

Abstract
Cytoscape is open-source software for integration, visualization and analysis of biological networks. It can be extended through Cytoscape plugins, enabling a broad community of scientists to contribute useful features. This growth has occurred organically through the independent efforts of diverse authors, yielding a powerful but heterogeneous set of tools. We present a travel guide to the world of plugins, covering the 152 publicly available plugins for Cytoscape 2.5–2.8. We also describe ongoing efforts to distribute, organize and maintain the quality of the collection.
High-throughput technologies allow enormous amounts of data to be collected on biological networks, including protein-protein interactions, protein-DNA interactions, kinase-substrate interactions, genetic interactions, gene coexpression and other functional relationships. One of the major computational platforms for analyzing these networks is Cytoscape, a general-purpose and freely available software platform for integration, visualization and statistical modeling of molecular networks together with other systems-level data1,2. To enable rapid prototyping and release of new methods, Cytoscape is implemented as an open-source software package with an accessible application programming interface (API) using the Java programming language.
One of the most powerful consequences of this design is that, through the Cytoscape API, software developers can write extensions called plugins that link Cytoscape with new code and provide access to new or alternative features. Plugins provide a flexible means by which any researcher can bring new concepts in network and systems biology to a broad user base of life scientists. Although some plugins come installed by default in the standard Cytoscape release, users optionally install most plugins to access the features they require (Box 1).
BOX 1. HOW TO FIND AND INSTALL PLUGINS FOR CYTOSCAPE.
The Cytoscape App Store (http://apps.cytoscape.org/) lists all publicly available plugins known to the Cytoscape development team. Once users finds a plugin, they download the plugin application file(s) and place it in the ‘plugins’ folder of the Cytoscape installation directory. After restarting Cytoscape, the plugin will be available along with the rest of Cytoscape.
The Cytoscape Plugin Manager can be launched from the Cytoscape ‘Plugins’ menu. The Plugin Manager allows the user to search for and install available plugins while Cytoscape is running without needing to restart.
This Perspective provides detailed information on many Cytoscape Plugins, including their functional tags, in the Supplementary Data.
Several publications1,3,59 give tutorials on the usage of Cytoscape. For the tutorial presented by Cline et al.1, the NatureProtocolsWorkflow plugin bundles together the set of plugins used in their tutorial.
In the past several years, the number of publicly available Cytoscape plugins has grown dramatically, from a few dozen in 2005 to 152 registered plugins in the beginning of April 2012. This growth greatly increases the power and versatility of network analysis. However, it has occurred organically across a heterogeneous community of researchers and software developers, consequently presenting the user with a diverse and sometimes bewildering array of choices. Although most plugins provide user documentation and many are described in peer-reviewed research papers, a summary evaluation of the entire collection of plugins is needed. That is the purpose of this paper.
Census of available plugins and initial validation
The Cytoscape website provides a mechanism for submitting plugins (http://www.cytoscape.org/plugin_submit.html), which keeps a copy of the plugin code and tracks information about each plugin: its authors and author affiliations, a brief description of its functionality, a link to the plugin homepage if one exists and the known compatible versions of Cytoscape. We used the plugin registry as our primary means of identifying plugins; as of April 2012, it contained a total of 152 publicly available plugins for Cytoscape v.2.5 or later (while this work was in review, 20 additional plugins were released, bringing the number up to 172). Laboratories contributing plugins are distributed worldwide, with the largest contributions coming from North America and Europe (Fig. 1a).
Figure 1.

Statistics for registered Cytoscape plugins. (a) Countries of origin for each plugin based on contact e-mail addresses and affiliations. (b) Bottom, number of downloads for each plugin, sorted by number of total downloads. Top, plugin names are shown for the top 20 plugins. The name and number of downloads for each plugin is in Supplementary Figure 6.
We first assessed the rate of use of each plugin by tabulating the number of downloads within the past year as well as the total number of downloads overall (Fig. 1b and Table 1). The former statistic indicates recent popularity and is directly comparable across plugins, whereas the latter statistic indicates all-time popularity but is skewed toward older plugins that have been consistently popular since their initial publication.
Table 1.
Tags and descriptions of top five most downloaded plugins
| Plugin | Functional tags | Description | Total downloads |
|---|---|---|---|
| BiNGO | Enrichment analysis, GO annotation, ontology analysis | Calculates overrepresented functions (GO terms) in the network and displays them as GO directed acyclic graphs | 43,641 |
| MCODE | Clustering, graph analysis | Clusters a given network on the basis of vertex weighting by local neighborhood density and outward traversal from a locally dense seed protein to isolate the dense regions | 16,260 |
| AgilentLiteratureSearch | Literature mining, network generation | Mines scientific literature to find publications related to search term and to create interaction network based on the search result | 15,432 |
| jActiveModules | Integrated analysis, functional module detection, graph analysis | Finds clusters where member nodes show significant changes in expression levels | 12,547 |
| MiMIplugin | Online data import, network generation, interaction database | Retrieves interactions associated with input IDs; user can add own annotations to genes, which can be viewed by different users | 10,108 |
The associated tags, description and total number of downloads are listed for each tag. The Supplementary Data file provides a complete list of plugins we tagged along with descriptions. Tags will be updated over time to gradually improve the classification.
