

Abstract
Animals harbor specialized neuronal systems that are used for sensing and coordinating responses to changes in oxygen (O2) and carbon dioxide (CO2). In Caenorhabditis elegans, the O2/CO2 sensory system comprises functionally and morphologically distinct sensory neurons that mediate rapid behavioral responses to exquisite changes in O2 or CO2 levels via different sensory receptors. How the diversification of the O2- and CO2-sensing neurons is established is poorly understood. We show here that the molecular identity of both the BAG (O2/CO2-sensing) and the URX (O2-sensing) neurons is controlled by the phylogenetically conserved SoxD transcription factor homolog EGL-13. egl-13 mutant animals fail to fully express the distinct terminal gene batteries of the BAG and URX neurons and, as such, are unable to mount behavioral responses to changes in O2 and CO2. We found that the expression of egl-13 is regulated in the BAG and URX neurons by two conserved transcription factors—ETS-5(Ets factor) in the BAG neurons and AHR-1(bHLH factor) in the URX neurons. In addition, we found that EGL-13 acts in partially parallel pathways with both ETS-5 and AHR-1 to direct BAG and URX neuronal fate respectively. Finally, we found that EGL-13 is sufficient to induce O2- and CO2-sensing cell fates in some cellular contexts. Thus, the same core regulatory factor, egl-13, is required and sufficient to specify the distinct fates of O2- and CO2-sensing neurons in C. elegans. These findings extend our understanding of mechanisms of neuronal diversification and the regulation of molecular factors that may be conserved in higher organisms.
Author Summary
During the development of an organism, certain neurons are programmed to perform specific tasks. For example, motor neurons coordinate locomotion and sensory neurons recognize specific environmental cues. The molecular mechanisms that generate specific neuronal classes are not fully understood. We investigated mechanisms that control the development of two distinct classes of neurons that are required for the nematode Caenorhabditis elegans to sense the respiratory gases O2 or CO2. In this study, we identified and characterized a conserved transcription factor, egl-13, that is required for the development of both of these classes of neurons. egl-13 is related to the SoxD family of transcription factor proteins in vertebrates. We found that egl-13 controls the production of specific proteins that provide these cells with the ability to sense both O2 and CO2. Further, we found that egl-13 works in conjunction with two additional factors, ahr-1 and ets-5, to regulate this developmental decision. This work provides new insight into how transcriptional regulatory networks specify different but related neuronal identities and provides a platform for future studies to understand how neuronal diversity is generated.
Introduction
The capacity of the nervous system to sense and respond to fluctuations in the external and internal environment is essential for homeostasis and survival. Neuronally controlled homeostatic buffering is delivered through cellular and systemic physiological adjustments and by seeking optimal environmental conditions through behavioral strategies [1]–[4]. A crucial homeostatic capacity of animals is the ability to sense and respond to changes in concentration of the respiratory gases oxygen (O2) and carbon dioxide (CO2) [5], [6]. O2 is essential for the generation of energy in the form of adenosine triphosphate (ATP); however, O2 also exerts toxicity through the production of reactive oxygen species (ROS) [1]–[4], [7]. CO2 is a by-product of oxidative metabolism and prolonged exposure leads to acidosis [5], [6], [8]. CO2 is also an environmental cue used in host- and mate- finding and can initiate both aversive or attractive behaviors [9]–[11]. The evolution of mechanisms required to sense and respond to O2 and CO2 is therefore paramount for survival.
In Drosophila, specific sensory systems respond to external O2 levels [1], [7], [12]. In addition, Drosophila uses specialized olfactory and gustatory neurons to detect CO2 changes via specialized chemosensory receptors called Gr21a/Gr63a [9], [13]. In humans, O2, CO2 and pH levels are monitored by specific regions of the brainstem and by specialized neurosecretory glomus cells of the carotid body [14], whereas in non-human mammals CO2 is also sensed by specific olfactory neurons that target the necklace glomeruli in the olfactory bulb via the guanylyl cyclase GC-D [15]. It is poorly understood how the specification of such specialized sensory neurons is regulated. However, recent work in Drosophila has shown that epigenetic mechanisms play an important role [16].
Respiratory gas sensing is a crucial modality for Caenorhabditis elegans whose natural environment, such as rotting fruit and compost, can have wide ranges of O2 and CO2 levels [17]. Previous work has shown that in the laboratory, worms have a behavioral preference for 5%–10% O2 and are exquisitely sensitive to minor changes in O2 concentration [18], [19]. In addition, worms mount avoidance responses to CO2 levels above 0.5% [4], [11]. Of the 302 neurons in the C. elegans nervous system, at least six neurons are specifically dedicated to the detection and response to changes in O2 and CO2 levels. These include the BAGL/R, URXL/R, AQR and PQR neurons. The BAG neurons are the primary CO2 sensors and they also respond to decrease in O2 concentration [11], [20]–[22]. The URX, AQR and PQR neurons are specialized for responding to increasing O2 concentrations [20]. In C. elegans, members of the guanylyl cyclase family of proteins are crucial factors required for O2 and CO2 sensing. Pioneering work revealed that the soluble guanylyl cyclases (sGCs) GCY-35 and GCY-36 mediate high O2 avoidance behavior via the URX, AQR and PQR neurons and that GCY-35 directly binds to molecular O2 [18]. In contrast, the sGCs, GCY-31 and GCY-33 function in the BAG neurons to sense decreases in O2 [20]. Recent work found that the membrane-bound receptor-type guanylyl cyclase GCY-9 acts specifically in the BAG neurons to mediate CO2 avoidance behavior [21]. Other molecules such as the Phe-Met-Arg-Phe-NH2 (FMRF-amide)-related peptides (FLP-8, FLP-13, FLP-17 and FLP-19) are expressed in either a subset or all of the O2- and CO2-sensing neurons; however their precise molecular functions in O2 and CO2 sensing are not known [23], [24].
