

Abstract
The evolution of the cancer cell into a metastatic entity is the major cause of death in cancer patients. Activation of epithelial to mesenchymal transition (EMT) endows invasive and metastatic properties upon cancer cells that favor successful colonization of distal target organs. The observation that in many cancers distant metastases resemble the epithelial phenotype of primary tumors has led to speculation that the disseminated tumor cells recruited to the target organs undergo mesenchymal to epithelial transition (MET). However, the MET cascade has not been recapitulated in vivo, and the cellular and molecular regulators that promote MET remain unknown. In a recent report, using a model of spontaneous breast cancer, we have shown that bone marrow (BM)-derived myeloid progenitor cells in the premetastatic lung secrete the proteoglycan versican, which induces MET of metastatic tumor cells and accelerates metastases. This review summarizes recent progress in MET research and outlines a unique paracrine cross talk between the microenvironment and the cancer cells that promotes tumor outgrowth in the metastatic organ and discusses opportunities for novel antimetastatic approaches for cancer therapy.
Epithelial – mesenchymal transitions in tumor metastasis
Transdifferentiation of polarized epithelial cells to mesenchymal cells (EMT), a key developmental program, is evoked during tumor invasion and metastasis and several molecular pathways that mediate EMT in cancer cells have been identified (1-4). In addition to promoting tumor cell invasion and metastasis, EMT leads to the generation of cancer cells with stem cell-like characteristics including increased self-renewal and tumor-initiating capabilities, and increased resistance to apoptosis and chemotherapy (3) However, while EMT has been demonstrated in cancer cell lines in culture, whether EMT occurs in vivo remains incompletely resolved (5, 6). Demonstration of EMT in vivo is beleaguered with both technical and biological challenges. First, the heterogeneity of the tumor makes it difficult to distinguish differentiated tumor cells that have undergone EMT from stromal cells that display a mesenchymal phenotype using morphological criteria and standard EMT markers. Second, the phenotypic complexity within the tumor cells such as existence of de-differentiated stem cell like pools makes it difficult to conclude if these are parental tumor cells or consequences of EMT. Third, EMT in tumor cell is transient; once a metastatic cell has invaded a new tissue its mesenchymal features disappear. Thus, the universality of EMT as a cardinal hallmark of metastasis has not been accepted by the clinical community (7, 8). To monitor EMT in vivo, it has become necessary to lineage trace individual cancer cells from the time they break off from a primary tumor to the point at which they colonize a new organ. Better markers are required to identify true tumor epithelial cells that have undergone EMT transition. Nonetheless, recent studies have begun to provide evidence of EMT in tumor progression in vivo. In a recent study, using cell-fate mapping strategies with stromal- and epithelial-specific cre-transgenic mice, oncogene Myc initiated breast tumors exhibited features of EMT (9). Using an intravital imaging approach, Giampieri et al. showed that single breast tumor motile cells that have an active TGF-β-Smad2/3 EMT promoting signaling were capable of hematogenous metastasis to distal organs, while those lacking this signaling pathway were prone to passive lymph metastasis (10). In a pancreatic cancer cell model, genetically labeled fluorescent tumor cells that invaded and entered the bloodstream were associated with EMT. Circulating pancreatic cells maintained a mesenchymal phenotype, exhibited stem cell properties, and seeded the liver (11). The ability to mark cell lineages and track their fate in transgenic mice suggests that EMT may occur in vivo, however, such a demonstration remains a challenge in human tumors.
In addition to the obstacles mentioned above, an apparent contradiction to the association between EMT and metastasis comes from clinical observations that distant metastases derived from a variety of primary carcinomas resemble an epithelial phenotype. For example, metastases in distal organs such as liver, lung, bone and brain derived from a variety of primary tumor types exhibit overtly epithelial phenotypes (7, 8, 12-14). These observations raised the possibility that tumor cells may disseminate without switching to a mesenchymal phenotype thereby casting doubts on the requirement of EMT for metastasis formation. On the other hand, if cancer cells must pass through an EMT in order to disseminate, an important question is why the resulting metastases closely resemble, at the histopathological level, the primary carcinomas from which they have arisen. This has led to a tantalizing possibility that the disseminated mesenchymal tumor cells recruited to the target organs may undergo a reverse phenotypic transition from mesenchymal back to epithelial by a process called mesenchymal to epithelial transition (MET).