Next, we validated each plugin by downloading and testing its basic function. The latest version of each plugin was installed on an appropriate version of Cytoscape as determined from the information in our plugin database. We briefly followed basic manipulations described in tutorials and documents provided by the plugin authors. Eighteen (12%) plugins did not pass the basic validation test and were marked ‘Did not work’. Eleven (7%) plugins passed validation but were missing some of the expected functions and were marked ‘Problem found’. Both types of errors were communicated by email to the plugin authors, nearly all of whom replied and are currently working with us to resolve the apparent difficulties. We expect that by the time this work is published, many of the issues will have been fixed. In the ‘Problem found’ category, many problems have been traced to errors or ambiguities in the user documentation, not errors in the code. The 20 plugins registered after April 2012 were not tested, but they are listed in the Supplementary Data.
Plugins as steps in a network analysis workflow
The utility of most Cytoscape plugins can be best understood within the larger context of how networks are analyzed (Fig. 2). A typical Cytoscape workflow begins by importing interactions (for example, protein-protein interactions) from a user’s own experiments or from public databases. Whereas experimental data on interactions are loaded directly into Cytoscape through standard file formats, public databases of interactions are accessed using plugins. Typically, the database is queried for interactions involving a list of genes of interest or for interactions among genes that have a certain attribute, such as a common molecular function or phenotype. Alternatively, interactions can be mined directly from the literature or through computational inference from non-interaction data such as expression profiles. Cytoscape has dozens of plugins for literature mining and for network inference.
Figure 2.

Network analysis workflow. Specific genes or attributes (blue) typically gathered in preparation for network analysis are imported and used for network generation (red). Many different types of networks are available (green) for import, after which Cytoscape visualization enables users to efficiently explore and biologically interpret the network65 (orange). Subsequent network analysis invokes computational algorithms or statistics to interpret and organize interactions (red). Commonly used plugins associated with each level are listed.
Following the import of networks and visualization in Cytoscape, a large repertoire of plugins is available for network analysis (Fig. 2). For instance, plugins for network topological analysis enable users to calculate statistics such as the distribution of network connectivity (that is, node degrees), and network clustering plugins allow users to extract densely connected network regions, which often correspond to functional modules such as protein complexes or pathways. Biological functions of these modules can be inferred with plugins that perform functional enrichment: identification of functional terms that are statistically enriched among the set of genes comprising the module. Functional modules can also be identified by integrating the network with expression data to identify regions that are coherently up- or downregulated, or by integrating networks across species to identify regions of the network with conserved interactions. Finally, plugins for scripting and programmatic access allow control over the workflow.
In what follows, we review Cytoscape plugins at each step of this workflow, with special focus on the plugins that are most widely used, that is, those that have the greatest numbers of total downloads. Further descriptive use-cases of plugins are available in previous reviews1,3. To enable users to find suitable plugins at each step, we have developed a plugin classification system based on a broad set of 41 tags and a companion plugin webstore (http://apps.cytoscape.org/) that organizes plugins by tag. As an example, Supplementary Table 1 shows the top ten tags according to the number of plugins annotated to each. This information can also be illustrated by a network (Fig. 3a,b and Supplementary Fig. 1) in which plugins are connected to tags, such that plugins having similar tag assignments fall close to one another in the network, and the overall popularity of a tag can be seen by its total number of connections (Fig. 3c).
Figure 3.

Relationships between Cytoscape plugins and tags. (a,b) Zoomed figure (a) and wide view (b) of relationship map. Orange diamonds, tags; blue rectangles, plugins. The full map of the plugin tags can be found in Supplementary Figure 1. (c) Percentages of plugins with indicated tags. A plugin is counted once for each tag assignment; it is counted multiple times if it is assigned multiple tags. Note that only plugins which passed our basic validation test are shown.
Network import
Cytoscape imports interaction data in various generic tabular formats including CSV (comma-separated values), TSV (tab-separated values) and Excel, along with network-specific formats such as SIF (simple interaction file, originally developed for Cytoscape), XGMML (Extensible Graph Markup and Modeling Language), GML (Graph Modelling Language), PSI MI (Proteomics Standards Initiative–Molecular Interaction format)4, BioPAX (Biological Pathway Exchange)5, OpenBEL (Open Biological Expression Language) and SBML (Systems Biology Markup Language)6. The generic tabular formats and SIF are especially useful when users wish to import their own experimental interaction data, which often consist of a simple list of gene pairs that have been found to interact. The network-specific formats can represent many additional details about each interaction when known, for example, the type, strength, mathematical details and functional consequence of interaction and, if applicable, the direction of information flow. Increasingly, the scientific community is beginning to use these more expressive formats, such as BioPAX, OpenBEL and SBML, to create and share models of biological networks among researchers.
