

Abstract
Changes in cellular phenotypes and identities are fundamentally regulated by epigenetic mechanisms including DNA methylation, post-translational histone modifications and chromatin remodeling. Recent genome-wide profiles of the mammalian DNA “methylome” suggest that hotspots of dynamic DNA methylation changes during cell fate transitions occur at distal regulatory regions with low or intermediate CpG densities. These changes are most prevalent early during the course of cellular differentiation and can be locally influenced by binding of cell-type specific transcription factors. With the advent of next-generation quantitative base-resolution maps of 5-methylcytosine and its oxidized derivatives and better coverage of the genome, we expect to learn more about the true significance of these DNA modifications in the regulation of cell fate choices.
Introduction
In mammalian genomes, the major epigenetic modification of DNA is methylation at the 5-position of the cytosine base, often at symmetrical CG dinucleotides (CpG). DNA methylation is implicated in numerous cellular processes during development including genomic imprinting, X-chromosome inactivation and transposon silencing [1]. DNA methylation at promoters is often associated with inhibition of transcriptional activity, but more recent genome-wide profiling of the DNA “methylome” in plants and animals is revealing new biological functions of this mark in gene regulation [2,3]. In this review, we discuss recent genome-wide location profiles of 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) in different mammalian cell types, focusing on models of cellular differentiation from pluripotent or multipotent states towards restricted somatic cell lineages. This subject has been recently reviewed [4-6] but continues to evolve with increasing depth and coverage of whole genome sequencing efforts. Due to space limitations, we will not discuss the role of DNA methylation in the specification of germ cells and in cancer, for which we refer the reader to other recent reviews [7-9].
Dnmts and Tet proteins in stem cells and development
In somatic cells, DNA methylation is generally stable because the maintenance DNA methyltransferase Dnmt1 faithfully restores methyl marks on newly-replicated DNA strands. In contrast, dynamic genome-wide changes in DNA methylation occur during early embryogenesis, most notably in the paternal pronucleus of the zygote where replication-independent demethylation occurs shortly after fertilization, and during reprogramming of primordial germ cells [10]. Subsequently, methylation profiles in the genome are re-established by the de novo methyltransferases Dnmt3a and Dnmt3b as cells develop into restricted lineages. The tight regulation of DNA methylation and demethylation is crucial since Dnmt-deficient mouse embryos are impaired in development [11,12]. Similarly, Dnmt-deficient (and therefore hypomethylated) mouse embryonic stem (ES) cells can be maintained in culture but are impaired in differentiation [13,14].
The recently discovered Tet family of 2-oxoglutarate (2OG)- and Fe(II)-dependent 5-methylcytosine oxygenases [15,16] alter DNA methylation status by converting 5mC to 5hmC and the further oxidation products 5-formylcytosine (5fC) and 5-carboxycytosine (5caC) in DNA [17-19] (Figure 1). The loss of 5mC in the mouse paternal pronucleus occurs concomitantly with the appearance of Tet3-mediated hydroxymethylation [20-22••], a finding initially thought to represent a process of active and global DNA demethylation. The caveat is that both 5fC and 5caC are deaminated and read as T after bisulfite treatment and PCR amplification, and so cannot be distinguished from unmodified cytosine (C). Thus the apparent demethylation in the zygote could reflect oxidation of 5mC through 5hmC (which cannot be distinguished from 5mC by bisulfite sequencing [23]) to 5fC and 5caC followed by passive dilution of these oxidized products during cell cleavage [24], active base excision repair of 5fC and 5caC by thymine-DNA glycosylase (TDG) [17], or removal of the carboxyl group from 5caC by a putative decarboxylase [25] (Figure 1). A recent study suggests that the de novo Dnmts are also redox-dependent DNA 5-dehydroxymethylases in vitro, adding another potential route for active DNA demethylation [26].
Figure 1.
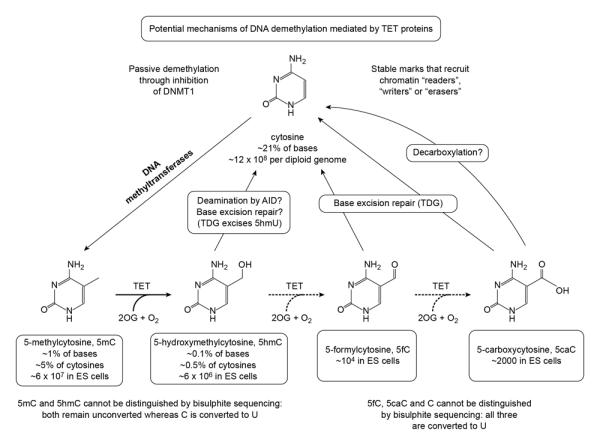
The potential pathways of DNA demethylation mediated by TET proteins. In addition to the active pathways shown (see text for details), demethylation may occur passively through inhibition of maintenance methylation. Moreover, the methylcytosine oxidation products generated by TET proteins, like 5hmC, may function as stable marks that recruit chromatin “readers”,“writers’ or “erasers”.
Whereas targeted knockout of Tet3 in mice results in perinatal or embryonic lethality, Tet1-deficient mice can be born viable but runted, with evidence of strain-dependent and partially penetrant embryonic lethality [22••,27-29•]. In contrast, Tet2-deficient mice are viable and fertile (reviewed in [30]). In mouse ES cells, Tet1 and Tet2 are highly expressed and together sustain steady state 5hmC levels [31]. Like Dmnt-deficient ES cells, Tet-deficient ES cells can be sustained in self-renewing culture and exhibit enhanced trans-differentiation potential towards the extra-embryonic fate [27•,31]. However, ES cells in which Tet1 is depleted by siRNA are still able to differentiate into all embryonic germ-layer derivatives, but exhibit skewed disposition towards mesendoderm relative to neuroectoderm fates in vitro [31]. Whereas the precise role of Tet1 in regulating the differentiative potential of ES cells remains to be clarified, the impact of Tet2 loss-of-function in hematopoietic stem cell differentiation has been reported by several groups (reviewed in [30]). Collectively, these studies suggest that loss of Dnmt or Tet proteins can be dispensable for the maintenance of stem cell character but moderate differentiation phenotypes to varying extents as stem cells exit from pluripotent or multipotent states.