Neuronal specialization within the O2/CO2-sensing system in C. elegans is an excellent model to study the control of neuron diversity. The O2-sensing (URX, AQR and PQR) and O2/CO2-sensing (BAG) neurons have overlapping and non-overlapping patterns of guanylyl cyclase and neuropeptide expression, which are reflected in their related, albeit distinct functionalities [20]. At present, it is unclear how the expression of these molecules is restricted to certain parts of the O2/CO2-sensing nervous system, and how such restrictions coordinate neuronal fate and function.
Here, we have identified the Sox transcription factor EGL-13 as an important regulator of the O2 and CO2-sensing neuron cell fate decision. EGL-13 is required for the expression of distinct proteins required for sensing both O2 and CO2 and as such, egl-13 mutant animals are unable to mount behavioral responses to changes in O2 and CO2. We found that the expression of EGL-13 is controlled by ETS-5 in the BAG neurons and by AHR-1 in the URX neurons, and acts in partially parallel pathways with these factors to drive neuronal fate. Finally, we found that EGL-13 is sufficient to drive O2- and CO2-sensing cell fates in certain cellular contexts. Therefore, EGL-13 is a core regulatory factor that is both required and sufficient to drive O2- and CO2-sensing neuron specification in C. elegans. As EGL-13 is a member of the SoxD family of transcription factors, we anticipate that the regulatory relationships described here will provide a paradigm for the control of neuronal fate specification by Sox proteins in other cellular contexts.
Results/Discussion
The SoxD transcription factor egl-13 specifies distinct O2- and CO2-sensing neurons
In order to identify molecules and pathways important for O2- and CO2-sensing neuron specification, we have taken a forward genetics approach in C. elegans. We isolated four independent allelic mutations (rp14, rp22, rp23 and rp26) that affect the expression of terminal differentiation markers in the O2 and/or CO2-sensing neurons (Figure 1 and Table S1) Mutant hermaphrodites of each of these alleles are severely egg-laying defective (Egl) and form a bag-of-worms where embryos hatch inside the mother (Figure S1). We investigated their vulval phenotype and found that the anchor cell fails to fuse with the uterine seam cell, causing a blockage of the uterus and the resultant Egl phenotype (Figure S1). This anchor cell fusion defect is reminiscent of that observed in egl-13(ku194) mutant animals [25] which we found to also exhibit defects in O2 and CO2 reporter expression (Figure 1 and Figure S1). Subsequent Sanger sequencing of rp14, rp22, rp23 and rp26 revealed genetic lesions in the egl-13 locus (Figure 1A). egl-13 encodes the C. elegans ortholog of the HMG-domain-containing SoxD family of transcription factors that has no previously reported role in the worm nervous system.
Figure 1. egl-13 is required for O2- and CO2-sensing neuron specification.

(A) Molecular identity of mutant alleles obtained from the forward genetic screens. egl-13 alleles first described in this article are shown in black (rp14, 22, 23, and 26) and the previously described ku194 [25] is shown in red. The nature of the molecular lesions is as follows: rp14 is a splice acceptor mutation between exons 8 and 9, causing a premature termination codon within the HMG domain. rp22 is an out-of-frame 317 bp deletion between exons 6 and 7 that leads to a predicted premature STOP codon in the EGL-13 protein that lacks the HMG domain. The rp23 allele is a promoter deletion, removing 1128 bp between −1700 and −572 upstream of the ATG codon. rp26 is a G-to-A transition that converts a highly conserved glycine to a glutamic acid in the HMG domain. The isolated egl-13 mutants are therefore predicted to either abrogate DNA binding (rp14, rp22 and rp26) or reduce/eliminate expression of egl-13 transcript (rp23). (B) Schematic of the O2- and CO2-sensing system. Anterior to the left. (C) Fluorescence reporter expression of the O2- and CO2-sensing neuron terminal gene batteries in wild-type (left) and egl-13(ku194) mutant (right) at the young adult stage. Quantification of data and information on reporter strains is shown in Table S1. Neuron positions are marked with dashed circles: BAG(red), URX(blue), AQR(yellow) and PQR(green). Fluorescent cells not marked with circles are non-O2/CO2-sensing neurons in the respective strains and their expression is unaffected by loss of egl-13. Scale bar, 20 µm. Anterior to the left.
Loss of egl-13 affects terminal fate of O2- and CO2-sensing neurons
The BAG, URX, AQR and PQR neurons in C. elegans are required for sensing and responding to fluctuations of O2 and CO2 levels in the environment [11], [18], [19]. Distinct batteries of genes are expressed in these neurons that are predicted to provide the optimal functionality required for O2 and CO2 sensing, however the role of only a subset of these genes has been analyzed in detail [20], [21], [26]. We used fluorescent reporter constructs to monitor expression of these gene batteries to understand how egl-13 controls O2 and CO2-sensing neuron cell fate (Figure 1). We analyzed the expression of guanylyl cyclases (gcy-9, gcy-31, gcy-33, gcy-35 and gcy-36) and Phe-Met-Arg-Phe-NH2 (FMRF-amide)-related peptides (flp-8, flp-13, flp-17 and flp-19) that are all terminal differentiation genes expressed in all or a subset of O2 and CO2-sensing neurons [18], [19], [21], [24]. We crossed these reporter transgenes into egl-13 mutant animals (ku194 allele) and found that none of the reporters were properly expressed in egl-13 mutants (Figure 1 and Table S1). We also found similar effects in the four egl-13 mutant alleles we isolated (rp14, rp22, rp23 and rp26) (Figure S1C). We noticed that some of the reporters were exquisitely sensitive to egl-13 loss whereas others exhibited partially penetrant defects (Table S1). This suggests that the expression of some terminal differentiation factors are under the collaborative control of additional factors that are able to compensate for the loss of egl-13.
