); or mAbs against CD41, CD40 or CD40L (eBioscience). Expression of phosphatidylserine was probed with APC-labelled annexin A5 (BD Biosciences). Expression of CD31 (PECAM-1) and CD36 was detected with mAbs from Sigma and ImmunoTools, respectively. Rat anti-human CX3CR1 antibody (MBL International) was used to identify the fractalkine receptor, CX3CR1. Isotype controls used were IgG1 (eBioscience) or rat IgG2b (MBL International). Antigen expression levels are presented as geometric means of fluorescence intensity, corrected for background fluorescence with isotype control immunoglobulins.
Binding and phagocytosis of PM by monocytic cells
FITC-labelled carboxylated polystyrene beads (1 μm diameter, 1 × 107/ml) were decorated with PM types by incubation at 1-5 × 108 PM/ml. The PM-coated beads were added to THP-1 cells (2 × 106/ml) for measurements of binding and phagocytosis. Unloaded beads served as a control. Interaction of the PM-coated beads with THP-1 cells was determined by flow cytometry. Inhibitors added were the integrin αIIbβ3 antagonist, eptifibatide (50 μg/ml, GlaxoSmithKline), the Ca2+/Mg2+ scavenger EDTA (5 mM), or phosphatidylserine-scavenging annexin A5 (1 μg/ml, BD Biosciences). Phagocytosis of beads was monitored by making z-stacks with confocal fluorescence microscopy, after immobilisation of the THP-1 cells on coverslips [20].
Monocytic cell responses
PM-induced release of hydrogen peroxide from THP-1 cells (2.5 × 106/ml) was measured after 2-24 hours. At indicated time points, cell supernatants were collected and assessed for oxidation of the hydrogen peroxide substrate, tetramethylbenzidine (TMB, Invitrogen). Increased absorbance was measured at 450 nm, after correction for absorbance at 570 nm. Calibration was performed with a concentration range of hydrogen peroxide.
Formation of filamentous actin in THP-1 cells was determined by flow cytometry. Cells were fixed with 4% paraformaldehyde, permeabilized with 5% Triton-X-100, and stained for 15 minutes with FITC-labelled phalloidin (1 mg/ml, Invitrogen), and washed twice with PBS.
PM-induced adhesion of THP-1 cells was measured as before [15]. Briefly, THP-1 cells (1.5 × 105/ml) were incubated with indicated amounts of PM or platelets, and allowed to adhere to fibronectin-coated wells at 37°C. After 44 hours, vital cell were labelled with BCECF acetoxymethyl ester (1 μg/ml, Sigma), and fluorescence from total and adherent cells was measured using a Spectra FluoPlus reader (Tecan).
Cytokine production was measured from THP-1 cells (3 × 105 cells/ml) or primary monocytes (1 × 106/ml), allowed to adhere to fibronectin-coated well plates. The cells were kept in RPMI-1640 medium containing L-glutamine and 10% foetal calf serum (37°C) for 24 and 44 hours, respectively. Supernatants were collected and analysed with ELISA kits for complement factor C5a (R&D Systems) and tumour necrosis factor-α (TNFα, eBioscience), according to the companies’ instructions.
Apoptosis assays
Apoptotic responses were measured in THP-1 cells (1 × 106/ml) incubated with indicated concentrations of PM or resting platelets for 3 hours (37°C). A centrifugation step was performed to remove unbound PM or platelets. Cellular apoptosis was induced with PMA (0.5 μg/ml) or with the Bcl-2 inhibitor, ABT737 (10 μM), for indicated times (37°C) [21]. Apoptosis was verified by measuring the execution protease, caspase-3, with a commercial fluorometric assay (R&D Systems). Control measurements were carried out with only PM or platelets in the absence of THP-1 cells. Because PM can be annexin A5-positive, apoptosis could not be assessed with this probe [22].
Measurement of Ca2+ fluxes in monocytes
Primary monocytes (2 × 106/ml) in RPMI-1640 medium supplemented with 5% foetal calf serum were loaded with the Ca2+ indicator, Fluo-4 acetoxymethyl ester (2 μM), for 30 minutes at room temperature [23]. Fluo-4-loaded cells were left to adhere on polylysine-coated coverslips for 15 minutes in RPMI-1640 medium with 5% foetal calf serum. Coverslips with adhered cells were mounted into an open incubation chamber, and placed onto the stage of an inverted fluorescence microscope (Diaphot, Nikon). Rises in cytosolic Ca2+ were monitored in single, adhered monocytes by high-frequency recording of fluorescence images using a shutter-controlled fluorescence microscope system, connected to a sensitive EM-CCD camera [20]. Fluorescence changes were measured upon addition of platelets (1.5 × 106/ml), PM types (4-7 × 108/ml) or ATP (10 μM) as a positive control stimulus. Series of images were analysed off-line, as described before [24]. Single cell cytosolic Ca2+ rises are expressed as changes in fluorescence (F) relative to the basal fluorescence per cell (F0). Normalised F/F0 levels higher than 1.2 were considered as relevant Ca2+ rises.
Statistical analysis
Data are presented as means ± SEM. Differences between groups were statistically analysed by the Student’s t-test (GraphPad).
Results
Apoptosis-, agonist- and sonication-induced PM
Based on earlier data that PMap derived from aging platelets can regulate the differentiation of THP-1 cells at longer time points [15], we now investigated the shorter-term, direct effects of various types of PM on monocytic cells, i.e. PMap produced by platelet aging, by platelet activation with collagen (PMcoll), thrombin (PMthr) or Ca2+ ionophore A23187 (PMiono), or by platelet ultrasonication (PMsonic). The PM formed in these ways were identified by flow cytometry, according to their small size (low forward scatter) and positive staining with FITC-labelled anti-CD61 mAb (detecting integrin αIIbβ3). Analysis of dot plots indicated that the formation rates of PM differed per condition (Figure 1A). In non-stimulated platelets stored at room temperature and neutral pH, PMap formed relatively slowly due to aging, being detectable after 24 hours but not after 1 hour (Figure 1B). Interestingly, the extent of formation of PMap was reduced, when the temperature was increased to 37°C or the pH was decreased to 6.6. On the other hand, platelet stimulation with thrombin, thrombin/collagen or A23187 at physiological pH gave considerable amounts of PM at shorter time periods. Stimulation with collagen alone resulted in no more than small numbers of PM.
Figure 1.