Although the ability to recognize interaction data in these formats is provided by the Cytoscape core application, in many cases the user does not have new data but instead seeks to access the large online databases of previously generated interactions. Therefore, to complement the core Cytoscape functionality, several plugins are available to import existing interaction data catalogued in public databases. For example, the BioGridPlugin can be used to import an entire interactome (that is, the full set of interactions mapped for a species to date) from BioGrid7, one of several large databases of molecular and genetic interactions. Alternatively, a user may wish to import interactions involving a defined subset of genes or proteins; many plugins have been developed for this purpose. Among these, MiMI8, ConsensusPathDB9 and APID2NET10 are established and robust examples with useful features. The MiMI plugin retrieves and displays interactions from the Michigan Molecular Interactions (MiMI) database (Fig. 1b and Table 1), which combines data from a variety of established primary-interaction databases. The ConsensusPathDB plugin allows users to computationally validate whether there is previous support for a set of interactions in their own data. APID2NET provides a sophisticated graphical user interface to extract interactions involving a set of genes from the APID server (Agile Protein Interaction DataAnalyzer, Fig. 1b) and to perform analyses including hub identification, protein motif annotation and Gene Ontology (GO) enrichment. For databases that provide a PSICQUIC11 web service (standardized programmatic access to molecular interaction databases over the Web), interactions can be imported into Cytoscape by the PSICQUICUniversalClient plugin.
Some specialized plugins have been designed to import and visualize metabolic networks in particular, which can consist of multiple types of nodes (enzymes, small molecules and cofactors) or edges (reversible or irreversible reactions). The Metscape plugin12 generates metabolic networks based on information in the Kyoto Encyclopedia for Genes and Genomes (KEGG)13 and the Edinburgh Human Metabolic Network database14 (Fig. 1b and Supplementary Fig. 2). This plugin is powerful for superposition of a metabolic network with user-defined data on enzyme expression levels or compound concentrations. As an alternative, the KGMLReader plugin imports KEGG metabolic networks and preserves their hand-drawn intuitive layout. However, some network information in KEGG cannot be imported by KGMLReader because of problems mapping between the KEGG and Cytoscape network representations. Other plugins for importing metabolic networks into Cytoscape include the BioCycPlugin, which provides access to the BioCyc metabolic network database (http://biocyc.org/), and ReConn, which provides access to Reactome (http://reactome.org/).
Specialized plugins have also been developed to import canonical signaling or regulatory networks curated from literature. The GPML (Fig. 1b) and Superpathways plugins import and visualize networks from WikiPathways15, an open platform for curation of biological networks by the scientific community. We also recommend the Pathway Commons16 website (http://www.pathwaycommons.org/), which is able to transfer a network of interest directly to Cytoscape by clicking a hyperlink that appears on the web page for that network.
Literature mining
The large corpus of published papers provides information about interactions that are not yet available in public databases. Thus, extraction of interactions based on computational literature mining has become an important activity. The chief means in Cytoscape (v.2.5 or later) of building networks from the literature is AgilentLiteratureSearch17, a plugin that mines literature abstracts from sources such as Medline, Online Mendelian Inheritance in Man (OMIM)18 and the US patent database to identify putative interactions and use them to automatically construct a network (Fig. 4a). After a user enters search terms, the plugin finds matching records, extracts genes and their associations described within the record and displays them as a network. Although interaction networks based on automatic literature mining usually contain substantial false positives, they allow users to visualize a draft set of protein interactions that may not be present in other databases. The sentences that support each interaction can be manually reviewed to eliminate false positives. Demand for AgilentLiteratureSearch is high: it is the number three plugin by total number of downloads (Fig. 1b and Table 1).
Figure 4.
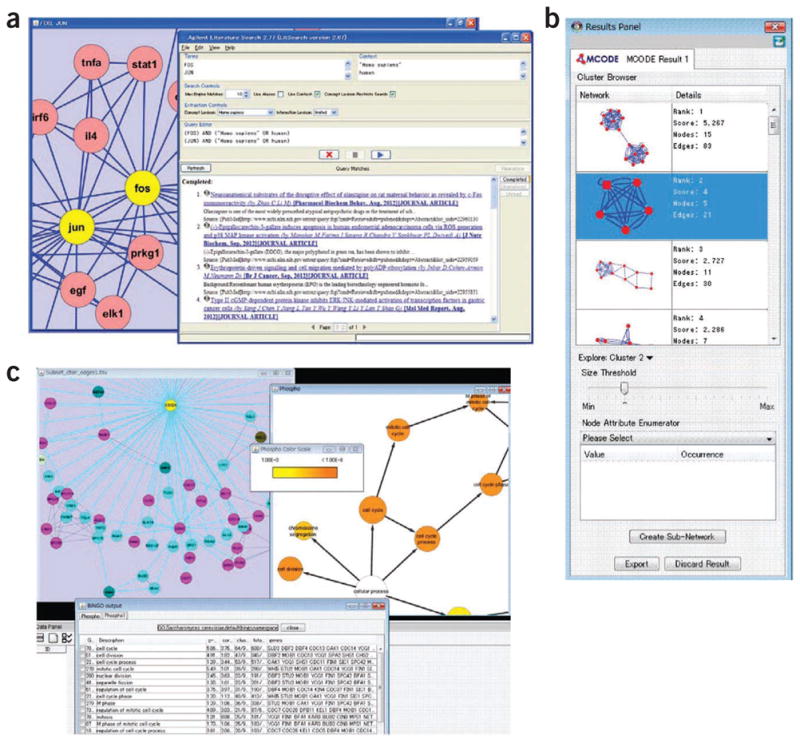
Examples of plugin outputs. (a) AgilentLiteratureSearch plugin. Human transcription factors FOS and JUN are input as an example. A network created by literature mining (left) and abstracts of scientific papers used to derive the network (right) are shown. (b) Application of MCODE to the mouse protein-protein interaction network in BioGRID. Modules extracted from the network are shown. (c) BiNGO plugin. A subnetwork containing dense kinase-substrate interactions is analyzed, with cell cycle related genes found to be enriched.