DNA methylation profiling in different cell states
Although promoter methylation contributes to repression of core pluripotency genes, such as Oct4 and Nanog, it is only a second-tier epigenetic change, occurring after histone H3(K9) methylation and heterochromatinization, to stably inhibit reactivation of these genes during differentiation [32] (Figure 2). Low CpG content promoters (LCP), which are generally associated with tissue-specific genes, tend to be constitutively highly methylated; on the other hand, high CpG content promoters (HCP), which often contain CpG “islands” (CGIs), remain unmethylated [33-35]. These findings imply that low concentrations of methylated cytosines at a promoter do not preclude gene activity; conversely, DNA demethylation is not sufficient for gene activation. Thus, a general view is that hypermethylation of HCPs, other than functioning as a secondary silencing mechanism for long-term stability and memory, is not a major mechanism of cell fate decision during development [35]. Nonetheless, a defined subset of CGIs exhibit de novo methylation during development. This occurs preferentially at intermediate CpG content promoters (ICPs), suggesting that weak CGIs are more prone to methylation during differentiation [33,34,36] (Figure 2).
Figure 2.
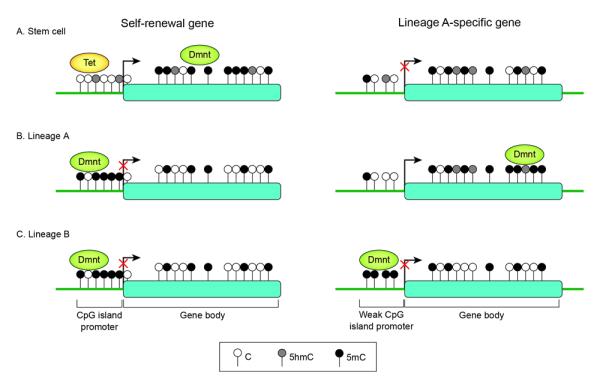
Schematic representation of DNA methylation status surrounding a self-renewal gene (left) or a tissue lineage-specific gene (right) in pluripotent stem cells (A), in the lineage in which the gene is expressed (B) and in an alternative lineage is which the gene is silent (C). Arrows indicate the transcription start sites (TSSs). Raised circles denote CpG sites. Black filled circles are 5-methylcytosine, grey filled circles are hydroxymethylcytosine and white dots circles are unmethylated cytosine. Promoters depicted are high CpG-content promoters (HCP) (left) typical of pluripotency genes (as well as germline-specific and X-inactivated genes) and intermediate or weak CpG –island promoters (right). Most HCP of housekeeping genes and silent but “poised” developmental regulators (not shown) remain unmethylated regardless of transcriptional states. In contrast, weak CpG island promoters are preferential targets for de novo methylation in somatic cells[33]. Gene silencing may result from lack of activators or alternative repressive mechanisms independent of DNA methylation.
In recent years, focus has been diverted to intra- and inter-genic regions as potential sites of dynamic DNA methylation changes during development. Analysis of the human methylome in three normal tissue types representing each of the embryonic lineages – liver (endodermal), spleen (mesodermal) and brain (ectodermal) – revealed that most tissue-specific differentially methylated regions (DMRs) occur not in promoters, and also not in CpG islands, but in regions located within 2 kb of islands with comparatively low CpG densities, termed “CpG island shores” [37]. The functional role of these tissue-specific DMRs has been proposed to regulate alternative transcription during differentiation. Another group identified numerous “orphan” CGIs as conserved features in intragenic regions of the mammalian genome, where they often mark alternative promoters but are frequently subject to DNA methylation during development to suppress promoter functions [38]. Subsequently, several genome-scale DNA methylome studies with more comprehensive coverage revealed low promoter methylation and high gene-body methylation in highly expressed genes in both plants and animals [39-43]. The association of gene body methylation with increased transcriptional activity has been accounted for by some examples of tissue-specific intragenic CGI methylation functioning to repress alternative promoters [44•]. However, a negative correlation between cell type-specific intragenic CGI methylation and expression of associated genes has been observed in immune cell types [45•]. Alternatively, intragenic methylation marks may repel polycomb components and functionally antagonize polycomb-mediated repression [46•].
A recent genome-scale quantitative analysis of cytosine methylation in mouse ES cells and neuronal progenitors defines cell-type specific low-methylated regions (LMRs) with an average methylation of 30%. These are typically CpG poor regulatory regions distal to promoters and are occupied by DNA-binding factors[47••]. The study suggests that by locally influencing DNA methylation, binding of cell-type-specific transcription factors is necessary and sufficient to create LMRs. Consistent with a model of dynamic DNA methylation and demethylation at these regions, the LMRs also exhibit a strong pre sence of 5hmC and Tet1 binding [47••] (Figure 3). The new evidence of cell-type specific distal LMRs is in timely agreement with new data from the Encyclopedia of DNA Elements (ENCODE) project, a consortia effort to delineate all functional elements in the human genome[48]. In particular, the integrative analysis of genome-scale DNase I hypersensitive sites (DHSs), DNA methylation and transcription factor expression in diverse cell and tissue types reveals novel relationships between chromatin accessibility, DNA methylation and cell-type-specific transcription regulation [49•]. The project reports distal DHS to be largely cell selective and to coincide with transcription-factor binding sites that are, on average, less frequently methylated in cell types that express those transcription factors. The data strongly argue that cell-type specific DNA-binding regulators are key drivers of the accessibility landscape of the chromatin; a corollary is that cell-type-specific methylation patterning results from passive methylation of sites left vacant by transcription factors[49•].
Figure 3.