The BAG and URX neurons are derived from the AB lineage and are posterior sisters of other neurons that have distinct fates [27] (Figure S2). We therefore asked whether egl-13 is also required for the specification of the sister cells of BAG or URX. We crossed egl-13(ku194) mutant animals into fluorescent reporter strains for the SMDV, zfIs2 (lgc-55::mCherry) and CEPD, vtIs1 (dat-1::gfp), sister cells for BAG and URX neurons respectively. We found that the expression of these reporters were unaffected by loss of egl-13 suggesting a specific role for egl-13 in the posterior branch of these lineages (Table S1 and Figure S2). Taken together, we conclude that egl-13 controls the expression of the distinct O2- and CO2-sensing neuron terminal gene batteries that distinguish them from lineage-related neurons.
egl-13 acts cell-autonomously in O2- and CO2-sensing neurons
To monitor egl-13 expression, we generated two promoter-driven fluorescent reporters (egl-13prom1::mCherry and egl-13prom1::gfp) that contain 3.5 kb of egl-13 upstream sequence (Figure 2A and Figure S3). Expression is first detected in 4 neuronal cells at around 350 min post-fertilization, which is the time at which the BAG and URX neurons are born (Figure S3). Expression is restricted to these 4 neurons during embryogenesis (Figure S3). At the first larval stage, egl-13 expression is observed in the BAG and URX neurons plus occasionally in a small number of unidentified cells in the head and tail (including the AQR and PQR neurons) (Figure S3). Later during larval development, egl-13 expression is observed in body wall muscle and vulval cells (data not shown). Neuronal expression is restricted to the O2 and CO2-sensing neurons in the adult (Figure 2A). Using the 3.5 kb egl-13 promoter (egl-13prom1) we transgenically expressed egl-13isoformAcDNA in egl-13(ku194) mutant animals and were able to rescue both the defect in O2 and CO2-sensing neuron fate marker expression and the Egl phenotype (Figure 2B–2C, Figure S4 and data not shown). To confirm that egl-13 acts cell autonomously to control O2 and CO2-sensing neuron fate, we used neuron-specific promoters to drive egl-13isoformAcDNA expression in the BAG or URX neurons (Figure 2D). We found that indeed neuron-specific expression of egl-13 rescued the O2 and CO2-sensing neuron fate defect of egl-13(ku194) mutant animals (Figure 2D). Therefore, we conclude that egl-13 acts autonomously in the BAG and URX neurons to direct their fate.
Figure 2. egl-13 functions cell autonomously to drive O2- and CO2-sensing neuron cell fate.

(A) Dorsal view of a young adult hermaphrodite expressing an egl-13prom1::mCherry transcriptional reporter transgene (top panels), a gcy-33prom::gcy-33::gfp translational reporter marking BAGL/R, URXL/R, AQR and PQR (center panels) and a merge of the two pictures showing co-localization in the O2- and CO2-sensing neurons (bottom panels). Left hand panels show the head region and right hand panels the tail region. We also observed expression of egl-13prom1::mCherry in muscle and vulval cells (not shown). A: anterior, L: left. The scale bar in lower panel is 20 µm. The egl-13prom1::mCherry transgene is rpEx272 and the gcy-33prom::GCY-33::gfp transgene is rpIs7. (B) Fluorescence micrographs of gcy-33prom::gcy-33::gfp expression in a wild-type animal (top panels), an egl-13(ku194) mutant animal (center panels) and an egl-13(ku194) mutant animal rescued by transgenic expression of egl-13isoformAcDNA driven by the endogenous 3.5 kb egl-13prom1 (bottom panels). Left hand panels show the head region and right hand panels the tail region. (C) Transgenic expression of egl-13isoformAcDNA under the control of egl-13prom1 rescues egl-13 mutant neuronal phenotypes, in the gcy-33prom::gcy-33::gfp strain. n = 52–55. ***P<0.001. Lines 1–3 = independent transgenic rescue lines. See materials and methods for neuronal scoring criteria used. (D) Transgenic expression of egl-13isoformAcDNA under the control of the gcy-331kb (BAG-specific) or unc-86(700bp) (URX-specific) promoters rescues egl-13 mutant neuronal phenotypes in the flp-19prom::gfp (BAG) and gcy-33prom::gcy-33::gfp (URX) strains. n = 39–55. ***P<0.001. Lines 1–2 = independent transgenic rescue lines. See materials and methods for neuronal scoring criteria used. (E) Transgenic expression of sox-2 cDNA under the control of egl-13prom1 is unable to rescue egl-13 mutant neuronal phenotypes, in the gcy-33prom::gcy-33::gfp strain. n = 56–60. (n.s.) indicates no significant difference from non transgenic egl-13 mutant animals. Lines 1–2 = independent transgenic rescue lines. See materials and methods for neuronal scoring criteria used.