Network inference
For many species, genome-wide interaction screens have not been conducted, and users thus cannot assemble networks for these species. Even in an organism such as budding yeast, in which large-scale genetic and physical interaction experiments have been performed, complete network coverage has not yet been achieved19. Accordingly, many methods have been developed to predict novel interactions and generate networks from currently available data. GeneMANIA20 is one of the more refined plugins for this purpose. For a defined set of genes or proteins, it integrates data from many sources, including physical interactions, genetic interactions, pairs of coexpressed genes, pairs of genes in the same pathway or pairs of genes with the same subcellular location, and then visualizes the possible molecular associations among the given genes and other genes (Supplementary Fig. 3), thus allowing users to predict functions of uncharacterized proteins on the basis of functions of proteins associated with them. ExpressionCorrelation and MONET21 are plugins that predict functionally interacting pairs of proteins from expression data. MONET also incorporates biological annotations of genes to predict a regulatory network. Finally, for inference of metabolic network models, the CytoSEED plugin interfaces Cytoscape with the Model SEED22 resource for automatic generation of metabolic models from prokaryotic genome sequences.
Topological analysis and clustering
Network topology refers to the arrangement or pattern of interactions within a network; several Cytoscape plugins have been developed to calculate topological properties. The NetworkAnalyzer23 plugin is installed in Cytoscape by default and calculates network metrics such as the distribution of node degrees (node degree refers to the number of interactions involving a node; it has been shown to correlate with the essential status of genes24). Users may also try CentiScaPe25,26 for this purpose (Fig. 1b) or the Interference plugin, which evaluates the topological effects of removing single or multiple nodes from a network.
A great deal of research has focused on mining networks for interaction clusters or ‘modules’, sets of interacting molecules that tightly associate with one another. Modules in a protein-protein interaction network, for instance, are suggestive of functional protein complexes. Plugins typically extract such modules by identifying densely connected subgraphs. MCL-new and APCluster implement the popular network clustering algorithms developed by Van Dongen27 and Frey et al.28, respectively, for clustering in general. MCODE29, one of the most popular Cytoscape plugins overall (Fig. 1b and Table 1), has been developed to perform network module identification specifically in biology. MCODE weights nodes by local neighborhood density, then performs an outward traversal from a locally dense seed protein node to isolate larger dense regions, and finally graphically displays extracted modules and associated information (Fig. 4b).
Several plugins improve on the basic MCODE algorithm or user interface. AllegroMCODE implements the MCODE algorithm using graphics-processing-unit–based parallelization to find clusters efficiently. NeMo30 identifies densely connected and bipartite network modules on the basis of the combination of a unique neighbor-sharing score with hierarchical agglomerative clustering. MINE31 clusters a given network via an agglomerative clustering algorithm similar to MCODE but using a modified vertex-weighting strategy.
Different network clustering plugins can yield quite different network modules from the same data. Plugin developers typically argue that more recently developed algorithms work better than older ones, with performance often measured by the ability to recapitulate known protein complexes or pathways. However, performance may also depend on particular characteristics of the input network: MINE was shown to outperform other algorithms including MCODE and NeMo specifically when analyzing the protein-protein interaction network of Caenorhabditis elegans, which has high interaction density31. Users should therefore test several different approaches to extract network modules and investigate which predicted modules make more biological sense. For this purpose, clusterMaker32 offers access to many different network clustering algorithms in one convenient interface (Fig. 1b). Also, literature comparing the performance of existing module identification algorithms is available33,34, which may help users to select appropriate plugins.
Functional enrichment and partitioning
Genes connected in a network are likely to have similar functions; as such, the function of a network module can be inferred by finding the enriched functions of its genes. Methods such as Gene Set Enrichment Analysis (GSEA)35 have been developed to find enriched functions in a given gene list. Cytoscape has several plugins that perform this task for sets of genes in a given network, most notably BiNGO36, Cytoscape’s most popular plugin (Fig. 1b and Table 1). BiNGO extracts enriched functional terms recorded in the GO37 database and visualizes them in a hierarchy (Fig. 4c). A sister plugin called PiNGO38 was recently released and works in the opposite way; it starts with user-defined GO categories of interest and then finds candidate genes in a given network associated with those categories.
The ClueGO39 plugin creates a functionally organized network of GO, KEGG and BioCarta pathway terms (Fig. 1b) that represents functional organization within a set of interacting genes or proteins. Similarly, EnrichmentMap40 organizes gene sets into a similarity network in which nodes represent gene sets, edges represent the overlap of member genes, and node color encodes the statistical significance of enrichment. WordCloud41 visually summarizes the gene functional descriptions associated with a set of selected nodes (that is, data attributes; see below) by generating a cloud of words sized by their frequency of occurrence in the selected nodes. WordCloud is useful for visually summarizing gene function annotation of a given set of nodes in a simple way.