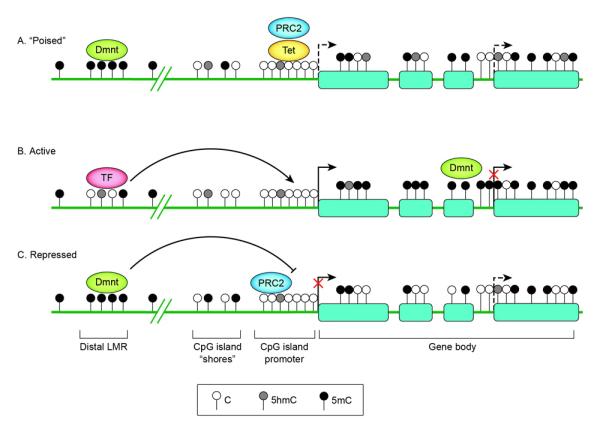
Schematic representation of DNA methylation status surrounding a gene encoding a developmental regulator in stem cells (A), in a cell displaying tissue-specific expression and differentiation towards the correct lineage (B) and in a cell differentiating towards an alternative lineage (C). At silent but “poised” promoters of genes encoding developmental regulators (A), PRC 2 mediates tri-methylation of lysine 27 of histone 3 (H3K27me3) at the promoters to keep the gene silent but “poised” for activation upon initiation of specific developmental pathways. The low-methylated regions (LMR) distal to promoters (the LMR shown here is depicted as intergenic but it can also be found at intragenic sites) form dynamically during differentiation in a cell-lineage specific manner upon binding by transcription factors (TF) [47••]. Tissue-specific DMRs have been described at CpG island (CGI) shores [37]. High-density CGI promoters generally stay unmethylated regardless of lineage fate; appropriate silencing may involve histone modifications by PRC2. Within gene bodies, methylation levels have been positively correlated with gene expression. The gene body methylation reflects a global reduction in DNA methylation observed to occur during differentiation [42,43]. Alternative TSSs are shown as dashed arrows. Full gene activation may require suppression of these alternative TSSs, possibly by intragenic CGI methylation [44•].
Tracking DNA methylation dynamics during cellular differentiation
Early studies suggested that DNA methylation can function in a highly locus-specific manner, involving in some instances changes at a single cytosine, to regulate lineage-specific development (reviewed in [4,5]). However, these studies have been obscured by genome-scale data that, as described above, illustrate more often than not that promoter methylation and transcriptional activity do not follow each other in an obligatory fashion.
The tracking of DNA methylation changes at genome-wide scale during the time-line of cellular differentiation is the next approach to identify new differentially methylated regions (DMRs) critical in lineage specification. The best established model to date is the derivation of neuronal progenitors and terminally differentiated neurons from ES cells, for which multipotent progenitors and terminal neurons can be obtained with high efficiency. Using a murine system of in vitro differentiation from stem cells to neuronal progenitors to terminally differentiated neurons, a methylated DNA immunoprecipitation (MeDIP)-array analysis of promoter methylome demonstrated that weak CpG islands are preferentially controlled by DNA methylation during somatic differentiation [50], in agreement with the aforementioned studies. However, most of these changes already occur during the early stage of commitment to a multipotent progenitor state. A similar conclusion has been drawn from methylome maps obtained during the progressive differentiation of mouse hematopoietic stem cells into restricted myeloid or lymphoid lineages [45•,51•,52•].
The ultimate challenge is to understand the most dynamic DNA methylation changes occurring during the earliest phases of mammalian development in vivo. The reduced representation bisulfite sequencing (RRBS) approach has been used to produce base-resolution DNA methylomes in gametes and through implantation in the early pre-specified mouse embryo [53•,54]. These studies reveal that large scale changes in DNA methylation specifically occur during two transitions in development: (i) upon fertilization, as a gross reduction in methylation during the sperm-to-zygote transition predominantly at intergenic DMRs enriched for retroelements of specific families and (ii) during the transition between pre-implantation to early gastrulation stage, as massive remethylation from the ICM to the early-streak embryo[53•]. The latter transition involves a multitude of complex changes in early development as the primary germ layers are formed from the pluripotent epiblast. While the RRBS approach is currently the only one applicable to very small numbers of cells, a comprehensive genome-wide dissection of the epigenetic changes in the early germ-layer lineages is warranted.
Mapping 5-hydroxymethylcytosine – a new methylation variant in disguise
A major limitation of bisulfite-based approaches to methylation profiling is the inability to discriminate between 5mC and 5hmC [23]. Thus, all current state-of-the-art high resolution DNA methylome maps still require reinterpretation since what has been previously called methylation is really a sum of 5mC and 5hmC. For instance, the high gene body methylation levels of expressed genes may indicate enrichment of 5hmC at the expense of 5mC, as evident in terminally differentiated neural cell types[55•]. To add to the complexities, even less is known about 5fC and 5caC, which are more rare than 5hmC and are read as unmethylated cytosines in traditional bisulfite sequencing.
Following the discovery of 5hmC as a bona fide constituent of the mammalian genome, multiple groups have obtained genome-wide maps of 5hmC, especially in ES cells, using primarily antibody-capture or selective chemical labeling methods (reviewed in [56]). The consensus view from these studies is that 5hmC is located predominantly in gene bodies, enhancers and intermediate CpG-density promoters of both active and repressed genes in ES cells. Where 5hmC is found at transcription start sites (TSS) of inactive genes, repression is likely mediated by co-bound chromatin modifiers such as Sin3a or the Polycomb Repressive Complex (PRC) 2 [57••-59]. Recently, two groups successfully mapped 5hmC at base-resolution in ES cells using either selective chemical oxidation (oxBS-Seq) or Tet enzyme-assisted bisulfite sequencing (TAB-Seq)[60•,61•]. With a higher resolution and quantitative analysis not previously attained by affinity-based methods, TAB-Seq reveals 5hmC as most abundant at regions of low CpG content and predominantly in the CpG context. Almost half of the 5hmCs reside in distal regulatory elements, where 5mC and 5hmC often coexist at nearly equal levels at the same cytosine [61•], suggesting that active demethylation is strongest outside of genes. Supporting this model, a kinetic study using hydroxyMeDIP (hMeDIP) revealed dynamic association of 5hmC with transcription factor binding at distal regulatory sites of genes activated during differentiation, indicating that DNA hydroxymethylation is an early event of tissue-specific enhancer activation [62•].