The egl-13 gene has 4 predicted isoforms, all of which contain the same HMG DNA/protein binding domain, however they each have varying lengths of amino terminal tail. Such tails in SoxD proteins can cooperate with other factors to control gene expression [28]. We therefore tested whether the long N-terminal region of EGL-13isoformA is required for its rescuing ability. We used egl-13prom1 to drive EGL-13isoformD (lacking 157 amino acids of the N-terminal tail of isoformA) in egl-13(ku194) animals and found that it fully rescued the defect in O2 and CO2-sensing neuron fate marker expression and the Egl phenotype (Figure S4 and data not shown). Thus, the EGL-13 N-terminal region is not required for its roles in vulval cell nor O2 and CO2-sensing neuron specification. We next asked whether SoxD proteins play specific roles in these decisions by attempting to rescue the egl-13 mutant defects with the SoxB family member, sox-2. We expressed sox-2 cDNA under the control of egl-13prom1 in egl-13(ku194) mutant animals. We found that sox-2 is unable to rescue O2 and CO2-sensing neuron fate marker expression (Figure 2E). These data indicate that the SoxD HMG domain plays a specific role in the specification of O2 and CO2-sensing neuron fate in C. elegans.
egl-13 is required and sufficient to induce O2-sensing neuron fate
We have shown that egl-13 is expressed throughout the life of the worm in the O2 and CO2-sensing neurons; and is required to induce terminal differentiation features. To ask whether egl-13 is required continuously to maintain the expression of the terminal gene battery of these neurons, we sought to postdevelopmentally remove egl-13 gene activity. egl-13 gene activity could not be removed by RNA-mediated interference in an RNAi sensitized background (data not shown) and there are no temperature-sensitive alleles of egl-13 available. Instead, we generated animals that lack endogenous EGL-13 protein but express heat-shock inducible egl-13 cDNA from an extrachromosomal array under the control of the hsp-16.2 promoter (Figure 3). We focused our analysis on the URX neurons and found that the loss of gcy-33prom::gcy-33::gfp reporter expression in egl-13(ku194) worms could be rescued through heat-shock induction of egl-13 during mid-larval stages (Figure 3A). This indicates that O2-sensing neurons generated during embryogenesis persist in an egl-13-responsive state. These neurons are, therefore, not converted into another fate when egl-13 is lost; however, they do not acquire the terminal O2-sensing neuron differentiation program. When egl-13 activity was supplied transiently, through removal of heat-shock stimulus, we observed a gradual loss of reporter expression during adulthood in the URX neurons (Figure 3A). Therefore, egl-13 gene activity is continuously required to maintain URX cell fate. To ask whether misexpression of egl-13 in other neurons is sufficient to induce O2 and CO2 terminal fate we expressed egl-13 under the control of an early neuronal promoter (Figure 3B). We found that egl-13 is indeed sufficient to induce expression of O2 and CO2 terminal fate markers in some cellular contexts (Figure 3B). This suggests that egl-13 is not only required but also sufficient to induce O2 and CO2-sensing neuron fate in specific contexts, which is similar to previous studies of terminal selector genes [29]–[31]. The restricted induction we observed may be dependent on the embryonic time-point of induction or the expression of other unknown co-factors that are required for induction of O2 and CO2-sensing neuron fate.
Figure 3. egl-13 is required and sufficient to induce O2- and CO2-sensing neuron fate.

(A) egl-13 is required to maintain O2/CO2-sensing neuron fate. Induction of egl-13 expression via transient heat-shock at the L2/3 stage restores gcy-33prom::gcy-33::gfp expression in the URX neurons of egl-13(ku194) mutant animals. A schematic describing the heat-shock protocol is shown at the top left. The number of positive neurons was assessed one day after heat-shock (L2/3+1 day). No increase in the number of positive neurons was observed in egl-13(ku194); gcy-33prom::gcy-33::gfp; Ex[hsp-16.2::egl-13] animals in the absence of heat shock. A significant decrease in the number of positive neurons was observed in egl-13(ku194); gcy-33prom::gcy-33::gfp; Ex[hsp-16.2::egl-13] animals 3 days after heat shock with growth at 15°C (L4+3 days) when compared to one day after heat shock (L4+1 day). Quantification of the number of gcy-33prom::gcy-33::gfp positive neurons is shown at the indicated time points. Error bars represent the standard error of the mean (SEM). Statistical model applied is a one-way ANOVA with Newman-Keuls multiple comparison test **P<0.05, ***P<0.005. Representative fluorescent micrographs on the right indicate the continual requirement for egl-13 to maintain O2/CO2-sensing neuron fate. Neuron positions are marked with dashed circles: BAG(red), URX(blue). Two different extrachromosomal lines were analyzed for this experiment and showed similar effects (only one is shown). 53–113 animals were used per time point per genotype. See materials and methods section for heat shock protocol and for neuronal scoring criteria used. Scale bar, 20 µm. Anterior to the left. (B) egl-13 is sufficient to induce O2/CO2-sensing cell fate markers. An unc-86 promoter was used to drive broad neuronal expression of egl-13 cDNA in a wild-type background. Graph indicates the percentage of animals that exhibit ectopic expression O2/CO2-sensing cell fate marker (+ array = transgenic animals expressing unc-86prom::egl-13cDNA array; − array = transgenic siblings not expressing the array). Representative fluorescent micrographs of ectopic expression of three O2/CO2-sensing cell fate markers: flp-8prom::gfp, flp-19prom::gfp and gcy-33prom::gcy-33::gfp. Neuron positions are marked with dashed circles: BAG(red), URX(blue) and ectopic expression of each reporter is marked with arrowheads. Scale bar, 20 µm. Anterior to the left.