Beyond looking for shared node attributes as described, there are also plugins that spatially partition a network layout on the basis of such attributes. BubbleRouter groups nodes having the same attribute by a rectangular box on the main network view window (Fig. 1b). It is useful for visualizing relationships between groups of nodes having similar functions or nodes that are localized in the same cellular compartment. A more sophisticated successor called Mosaic has recently been released. Mosaic will retrieve GO annotations for nodes in any network with standard gene identifiers and then systematically partition, lay out and color the nodes as they relate to each of the three branches of GO. Mosaic thus provides a way to visualize molecular interaction networks in a known biological context.
Integrating networks with other data
A powerful feature of the core Cytoscape application is the ability to integrate biological networks with other types of data, including gene and protein sequences, functions, alternative identifiers and gene expression and other omics measurements. These other data sets are handled by what Cytoscape calls ‘Data Attributes’: tables that associate nodes and edges with columns of additional data values (of arbitrary type). As for networks, tables of biological data can be imported into Cytoscape from a user-supplied file or fetched from online sources using plugins. For instance, the BiomartClient and NCBIClient plugins (Fig. 1b) import basic gene and protein information into Cytoscape from the Biomart42 and NCBI databases, respectively. BioMartClient is also useful for retrieving or converting gene identifiers (IDs) so that newly imported information will match the IDs used in the current network. Another plugin that helps with identifier mapping is CyThesaurus, which converts gene, protein or metabolite IDs for one database to another via BridgeDb software43.
Gene and protein expression data can add information about which parts of a network are active in a given condition. The VistaClara plugin44 integrates expression data with network visualization. It provides a heat map view of gene expression data, colors genes in the network according to their expression levels (Fig. 1b and Supplementary Fig. 4) and can play a movie that animates expression changes over multiple conditions. NetAltas45 and OmicsAnalyzer46 are also available to visually investigate expression patterns of genes in a network. A unique feature of OmicsAnalyzer is that it can overlay a chart of the relevant gene expression data and statistics directly on each node.
Beyond visualization of gene expression data, some plugins enable a user to identify regions of a network that are enriched for highly or lowly expressed genes (network hot or cold spots). A popular plugin of this type is jActiveModules, which identifies and returns subnetworks in which the average gene expression level is significantly high or low in particular conditions47 (Fig. 1b and Table 1). Users may also want to try KeyPathwayMiner48, which tries to find densely connected networks in which genes have similar expression patterns by using a maximal-connected-subnetwork–finding algorithm. Alternatively, clusterMaker32 implements various algorithms for node clustering on the basis of not only graph structure but also gene expression patterns and may also be useful for finding network hot or cold spots (Fig. 1b). Finally, the PinnacleZ plugin49 identifies subnetworks for which the average expression level is diagnostic for clinical cases versus controls.
Other plugins such as BioQualiPlugin50, ExprEssence51 and PerturbationAnalyzer52 can be used to investigate the relationships between gene expression patterns and network structure. BioQualiPlugin checks global consistency between a regulatory network model (linking regulators to targets) and a set of expression data. ExprEssence compares gene expression levels in two experiments and highlights possible regulatory links that cause expression changes. PerturbationAnalyzer investigates the effects of perturbing protein concentration on protein interaction networks. DomainGraph53 allows users to combine full-length mRNA and exon expression data with interaction networks to analyze the effects of alternative splicing on pathways, protein-protein and domain-domain interaction networks.
Finally, one of the most integrative Cytoscape plugins to date (in terms of the number of layers of data being addressed) is the iCTNet plugin54, which was recently developed to integrate genome-wide association data (associations between single-nucleotide polymorphisms and phenotypes) with protein-protein, disease-tissue, tissue-gene and drug-gene interactions. It may assist users in elucidating a new trait classification, pathogenic mechanism or treatment for human disease traits.
Network comparison and merging
Several plugins have been developed to compare or integrate multiple networks. One of the simplest examples is AdvancedNetworkMerge, which comes pre-installed with Cytoscape. This plugin performs defined operations (union, intersection and difference) on the sets of interactions in multiple networks loaded into Cytoscape. The Venndiagrams and VennDiagramGenerator plugins can compare two networks and draw a Venn or Euler diagram showing the overlap of nodes or edges between them. CABIN55 is a more refined plugin which has been used to integrate interaction data sets from different resources and to help explore the integrated network56. A user can conduct confidence analysis of the interactions with the integrated network.
Several plugins with more specialized comparison functions have also been developed. Based on the idea that interactions (known as interologs) are conserved to some extent across multiple species, the plugins NetworkEvolution57 and OrthoNets58 were developed to allow users to integrate interactions from multiple species to build conserved networks. Finally, because high-throughput genetic interaction screens have become feasible, integrating genetic interactions with other types of networks has been an important issue: the PanGIA plugin59 has recently been developed to integrate physical and genetic interactions to create hierarchical module maps (Supplementary Fig. 5).