MET is an essential developmental process that has been studied in kidney organogenesis and somatogenesis (15), and it has been considered that MET may be associated with tumor metastasis (3, 16, 17). Evidence for MET has been limited to in vitro and xenograft experiments (12, 18, 19). In one study, disseminated breast tumor cells expressed fibroblastic/mesenchymal marker Fsp-1, suggesting that EMT had occurred, which could shift back to Fsp-1 negative phenotype suggesting MET (20). In another study, increased epithelial phenotype was observed during the formation of bone metastases from bladder carcinoma cells (12). Consistent with this observation differentiated metastases have been observed in lung, prostate, and breast, colorectal and hepatic carcinoma models. These findings suggest that cancer cells may undergo MET in the secondary organ, nonetheless, the likelihood that metastases may have arisen from minor population of E-cadherin+ cells cannot be completely excluded (21). Two photon intravital microscopy (TPIVM) has been useful in imaging intravasation and extravasation (22, 23), however, two major obstacles exist in observing MET. First, the sporadic nature of invasion and colonization makes observation statistically improbable. The use of two-photon excitation, which relies on simultaneous absorption of two near infrared photons in a small excitation volume provides imaging depths around 500 μm with high resolution and low phototoxicity and photobleaching (24), expanding the volume of tissue that can be surveyed up to ten-fold. TPIVM observation of the tissues where metastasis is most likely to occur, liver, brain, and lung, greatly increases the probability for success. Second, there is no easily observable anatomical phenotype to distinguish between mesenchymal and epithelial cells. One potential solution to this problem would be to create tumor cell lines that express different fluorescent proteins under promoters that specifically express in epithelial or mesenchymal cells.
Bone marrow-derived pre-metastatic niche mediates mesenchymal to epithelial transition
We reasoned that to demonstrate MET in vivo, it would be imperative to identify critical mediators of MET as this would allow us to block these mediators and evaluate the impact on MET and overall metastasis. We considered cancer cell intrinsic mediators of MET, however, genomic analysis of primary tumors and distant metastases have indicated a surprisingly high degree of similarity at the level of global gene copy number alterations, loss of heterozygosity and single nucleotide variation (25-28). Furthermore, gene expression profiling has shown that human primary breast tumors are strikingly similar to the distant metastases of the same patient (29). These studies suggested that MET of the disseminated tumor cell may not be driven by cell intrinsic evolving mutations, but may be under the keen influence of the microenvironment prevailing in the metastatic organ of the host. Indeed, bone marrow (BM) and other cancer associated stromal cells influence patient outcomes through paracrine and endocrine effect (30, 31). Based on these studies, we posited that tumor-induced pre-metastatic niches in distant organs, which serve as permissive hubs for future metastases, may also contribute to MET of disseminated tumor cells (32, 33). To explore these putative MET promoting niches, we used a spontaneous breast cancer model (MMTV-PyMT), which is metastatic to the lungs. As expected, cancer cells in the mammary gland and in the metastatic lesions in the lungs showed epithelial phenotypes as revealed by increased expression of E-cadherin compared to vimentin. Importantly, genetically tagged single disseminated breast tumor cells that had colonized the lung showed a mesenchymal phenotype as determined by elevated vimentin levels (unpublished data), suggesting that epithelial tumor cells had undergone EMT at the primary tumor site followed by MET in the metastatic organ. To identify mediators of MET, BM-derived hematopoietic cells that comprise the pre-metastatic niche were profiled for gene expression, and a subset of the BM-derived myeloid cells comprising the monocytic CD11b+Ly6C+ fraction was found to express elevated versican, an extracellular matrix chondroitin sulfate proteoglycan. Previous studies have shown that versican expressed by intratumoral stromal cells is associated with a worse prognosis in cancer patients (34, 35). Notably, during kidney development, the conversion of metanephric mesenchyme to epithelium involves MET that is regulated by versican, (36) (37) suggesting that BM-derived versican may also induce MET of tumor cells in a paracrine fashion.
Versican expressed by the myeloid progenitor cells promoted metastatic tumor outgrowth by enhancing cell proliferation, and specific knockdown of versican in the BM cells did not impact primary tumors, but resulted in a severe reduction in macrometastases without affecting micrometastases (38). This data was further supported by the observation that depletion of versican-producing BM myeloid progenitor cells produced vimentin-positive micrometastatic lesions. Versican suppression did not perturb the recruitment of myeloid progenitor cells in the lung microenvironment; neither did it change the immune microenvironment, suggesting that the immunosuppressive arm of the myeloid cell function was not affected.