egl-13 mutants are defective in O2 and CO2 sensing
The crucial role for egl-13 in O2 and CO2-sensing neuron fate determination suggested that egl-13 mutant animals would be defective in O2 and CO2 sensing. We applied three behavioral paradigms that have been previously reported to be specific to either one of these neuron classes: BAG neurons modulate the animals' locomotion speed in response to an oxygen downshift from 21% O2 towards 10% O2 (Figure 4A, 4E) [20]. In addition, BAG neurons detect increases in CO2 concentrations, which trigger reorientation movements (omega turns) (Figure 4G) [11], [21]. URX neurons modulate the animals' locomotion speed in response to O2 upshifts towards 21% O2 (Figure 4A, 4F) [20]. We applied these behavioral assays to test how BAG and URX neurons are functionally affected in egl-13 mutants. We tracked animals in a chamber without food, in an air-flow that switched between 21% O2 and 10% O2, or between 0% CO2 and 1% CO2. In contrast to wild-type animals, egl-13(ku194) mutant animals do not slow their locomotion in response to O2 upshift or downshift (Figure 4A, 4C, 4E, 4F). We found that egl-13(ku194) mutants are also defective in CO2 sensing since they fail to slow or perform omega turns in response to CO2 (Figure 4G). O2 and CO2 behavioral defects of egl-13(ku194) mutants are fully rescued when egl-13 cDNA is resupplied under the control of egl-13prom1 (Figure 4D–4G). These data confirm that egl-13 is crucial for the specification and function of O2 and CO2-sensing system in C. elegans.
Figure 4. egl-13 is essential for locomotion responses to O2 and CO2 concentration shifts.

(A-D) Locomotion speed of C. elegans during O2 concentration shifts. Traces show average forward speed and dark shading indicates standard error of the mean (SEM). O2 concentrations were switched between 21% and 10%. Shading represents intervals at 21%. (A) Wild-type animals. (B) egl-13(rp23) mutants. (C) egl-13(ku194) mutants. (D) Rescue of egl-13(ku194) mutant phenotype by transgenic expression of egl-13 cDNA under control of its own promoter. Transgene rpEx401. Note the respective reduction of speed levels in N2 after up- and downshift, which are abolished in egl-13(ku194) and restored in the transgenic line. egl-13(rp23) animals are affected mostly in their response to O2 upshift. (E, F) Quantification of data in A–D. Average speed changes in percent from basal speed to O2 downshift (E) and upshift (F) of animals with indicated genotypes. Transgenic rescue lines are significantly different from egl-13(ku194) mutant animals. (G) Average changes in omega turn frequency of animals with indicated genotypes, in response to 1% CO2. The defect in omega turn responses seen in egl-13(ku194) animals is restored in the transgenic lines (rpEx399 and rpEx401). egl-13(rp23) animals only exhibit a partial defect. Error bars = SEM. Symbols indicate all significant differences one-way ANOVA with Bonferroni's Multiple Comparison Test (*/◊ p = 0.01–0.05, **/◊◊ p = 0.001–0.01, ***/◊◊◊ p<0.001). Asterisks indicate significant difference compared to wild-type, while diamonds indicate significant difference compared to egl-13(ku194) mutants. Data were calculated from n = 3 independent experiments for each mutant and transgenic rescue strain, and n = 6 independent experiments for wild-type. Each individual experiment was performed on 60–70 animals.
The egl-13 promoter contains neuron-specific regulatory modules
One of the egl-13 mutant alleles retrieved from our screen was a promoter deletion mutant (rp23). The rp23 deletion removes 1128 bp of egl-13 promoter from −1700 to −572 upstream of the translational start site (Figure 1A and Figure 5A). Intriguingly, the rp23 mutation affects terminal marker expression in the URX but not BAG sensory neurons and is mostly defective in URX and less affected in BAG regulated behaviors (Figure 4B, 4E–4G; Figure S4C; and Table S1). This suggests that the rp23 promoter deletion removes element(s) required to drive egl-13 in the URX neurons while leaving the BAG-specific element(s) intact. To identify which upstream factors drive expression of egl-13 in the molecularly and functionally distinct BAG and URX neurons we performed promoter deletion analysis, using the 3.5 kb upstream element (egl-13prom1) as a template. We generated transgenic worms expressing truncated versions of egl-13prom1 driving mCherry or gfp protein and focused our expression analysis on BAG and URX regulation (Figure 5A). A 900 bp fragment (egl-13prom3), which includes 360 bp corresponding to the 3′ end of the rp23 deletion, drove expression in BAG and URX. However, a 691 bp fragment (egl-13prom4), which lacks the missing region in the rp23 deletion, only drove expression in the BAG neurons. Therefore, an important element required for egl-13 expression specifically in the URX neurons lies within the 200 bp region included in egl-13prom3. Bioinformatic analysis of this region revealed that there are two conserved motifs that are potential binding sites for EGL-13/SOX5 itself and AHR-1, an aryl hydrocarbon receptor bHLH protein. Interestingly, ahr-1 was previously shown to be required for the expression of some URX terminal fate markers [32]. Site-directed mutagenesis of the predicted AHR-1 binding site significantly reduced egl-13prom1::mCherry expression and a subsequent mutation in the putative EGL-13 binding site further reduced expression (Figure 5A). This suggests that both AHR-1 and EGL-13 regulate egl-13 expression. To test this, we crossed egl-13prom1mCherry/gfp-expressing animals into egl-13(ku194) and ahr-1(ia3) mutants and found that URX expression was reduced in both cases (Figure 5B). Therefore, AHR-1 and EGL-13 both contribute to the control of egl-13 expression in the URX neurons.
Figure 5. Independent regulatory modules drive egl-13 expression in O2 versus O2/CO2-sensing neurons.