Scripting and programmatic access
There is a rich set of plugins for scripting—that is, for control of Cytoscape using short commands. These include the use of programming languages other than Java (the language in which Cytoscape is written): scripting plugins are available for JavaScript, Python, Ruby and Clojure (JavaScriptEngine, PythonScriptingEngine, RubyScriptingEngine and ClojureEngine, respectively). They are managed by ScriptEngineManager, another plugin. There are also several plugins that can control Cytoscape through APIs such as CytoscapeRPC, which enables Cytoscape to be controlled from other programs and languages using the XML-RPC protocol. For instance, one of the packages of Bioconductor, RCytoscape, uses XML-RPC to communicate between R and Cytoscape. CyGoose allows Cytoscape to route data sets from one application to another using the Gaggle Framework60. Finally, commandTool is a plugin that provides access to a core set of commands built into Cytoscape; using commandTool, these commands can be scripted and executed in batch mode.
Additional plugins
The remaining Cytoscape plugins do not cluster tightly with others. They do, however, fall under general high-level categories that help convey their functions. We have added the tag ‘Utility’ for plugins that enhance the basic functionality of Cytoscape. This tag covers plugins that deal with selecting multiple nodes and processing them in different ways. For example, NamedSelection assigns a label to selected nodes and, after de-selection, enables users to reselect the nodes according to the label. Other plugins extend the basic definition of a network graph, nominally defined as a set of nodes and a set of edges connecting these nodes. For example, GroupTool enables a user to define groups of nodes and, for each group, to display basic information on the Cytoscape panel. MetaNodePlugin2 enables a user to define a ‘meta-node’ as a set of nodes that can be collapsed into a single node and then expanded back to the original set (Fig. 1b). These two plugins were tagged as ‘Grouping’. Seven plugins were tagged as ‘Layout’ because they are related to layout of nodes in the network. For example, ReOrientPlugin lays out nodes according to positions saved in a user-created Cytoscape session file. Three plugins, TransClust, BLAST2SimilarityGraph and clusterExplorerPlugin61, were tagged as ‘Sequence similarity’. They enable a user to visualize sequence similarity (for example, BLAST) results as networks of edges connecting genes that have high-scoring similar sequences. Another three plugins, ChemViz, structureViz62 (Fig. 1b) and RINalyzer63, were labeled with the ‘Molecular structure’ tag, as they visualize chemical and protein structures as networks on Cytoscape. FERN64 has the ‘Network simulation’ functional tag because it performs stochastic simulation of chemical reaction networks. In the future, we will allow developers and users to suggest tags for plugins to enable the community to maintain and extend our categorization system. The number of downloads for all plugins is shown in Supplementary Figure 6.
Cytoscape community and future directions
We are developing a number of community resources and improvements to Cytoscape to help make the plugin development process more fun and efficient. First, we are developing the next version of Cytoscape, version 3.0, to address the problem of maintaining backwards compatibility between Cytoscape and plugin versions. Cytoscape 3.0 uses the modular OSGi (Open Services Gateway initiative) framework (http://www.osgi.org/), which means that plugins will be less sensitive to changes in the software code as Cytoscape evolves and will be fully interoperable with other plugins. In the meantime, all of the plugins we review here will continue to work with v.2.8 and will be migrated to Cytoscape 3.0 soon after its release.
Second, we are developing the Cytoscape AppStore (http://apps.cytoscape.org/), a new online community forum centered on Cytoscape plugins that will promote the development, testing and distribution of plugins. Users can interactively tag, rate, review, document and install plugins via the web or from within Cytoscape.
Third, each year a different group of Cytoscape developers hosts an annual Cytoscape symposium to coordinate the use and development of Cytoscape and its plugins and to facilitate the exchange of ideas and research on network analysis. Information on the next Cytoscape symposium is available at http://www.cytoscape.org/.
Since Cytoscape was released and published a decade ago, a large number of plugins have been developed. This contribution by highly motivated users, developers and organizers has been crucial to the success and utility of the Cytoscape platform. If you are interested in participating in the Cytoscape community, we invite you to attend the symposium, develop a plugin, join our mailing list or simply try out Cytoscape.
Supplementary Material
Acknowledgments
Work on this review was funded by the National Resource for Network Biology (P41 GM103504) and the San Diego Center for Systems Biology (P50 GM085764). We thank J. Dutkowski, D. Emig and G. Hannum for advice and critical reading of the manuscript. Finally, the greatest thanks go to all of the plugin developers who have enriched the Cytoscape user experience with their ideas. We apologize to those plugin authors whose excellent work was not covered here because of space limitations.