The three Tet family genes exhibit marked differential expression patterns in different tissue types and cellular states and are likely recruited to different loci in the genome to orchestrate a diverse set of epigenetic landscapes (reviewed in [63]). The relevance of 5hmC in neurodevelopment has received attention since the base was also discovered at abundant levels in Purkinje neurons of the rodent brain [64]. Subsequently, 5hmC has been mapped as a stable and dynamically acquired mark at developmentally activated genes in neuronal cells during postnatal development through adulthood [55•,65•].
More recent examples of cell fate control by 5mC-5hmC conversion at lineage-specific genes have been attributed to Tet2: at the Hoxa cluster of developmental genes during retinoic acid-induced differentiation [66] and at gene promoters of specific myeloid genes during CEBPα-induced transdifferentation of pre-B cells into macrophages [67•]. Future studies are likely to illuminate more cases of Tet-dependent 5hmC marks at critical cell fate transitions, including the role of these marks in differentiated cells.
When normal tissues adapt to cell culture, global 5hmC content decreases rapidly whereas global methylation state increases over passaging, suggesting that culture conditions modulate the cellular DNA hydroxylation machinery and phenotype [35,68]. Thus, a full understanding of the complexities of 5mC and 5hmC function will require analyses with primary cells from human and mouse.
Conclusions and Perspectives
The recent advances in DNA methylome studies have rapidly changed previous perceptions of DNA methylation. We summarize with the following points (Figure 4):
DNA methylation/hydroxymethylation changes at distal elements with low to intermediate CpG densities are likely more informative than changes at promoters/gene bodies in regulating cell fate.
DNA methylation changes are most prevalent during the early stages of lineage commitment.
Cell-type specific transcription factors can influence DNA methylation locally to cause tissue-specific expression, replacing the old dogma that DNA methylation blocks binding of transcription factors during differentiation.
DNA methylation can be acquired during culture adaptation, so that what we know from in vitro cellular differentiation models may underestimate the changes occurring in vivo.
Figure 4.
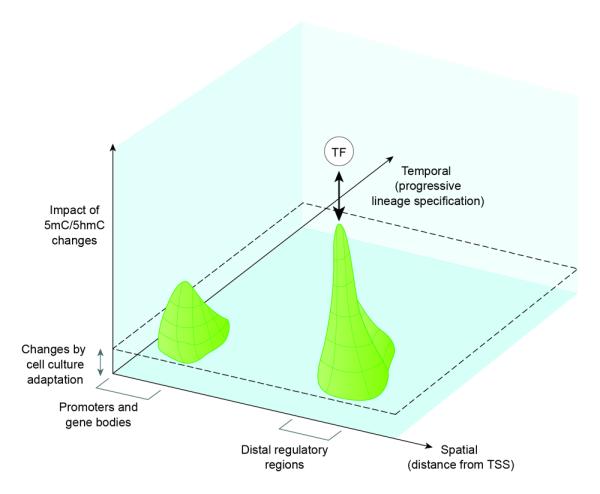
Schematic plot of the significance of 5mC/5hmC changes against a spatial axis of genome location and against a temporal axis of progressive lineage specification. The contour plot depicts a tall peak at distal regulatory regions and a lower peak at promoter and gene bodies along the spatial axis, indicating that the influence of cell type-specific dynamic changes in DNA methylation on lineage-specific gene expression is strongest at distal sites. These peaks align at an early time window along the temporal axis of increasing lineage commitment (or decreasing multilineage potency), indicating that most DNA methylation changes occur early when cells exit pluri- or multi-potency during the course of differentiation. In the “fourth” dimension, cell type specific transcription factors are shown above the DNA methylation contours to indicate that binding of transcription factors at distal regulatory elements influences DNA methylation status locally. The raised level indicated by the dotted lines denotes possible DNA methylation “artifacts” introduced by culture adaptation of cells and long-term passaging in vitro.
Collectively, the multiple efforts to profile 5mC and 5hmC in various cell states has generated a blueprint for further mechanistic understanding of these epigenetic marks in mammalian development. However, the variety of methods used, each with its strength and limitation, necessitates more fine-tuning of the landscape. With more advanced whole-genome sequencing studies unraveling regions of low CpG densities as hot spots of dynamic DNA changes critical in cell fate decisions, our current understanding of this fundamental epigenetic process may still represent only the tip of the iceberg. In the near future, the ideal approach is to obtain unbiased genome-scale base-resolution maps of 5mC and 5hmC (and even 5fC and 5caC) to venture beyond the CpG islands and shores to discover novel DMRs during differentiation.
Despite the allure of genome-wide approaches, the utility of candidate gene loci-specific studies should nonetheless remain relevant. As shown in the case of Tet2-regulated myeloid target genes, the critical switch in lineage-specific gene expression is accompanied by fairly subtle changes in DNA methylation [67•]. Thus, the dynamic range of DNA methylation changes during lineage specification appears narrow, yet may exert a threshold effect sufficient to trigger more widespread chromatin remodeling to affect key cell fate decisions. In this regard, candidate loci-specific probing of DNA methylation changes can be more powerful than genome-wide overviews to reveal the subtle effects.
Beyond mapping these epigenetic marks, the ultimate goal is to harness these insights to devise epigenetic-based strategies to skew in vitro differentiation of stem cells into desired cell types for multiple applications in drug testing, disease modeling and cellular replacement therapies. This may require precise definition of the cause-and-effect relationship of loci-specific changes (or DMRs) and cellular phenotype, perhaps the most elusive question in the field of epigenetics. Unlike single gene studies in which loss-of and gain-of function approaches are available to confer precise functions, histone and DNA modifications are not readily amenable to precise targeting. In view of the tight association between DNA binding of lineage-specific transcription factors and methylation states of regulatory elements, one can postulate testing such hypothesis using fusion constructs of sequence-specific binding factors and chromatin-modifying catalytic domains to enhance skewing towards specific lineages. The revolutionary reprogramming of somatic cells to induced pluripotent stem cells as well as directed the interconversion between lineages by forced introduction of key transcription factors already demonstrate that cellular identity is inherently more plastic than previously thought. Future endeavor promises to define a limited set of general rules and key factors sufficient to modulate the epigenome to attain the correct cell fate.