Mesenchymal to epithelial transition is associated with enhanced proliferation and accelerated metastases
To better understand the mechanism by which versican promotes metastases in vivo, an experimental metastases system was used, to enable exclusive analysis of post-EMT events in the metastatic organ. A metastatic human breast cancer cell line MDA-MB-231 was used as it exhibits a E-cadherin- vimentin+ mesenchymal (post EMT) phenotype and is therefore amenable to MET analysis. Administration of these cells in immunocompromised SCID mice gave rise to E-cadherin+ metastatic lesions in the lung, consistent with previous studies (39). In this setting, specific depletion of versican-producing BM myeloid progenitor cells blocked formation of E-cadherin+ metastases and resulted in vimentin+ micrometastatic lesions implicating a role of versican in inducing MET. Versican inhibited the TGF-β-Smad2/3 signaling pathway to stimulate MET. Versican-induced MET resulted in increased cell proliferation in agreement with published studies demonstrating that TGF-β inhibits the proliferation of breast cancer lines including MDA-MB-231 by regulating expression of cytostatic genes (40, 41). Consistently, versican-mediated blockade of the TGF-β-Smad2/3 pathway resulted in increased cell proliferation and notably, expression of constitutively activated TGF-β-R1 rescued versican-mediated blockade of TGF-β-Smad2/3 pathway, reversed MET and proliferation, and suppressed metastases. In a similar fashion, microRNAs (miRNAs), particularly the miR-200 family, have been implicated in EMT/MET transitions in cancer (42). miR-200c inhibited EMT and induced an epithelial phenotype (43), and was sufficient to prevent TGFβ–dependent EMT induction. Furthermore, by virtue of targeting ZEB1/2 in mesenchymal cells, miR-200 induced MET (44). Importantly, in a negative feedback fashion, ZEB1/2 regulated miR-200 expression via the possession of conserved ZEB-binding sites in the miR-200 promoter (45, 46). This feedback loop explains the loss of miR-200 expression in invasive breast cancer cells that exhibit a mesenchymal phenotype (44). On the other hand miR-200 expression is also associated with increased metastatic potential and survival in breast cancer patients due to miR-200-mediated direct suppression of Sec23a, a regulator of metastasis-suppressive proteins (47). In this context, recent studies have identified a potential second loop comprised of miR34-Snail1 as a driver of epithelial-mesenchymal plasticity (48, 49). Thus the ZEB/miR-200 and Snail1/mir-34 feedback loops appears to regulate the reversible phenotypic switch that allows the cancer cell to exhibit EMT/MET plasticity in response to the changing microenvironment at the primary tumor site and the distant metastatic site (50). Notably, tumor suppressor p53 has been shown to regulate expression of these miRNAs in many cancer types, and p53-miR200-ZEB1/2 and p53-miR34-Snail1 axis have emerged as important regulators of cancer cell EMT/MET programs (48, 51).
The host microenvironment regulates epithelial- mesenchymal plasticity of tumor cells
Interactions of cancer cells with the tumor microenvironment are important determinants of cancer progression toward metastasis. Thus, tumor cells exhibit EMT/MET plasticity to adapt to the changing microenvironment that they encounter both at the primary and distant sites (Figure 1). Diverse interactions between the host microenvironment and cancer cells determine the course of tumor progression and metastasis (52). For example, in a spontaneous murine model of melanoma, recruited BM-derived inflammatory myeloid-derived suppressor cells (MDSC) induced EMT via the TGF-β, EGF, and HGF signaling pathways (53). In a recent study, interaction between platelets and tumor cells in the circulation conferred a prometastatic potential upon tumor cells (54). Platelet-derived TGFβ activated the TGFβ/Smad and NF-κB pathways in cancer cells, resulting in EMT and enhanced metastasis in vivo. Inhibition of NF-κB signaling in cancer cells or ablation of TGFβ1 expression in platelets protected against lung metastasis in vivo. Significant upregulation of EMT specific markers was also observed following co-culture of cancer cells with BM-derived mesenchymal cells (MSCs) with a reciprocal downregulation of E-cadherin protein expression, suggesting that MSCs may promote breast cancer metastasis by promoting EMT (55). Tumor associated macrophages (TAMs), represent a major component of the tumor microenvironment which confer key protumorigenic functions including angiogenesis and immune suppression (56, 57). In addition, TAMs also promote EMT of tumor cells by producing TGF-β, and analysis of 491 non-small cell lung cancer (NSCLC) patients revealed a positive correlation between intratumoral macrophage densities, EMT markers, intraepithelial TGF-β levels, and tumor grade (58). Consistent with these observations, we have observed that differential recruitment of BM cell subsets in the primary tumors and in the metastatic organ may regulate EMT and MET respectively. In the primary breast tumors of MMTV-PyMT mice, enhanced recruitment of TAMs generated EMT-promoting microenvironment by increasing expression of TGF, PDGF and EGF. In contrast, the metastatic organ showed relatively fewer TAMs and lower levels of EMT-promoting factors, and elevated recruitment of myeloid progenitor cells via expression of versican was able to promote MET. Other components of the tumor microenvironment, such as carcinoma-associated fibroblasts (CAFs) have been implicated in EMT. For example, CAFs obtained from lung cancer tissue produced HGF, thereby activating the EMT-related c-Met pathway in cancer cells. Of note, through the EMT induction by CAFs, tumor cells acquired resistance to conventional tyrosine kinase inhibitors against the EGF receptor (59). Recently, Giannoni et al. have reported that CAFs isolated from prostate carcinoma specimens activated the EMT programing of prostate cancer cells by producing MMPs (60).