Footnotes
Supplementary information is available in the online version of the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Cline MS, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yeung N, Cline MS, Kuchinsky A, Smoot ME, Bader GD. Exploring biological networks with Cytoscape software. Curr Protoc Bioinformatics. 2008;23:8.13. doi: 10.1002/0471250953.bi0813s23. [DOI] [PubMed] [Google Scholar]
- 4.Hermjakob H, et al. The HUPO PSI’s molecular interaction format—a community standard for the representation of protein interaction data. Nat Biotechnol. 2004;22:177–183. doi: 10.1038/nbt926. [DOI] [PubMed] [Google Scholar]
- 5.Demir E, et al. The BioPAX community standard for pathway data sharing. Nat Biotechnol. 2010;28:935–942. doi: 10.1038/nbt.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hucka M, et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics. 2003;19:524–531. doi: 10.1093/bioinformatics/btg015. [DOI] [PubMed] [Google Scholar]
- 7.Stark C, et al. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011;39:D698–D704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao J, et al. Integrating and annotating the interactome using the MiMI plugin for cytoscape. Bioinformatics. 2009;25:137–138. doi: 10.1093/bioinformatics/btn501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pentchev K, Ono K, Herwig R, Ideker T, Kamburov A. Evidence mining and novelty assessment of protein-protein interactions with the ConsensusPathDB plugin for Cytoscape. Bioinformatics. 2010;26:2796–2797. doi: 10.1093/bioinformatics/btq522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hernandez-Toro J, Prieto C, De las Rivas J. APID2NET: unified interactome graphic analyzer. Bioinformatics. 2007;23:2495–2497. doi: 10.1093/bioinformatics/btm373. [DOI] [PubMed] [Google Scholar]
- 11.Aranda B, et al. PSICQUIC and PSISCORE: accessing and scoring molecular interactions. Nat Methods. 2011;8:528–529. doi: 10.1038/nmeth.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao J, et al. Metscape: a Cytoscape plug-in for visualizing and interpreting metabolomic data in the context of human metabolic networks. Bioinformatics. 2010;26:971–973. doi: 10.1093/bioinformatics/btq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma H, et al. The Edinburgh human metabolic network reconstruction and its functional analysis. Mol Syst Biol. 2007;3:135. doi: 10.1038/msb4100177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pico AR, et al. WikiPathways: pathway editing for the people. PLoS Biol. 2008;6:e184. doi: 10.1371/journal.pbio.0060184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cerami EG, et al. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011;39:D685–D690. doi: 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vailaya A, et al. An architecture for biological information extraction and representation. Bioinformatics. 2005;21:430–438. doi: 10.1093/bioinformatics/bti187. [DOI] [PubMed] [Google Scholar]
- 18.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cusick ME, et al. Literature-curated protein interaction datasets. Nat Methods. 2009;6:39–46. doi: 10.1038/nmeth.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montojo J, et al. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics. 2010;26:2927–2928. doi: 10.1093/bioinformatics/btq562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee PH, Lee D. Modularized learning of genetic interaction networks from biological annotations and mRNA expression data. Bioinformatics. 2005;21:2739–2747. doi: 10.1093/bioinformatics/bti406. [DOI] [PubMed] [Google Scholar]
- 22.Henry CS, et al. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol. 2010;28:977–982. doi: 10.1038/nbt.1672. [DOI] [PubMed] [Google Scholar]
- 23.Assenov Y, Ramirez F, Schelhorn SE, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2008;24:282–284. doi: 10.1093/bioinformatics/btm554. [DOI] [PubMed] [Google Scholar]
- 24.Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 25.Ladha J, et al. Glioblastoma-specific protein interaction network identifies PP1A and CSK21 as connecting molecules between cell cycle-associated genes. Cancer Res. 2010;70:6437–6447. doi: 10.1158/0008-5472.CAN-10-0819. [DOI] [PubMed] [Google Scholar]
- 26.Scardoni G, Petterlini M, Laudanna C. Analyzing biological network parameters with CentiScaPe. Bioinformatics. 2009;25:2857–2859. doi: 10.1093/bioinformatics/btp517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frey BJ, Dueck D. Clustering by passing messages between data points. Science. 2007;315:972–976. doi: 10.1126/science.1136800. [DOI] [PubMed] [Google Scholar]
- 29.Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rivera CG, Vakil R, Bader JS. NeMo: Network Module identification in Cytoscape. BMC Bioinformatics. 2010;11 (suppl 1):S61. doi: 10.1186/1471-2105-11-S1-S61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rhrissorrakrai K, Gunsalus KC. MINE: Module Identification in Networks. BMC Bioinformatics. 2011;12:192. doi: 10.1186/1471-2105-12-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris JH, et al. clusterMaker: a multi-algorithm clustering plugin for Cytoscape. BMC Bioinformatics. 2011;12:436. doi: 10.1186/1471-2105-12-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, Wu M, Kwoh CK, Ng SK. Computational approaches for detecting protein complexes from protein interaction networks: a survey. BMC Genomics. 2010;11 (suppl 1):S3. doi: 10.1186/1471-2164-11-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moschopoulos CN, et al. Which clustering algorithm is better for predicting protein complexes? BMC Res Notes. 2011;4:549. doi: 10.1186/1756-0500-4-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 37.Ashburner M, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smoot M, Ono K, Ideker T, Maere S. PiNGO: a Cytoscape plugin to find candidate genes in biological networks. Bioinformatics. 