Highlights.
DNA methylation changes at distal elements of low CpG densities regulate cell fate.
These changes are most prevalent during the early stages of lineage commitment.
Cell-type specific transcription factors locally influence DNA methylation.
Acknowledgements
We apologize to those whose work could not be cited due to space limitations. Research in the laboratory of Kian Koh is supported by the Fonds voor Wetenschappelijk Onderzoek Research Foundation – Flanders (G.0C56.13N and G.0632.13), the Ministerie van de Vlaamse Gemeenschap and the Marie Curie Career Integration Grant (PCIG-GA-2012-321658). Research in the lab of Anjana Rao is supported by NIH R01 grants HD065812, AI44432 and CA151535, grant RM1-01729 from the California Institute for Regenerative Medicine, and Translational Research Program Award 6187-12 from the Leukemia Society of America.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest.
- 1.Ooi SKT, O’Donnell AH, Bestor TH. Mammalian cytosine methylation at a glance. J Cell Sci. 2009;122:2787–2791. doi: 10.1242/jcs.015123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11:204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 4.Weber M, Schubeler D. Genomic patterns of DNA methylation: targets and function of an epigenetic mark. Curr Opin Cell Biol. 2007;19:273–280. doi: 10.1016/j.ceb.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 5.Meissner A. Epigenetic modifications in pluripotent and differentiated cells. Nat Biotech. 2010;28:1079–1088. doi: 10.1038/nbt.1684. [DOI] [PubMed] [Google Scholar]
- 6.Ndlovu MN, Denis H, Fuks F. Exposing the DNA methylome iceberg. Trends in Biochemical Sciences. 2011;36:381–7. doi: 10.1016/j.tibs.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Esteller M. Epigenetics in Cancer. New England Journal of Medicine. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 8.Smallwood SA, Kelsey G. De novo DNA methylation: a germ cell perspective. Trends in Genetics. 2012;28:33–42. doi: 10.1016/j.tig.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 9.Seisenberger S, Peat JR, Hore TA, Santos F, Dean W, Reik W. Reprogramming DNA methylation in the mammalian life cycle: building and breaking epigenetic barriers. Philosophical Transactions of the Royal Society B: Biological Sciences. 2013;368 doi: 10.1098/rstb.2011.0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science. 2010;330:622–627. doi: 10.1126/science.1190614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 12.Okano M, Bell DW, Haber DA, Li E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 13.Jackson M, Krassowska A, Gilbert N, Chevassut T, Forrester L, Ansell J, Ramsahoye B. Severe Global DNA Hypomethylation Blocks Differentiation and Induces Histone Hyperacetylation in Embryonic Stem Cells. Mol Cell Biol. 2004;24:8862–8871. doi: 10.1128/MCB.24.20.8862-8871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ng RK, Dean W, Dawson C, Lucifero D, Madeja Z, Reik W, Hemberger M. Epigenetic restriction of embryonic cell lineage fate by methylation of Elf5. Nat Cell Biol. 2008;10:1280–1290. doi: 10.1038/ncb1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iyer LM, Tahiliani M, Rao A, Aravind L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle. 2009;8:1698–1710. doi: 10.4161/cc.8.11.8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfaffeneder T, Hackner B, Truβ M, Münzel M, Müller M, Deiml CA, Hagemeier C, Carell T. The Discovery of 5-Formylcytosine in Embryonic Stem Cell DNA. Angewandte Chemie International Edition. 2011;50:7008–7012. doi: 10.1002/anie.201103899. [DOI] [PubMed] [Google Scholar]
- 20.Iqbal K, Jin S-G, Pfeifer GP, Szabo PE. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc Natl Acad Sci USA. 2011;108:3642–3647. doi: 10.1073/pnas.1014033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, Boiani M, Arand J, Nakano T, Reik W, Walter J. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun. 2011;2:241. doi: 10.1038/ncomms1240. [DOI] [PubMed] [Google Scholar]
- 22••.Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, Xie ZG, Shi L, He X, Jin SG, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–610. doi: 10.1038/nature10443. Using conditional knockout mice, the authors present definitive evidence that DNA hydroxylation mediated by maternal Tet3 accounts for epigenetic reprogramming of the zygotic paternal genome.
- 23.Huang Y, Pastor WA, Shen Y, Tahiliani M, Liu DR. Rao A: The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One. 2010;5:e8888. doi: 10.1371/journal.pone.0008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inoue A, Shen L, Dai Q, He C, Zhang Y. Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Research. 2011;21:1670–1676. doi: 10.1038/cr.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schiesser S, Hackner B, Pfaffeneder T, Muller M, Hagemeier C, Truss M, Carell T. Mechanism and stem-cell activity of 5-carboxycytosine decarboxylation determined by isotope tracing. Angewandte Chemie. 2012;51:6516–6520. doi: 10.1002/anie.201202583. [DOI] [PubMed] [Google Scholar]
- 26.Chen CC, Wang KY, Shen CK. The Mammalian de Novo DNA Methyltransferases DNMT3A and DNMT3B Are Also DNA 5-Hydroxymethylcytosine Dehydroxymethylases. J Biol Chem. 2012;287:33116–33121. doi: 10.1074/jbc.C112.406975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27•.Dawlaty Meelad M, Ganz K, Powell Benjamin E, Hu Y-C, Markoulaki S, Cheng Albert W, Gao Q, Kim J, Choi S-W, Page David C, et al. Tet1 Is Dispensable for Maintaining Pluripotency and Its Loss Is Compatible with Embryonic and Postnatal Development. Cell Stem Cell. 2011;9:166–175. doi: 10.1016/j.stem.2011.07.010. This study reports the generation of Tet1 knockout mice and ES cells by targeting deletion of exon 4 of the coding sequence. The Tet1−/− mice are viable and fertile with only mild signs of developmental delay. Tet1−/− ES cells are pluripotent and support development of live-born mice in tetraploid complementation assay, but display skewed differentiation toward trophectoderm in vitro. The authors conclude that loss of Tet1 is compatible with embryonic and postnatal development.