(A) egl-13 promoter analysis. Schematic representation of the egl-13 locus with its 3.5 kb upstream region. The ATG codon is marked with an arrow and the exons are represented as black blocks. The upstream region deleted in the rp23 allele is indicated with a blue horizontal line and a grey dashed vertical line. Below is a representation of cloned and injected constructs, and their expression pattern in the URX and BAG neurons. Black lines denote the promoter fragment placed in front of mCherry fluorescent protein (red boxes). Orange, blue and green crosses represent mutated Sox5, AHR-1 and ETS binding sites respectively. “+” indicates consistent reporter expression in at least 50% of animals in all lines. “+/−” indicates less than 50% of animals expressing the reporter in all lines. “−” indicates loss of expression. Three independent transgenic lines were analyzed for each promoter as indicated. (B) Expression of egl-13 and ets-5 reporter transgenes in the BAG neurons in the reciprocal mutant backgrounds (left). egl-13(ku194) and ets-5(tm1734) alleles were used. Expression of egl-13 and ahr-1 reporter transgenes in the URX neurons in the reciprocal mutant backgrounds (right). egl-13(ku194) and ahr-1(ia3) alleles were used. n>50 per strain. Error bars represent the standard error of the mean (SEM) **P<0.05, ***P<0.005. (n.s.) indicates no significant difference from wild-type. See materials and methods for neuronal scoring criteria used. (C) In the BAG neurons (left), the ETS transcription factor, ETS-5 regulates the expression of egl-13 in the BAG neurons via conserved ETS binding sites located in the egl-13 promoter. In addition, egl-13 and ets-5 autoregulate in the BAG neurons to generate BAG fate. In the URX neurons (right), egl-13 and ahr-1 autoregulate, in addition to regulating the expression of each other. Other unknown factors act in parallel to and in combination with these factors to drive BAG and URX neuronal fate.
To identify the regulatory module(s) that control egl-13 expression in the BAG neurons, we continued to dissect the egl-13 promoter. We identified a 432 bp region (egl-13prom5.1), immediately upstream of the ATG codon, which is sufficient to drive expression in the BAG neurons. Intriguingly, we found two conserved ETS-5/Pet1 binding sites in this region (Figure 5). Previous work identified ETS-5 as a crucial factor required for the specification of the BAG neurons, suggesting that ETS-5 may regulate egl-13 expression in these neurons [26], [33]. We used site-directed mutagenesis to eliminate the ETS-5/Pet1 binding sites individually and in combination, and found that when both ETS-5/Pet1 binding sites are mutated the expression of egl-13 is abrogated in the BAG neurons (Figure 5A). This suggests that ETS-5 directly regulates the expression of egl-13 in the BAG neurons via conserved binding sites. We crossed the ets-5(tm1734) mutant into the egl-13prom1::mCherry strain and indeed found that BAG expression was affected (Figure 5B). In addition, we found that egl-13 can regulate its own expression in the BAG neurons independently of ets-5 via an, as yet, unidentified mechanism (Figure 5B).
Taken together, these data indicate that control of egl-13 expression is coordinated by two independent regulatory mechanisms. In the O2-sensing URX neurons, egl-13 expression is predominantly regulated by AHR-1 (Figure 5C). In contrast, an independent promoter module controlled by ETS-5 regulates egl-13 expression in the O2/CO2-sensing BAG neurons (Figure 5C). In addition, egl-13 is able to autoregulate in both the URX and BAG neurons (Figure 5C).
egl-13 acts in partially parallel pathways with ets-5 and ahr-1
Previous work has identified ets-5 and ahr-1 as regulators of BAG and URX specification respectively [26], [32], [33] and we have shown that these factors are predominantly required to drive egl-13 expression in these cells. To understand how these factors function together to coordinate BAG and URX specification, we analyzed the expression of terminal fate markers in single and double mutant combinations, where appropriate. We analyzed three URX markers (flp-8::gfp, flp-19::gfp and gcy-33prom::gcy-33::gfp) and six BAG markers (flp-13::gfp, flp-17::gfp, flp-19::gfp, gcy-9::gfp, gcy-31::gfp and gcy-33prom::gcy-33::gfp) and compared the effect of the individual loss of egl-13, ets-5 and ahr-1 (Figure S5).
The first observation from this analysis was that the expression of a subset of terminal differentiation markers is completely dependent on egl-13 and one of the other factors acting in a linear pathway. For example, we find that BAG expression of flp-13::gfp and flp-19::gfp is almost 100% affected in both the egl-13 and ets-5 single mutants (Figure S5). This suggests that for these markers egl-13 and ets-5 act in the same pathway to drive marker expression. In contrast, expression of gcy-9::mCherry is completely dependent on ets-5 with egl-13 playing a minor role in its regulation (Figure S5). At the other end of the spectrum, ets-5 and egl-13 are minimally required to drive gcy-31::mCherry expression in the BAG neurons suggesting other factor(s) control the expression of this terminal fate marker (Figure S5). Taken together, these data indicate that egl-13 and ets-5 act in partially parallel pathways to drive BAG cell fate and that other unknown factors possibly act in a combinatorial manner to drive specific aspects of BAG fate.
We also observed differential effects of egl-13 loss with URX terminal fate markers. Expression of the flp-8::gfp reporter is partially affected by single loss of egl-13 and ahr-1, whereas loss of both genes totally abrogates expression, suggesting that egl-13 and ahr-1 act in parallel pathways to regulate flp-8::gfp expression (Figure S5). However, in the case of flp-19::gfp, loss of egl-13 causes complete loss of expression and ahr-1 plays a minor role in its regulation (Figure S5).
To further investigate the regulatory relationship between egl-13, ets-5 and ahr-1 we analyzed how they affect the expression of each other. We have already shown that ets-5 positively regulates the expression of egl-13 in the BAG neurons (Figure 5B). In a reciprocal experiment, we found that ets-5::gfp expression is unaffected in egl-13(ku194) mutant animals (Figure 5B). These data and other work [26], [33] suggest that ets-5 acts upstream and in parallel to egl-13 to direct BAG cell fate (Figure 5C). In addition, we found that egl-13 is able to regulate its own expression in the BAG neurons, in parallel to ets-5; however, the mechanistic basis of this regulation is unclear (Figure 5B). In the URX neurons, we found that egl-13 and ahr-1 regulate the expression of each other in addition to having autoregulatory capabilities (Figure 5B,C and Figure S5).