2011;27:1030–1031. doi: 10.1093/bioinformatics/btr045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bindea G, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS ONE. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oesper L, Merico D, Isserlin R, Bader GD. WordCloud: a Cytoscape plugin to create a visual semantic summary of networks. Source Code Biol Med. 2011;6:7. doi: 10.1186/1751-0473-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haider S, et al. BioMart Central Portal—unified access to biological data. Nucleic Acids Res. 2009;37:W23–W27. doi: 10.1093/nar/gkp265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Iersel MP, et al. The BridgeDb framework: standardized access to gene, protein and metabolite identifier mapping services. BMC Bioinformatics. 2010;11:5. doi: 10.1186/1471-2105-11-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kincaid R, Kuchinsky A, Creech M. VistaClara: an expression browser plug-in for Cytoscape. Bioinformatics. 2008;24:2112–2114. doi: 10.1093/bioinformatics/btn368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang L, Walker JR, Hogenesch JB, Thomas RS. NetAtlas: a Cytoscape plugin to examine signaling networks based on tissue gene expression. In Silico Biol. 2008;8:47–52. [PubMed] [Google Scholar]
- 46.Xia T, Hemert JV, Dickerson JA. OmicsAnalyzer: a Cytoscape plug-in suite for modeling omics data. Bioinformatics. 2010;26:2995–2996. doi: 10.1093/bioinformatics/btq583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ideker T, Ozier O, Schwikowski B, Siegel AF. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002;18 (suppl 1):S233–S240. doi: 10.1093/bioinformatics/18.suppl_1.s233. [DOI] [PubMed] [Google Scholar]
- 48.Alcaraz N, Kücük H, Weile J, Wipat A, Baumbach J. KeyPathwayMiner: detecting case-specific biological pathways using expression data. Internet Math. 2011;7:299–313. [Google Scholar]
- 49.Chuang HY, Lee E, Liu YT, Lee D, Ideker T. Network-based classification of breast cancer metastasis. Mol Syst Biol. 2007;3:140. doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guziolowski C, Bourde A, Moreews F, Siegel A. BioQuali Cytoscape plugin: analysing the global consistency of regulatory networks. BMC Genomics. 2009;10:244. doi: 10.1186/1471-2164-10-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Warsow G, et al. ExprEssence—revealing the essence of differential experimental data in the context of an interaction/regulation net-work. BMC Syst Biol. 2010;4:164. doi: 10.1186/1752-0509-4-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li F, et al. PerturbationAnalyzer: a tool for investigating the effects of concentration perturbation on protein interaction networks. Bioinformatics. 2010;26:275–277. doi: 10.1093/bioinformatics/btp634. [DOI] [PubMed] [Google Scholar]
- 53.Emig D, et al. AltAnalyze and DomainGraph: analyzing and visualizing exon expression data. Nucleic Acids Res. 2010;38:W755–W762. doi: 10.1093/nar/gkq405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang L, Khankhanian P, Baranzini SE, Mousavi P. iCTNet: a Cytoscape plugin to produce and analyze integrative complex traits networks. BMC Bioinformatics. 2011;12:380. doi: 10.1186/1471-2105-12-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singhal M, Domico K. CABIN: collective analysis of biological interaction networks. Comput Biol Chem. 2007;31:222–225. doi: 10.1016/j.compbiolchem.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 56.Petyuk VA, et al. Characterization of the mouse pancreatic islet proteome and comparative analysis with other mouse tissues. J Proteome Res. 2008;7:3114–3126. doi: 10.1021/pr800205b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.WoŸniak M, Tiuryn J, Dutkowski J. MODEVO: exploring modularity and evolution of protein interaction networks. Bioinformatics. 2010;26:1790–1791. doi: 10.1093/bioinformatics/btq274. [DOI] [PubMed] [Google Scholar]
- 58.Hao Y, et al. OrthoNets: simultaneous visual analysis of orthologs and their interaction neighborhoods across different organisms. Bioinformatics. 2011;27:883–884. doi: 10.1093/bioinformatics/btr035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Srivas R, et al. Assembling global maps of cellular function through integrative analysis of physical and genetic networks. Nat Protoc. 2011;6:1308–1323. doi: 10.1038/nprot.2011.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shannon PT, Reiss DJ, Bonneau R, Baliga NS. The Gaggle: an open-source software system for integrating bioinformatics software and data sources. BMC Bioinformatics. 2006;7:176. doi: 10.1186/1471-2105-7-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wittkop T, et al. Comprehensive cluster analysis with Transitivity Clustering. Nat Protoc. 2011;6:285–295. doi: 10.1038/nprot.2010.197. [DOI] [PubMed] [Google Scholar]
- 62.Morris JH, Huang CC, Babbitt PC, Ferrin TE. structureViz: linking Cytoscape and UCSF Chimera. Bioinformatics. 2007;23:2345–2347. doi: 10.1093/bioinformatics/btm329. [DOI] [PubMed] [Google Scholar]
- 63.Doncheva NT, Klein K, Domingues FS, Albrecht M. Analyzing and visualizing residue networks of protein structures. Trends Biochem Sci. 2011;36:179–182. doi: 10.1016/j.tibs.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 64.Erhard F, Friedel CC, Zimmer R. FERN – a Java framework for stochastic simulation and evaluation of reaction networks. BMC Bioinformatics. 2008;9:356. doi: 10.1186/1471-2105-9-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Merico D, Gfeller D, Bader GD. How to visually interpret biological data using networks. Nat Biotechnol. 2009;27:921–924. doi: 10.1038/nbt.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.