- 28•.Yamaguchi S, Hong K, Liu R, Shen L, Inoue A, Diep D, Zhang K, Zhang Y. Tet1 controls meiosis by regulating meiotic gene expression. Nature. 2012 doi: 10.1038/nature11709. Using a gene-trap mouse line in which splicing of the first exon of Tet1 to the inserted β-geo cassette nullifies the expression of full-length Tet1 protein, the authors show that Tet1 deficiency reduces female germ-cell numbers and fertility by causing defective DNA demethylation and decreased expression of a subset of meiotic genes in primordial germ cells. Intriguingly, the Tet1Gt/Gt mutants are embryonic lethal at early backcross generations but become viable at further backcrosses, although viable homozygous mutant mice are still born at less than the expected Mendelien ratios. The discrepancy with [27•] remains to be resolved.
- 29•.Dawlaty Meelad M, Breiling A, Le T, Raddatz G, Barrasa MI, Cheng Albert W, Gao Q, Powell Benjamin E, Li Z, Xu M, et al. Combined Deficiency of Tet1 and Tet2 Causes Epigenetic Abnormalities but Is Compatible with Postnatal Development. Developmental Cell. 2013;24:310–323. doi: 10.1016/j.devcel.2012.12.015. By intercrossing Tet1 [27] and Tet2-knockout mice, the authors generated double knockout (DKO) ES cells and mice. Although DKO mutants can survive to become overtly normal and fertile adults, a fraction of DKO embryos develop midgestation abnormalities with perinatal lethality. Imprinting is also partially compromised in DKO mice.
- 30.Cimmino L, Abdel-Wahab O, Levine RL, Aifantis I. TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell. 2011;9:193–204. doi: 10.1016/j.stem.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, et al. Tet1 and Tet2 Regulate 5-Hydroxymethylcytosine Production and Cell Lineage Specification in Mouse Embryonic Stem Cells. Cell Stem Cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feldman N, Gerson A, Fang J, Li E, Zhang Y, Shinkai Y, Cedar H, Bergman Y. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat Cell Biol. 2006;8:188–194. doi: 10.1038/ncb1353. [DOI] [PubMed] [Google Scholar]
- 33.Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 34.Fouse SD, Shen Y, Pellegrini M, Cole S, Meissner A, Van Neste L, Jaenisch R, Fan G. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG complex, and histone H3 K4/K27 trimethylation. Cell Stem Cell. 2008;2:160–169. doi: 10.1016/j.stem.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farthing CR, Ficz G, Ng RK, Chan CF, Andrews S, Dean W, Hemberger M, Reik W. Global mapping of DNA methylation in mouse promoters reveals epigenetic reprogramming of pluripotency genes. PLoS Genet. 2008;4:e1000116. doi: 10.1371/journal.pgen.1000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr ARW, James KD, Turner DJ, Smith C, Harrison DJ, Andrews R, Bird AP. Orphan CpG Islands Identify Numerous Conserved Promoters in the Mammalian Genome. PLoS Genet. 2010;6:e1001134. doi: 10.1371/journal.pgen.1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, et al. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci USA. 2010;107:8689–8694. doi: 10.1073/pnas.1002720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- 42.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo Q-M, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al. Dynamic changes in the human methylome during differentiation. Genome Research. 2010;20:320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44•.Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. The authors used two complementary next-generation sequencing-based approaches for detecting methylated and unmethylated DNA to generate high-resolution methylome maps of the human brain. The extensive coverage reveals that the majority of methylated CpG islands are in intragenic and intergenic regions. By an in-depth analysis of the human SHANK3 locus and the mouse homologue, the authors demonstrated that intragenic DNA methylation in gene bodies regulates intragenic promoter activity and differential expression of alternative transcripts in a tissue- and cell type-specific manner.
- 45•.Deaton AM, Webb S, Kerr ARW, Illingworth RS, Guy J, Andrews R, Bird A. Cell type–specific DNA methylation at intragenic CpG islands in the immune system. Genome Research. 2011;21:1074–1086. doi: 10.1101/gr.118703.110. Using MBD affinity purification coupled to high-throughput sequencing to assess DNA methylation in cells of the hematopoietic lineages, this study shows that large differences in gene expression between immune cells are accompanied by relatively few DNA methylation changes. Altered DNA methylation occurs predominantly at CGIs within gene bodies and arises early in lineage commitment, while only a few are acquired during the later stages of terminal differentiation. Unexpectedly, elevated intragenic CGI methylation correlates with silencing of the associated gene. Moreover, the majority of intragenic CGIs showing cell type-specific methylation are deficient in H3K4me3 in immune cells even when unmethylated, suggesting that these CGIs are not active promoters in immune cells. However, the authors suggest that these differentially methylated intragenic CGIs may represent promoters active in other cell types.
- 46•.Wu H, Coskun V, Tao J, Xie W, Ge W, Yoshikawa K, Li E, Zhang Y, Sun YE. Dnmt3a-Dependent Nonpromoter DNA Methylation Facilitates Transcription of Neurogenic Genes. Science. 2010;329:444–448. doi: 10.1126/science.1190485. Dmnt3a is expressed in postnatal neural stem cells (NSCs) and is required for neurogenesis in the mouse. This study mapped genome-wide Dmnt3a location in postnatal NSCs by ChIP-chip and makes a surprising finding that Dmnt3a-dependent nonproximal promoter methylation promotes expression of the associated neurogenic genes. The results support a mechanism in which Dnmt3a-dependent DNA methylation antagonizes binding of Polycomb components at these sites to maintain active chromatin states of genes criticial for development.