Our studies have elucidated a novel function for egl-13, the SoxD homolog, in the specification of distinct classes of O2 and CO2 sensory neurons in C. elegans. We show that egl-13 is expressed in the O2- and CO2-sensing neurons and acts cell-autonomously to regulate their distinct cell fates. We further show that egl-13 is continuously expressed in the O2- and CO2-sensing system to maintain the expression of terminal features of these neurons. In certain cellular contexts, egl-13 is also sufficient to induce O2- and CO2-sensing neuron cell fate. We found that the regulatory inputs controlling the expression of egl-13 in the O2- and CO2-sensing system are mechanistically distinct. Independent regulatory modules control egl-13 expression in the BAG neurons (CO2 and O2 downshift sensors) versus the URX neurons (O2 upshift sensors). Interestingly, we found that egl-13 expression in the BAG neurons is controlled by the ETS-5 transcription factor via conserved ETS binding sites. In contrast, in the URX neurons, egl-13 expression is controlled by the bHLH transcription factor AHR-1 via a conserved AHR1 binding site.
The influence EGL-13 exerts on the expression of the terminal gene batteries of the distinct O2- and CO2-sensing neurons is diverse. Particular factors are exquisitely sensitive to loss of egl-13, whereas others are only partially affected. These findings suggest that alternative unknown modes of regulation are in place to ensure that particular molecules are faithfully expressed in the O2 and CO2 sensory neurons, which work in conjunction with and/or in parallel to egl-13.
Sox transcription factors have diverse functions during development and play crucial roles in regulating neuronal fate [34]–[37]. In addition, Sox proteins act at different levels to preselect neuronal genes in embryonic stem cells and to direct the activation of these genes in neuronal precursors and fully differentiated neurons [38]. Here we describe a novel role for EGL-13, the SoxD transcription factor in C. elegans, in driving the specification of different but related sensory neuron identities. Closely related orthologs of EGL-13 are found in vertebrates, some of which are expressed in sensory neurons [39], therefore; SoxD proteins may have a previously unrecognized conserved function in the specification of gas-sensing neurons in higher organisms.
Materials and Methods
Strains used in this study
Strains were grown using standard growth conditions on NGM agar at 20°C on Escherichia coli OP50 [8], [40]. Transgenic animals were created according to [41]. Strain information is detailed in Table S2.
Forward genetic screening approaches
In all screens, animals were mutagenized with EMS (ethyl methanesulfonate) according to standard protocols [42]. Worms were incubated at 25°C at all times. In the manual screens, 5 parental (P0) mutagenized animals were placed in each of 10 founder plates. Three days later, 400 F1 progeny of the mutagenized P0 animals were singled. Their ensuing F2 progeny were screened under a fluorescence stereomicroscope.
In the automated worm sorter screen, around 100,000 synchronized larval stage L4 animals were mutagenized with EMS, the following day the P0 young adult animals were bleached and their F1 progeny synchronized at larval stage L1 by starvation (approximately 1,000,000 animals). F1 animals were grown to the young adult stage, bleached and their F2 progeny synchronized at larval stage L1 by starvation (approximately 10,000,000 animals). The F2 progeny were grown until larval stage L4 and 10% of the population (approximately 1,000,000) was passed through a COPAS biosorter (Karolinska Institute, Stockholm, Sweden).
C. elegans expression constructs and generation of transgenic worms
Reporter gene constructs were generated by PCR amplifying promoter elements and cloning into the pPD95.75-mCherry and gfp vectors (Fire Vector Kit). Mutagenesis was performed using the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene). Rescue constructs were generated by cloning promoter and cDNA sequences into the pPD49.26 expression vector (Fire Vector Kit). Constructs were injected into young adult hermaphrodites as either simple arrays (gcy-33prom::gcy-33::gfp (50 ng ul−1) and pRF4 (50 ng ul−1) as injection markers) or as complex arrays using 1–10 ng ul−1 of linearized plasmid, 150 ng ul−1 of PvuII-digested bacterial genomic DNA and myo-2prom::dsRed (3–5 ng ul−1), elt-2prom::gfp (3–15 ng ul−1) as injection markers.
Behavioral assays
Animals were transferred without food to 14 cm NGM assay plates containing a cut out arena of Whatman filter paper soaked in 20 mM CuCl2 to prevent them from leaving a 56 mm×56 mm center area. Sixty to seventy animals were used in a single experiment and starved for one hour prior to examination. Each experiment was carried out three times, except for wild-type, which was performed six times. A custom-made transparent plexiglass device with a flow arena of 60 mm×60 mm×0.7 mm was placed onto the assay arena and animals were accustomed to a gas flow of 100 ml/min containing 21% (v/v) oxygen for 5 minutes. During the assays animals were exposed for 6 minutes to 21% O2 before and after a 6 minute stimulus interval of either 10% O2 or 1% CO2 (+21% O2). All gas mixtures were balanced with N2. Gases were mixed with a static mixing element connected to mass flow controllers (Vögtlin Instruments) that were operated by LabView software. Recordings of freely behaving animals illuminated with flat red LED lights were made at 3 fps on a 4 megapixel CCD camera (Jai) using Streampix software (Norpix). Movies were analyzed by MatLab-based image processing and tracking scripts as previously described [43], [44]. The resulting trajectories were used to calculate instantaneous speed during continuous forward movements (1 second binning). Omega turns were detected based on characteristic changes in object eccentricity and their frequency was calculated in 15 second bins. For quantifications, relative speed changes were calculated between representative intervals of 120 seconds before (basal level) and 4 seconds after the stimulus, capturing the minimum speed levels (4–8 seconds post stimulus). Data were normalized to the basal level. Changes in omega turn frequency were calculated between representative intervals of 180 seconds before (basal level) and 60 seconds after the stimulus, to capture the maximum rise phase (55–115 seconds post stimulus).