Figure 1. Schematic depicting the contribution of bone marrow-derived cells to the formation of lung metastases from a primary breast tumor.

Bone marrow contributes F4/80+ macrophages that express EMT promoting factors in the primary tumor microenvironment, and Ly6C+ myeloid progenitor cells in the metastatic lungs that express versican to stimulate MET of disseminated tumor cells. EMT, epithelial to mesenchymal transition; MET, mesenchymal to epithelial transition.
Mathematical Modeling of EMT
Mathematical modeling is becoming a powerful tool for understanding and predicting the complex biological progression of tumor (61, 62). In spite of many studies on intracellular signaling pathways, there are limited reports on mathematical modeling and systematical simulation of the EMT and MET progression in cancer metastasis. Neagu et al reported a mathematical model of EMT in the formation of cardiac cushions (63), which can be potentially adapted to simulate the EMT and MET processes in cancer metastasis. The proposed mathematical model is a discrete model, which describes the cells, e.g. epithelial and mesenchymal cells, and extra-cellular matrix (ECM) by using a set of lattices in a 2D plane. The dynamic behavior of cells, including the migration, proliferation and transformation from epithelial to mesenchymal cells, are guided by an energy function describing the cell-cell and cell-ECM adhesion abilities, defined as follows.
where i,j denote the types of two adjacent objects, i.e. cells or ECM, and N is the number of types of objects. Four objects (N = 4) are defined in (63), and i = 1, 2, 3, and 4 represent Medium, ECM, Endothelial, and mesenchymal cells. Function Aij means the number of adjacent objects with different (adhesion βij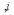 ≠jd) or same types (cohesion βii), and . The n denotes the number of interactions between the nearest, next nearest and second nearest neighbors. Finding the stable cell organization by minimizing the aforementioned equation, the first term prefers the mixture of different objects, whereas the second term tries to keep the anisotropy (i.e., reduce the mixture of different objects). Such a model can be extended to the study of EMT and MET processes in cancer metastasis under different conditions. For example, we can integrate the signaling pathways into the model, as EMT or MET are regulated by the ligands or molecules outside cells to activate related signaling inside the cells. The secretion and diffusion of the ligands can be described by partial differential equations (PDEs) (61), and the dynamics of signaling cascades could be formulated by ordinary differential equations (ODEs) (64). Moreover, the cell growth can be described by increasing the size of cells over time, which is also regulated by the availability of nutrients around them. Consequently, the cell-cell adhesion ability can be updated by considering the boundaries of cells (larger cells should have stronger adhesion ability). These adoptions will improve the predictive accuracy of the mathematical modeling of EMT in cancer metastasis and shall offer insight into the in vivo EMT and MET processes, which remain unclear.