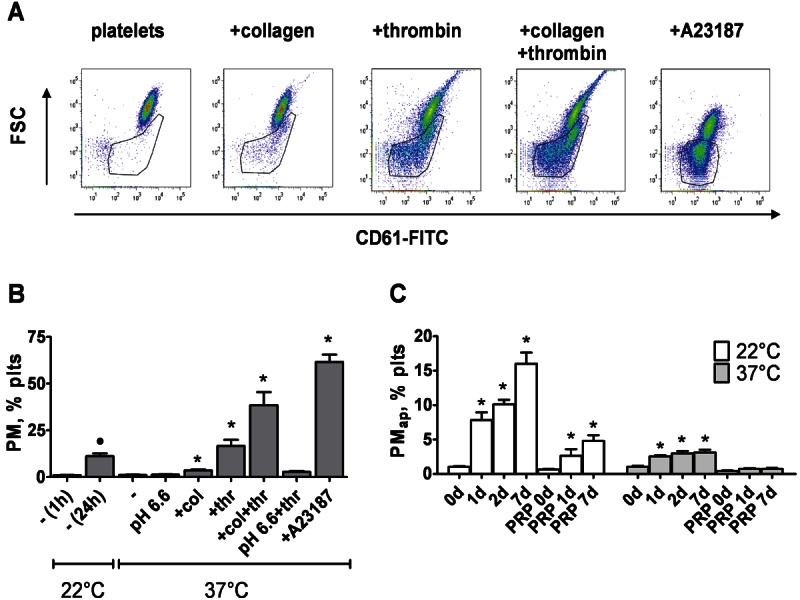
Formation of PM from stimulated and aging platelets. Suspensions of human platelets were unstimulated or stimulated with thrombin (thr, 1 U/ml) and/or collagen (col, 5 μg/ml), or A23187 (15 μM), as indicated. Platelets were also sonicated by ultrasound. After indicated times, formation of PM was determined by flow cytometry and staining with fluorescently-labelled anti-CD61 mAb. For calibration, fixed amounts of 1 and 6 μm beads were added to all samples. A: Representative dot plots of (activated) platelet suspensions after 1 hour. Gated regions represent PM. B: Quantification of PM formed after 1-24 hours of stimulation at 22°C, or 1 hour at 37°C. C: Quantification of PM formed in washed platelets or in PRP after indicated time points. Mean ± SEM (n = 3); *p < 0.05 vs. control at 37°C; ●p < 0.05 vs. control at 22°C.
Further experiments were conducted to compare the effects of plasma presence and temperature on the formation of PMap (Figure 1C). The accumulation rate of these PM in general was lower with plasma present, but still - independently of the presence of plasma appeared to reduce when raising the storage temperature from 22 to 37°C.
Characterisation of different PM types
The PM generated from the stored or activated platelets were isolated from the parent platelet suspension by a two-step centrifugation procedure, firstly removing all platelets by slow centrifugation, then spinning down the PM by ultraspeed centrifugation. By filtration of the resuspended pellet through a 0.8 μm diameter pore filter, residual platelets and larger platelet fragments were removed. The resulting filtered PM suspensions were characterised by flow cytometry, again detecting events staining for integrin αIIbβ3 (CD61). Interestingly, several of the PM types distributed over two subpopulations, i.e. smaller events being single microparticles, and larger size events with high integrin levels, which were considered to be PM aggregates (Figure 2A). Quantification of the two populations indicated that especially the PMap and PMsonic preparations were rich in larger size events, whereas the PMthr and PMiono (derived from activated platelets) mostly consisted of smaller events (Figure 2B).
Figure 2.
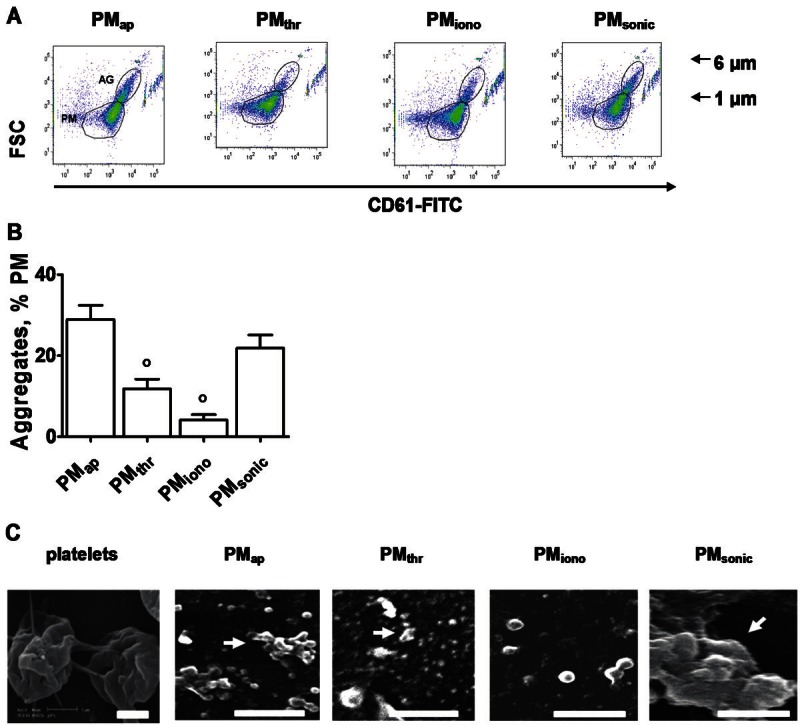
Morphological characterisation of different PM types. A: Flow-cytometry dot plots of PM types, obtained after ultracentrifuge isolation and filtering. Gated regions indicate aggregated (AG) and non-aggregated microparticles. B: Quantitative analysis of the extent of PM aggregation, determined by flow cytometry. Mean ± SEM (n = 4-6); °p < 0.05 vs. PMap. C: Scanning electron microscopic images of preparations of different PM types. Centrifuged PM types were fixed and immobilised on polylysine for electron microscopy. Shown are representative electron micrographs. Arrows indicate PM aggregates (bars = 1 μm).
In each preparation of PM types, concentrations were at first determined by calibrated flow cytometry, i.e. by counting the detectable microparticle events in comparison to the events from added fluorescently-labelled beads (Figure 2A). Because conventional flow cytometry is notoriously insensitive in detecting the smallest size microparticles, we also quantified the PM in a different way. The method relied on measurement of the concentration of procoagulant membrane phospholipids present in the PM preparations, using an in-house developed thrombin generation assay. All preparations were first pre-sonicated to achieve full phospholipid scrambling and, hence, full exposure of procoagulant phosphatidylserine. For the different types of PM preparations, adjusted to a count of 3,300/μl (assessed by flow cytometry), we obtained nanomolar concentrations of phospholipids of 451 (PMap), 459 (PMthr), 229 (PMiono) and 433 (PMsonic) with a variation of ~15%. Hence, we concluded that the flow cytometric test gave a reasonable estimate of the total membrane content of most of the PM types. Only the preparations of PMiono were somewhat lower in phospholipid content.
Scanning electron microscopy was used to check the ability of PM types to form aggregates. Electro-micrographs indicated that all preparations consisted of identifiable features smaller than 1 μm, as expected from the filtration procedure (Figure 2C). In agreement with the flow-cytometric data, a proportion of the PM prepared from stored platelets (PMap) and sonicated platelets (PMsonic) appeared as aggregated features, which could be up to 6 μm in size. Few aggregates were also observed in PM from thrombin-activated platelets (PMthr), while the PMiono did only appear as single features. Hence, we concluded that the different preparation methods resulted in PM types with a different tendency to aggregation.