- 47••.Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. doi: 10.1038/nature10716. The authors use whole-genome bisulfite sequencing to generate quantitative base-resolution mouse methylomes in ES cells and in neuronal progenitors and identify novel epigenome features defined by localized reduced levels of methylation. These low-methylated regions (LMRs) are often CpG-poor regions distal to promoters and are formed dynamically during differentiation through the binding of cell-type-specific transcription factors. This study reveals that the mouse methylome is more dynamic during cellular differentiation than anticipated and that DNA binding factors actively influence localized changes at distal regulatory regions linked to lineage-specific gene regulation.
- 48.Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49•.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. As part of the ENCODE project, the authors present an extensive map of human DNase1 hypersensitive sites (DHS) composed from genome-wide profiling in 125 diverse cell and tissue types. The study reveals a large pool of cell-selective distal DHSs as well as novel relationships between chromatin accessibility, transcription, DNA methylation and regulatory factor occupancy patterns. The results suggest that transcription factors are the key drivers of chromatin accessibility and that cell-selective site-specific DNA methylation patterning passively mirrors vacation of differentially expressed transcription factors from regulatory DNA.
- 50.Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schübeler D. Lineage-Specific Polycomb Targets and De Novo DNA Methylation Define Restriction and Potential of Neuronal Progenitors. Molecular Cell. 2008;30:755–766. doi: 10.1016/j.molcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 51•.Ji H, Ehrlich LIR, Seita J, Murakami P, Doi A, Lindau P, Lee H, Aryee MJ, Irizarry RA, Kim K, et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–342. doi: 10.1038/nature09367. The authors track methylome changes during hematopoiesis as mouse multipotent progenitors differentiate into progressively restricted myeloid or lymphoid progenitors. Hematopoietic stem cells were not included in the study. In their method using comprehensive high-throughput arrays for relative methylation (CHARM) (also used in [37]), DNA is digested with McrBC, an enzyme that cuts methylated DNA promiscuously, then size-fractionated to enrich for unmethylated DNA and hybridized to an optimized custom-designed microarray that interrogates ~ 4.6 million CpG sites including all CpG islands in the genome. The analysis revealed striking epigenetic plasticity as multipotent progenitors differentiate progressively into restricted myeloid or lymphoid progenitors, observed as an increase in overall methylation upon lymphoid relative to myeloid commitment. Inhibiting DNA methylation using the Dnmt inhibitor 5-aza-2′-deoxycytidine promoted myeloid versus lymphoid specification, suggesting that lymphopoiesis is much more dependent on acquisition of DNA methylation marks whereas myelopoiesis more dependent on their loss. Differential DNA methylation between hematopoietic populations correlated with gene expression more strongly at CpG island shores than at CpG islands.
- 52•.Bock C, Beerman I, Lien W-H, Smith Zachary D, Gu H, Boyle P, Gnirke A, Fuchs E, Rossi Derrick J, Meissner A. DNA Methylation Dynamics during In Vivo Differentiation of Blood and Skin Stem Cells. Molecular Cell. 2012 doi: 10.1016/j.molcel.2012.06.019. The authors analysed the DNA methylome in 19 purified cell populations isolated from the mouse in different stages of development progressing from hematopoietic and skin stem cells towards the blood and skin lineages respectively. In a reduced representation bisulfite sequencing (RRBS) approach, genomic DNA is first digested with the methylation-insensitive restriction enzyme MspI, size-selected, treated with sodium bisulfphite and then 5′end-sequenced. This method provides high resolution DNA methylation maps covering ~4.8% (~ 1 million) of all CpGs in the mouse genome and significantly enriches for high-CpG-density promoters and other CpG-rich features. The results demonstrate that DNA methylation changes are highly lineage-specific, relative modest in magnitude but informative during in vivo differentiation of adult stem cells. In agreement with [51•], the study showed that methylation of gene-regulatory elements associated with alternative lineages is crucial for lymphoid differentiation, particularly to protect loci from aberrant activation by myeloid specification factors, but largely dispensable for myeloid differentiation. However, genomic regions with intermediate DNA methylation levels are even more powerful predictors of cellular identity than CpG islands and shores. The consistent observation is that gene-regulatory regions associated with other lineages and with stemness become increasingly methylated, whereas regions regulating the chosen lineage lose methylation in committed progenitors and in terminally differentiated cells. maintained downstream of adult stem cells, suggesting that the epigenetic switch only needs to occur once in the course of differentiation to define cellular identity.
- 53•.Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A. The gain of DNA methylation and loss of gene expression are robustly A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012;484:339–44. doi: 10.1038/nature10960. Using the RRBS method [see 52•], the authors provide a genome-scale base-resolution time-line profile of DNA methylation in mouse gametes and from the zygote through post-implantation. In agreement with the classical model of DNA methylation dynamics during early embryogenesis, the study shows that the most dramatic changes in DNA methylation occur during two developmental transitions: between sperm and the zygote and between the early ICM and the post-implantation embryo.
- 54.Smallwood SA, Tomizawa S-i, Krueger F, Ruf N, Carli N, Segonds-Pichon A, Sato S, Hata K, Andrews SR, Kelsey G. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat Genet. 2011;43:811–814. doi: 10.1038/ng.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55•.Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 Binds to 5hmC Enriched within Active Genes and Accessible Chromatin in the Nervous System. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. 5hmC is approximately 10-fold more abundant in neurons than in peripheral tissues or ES cells. By genome-wide analysis of 5hmC, 5mC and gene expression in differentiated neuronal cell types, the authors report that 5hmC is enriched within the gene bodies of active genes and that, surprisingly, 5mC is depleted over these regions. Moreover, they identify methyl-CpG-binding protein 2 (MeCP2) as the major 5hmC-binding protein in the brain. Although MeCP2 binds 5hmC- and 5mC-containing DNA with similar affinities, the data suggest that MeCP2 regulates the chromatin accessibility of 5hmC-containing DNA. The authors propose that binding of MeCP2 to 5hmC in expressed genes facilitates transcription through regulation of chromatin structure, explaining how MeCP2 can also activate gene expression in the nervous system.