≠jd) or same types (cohesion βii), and . The n denotes the number of interactions between the nearest, next nearest and second nearest neighbors. Finding the stable cell organization by minimizing the aforementioned equation, the first term prefers the mixture of different objects, whereas the second term tries to keep the anisotropy (i.e., reduce the mixture of different objects). Such a model can be extended to the study of EMT and MET processes in cancer metastasis under different conditions. For example, we can integrate the signaling pathways into the model, as EMT or MET are regulated by the ligands or molecules outside cells to activate related signaling inside the cells. The secretion and diffusion of the ligands can be described by partial differential equations (PDEs) (61), and the dynamics of signaling cascades could be formulated by ordinary differential equations (ODEs) (64). Moreover, the cell growth can be described by increasing the size of cells over time, which is also regulated by the availability of nutrients around them. Consequently, the cell-cell adhesion ability can be updated by considering the boundaries of cells (larger cells should have stronger adhesion ability). These adoptions will improve the predictive accuracy of the mathematical modeling of EMT in cancer metastasis and shall offer insight into the in vivo EMT and MET processes, which remain unclear.
Conclusions
In advanced cancer patients, widespread manifestation of distant metastases is a major cause of cancer-related deaths. Despite this important clinical problem, little is known about the mediators that promote tumor outgrowth in the metastatic organ. As early as 1889, Steven Paget proposed his ‘seed and soil’ hypothesis establishing the concept that a fertile “soil” (the microenvironment) is critical for the disseminated tumor cell (the “seed”) to grow (65, 66). Experimental support for this hypothesis has begun to emerge from recent studies, which show that primary tumors are able to systematically generate fertile “soil” or a premetastatic niche in the distant metastatic organs, and that these niches pave the way for future metastases (32, 33). By dissecting the components of the premetastatic niche further, our study highlights the role of the cancer cell extrinsic microenvironment prevailing in the metastatic organ as a major promoter of outgrowth of disseminated tumor cells by induction of MET.
From a therapeutic perspective, these findings support the idea that in addition to targeting the cancer cell, targeting the stromal cell or the stroma-tumor crosstalk as a potential antimetastatic approach should be considered. Indeed, recent studies including our own have begun to show that the stroma mediates important hallmarks of cancer such as angiogenesis, inflammation, immunity and EMT, establishing a concept of stromal therapeutics (67-69). From a mechanistic perspective, BM-derived versican, in a paracrine fashion, stimulated MET and increased tumor proliferation by blocking the canonical TGF-β–Smad2/3 signaling pathway in cancer cells, suggesting that in addition to targeting versican strategies to induce the TGF-β pathway may have antimetastatic consequences. However, targeting the TGF-β pathway is problematic as TGF-β is considered a double-edged sword in cancer (70). TGF-β inhibits primary tumor growth in early stages but promotes metastasis by enhancing EMT. Thus, in distant organs, if the same pathway is inhibitory again for tumor outgrowth and versican-mediated blockade promotes metastases progression, targeting versican may not impact primary tumor dissemination and micrometastases formation, but will impair the critical progression of micrometastases to lethal macrometastases.
Several critical questions need to be addressed before we can translate the findings that both EMT and MET play equivocal roles in tumor metastasis, into efficient therapeutic approaches. First, lineage-tracing strategies coupled with intravital imaging are necessary to follow epithelial-mesenchymal transitions in vivo, so that the mechanisms by which lethal macrometastatic lesions develop are understood. Second, it is not clear how universal the EMT/MET cascade is in tumor metastasis, and analysis of EMT/MET in many individual tumors and tumor types is necessary. For example, Trimboli et al. observed that EMT only occurred in Myc-initiated but not T antigen or Neu-initiated spontaneous breast cancers in mice (9). Furthermore, in some cases EMT was not required for metastasis, since many mice bearing neu- and PyMT-initiated tumors that lacked any evidence of EMT had significant amounts of lung metastases. Therefore, if the EMT/MET cascade is only required for metastasis by certain types of tumors, specific markers to identify theses tumors will be critical for the development of therapeutic strategies. Third, since both EMT and MET promote metastasis; therapeutic approaches to inhibit one of them may result in the activation of the other. This dual role may explain the potential controversies associated with EMT or MET targeting strategies. For example, the bone morphogenetic protein 7 (BMP7), by antagonizing TGF-β-induced EMT in vivo, has been shown to inhibit metastasis in some tumor models (71), while it promotes metastasis by increasing the anchorage-independent cell growth in others (72, 73) and higher BMP7 expression has been associated with worse prognosis of cancer patients (73).
Thus identification of specific mediators of EMT and MET signaling cascades is likely to lead to a rational approach to targeting these critical protumorigenic pathways. Our new finding that EMT and MET are regulated by selected components in the tumor microenvironment (38) provides unique opportunities to target EMT and MET individually or simultaneously, which will benefit cancer patients who are diagnosed with either early cancer lesions or already-established metastases. However, significant developments will be required in this field before this vision can become a clinical reality.