Surface receptor properties of different PM types
The most abundantly formed types of microparticles (PMap, PMthr, PMiono and PMsonic) were characterised for the expression of platelet membrane glycoproteins (Figure 3A). Most PM types showed similar levels of the glycoproteins CD61/CD41 (integrin αIIbβ3), CD31 (PECAM-1), CD36 (thrombospondin-1 receptor), and CD40 (TNFα receptor superfamily), but PMsonic displayed relatively high expression levels of CD61/CD41. Levels of CD62P (P-selectin, marker of α-granule secretion) were increased in all PM in comparison to (larger size) resting platelets. However, the various PM types differed considerably in the presence of the activated αIIbβ3 integrin conformation (detected with PAC-1 mAb). PMsonic followed by PMap were highest in PAC-1 mAb binding, which is in agreement with the tendency of these PM types to form aggregates. In contrast, PM formed by platelet activation with thrombin (PMthr) or Ca2+ ionophore (PMiono) were low in activated αIIbβ3, while these types displayed increased CD63 expression (marker of dense-granule and lysosome secretion). In marked contrast to PMap, the types PMthr, PMiono and PMsonic showed high levels of phosphatidylserine at their surface (detected with annexin A5). In comparison to PMthr, the PMiono showed low expression levels of CD31, CD40L, CD61 and CD62P. On the other hand, only PMsonic were high in the signalling receptors CD40L and CX3CR1 (fractalkine receptor). Overall, this pointed to strikingly different profiles of platelet surface activation markers for the PM types. The PMap from aging platelets characteristically were high in the activated αIIbβ3 conformation, while PMthr and PMiono from activated platelets were typically high in surface exposure of CD63 and phosphatidylserine.
Figure 3.
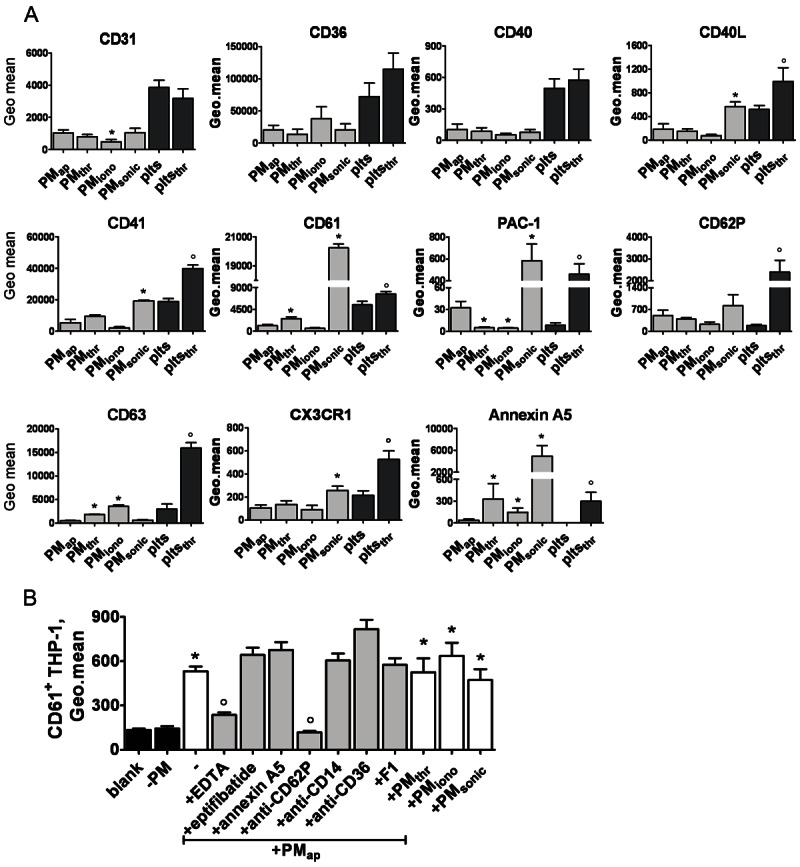
Surface characteristics of PM types and ability to bind to monocytic cells. A: Flow cytometric staining of PM types and platelets (plts) for different glycoprotein markers. Platelets were resting or stimulated with thrombin (thr, 1 U/ml). Data represent geometric means of fluorescence intensity, after correction for staining with control IgG. B: Flow cytometry detection of PM binding to monocytic THP-1 cells, treated with indicated PM for 30 minutes in the presence or absence of either: neutralising antibodies (20 μg/ml), EDTA (5 mM), eptifibatide (50 μg/ml), or CX3CR1 inhibitor F1 (0.5 μg/ml). PM complexes with THP-1 cells were detected as THP-1 gated events staining for FITC anti-CD61 mAb. Blank represents control with unstained cells. Means ± SEM (n = 3-5); *p < 0.05 vs. PMap; °p < 0.05 vs. Plts.
Binding of PM types to monocytic cells and cell stimulation
We subsequently determined the capability of these PM types to interact with monocytic cells. Flow cytometry demonstrated that, in spite of the differences in receptor expression profile, all PM types stably bound to THP-1 cells (Figure 3B). Using a panel of receptor antagonists, we examined the PMap binding in more detail. It was greatly diminished by Mg2+/Ca2+ chelation with EDTA as well as with a blocking mAb against P-selectin (20 μg/ml), which suggested a key role of the latter receptor in the interaction with PM. Other blocking agents were without effect. For instance, blockage of integrin αIIbβ3 with eptifibatide (50 μg/ml) or of phosphatidylserine with annexin A5 (1 μg/ml) did not impair PM binding to monocytic cells. Similarly, blocking antibodies against CD14, CD36 or inhibition of CX3CR1 with the blocking compound F1 were ineffective. Concerning PMthr, as the most physiological type of platelet activation-induced PM form, EDTA and P-selectin blocking diminished the binding to THP-1 cells (data not shown).
For more detailed study of the consequences of PM-monocytic cell interaction, we used FITC-labelled carboxylated polystyrene beads that were coated with the different microparticles. Incubation of the beads with increasing concentrations of PMap or PMthr (0.3-3 × 108/ml ) resulted in a gradually increasing but saturated staining with APC-labelled anti-CD61 mAb (Figure 4A). This indicated that the FITC-labelled beads were able to bind saturating numbers of αIIbβ3-expressing PM. Similar results were obtained with beads incubated with either PMiono or PMsonic (data not shown). Given the comparable APC labelling of beads with all PM types (Figure 4A), it was concluded that all of these coated the beads at similar amounts.
Figure 4.

Binding and uptake of PM types by monocytic cells. A: Flow cytometric detection of FITC-labelled carboxylated beads coated with PMap or PMthr. Beads were incubated with 3 × 107 PM/ml, or otherwise as indicated. PM coating was verified by co-labelling with APC anti-CD61 mAb. B: Interaction of PM-coated beads with THP-1 cells, determined by flow cytometry. Cells were incubated with beads for 40 minutes at 22°C. Note abolished binding in the presence of EDTA (5 mM), but not with annexin A5 (1 μg/ml). C: Numbers of bound and internalised PM-coated beads per THP-1 cell, determined by confocal microscopy. D: Confocal microscopy of PM-coated beads (FITC label, green) bound and internalised into TPH-1 cells (APC anti-CD45 mAb, red). Shown are representative 3-dimensional reconstructions of recorded z-stacks (bars, 10 μm). Means ± SEM (n = 3-4); *p < 0.05 vs. control; °p < 0.05 vs. no inhibitor.