- 56.Wu H, Zhang Y. Tet1 and 5-hydroxymethylation: A genome-wide view in mouse embryonic stem cells. Cell Cycle. 2011;10:0–1. doi: 10.4161/cc.10.15.16930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57••.Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PAC, Rappsilber J, Helin K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348. doi: 10.1038/nature10066. This is one of several concurrent studies (reviewed in [56]) reporting Tet1 binding sites and 5hmC location in mouse ES cells. In this study, the authors show that the majority of Tet1 binding sites are located at TSSs of CpG-rich promoters and within genes, whereas 5hmC is found in gene bodies and also enriched at CpG-rich TSSs. The data suggest a role of Tet1 in transcriptional repression. A significant proportion of Polycomb group target genes are also Tet1 targets, in agreement with the colocalization of these target genes with 5hmC in [58] and [59]. Furthermore, the authors show an association of Tet1 with the SIN3A co-repressor complex. Interestingly, the study shows that several genes up-regulated or down-regulated by Tet1 depletion in normal ES cells are similarly affected by Tet1 depletion in Dnmt TKO (triple knockout) ES cells, which are deficient in both 5mC and 5hmC. Thus, the majority of Tet1-mediated transcriptional effects may be independent of conversion of 5mC to 5hmC. The authors propose that TET1 fine-tunes transcription, opposes aberrant DNA methylation at CpG-rich sequences and thereby contributes to the regulation of DNA methylation fidelity.
- 58.Wu H, D’Alessio AC, Ito S, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25:679–684. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, Ko M, McLoughlin EM, Brudno Y, Mahapatra S, Kapranov P, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60•.Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, Balasubramanian S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–937. doi: 10.1126/science.1220671. This study introduces oxidative bisulfite sequencing (oxBS-seq), a novel modification of the traditional bisulfite method that utilizes potassion perruthenate (KRuO4) to chemically oxidize 5hmC to 5fC followed by bisulfite conversion of 5fC to uracil. The combination of BS-Seq and oxBS-Seq enables discrimination between 5hmC and 5mC and quantitative mapping of 5hmC at single nucleotide resolution. Using these methods, the authors performed deep sequencing of a fraction of the mouse ESC genome that is highly enriched for CpG islands and identified high levels of 5hmC in CGIs associated with transcriptional regulators and in LINE1 elements.
- 61•.Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the Mammalian genome. Cell. 2012;149:1368–1380. doi: 10.1016/j.cell.2012.04.027. This study introduces a second method to map 5hmC at single base resolution, by Tet-assisted bisulfite sequencing (TAB-Seq). In this enzymatic approach, 5hmCs in genomic DNA are protected by glucosylation before oxidation of 5mCs to 5caCs by recombinant Tet1 catalytic domain. After bisulfite treatment, both 5caC (generated from 5mC) and C display as T, whereas glycosylated 5hmC displays as C. In conjunction with traditional bisulfite sequencing, TAB-Seq shows that 5hmC is more abundant at regions of low CpG content in mouse and human ES cells. Together with [60,•], these studies suggest that it is now possible to derive genome-wide quantitative maps of 5hmC in cells at single nucleotide resolution.
- 62•.Serandour AA, Avner S, Oger F, Bizot M, Percevault F, Lucchetti-Miganeh C, Palierne G, Gheeraert C, Barloy-Hubler F, Peron CL, et al. Dynamic hydroxymethylation of deoxyribonucleic acid marks differentiation-associated enhancers. Nucleic acids research. 2012 doi: 10.1093/nar/gks595. Using murine P19 and 3T3-L1 cells that undergo neural or adipocyte differentiation in culture respectively, this study characterizes the dynamics of 5hmC and its correlation with those of active enhancer chromatin marks during cellular differentiation. To map 5hmC, the authors use deep sequencing of hydroxymethylated DNA recovered from cells by immunoprecipitation with a 5hmC antibody and by selective chemical labelling asssays. They show that regions gaining 5hmC are associated with lineage-related genes expressed during differentiation and located in distal enhancer regions that binds lineage-specific transcription factors. The results suggest that DNA hydroxymethylation is an early event of enhancer activation required for selective activation of tissue-specific genes.
- 63.Tan L, Shi YG. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development. 2012;139:1895–1902. doi: 10.1242/dev.070771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65•.Szulwach KE, Li X, Li Y, Song CX, Wu H, Dai Q, Irier H, Upadhyay AK, Gearing M, Levey AI, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nature neuroscience. 2011;14:1607–1616. doi: 10.1038/nn.2959. This study maps 5hmC genome-wide in mouse hippocampus and cerebullum at three different ages using a method of specific chemical labeling and capture. The authors found that 5hmC markedly increased from the early postnatal stage to adulthood, despite no significant increase in Tet family gene expression. 5hmC marks are acquired in developmentally activated genes, are relatively stable, but still exhibiting dynamic changes particularly in intragenic regions and intergenic CGIs of intermediate CpG and GC content.
- 66.Bocker MT, Tuorto F, Raddatz G, Musch T, Yang FC, Xu M, Lyko F, Breiling A. Hydroxylation of 5-methylcytosine by TET2 maintains the active state of the mammalian HOXA cluster. Nature communications. 2012;3:818. doi: 10.1038/ncomms1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67•.Kallin EM, Rodriguez-Ubreva J, Christensen J, Cimmino L, Aifantis I, Helin K, Ballestar E, Graf T. Tet2 Facilitates the Derepression of Myeloid Target Genes during CEBPalpha-Induced Transdifferentiation of Pre-B Cells. Molecular cell. 2012 doi: 10.1016/j.molcel.2012.08.007. Although multiple studies have linked Tet2 loss-of-function with hematopoietic disorders and effects on commitment to the myeloid lineage, the target genes remain elusive. By knockdown of Tet2 during CEBPalpha-induced transdifferentiation of pre-B cells into macrophages, this study identifies a small subset of myeloid genes that are activated upon Tet2-mediated promoter hydroxymethylation.
- 68.Nestor CE, Ottaviano R, Reddington J, Sproul D, Reinhardt D, Dunican D, Katz E, Dixon JM, Harrison DJ, Meehan RR. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Research. 2011 doi: 10.1101/gr.126417.111. [DOI] [PMC free article] [PubMed] [Google Scholar]