Acknowledgments
We thank Anna Durrans for critically reading this manuscript, Fuhai Li and Ming Zhan, for discussions on mathematical modeling of EMT, as well as Xiaoping Xu and James Mancuso on intravital cancer imaging. VM is supported by NIH grants CA135417, CA107429, RCA146065, and by Cornell Center on the Microenvironment and Metastasis through Award Number U54CA143876 from the National Cancer Institute and the Neuberger Berman Lung Cancer Center. The authors apologize for studies that could not be included due to space limitations.
References
- 1.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–73. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 3.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 4.Dave B, Mittal V, Tan NM, Chang JC. Epithelial-mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res. 2012;14:202. doi: 10.1186/bcr2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ledford H. Cancer theory faces doubts. Nature. 2011;472:273. doi: 10.1038/472273a. [DOI] [PubMed] [Google Scholar]
- 6.Bastid J. EMT in carcinoma progression and dissemination: Facts, unanswered questions, and clinical considerations. Cancer Metastasis Rev. 2012 doi: 10.1007/s10555-011-9344-6. [DOI] [PubMed] [Google Scholar]
- 7.Tarin D, Thompson EW, Newgreen DF. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005;65:5996–6000. doi: 10.1158/0008-5472.CAN-05-0699. discussion -1. [DOI] [PubMed] [Google Scholar]
- 8.Thompson EW, Newgreen DF, Tarin D. Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer Res. 2005;65:5991–5. doi: 10.1158/0008-5472.CAN-05-0616. discussion 5. [DOI] [PubMed] [Google Scholar]
- 9.Trimboli AJ, Fukino K, de Bruin A, Wei G, Shen L, Tanner SM, et al. Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res. 2008;68:937–45. doi: 10.1158/0008-5472.CAN-07-2148. [DOI] [PubMed] [Google Scholar]
- 10.Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol. 2009;11:1287–96. doi: 10.1038/ncb1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–61. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, Williams ED. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res. 2006;66:11271–8. doi: 10.1158/0008-5472.CAN-06-2044. [DOI] [PubMed] [Google Scholar]
- 13.Yates C. Prostate tumor cell plasticity: a consequence of the microenvironment. Adv Exp Med Biol. 2011;720:81–90. doi: 10.1007/978-1-4614-0254-1_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prudkin L, Liu DD, Ozburn NC, Sun M, Behrens C, Tang X, et al. Epithelial-to-mesenchymal transition in the development and progression of adenocarcinoma and squamous cell carcinoma of the lung. Mod Pathol. 2009;22:668–78. doi: 10.1038/modpathol.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vainio S, Lin Y. Coordinating early kidney development: lessons from gene targeting. Nat Rev Genet. 2002;3:533–43. doi: 10.1038/nrg842. [DOI] [PubMed] [Google Scholar]
- 16.Chaffer CL, Thompson EW, Williams ED. Mesenchymal to epithelial transition in development and disease. Cells Tissues Organs. 2007;185:7–19. doi: 10.1159/000101298. [DOI] [PubMed] [Google Scholar]
- 17.Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, et al. Epithelial--mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374–83. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- 18.Olmeda D, Jordá M, Peinado H, Fabra A, Cano A. Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene. 2007;26:1862–74. doi: 10.1038/sj.onc.1209997. [DOI] [PubMed] [Google Scholar]
- 19.Bonnomet A, Syne L, Brysse A, Feyereisen E, Thompson EW, Noël A, et al. A dynamic in vivo model of epithelial-to-mesenchymal transitions in circulating tumor cells and metastases of breast cancer. Oncogene. 2011 doi: 10.1038/onc.2011.540. [DOI] [PubMed] [Google Scholar]
- 20.Xue C, Plieth D, Venkov C, Xu C, Neilson EG. The gatekeeper effect of epithelial-mesenchymal transition regulates the frequency of breast cancer metastasis. Cancer Res. 2003;63:3386–94. [PubMed] [Google Scholar]
- 21.Tsuji T, Ibaragi S, Hu GF. Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Res. 2009;69:7135–9. doi: 10.1158/0008-5472.CAN-09-1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wyckoff JB, Jones JG, Condeelis JS, Segall JE. A critical step in metastasis: in vivo analysis of intravasation at the primary tumor. Cancer Res. 2000;60:2504–11. [PubMed] [Google Scholar]