The PM-coated FITC-labelled beads were then co-incubated with THP-1 cells, and binding to the cells was examined by flow cytometry. As indicated in Figure 4B, the THP-1 cells readily interacted with beads coated with any type of PM. For beads coated with PMap or PMthr, the binding properties were characterised in more detail. In either case, binding was reversed in the presence of EDTA or by blocking P-selectin with a mAb, but not with annexin A5. More detailed examination by confocal microscopy showed that, after 2 hours of incubation, part of the beads (~35%) was internalised into the cells, regardless of the type of PM present on the beads (Figure 4C, 4D). Control beads, not coated with PM, hardly bound to THP-1 cells and were not internalised.
Secretion of reactive oxygen species like hydrogen peroxide is an established activation marker of monocytes, e.g. in response to chemokines or when interacting with platelets [25,26]. We therefore examined the capability of PM to stimulate hydrogen peroxide production after interaction with THP-1 cells. After an incubation period of 24 hours, all PM types provoked significant release of peroxide (Figure 5A). At this time point, the cells had undergone major changes in actin cytoskeleton structure, as apparent from a marked increase in F-actin content in the presence of PMap, PMthr or PMsonic (Figure 5B). With PMiono, a smaller increase in F-actin was observed, but the difference was not statistically significant. In agreement with earlier results using PMap [15], we found that longer-term 48-hour incubations with both PMap and PMthr provoked adhesion of the THP-1 cells (1.5 × 105/ml) to a fibronectin surface, an effect that was maximal at ≥ 3 × 105 PM/ml (data not shown). Collectively, these results indicated that the various PM types are capable of binding and becoming internalised into monocytic cells, while causing major changes in cell activation properties.
Figure 5.
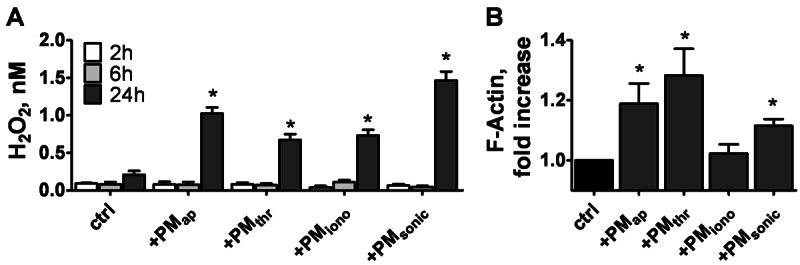
Effects of PM types on peroxide secretion and actin cytoskeleton rearrangement in monocytic cells. THP-1 cells (2.5 × 106/ml) were treated with vehicle (ctrl), PMap, PMthr, PMiono or PMsonic (2.5 × 107/ml) for 2-24 hours. A: Hydrogen peroxide secretion measured after indicated treatment times. B: Filamentous actin (F-actin) formation after 24 hours, as assessed by staining with FITC-phalloidin and flow cytometry. Means ± SEM (n = 3-5); *p < 0.05 vs. control.
Anti-apoptotic effects of PM types on monocytic cells
Our recent findings of PMap-induced monocytic cell activation and differentiation [15], may also point to pro-survival effects of PM on these cells. A possible mechanism to stimulate survival is by inhibiting the apoptotic program. We therefore checked the ability of aging- and activation-induced PM to influence apoptosis in THP-1 cells, induced by the commonly used agonist, PMA. Activity of the executioner caspase-3 was determined as a marker of apoptosis. Strikingly, PMap, PMiono, PMsonic and to a lesser extent PMthr were found to greatly suppress the PMA-induced caspase-3 activity (Figure 6A, left panel). In addition, cells were treated with the compound ABT737 (a specific antagonist of the anti-apoptotic Bcl-family proteins), which induces apoptosis via a mitochondrial pathway with massive caspase activation [27,28]. Again, all types of PM suppressed ABT737-induced caspase-3 activation in THP-1 cells (Figure 6A, right panel). Supernatants from MP preparations were ineffective, while also resting platelets did not influence the caspase-3 activation in monocytic cells. Control experiments showed that PM or platelets alone had no more than negligible caspase-3 activity (not shown). These data thus indicated that the different types of PM have a significant anti-apoptotic potential on monocytic cells.
Figure 6.
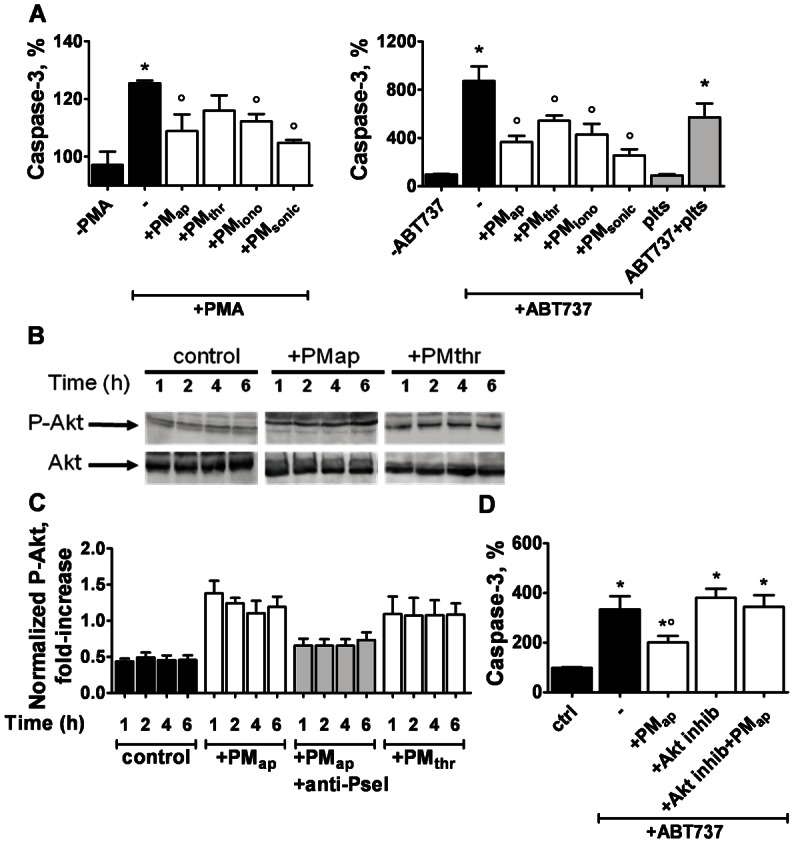
Anti-apoptotic effect of PM types on monocytic cells. THP-1 cells (1.25 × 106/ml) were treated with vehicle (control), resting platelets or PM types (each 1.25 × 107/ml) for 1-6 hours. The Akt inhibitor, AKT124005 (10 μM), was present as indicated. Cells were then stimulated with PMA (0.5 μg/ml) or ABT737 (10 μM) for 1 hour to start apoptosis. A: PM effect on PMA- or ABT737-induced caspase-3 activation. Caspase-3 activity is expressed as increased fluorescence compared to non-stimulated control cells. B, C: PM effect on Akt phosphorylation. B: Representative western blots of phosphorylated (P)-Akt or total Akt from cells incubated with PMap or PMthr for 1-6 hours. C: Densitometric analysis of P-Akt bands, normalised to Akt staining. D: Effect of Akt inhibitor on PM-regulated caspase-3 activity. Means ± SEM (n = 3-7); *p < 0.05 vs. vehicle control; °p < 0.05 vs. PMA or ABT737.