- 23.Pang H, Flinn R, Patsialou A, Wyckoff J, Roussos ET, Wu H, et al. Differential enhancement of breast cancer cell motility and metastasis by helical and kinase domain mutations of class IA phosphoinositide 3-kinase. Cancer Res. 2009;69:8868–76. doi: 10.1158/0008-5472.CAN-09-1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–6. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 25.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–4. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu W, Laitinen S, Khan S, Vihinen M, Kowalski J, Yu G, et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med. 2009;15:559–65. doi: 10.1038/nm.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turajlic S, Furney SJ, Lambros MB, Mitsopoulos C, Kozarewa I, Geyer FC, et al. Whole genome sequencing of matched primary and metastatic acral melanomas. Genome Res. 2012;22:196–207. doi: 10.1101/gr.125591.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weigelt B, Glas AM, Wessels LF, Witteveen AT, Peterse JL, van’t Veer LJ. Gene expression profiles of primary breast tumors maintained in distant metastases. Proc Natl Acad Sci U S A. 2003;100:15901–5. doi: 10.1073/pnas.2634067100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim PK, Bliss SA, Patel SA, Taborga M, Dave MA, Gregory LA, et al. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011;71:1550–60. doi: 10.1158/0008-5472.CAN-10-2372. [DOI] [PubMed] [Google Scholar]
- 31.McAllister SS, Gifford AM, Greiner AL, Kelleher SP, Saelzler MP, Ince TA, et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell. 2008;133:994–1005. doi: 10.1016/j.cell.2008.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009;9:285–93. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carlini MJ, De Lorenzo MS, Puricelli L. Cross-talk between Tumor Cells and the Microenvironment at the Metastatic Niche. Curr Pharm Biotechnol. 2011 doi: 10.2174/138920111798377058. [DOI] [PubMed] [Google Scholar]
- 34.Ricciardelli C, Brooks JH, Suwiwat S, Sakko AJ, Mayne K, Raymond WA, et al. Regulation of stromal versican expression by breast cancer cells and importance to relapse-free survival in patients with node-negative primary breast cancer. Clin Cancer Res. 2002;8:1054–60. [PubMed] [Google Scholar]
- 35.Ricciardelli C, Sakko AJ, Ween MP, Russell DL, Horsfall DJ. The biological role and regulation of versican levels in cancer. Cancer Metastasis Rev. 2009;28:233–45. doi: 10.1007/s10555-009-9182-y. [DOI] [PubMed] [Google Scholar]
- 36.Horster MF, Braun GS, Huber SM. Embryonic renal epithelia: induction, nephrogenesis, and cell differentiation. Physiol Rev. 1999;79:1157–91. doi: 10.1152/physrev.1999.79.4.1157. [DOI] [PubMed] [Google Scholar]
- 37.Sheng W, Wang G, La Pierre DP, Wen J, Deng Z, Wong CK, et al. Versican mediates mesenchymal-epithelial transition. Mol Biol Cell. 2006;17:2009–20. doi: 10.1091/mbc.E05-10-0951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res. 2012;72:1384–94. doi: 10.1158/0008-5472.CAN-11-2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chao YL, Shepard CR, Wells A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol Cancer. 2010;9:179. doi: 10.1186/1476-4598-9-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mur C, Martínez-Carpio PA, Fernández-Montolí ME, Ramon JM, Rosel P, Navarro MA. Growth of MDA-MB-231 cell line: different effects of TGF-beta(1), EGF and estradiol depending on the length of exposure. Cell Biol Int. 1998;22:679–84. doi: 10.1006/cbir.1998.0306. [DOI] [PubMed] [Google Scholar]
- 41.Zugmaier G, Ennis BW, Deschauer B, Katz D, Knabbe C, Wilding G, et al. Transforming growth factors type beta 1 and beta 2 are equipotent growth inhibitors of human breast cancer cell lines. J Cell Physiol. 1989;141:353–61. doi: 10.1002/jcp.1041410217. [DOI] [PubMed] [Google Scholar]
- 42.Bullock MD, Sayan AE, Packham GK, Mirnezami AH. MicroRNAs: critical regulators of epithelial to mesenchymal (EMT) and mesenchymal to epithelial transition (MET) in cancer progression. Biol Cell. 2012;104:3–12. doi: 10.1111/boc.201100115. [DOI] [PubMed] [Google Scholar]
- 43.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–4. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 45.Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68:7846–54. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- 46.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582–9. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celià-Terrassa T, et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011;17:1101–8. doi: 10.1038/nm.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim NH, Kim HS, Li XY, Lee I, Choi HS, Kang SE, et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol. 2011;195:417–33. doi: 10.1083/jcb.201103097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siemens H, Jackstadt R, Hünten S, Kaller M, Menssen A, Gö1tz U, et al. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle. 2011;10:4256–71. doi: 10.4161/cc.10.24.18552. [DOI] [PubMed] [Google Scholar]
- 50.Brabletz S, Brabletz T. The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO Rep. 2010;11:670–7. doi: 10.1038/embor.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317–23. doi: 10.1038/ncb2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tse JC, Kalluri R. Mechanisms of metastasis: epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J Cell Biochem. 2007;101:816–29. doi: 10.1002/jcb.21215. [DOI] [PubMed] [Google Scholar]
- 53.Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong WC, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011;9:e1001162. doi: 10.1371/journal.pbio.1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20:576–90. doi: 10.1016/j.ccr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martin FT, Dwyer RM, Kelly J, Khan S, Murphy JM, Curran C, et al. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: stimulation of epithelial to mesenchymal transition (EMT) Breast Cancer Res Treat. 2010;124:317–26. doi: 10.1007/s10549-010-0734-1. [DOI] [PubMed] [Google Scholar]
- 56.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–6. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 58.Bonde AK, Tischler V, Kumar S, Soltermann A, Schwendener R. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer. 2012;12:35. doi: 10.1186/1471-2407-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang W, Li Q, Yamada T, Matsumoto K, Matsumoto I, Oda M, et al. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 2009;15:6630–8. doi: 10.1158/1078-0432.CCR-09-1001. [DOI] [PubMed] [Google Scholar]
- 60.Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010;70:6945–56. doi: 10.1158/0008-5472.CAN-10-0785. [DOI] [PubMed] [Google Scholar]
- 61.Anderson AR. A hybrid mathematical model of solid tumour invasion: the importance of cell adhesion. Math Med Biol. 2005;22:163–86. doi: 10.1093/imammb/dqi005. [DOI] [PubMed] [Google Scholar]
- 62.Macklin P, McDougall S, Anderson AR, Chaplain MA, Cristini V, Lowengrub J. Multiscale modelling and nonlinear simulation of vascular tumour growth. J Math Biol. 2009;58:765–98. doi: 10.1007/s00285-008-0216-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Neagu A, Mironov V, Kosztin I, Barz B, Neagu M, Moreno-Rodriguez RA, et al. Computational modeling of epithelial-mesenchymal transformations. Biosystems. 2010;100:23–30. doi: 10.1016/j.biosystems.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laise P, Fanelli D, Lio P, Arcangeli A. Modeling TGF-β signaling pathway in epithelial-mesenchymal transition. AIP Advances. 2012;2 [Google Scholar]
- 65.Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98–101. [PubMed] [Google Scholar]
- 66.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–8. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 67.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–52. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao D, Nolan DJ, Mellick AS, Bambino K, McDonnell K, Mittal V. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science. 2008;319:195–8. doi: 10.1126/science.1150224. [DOI] [PubMed] [Google Scholar]
- 69.Gao D, Mittal V. The role of bone-marrow-derived cells in tumor growth, metastasis initiation and progression. Trends Mol Med. 2009;15:333–43. doi: 10.1016/j.molmed.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 70.Akhurst RJ, Derynck R. TGF-beta signaling in cancer--a double-edged sword. Trends Cell Biol. 2001;11:S44–51. doi: 10.1016/s0962-8924(01)02130-4. [DOI] [PubMed] [Google Scholar]
- 71.Na YR, Seok SH, Kim DJ, Han JH, Kim TH, Jung H, et al. Bone morphogenetic protein 7 induces mesenchymal-to-epithelial transition in melanoma cells, leading to inhibition of metastasis. Cancer Sci. 2009;100:2218–25. doi: 10.1111/j.1349-7006.2009.01301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kobayashi A, Okuda H, Xing F, Pandey PR, Watabe M, Hirota S, et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med. 2011;208:2641–55. doi: 10.1084/jem.20110840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sakai H, Furihata M, Matsuda C, Takahashi M, Miyazaki H, Konakahara T, et al. Augmented autocrine bone morphogenic protein (BMP) 7 signaling increases the metastatic potential of mouse breast cancer cells. Clin Exp Metastasis. 2012;29:327–38. doi: 10.1007/s10585-012-9453-9. [DOI] [PubMed] [Google Scholar]