In a previous study, it was shown that PM in endothelial progenitor cells can trigger the signalling pathway of phosphoinositide 3-kinase (PI3K) and protein kinase Akt [10]. In various cell types, this pathway is considered to be antagonistic to apoptosis [29]. The proposed molecular mechanism is that PI3K/Akt activation suppresses the pro-apoptotic proteins Bak/Bax [30]. To investigate a role of this pathway in PM-mediated inhibition of apoptosis in THP-1 cells, we determined whether the various PM types can stimulate phosphorylation of the Ser473 site of Akt, as an established downstream effect of PI3K [31]. We indeed detected marked phosphorylation of Akt in response to both PMap and PMthr already after 1 hour of incubation, which persisted for up to 6 hours (Figure 6B, 6C). Blocking of the interaction of PMap with THP-1 cells with an anti-P-selectin mAb abolished the Akt phosphorylation (Figure 6C), indicating that PM-cell contact is required for this signalling event. Subsequent measurements of caspase-3 activity showed that the inhibitory effect of PM on ABT737-induced caspase was fully antagonised in the presence of the specific Akt inhibitor, AKT124005 (Figure 7D). Taken together, these data indicate that both aging- and activation-induced PM suppress Bak/Bax-dependent apoptosis, likely via activation of the PI3K/Akt pathway.
Figure 7.
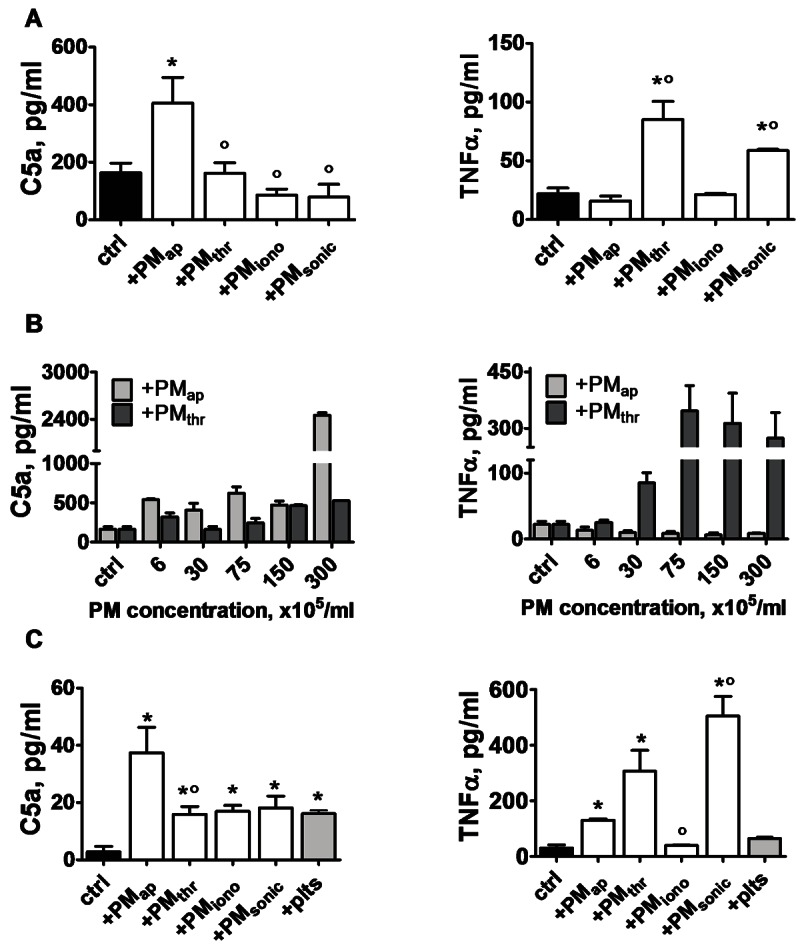
Different effects of PM types on cytokine release by monocytic cells and monocytes. THP-1 cells (3 × 105/ml) or monocytes (1 × 106/ml) were treated with vehicle solution (ctrl), indicated PM types, or platelets for 24-44 hours at 37°C. A: Levels of complement factor C5a and TNFα, measured in supernatants after 24-hours treatment of THP-1 cells with PM (3 × 106/ml). B: Dose effect of PM types (0.6-30 × 106/ml) on C5a and TNFα release by THP-1 cells. C: Cytokine levels in supernatants after treatment of monocytes (44 hours) with PM types (1 × 107/ml) or resting platelets (1 × 107/ml). Means ± SEM (n = 3-4); *p < 0.05 vs. control; °p < 0.05 vs. PMap.
Mediator release induced by PM types in monocytes
To determine the functional consequences of PM interactions with THP-1 cells, we assessed the release of chemotactic, inflammatory mediators, known to be produced by monocytes in contact with platelets. Interestingly, only PMap stimulated the release of complement factor C5a, while PMthr and PMsonic preferentially stimulated release of the systemic inflammatory cytokine TNFα (Figure 7A). Dose-response studies confirmed that the differential release of C5a and TNFα with PMap and PMthr, respectively, was maintained at high PM concentrations (Figure 7B).
Given the physiological consequences of release of these immune and inflammatory mediators, similar experiments were performed with primary monocytes isolated from peripheral human blood. Dual-labelling flow cytometry indicated that all types of PM interacted with monocytes, thus confirming the data for monocytic THP-1 cells (not shown). Treatment of the monocytes with PMap resulted in considerable release of complement factor C5a, while other PM types or resting platelets caused only minimal C5a release (Figure 7C). Also in monocytes, PMthr and PMsonic most strongly stimulated the release of TNFα, with the other PM types being less active.
To explain the differential effects of PM types, Ca2+ responses were measured as an initial signalling event. In Fluo-4-loaded monocytes adhered to coverslips, single-cell rises in cytosolic Ca2+ were determined by microscopic fluorescence imaging. Monocytes remained low in cytosolic Ca2+ in the absence of stimuli, but responded by a prolonged Ca2+ rise when treated with ATP as a control agonist (Figure 8A). When comparing the effects of various types of PM, it appeared that only addition of PMap evoked repetitive and transient Ca2+ spikes in the majority of the cells, with an averaged spiking frequency of 0.26/minute (Figure 8A, 8B). Addition of PMthr resulted in only low-amplitude spikes with a frequency of 0.13/minute. Addition of PMiono or platelets did not result in Ca2+ signalling events. Taken together, these results indicated that aging- and activation-induced PM markedly differed in inducing early signalling events in monocytes.
Figure 8.
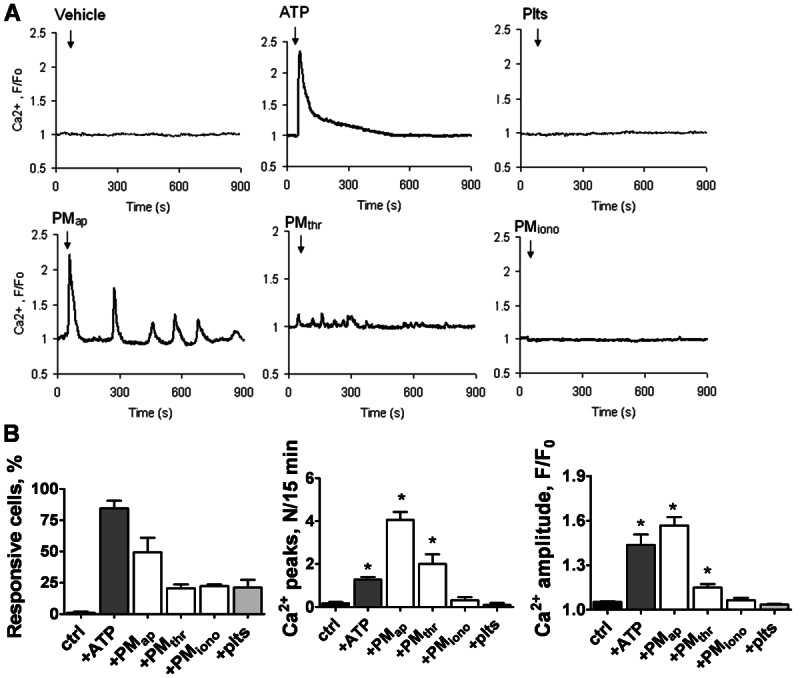
Different effects of PM types on Ca2+ responses in monocytic cells. Fluo-4-loaded CD14-positive monocytes on coverslips were left untreated (control) or treated with indicated types of PM or resting platelets (each 2 × 108/ml). The agonist ATP (10 μM) was used as a positive control. Rises in Ca2+ in single cells were monitored by high-frequency fluorescence image recording. A: Time traces of pseudo-ratio Ca2+ responses (F/Fo) from 3 representative cells per condition. B: Average Ca2+ responses from > 25 cells. Means ± SEM; *p < 0.05 vs. control.
Discussion
Characterisation experiments demonstrated that the PM produced from platelets in various ways, i.e. from aging platelets (PMap), from agonist-stimulated platelets (PMthr, PMiono) or by platelet sonication (PMsonic), strikingly differ in surface receptor expression levels. Flow cytometry showed that all PM types expressed P-selectin, but varied in the expression of other platelet activation markers. The activated conformation of integrin αIIbβ3 was highly present on PMap and PMsonic, which PM types also tend to cluster into aggregates. Activation-induced PM types, i.e. PMthr and PMiono, were low in the activated conformation of αIIbβ3, but displayed high expression of CD63 along with phosphatidylserine. The aging produced PMap characteristically were high in integrin αIIbβ activation, but low in phosphatidylserine exposure. Especially PMsonic were enriched in the signalling receptors, CD40L and CX3CR1. Accordingly, the circumstance of PM shedding from platelets - aging or activation - seems to result in microparticles with a different capacity to interact with blood cells and plasma (coagulation) factors.
In studying the functional and signalling effects of PM on monocytes, we in particular concentrated on PMap and PMthr as the most prominent types likely to be formed under (patho)physiological conditions in vivo. Using flow cytometry and electron microscopy, we found that, unlike resting platelets, these two PM types interacted with monocytic cells, despite the differences in surface properties. In agreement with the expression of P-selectin on these two PM types, blocking antibodies against this receptor impaired this interaction. This finding is in line with the established role of P-selectin in platelet-leukocyte interaction [32]. Furthermore, the interaction of these PM types with monocytic cells was dependent on divalent cations, i.e. blocked with EDTA. The same interactions - via divalent cations and P-selectin - were also required for the binding of fluorescent beads coated with PMap or PMthr to monocytic cells. In this respect, others have previously indicated that interaction of activated platelets with monocytes via P-selectin prepares for cell activation via RANTES [33].
Experiments with the PM-coated fluorescently-labelled beads furthermore showed that various types of PM (PMap, PMthr, PMiono and PMsonic) are taken up by the cells via a phagocytosis process. Remarkably, inhibitor studies indicated that CD14, CD36 or phosphatidylserine were not involved in PMap binding (and uptake), in spite of the established role of these membrane components in apoptotic body clearance [34,35]. Uptake of PM has also been described for other cells, in particular (progenitor) endothelial cells [26,36]. Surface-expressed P-selectin - a feature shown by all PM types - may directly or indirectly be involved in the uptake mechanism. Interestingly, the phagocytic response with all PM types was accompanied by cytoskeleton changes and production of the reactive oxygen species H2O2. How PM binding and uptake causes these monocytic responses is still unclear.
Another key finding was that the various types of PM (PMap, PMthr, PMiono and PMsonic) had a clear anti-apoptotic effect on THP-1 cells, regardless of the apoptotic trigger, i.e. PMA causing continuous protein kinase C stimulation, or ABT737 inhibiting the Bcl-family proteins. Thus, these PM types stimulated caspase-3 activity induced by these stimuli. For PMap, it could be demonstrated that this stimulating effect was antagonised by inhibition of the protein kinase Akt. Furthermore, both PMap and PMthr were able to activate the PI3K/Akt pathway, as was apparent from the PI3K-dependent phosphorylation of Akt at Ser473. At least in case of PMap, the Akt activation relied on P-selectin-dependent interaction with the monocytic cells. These data are in good agreement with published findings that platelet-derived microparticles stimulate Akt phosphorylation and activation in endothelial and neuronal progenitor cells [36,37]. Mechanistically, this feeds well into the known anti-apoptotic role of PI3K/Akt stimulation, likely acting through Akt-dependent inactivation of the pro-apoptotic Bad proteins [29,38].
Elevated cytosolic Ca2+ is a common initial signal for many blood cell responses, including phagocytosis, oxidative burst and paracrine mediator release [39-42]. We demonstrate for the first time that particularly platelet-derived microparticles evoke Ca2+ rises when interacting with monocytes. Interestingly, PMap were most efficient in this response, with PMthr triggering only minor Ca2+ transients, and PMiono or intact platelets not having any effect. Similarly, PMap and PMthr were most effective in increasing levels of filamentous actin, which is a Ca2+-dependent event. The precise downstream effector pathways of the Ca2+ rises need to be explored further, but in general may include Ca2+-dependent operation of transcription factors such as NFAT [43] and the release of certain mediators. In this context, it should be noted that monocytes, in contrast to other leukocytes, are cells poor in secretion granules, showing no or limited exocytosis.
A typical and likely relevant difference between aging- (PMap) and activation- (PMthr) induced microparticles was their diverse ability to stimulate the formation of bioactive mediators in THP-1 cells and monocytes, with only the former causing release of complement factor C5a, and only the latter releasing proinflammatory TNFα. A recent study with macrophages also indicates that microparticles from platelets influence the release of TNFα [44]. Our previous data indicated that PMap stimulate monocyte differentiation into a resident macrophage phenotype [15]. It now appears that rather PM from activated platelets stimulate the development to a more inflammatory type of monocytes, producing the reactive cytokine TNFα. The latter PM types also have a high procoagulant potential because of the increased surface exposure of phosphatidylserine.
The activation-induced PMiono differed in various aspects from PMthr in triggering responses in monocytic cells, i.e. they were unable to trigger Ca2+ responses and TNFα release. Interestingly, most likely due to their exposure to prolonged elevation in Ca2+ in response to A23187, the PMiono showed low expression levels of several surface receptors known to be substrates of ADAM proteases (i.e. CD31, CD40L, CD61, CD62P) [45]. Hence, we may speculate that surface shedding of these or other receptors explains the lack of PMiono to evoke TNFα release in THP-1 cells and monocytes.
Taken together, our results suggest that in vivo not only the amount but also the type of PM produced can be important for the fate and the responsiveness of circulating monocytes for instance in inflammation. Stated otherwise, aging or activated platelets seem to be able to disseminate and extend their roles in the stimulation of leukocytes [46] in different ways via the production of different types of PM. The type of circulating PM under pathological conditions of thrombosis thus may direct the differentiation program and repertoire of released mediators by patients’ monocytes.
Acknowledgements
Deutsche Forschungsgemeinschaft (DFG FOR809: TP2, TP4, TP6, Ko2948/1-2 and Hu1618/1-2), the Euregio Cardiovascular International Research Training Group (EuCAR) GRK1508 ‘Arterial Remodeling’, the European Research Council (Advanced Grant 249929 - Atheroprotect (C.W), ZonMW VIDI grant 016.126.358 (R.R.K.). We thank Dr. S. Mause for helpful discussions.
Conflict of interests
The authors declare no relevant conflicts of interest.
References
- 1.Berckmans RJ, Nieuwland R, Boing AN, Romijn FP, Hack CE, Sturk A. Cell-derived microparticles circulate in healthy humans and support low grade thrombin generation. Thromb Haemost. 2001;85:639–646. [PubMed] [Google Scholar]
- 2.Morel O, Toti F, Hugel B, Freyssinet JM. Cellular microparticles: a disseminated storage pool of bioactive vascular effectors. Curr Opin Hematol. 2004;11:156–164. doi: 10.1097/01.moh.0000131441.10020.87. [DOI] [PubMed] [Google Scholar]
- 3.Koga H, Sugiyama S, Kugiyama K, Fukushima H, Watanabe K, Sakamoto T, Yoshimura M, Jinnouchi H, Ogawa H. Elevated levels of remnant lipoproteins are associated with plasma platelet microparticles in patients with type-2 diabetes mellitus without obstructive coronary artery disease. Eur Heart J. 2006;27:817–823. doi: 10.1093/eurheartj/ehi746. [DOI] [PubMed] [Google Scholar]
- 4.Nomura S, Ozaki Y, Ikeda Y. Function and role of microparticles in various clinical settings. Thromb Res. 2008;1:8–23. doi: 10.1016/j.thromres.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 5.Vasina E, Heemskerk JW, Weber C, Koenen RR. Platelets and platelet-derived microparticles in vascular inflammatory disease. Inflamm Allergy Drug Targets. 2010;9:346–354. doi: 10.2174/187152810793938008. [DOI] [PubMed] [Google Scholar]
- 6.Nomura S, Imamura A, Okuno M, Kamiyama Y, Fujimura Y, Ikeda Y, Fukuhara S. Platelet-derived microparticles in patients with arteriosclerosis obliterans: enhancement of high shear-induced microparticle generation by cytokines. Thromb Res. 2000;98:257–268. doi: 10.1016/s0049-3848(00)00186-9. [DOI] [PubMed] [Google Scholar]
- 7.Cauwenberghs S, Feijge MA, Harper AG, Sage SO, Curvers J, Heemskerk JW. Shedding of procoagulant microparticles from unstimulated platelets by integrin-mediated destabilization of actin cytoskeleton. FEBS Lett. 2006;580:5313–5320. doi: 10.1016/j.febslet.2006.08.082. [DOI] [PubMed] [Google Scholar]
- 8.Owens AP, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. 2011;108:1284–1297. doi: 10.1161/CIRCRESAHA.110.233056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia BA, Smalley DM, Cho H, Shabanowitz J, Ley K, Hunt DF. The platelet microparticle proteome. J Proteome Res. 2005;4:1516–1521. doi: 10.1021/pr0500760. [DOI] [PubMed] [Google Scholar]
- 10.Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C. Platelet microparticles: a transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol. 2005;25:1512–1518. doi: 10.1161/01.ATV.0000170133.43608.37. [DOI] [PubMed] [Google Scholar]
- 11.Heemskerk JW, Kuijpers MJ, Munnix IC, Siljander PR. Platelet collagen receptors and coagulation. A characteristic platelet response as possible target for antithrombotic treatment. Trends Cardiovasc Med. 2005;15:86–92. doi: 10.1016/j.tcm.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 12.Dean WL, Lee MJ, Cummins TD, Schultz DJ, Powell DW. Proteomic and functional characterisation of platelet microparticle size classes. Thromb Haemost. 2009;102:711–718. doi: 10.1160/TH09-04-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sims J, Wiedmer T, Esmon CT, Weiss HJ, Shattil SJ. Assembly of the platelet prothrombinase complex is linked to vesiculation of the platelet plasma membrane. Studies in Scott syndrome, an isolated defect in platelet procoagulant activity. J Biol Chem. 1989;264:17049–17057. [PubMed] [Google Scholar]
- 14.Schwertz H, Koster S, Kahr WH, Michetti N, Kraemer BF, Weitz DA, Blaylock RC, Kraiss LW, Greinacher A, Zimmerman GA, Weyrich AS. Anucleate platelets generate progeny. Blood. 2010;115:3801–3809. doi: 10.1182/blood-2009-08-239558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vasina EM, Cauwenberghs S, Feijge MA, Heemskerk JW, Weber C, Koenen RR. Microparticles from apoptotic platelets promote resident macrophage differentiation. Cell Death Disease. 2011;2:e210. doi: 10.1038/cddis.2011.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feijge MA, van Pampus EC, Lacabaratz-Porret C, Hamulyak K, Lévy-Toledano S, Enouf J, Heemskerk JW. Inter-individual varability in Ca2+ signalling in platelets from healthy volunteers, relation with expression of endomembrane Ca2+-ATPases. Br J Haematol. 1998;102:850–859. doi: 10.1046/j.1365-2141.1998.00844.x. [DOI] [PubMed] [Google Scholar]
- 17.Tans G, Rosing J, Thomassen MC, Heeb MJ, Zwaal RF, Griffin JH. Comparison of anticoagulant and procoagulant activities of stimulated platelets and platelet-derived microparticles. Blood. 1991;77:2641–2648. [PubMed] [Google Scholar]
- 18.Cauwenberghs S, Feijge MA, Theunissen E, Heemskerk JW, van Pampus EC, Curvers J. Novel methodology for the assessment of platelet transfusion therapy by measuring increased thrombus formation and thrombin generation. Br J Haematol. 2007;136:480–490. doi: 10.1111/j.1365-2141.2006.06453.x. [DOI] [PubMed] [Google Scholar]
- 19.Siljander P, Farndale RW, Feijge MA, Comfurius P, Kos S, Bevers EM, Heemskerk JW. Platelet adhesion enhances the glycoprotein VI-dependent procoagulant response: involvement of p38 MAP kinase and calpain. Arterioscler Thromb Vasc Biol. 2001;21:618–627. doi: 10.1161/01.atv.21.4.618. [DOI] [PubMed] [Google Scholar]
- 20.Nergiz-Unal R, Lamers MM, van Kruchten R, Luiken JJ, Cosemans JM, Glatz JF, Kuijpers MJ, Heemskerk JW. Signaling role of CD36 in platelet activation and thrombus formation on immobilized thrombospondin or oxidized low density lipoprotein. J Thromb Haemost. 2011;9:1835–1846. doi: 10.1111/j.1538-7836.2011.04416.x. [DOI] [PubMed] [Google Scholar]
- 21.Gurtu V, Kain SR, Zhang G. Fluorometric and colorimetric detection of caspase activity associated with apoptosis. Anal Biochem. 1997;251:98–102. doi: 10.1006/abio.1997.2220. [DOI] [PubMed] [Google Scholar]
- 22.Vermes I, Haanen C, Reutelingsperger C. Flow cytometry of apoptotic cell death. J Immunol Methods. 2000;243:167–90. doi: 10.1016/s0022-1759(00)00233-7. [DOI] [PubMed] [Google Scholar]
- 23.Den Dekker E, Molin DG, Breikers G, van Oerle R, Akkerman JW, van Eys GJ, Heemskerk JW. Expression of transient receptor potential mRNA isoforms and Ca2+ influx in differentiating human stem cells and platelets. Biochim Biophys Acta. 2001;1539:243–255. doi: 10.1016/s0167-4889(01)00112-4. [DOI] [PubMed] [Google Scholar]
- 24.Heemskerk JW, Willems GM, Rook MB, Sage SO. Ragged spiking in free calcium in ADP-stimulated platelets: regulation of puff-like calcium signal in vitro and ex vivo. J Physiol. 2001;535:625–635. doi: 10.1111/j.1469-7793.2001.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakagawara A, Nathan FC, Cohn ZA. Hydrogen peroxide metabolism in human monocytes during differentiation in vitro. J Clin Invest. 1981;68:1243–1252. doi: 10.1172/JCI110370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Terrisse AD, Puech N, Allart S, Gourdy P, Xuereb JM, Payrastre B, Sie P. Internalization of microparticles by endothelial cells promotes platelet/endothelial cell interaction under flow. J Thromb Haemost. 2010;8:2810–9. doi: 10.1111/j.1538-7836.2010.04088.x. [DOI] [PubMed] [Google Scholar]
- 27.Kline MP, Rajkumar SV, Timm MM, Kimlinger TK, Haug JL, Lust JA, Greipp PR, Kumar S. ABT-737, an inhibitor of Bcl-2 family proteins, is a potent inducer of apoptosis in multiple myeloma cells. Leukemia. 2007;21:1549–1560. doi: 10.1038/sj.leu.2404719. [DOI] [PubMed] [Google Scholar]
- 28.Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires Bcl2 to sequester prodeath Bim, explaining sensitivity to Bcl2 antagonist ABT-737. J Clin Invest. 2007;117:112–121. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 30.Zhang S, Ye J, Zhang Y, Xu X, Liu J, Zhang SH, Kunapuli SP, Ding Z. P2Y12 protects platelets from apoptosis via PI3K-dependent Bak/Bax inactivation. J Thromb Haemost. 2013 Jan;11:149–60. doi: 10.1111/jth.12063. [DOI] [PubMed] [Google Scholar]
- 31.Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 32.Forlow SB, McEver RP, Nollert MU. Leukocyte-leukocyte interactions mediated by platelet microparticle under flow. Blood. 2000;95:1317–1323. [PubMed] [Google Scholar]
- 33.Weyrich AS, Elstad MR, McEver RP, McIntyre TM, Moore KL, Morrissey JH, Prescott SM, Zimmerman GA. Activated platelets signal chemokine synthesis by human monocytes. J Clin Invest. 1996;97:1525–1534. doi: 10.1172/JCI118575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. 2010;189:1059–1070. doi: 10.1083/jcb.201004096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee SJ, Park SY, Jung MY, Bae SM, Kim IS. Mechanism for phosphatidylserine-dependent erythrophagocytosis in mouse liver. Blood. 2011;117:5215–5223. doi: 10.1182/blood-2010-10-313239. [DOI] [PubMed] [Google Scholar]
- 36.Mause SF, Ritzel E, Leihn EA, Hristov M, Bidzhekov K, Müller-Newen G, Soehnlein O, Weber C. Platelet microparticles enhance the vasoregenerative potential of angiogenic early outgrowth cells after injury. Circulation. 2010;122:495–506. doi: 10.1161/CIRCULATIONAHA.109.909473. [DOI] [PubMed] [Google Scholar]
- 37.Hayon Y, Dashevsky O, Shai E, Varon D, Leker RR. Platelet microparticles promote neural stem cell proliferation, survival and differentiation. J Mol Neurosci. 2012;47:659–665. doi: 10.1007/s12031-012-9711-y. [DOI] [PubMed] [Google Scholar]
- 38.Gajewski TF, Thompson CB. Apoptosis meets signal transduction: elimination of a BAD influence. Cell. 1996;87:589–592. doi: 10.1016/s0092-8674(00)81377-x. [DOI] [PubMed] [Google Scholar]
- 39.Lund-Johansen F, Olweus J. Signal transduction in monocytes and granulocytes measured by multiparameter flow cytometry. Cytometry. 1992;13:693–702. doi: 10.1002/cyto.990130705. [DOI] [PubMed] [Google Scholar]
- 40.Mattson MP, Chan SL. Calcium orchestrates apoptosis. Nat Cell Biol. 2003;5:1041–1043. doi: 10.1038/ncb1203-1041. [DOI] [PubMed] [Google Scholar]
- 41.Melendez AJ, Tay HK. Phagocytosis: a repertoire of receptors and Ca2+ as a key second messenger. Biosci Rep. 2008;28:287–298. doi: 10.1042/BSR20080082. [DOI] [PubMed] [Google Scholar]
- 42.Versteeg HH, Reitsma PS, Heemskerk JW, Levi MM. New fundamentals in hemostasis. Physiol Rev. 2013;13:327–358. doi: 10.1152/physrev.00016.2011. [DOI] [PubMed] [Google Scholar]
- 43.Vukcevic M, Zorzato F, Spagnoli G, Treves S. Frequent calcium oscillations lead to NFAT activation in human immature dendritic cells. J Biol Chem. 2010;285:16003–16011. doi: 10.1074/jbc.M109.066704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sadallah S, Eken C, Martin PJ, Schifferli JA. Microparticles (ectosomes) shed by stored human platelets downregulate macrophages and modify the development of dendritic cells. J Immunol. 2011;186:6543–6552. doi: 10.4049/jimmunol.1002788. [DOI] [PubMed] [Google Scholar]
- 45.Fong KP, Barry C, Tran AN, Traxler EA, Wannemacher KM, Tang HY, Speichler KD, Blair IA, Speicher DW, Grosser T, Brass LF. Deciphering the human platelet sheddome. Blood. 2011;117:e15–e26. doi: 10.1182/blood-2010-05-283838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Passacquale G, Vamadevan P, Pereira L, Hamid C, Corrigall V, Ferro A. Monocyte-platelet interaction induces a pro-inflammatory phenotype in circulating monocytes. Plos One. 2011;6:e25595. doi: 10.1371/journal.pone.0025595. [DOI] [PMC free article] [PubMed] [Google Scholar]