Heat-shock experiments
Two transgenic lines for hsp-16.2::egl-13 were used for the heat-shock experiments. For the rescue and maintenance experiments, third larval stage (L3) worms were heat shocked at 37°C two times for 30 min. After heat shock, worms were kept at 25°C overnight and then transferred to 15°C for 2 days.
Microscopy
Worms were mounted on 5% agarose on glass slides and images were taken using an automated fluorescence microscope (Zeiss, AXIO Imager M2) and MicroManager software (version 3.1).
Neuronal scoring
Neurons were given a numerical value according to their expression levels. Wild-type expression scored 1, decreased expression scored 0.5 and abolished expression scored 0. Percentage of GFP expressing animals was then correlated to the theoretical maximum score using the equation below.
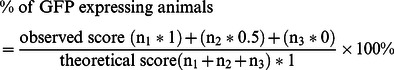 |
Bioinformatic analysis
The Jaspar program (http://jaspar.genereg.net/) was used to predict the transcription factor binding sites in the egl-13 upstream regulatory sequence.
Statistical analysis
Statistical analysis was performed in GraphPad Prism 5 using one-way ANOVA with Newman-Keuls Multiple Comparison Test. Values are expressed as mean ± s.d. Differences with a P value<0.05 were considered significant. For the behavioral assays statistical significance was determined using one-way ANOVA with Bonferroni's Multiple Comparison Test.
Supporting Information
egl-13 mutants have anchor cell fusion defects. (A) egl-13(ku194) mutant hermaphrodites are severely egg-laying defective due to a defect in anchor cell fusion (right, compared to the wild-type adult on the left). (B) egl-13 mutant alleles isolated from our forward genetic screens (rp14, 22, 23 and 26) all have anchor cell fusion defects comparable to the ku194 allele. In the wild-type vulva, the anchor cell fuses to the utse cells to form the mature uterine-vulval connection (green arrowhead, top left). In egl-13 mutant animals, the anchor cell fails to fuse to the utse (red arrowheads) and blocks egg-laying. (C) Scoring of neuronal phenotypes in egl-13 mutants. rp13 was isolated with the BAG marker, gcy-33prom::gfp. rp22 and rp23 were isolated with the URX marker, flp-8prom::GFP and rp26 was isolated with the BAG, URX, AQR and PQR marker, gcy-33prom::GCY-33::gfp. Expression of each marker in wild-type animals is stated with a +. Scorings were conducted as stated in the materials and methods section.
(EPS)
BAG and URX lineages. Lineage diagrams of the BAG and URX neurons. Neurons whose fate are affected in egl-13(ku194) mutants are indicated in red, unaffected in green and untested in black. See Table S2 for fate markers used.
(EPS)
egl-13 expression pattern. Expression pattern of egl-13 at different stages (330–360 mins, 550 mins and L1 larva), using the egl-13prom1::gfp reporter transgene. Fluorescence micrographs (right) and differential interference contrast (DIC) microscopy images (left). Scale bar, 10 µm.
(EPS)
egl-13 acts cell autonomously to control O2- and CO2-sensing neuron fate. (A) Protein domain structure of EGL-13 isoforms A+D. (B) Extrachromosomal transgenic rescue of egl-13(ku194) mutant phenotypes, in the gcy-33prom::gcy-33::gfp strain, using egl-13prom1-driven egl-13isoformAcDNA (left) and egl-13isoformDcDNA (right). Both isoforms equally rescue the egl-13(ku194) mutant phenotype. n = 46–61. *P<0.05, ***P<0.001 indicates significant difference from non-transgenic egl-13 mutant animals. (C) Transgenic lines expressing egl-13 cDNA under the control of egl-13prom1 rescues all the egl-13 alleles retrieved from the genetic screens. Rescue of rp14 in BAG (gcy-33prom::gfp), rp22 in URX (flp-8prom::gfp), rp23 in URX (flp-8prom::gfp) and rp26 in BAG, URX, AQR and PQR (gcy-33prom::GCY-33::gfp). n = 47–87. **P<0.01, ***P<0.001 indicates significant difference from non-transgenic egl-13 mutant animals. See materials and methods for neuronal scoring criteria used.
(EPS)
Differential regulation of O2- and CO2-sensing neuron terminal differentiation markers by egl-13, ets-5, and ahr-1. (A) Requirement of egl-13 and ets-5 for the expression of BAG terminal differentiation markers: flp-13, flp-17, flp-19, gcy-9, gcy-31 and gcy-33. egl-13 and ets-5 are both absolutely required for the expression of flp-13 and flp-19 suggesting that they act in a common pathway. gcy-9 is predominantly regulated by ets-5 with egl-13 playing a lesser role. flp-17 is regulated by both egl-13 and ets-5 acting in parallel pathways. Finally, gcy-31 and gcy-33 expression is predominantly regulated by egl-13. + = wt, red bars = mutant backgrounds. (B) Requirement of egl-13 and ahr-1 for the expression of URX terminal differentiation markers: flp-8, flp-19 and gcy-33. egl-13 and ahr-1 act in parallel pathways to regulate flp-8 expression. egl-13 is absolutely required for the expression of flp-19 whereas ahr-1 is less important. egl-13 and ahr-1 are both required for gcy-33 expression, probably via the same pathway. + = wt, blue bars = mutant backgrounds. Statistical model applied is a one-way ANOVA with Newman-Keuls multiple comparison test **P<0.05, ***P<0.005. Statistical model applied is a one-way ANOVA with Newman-Keuls multiple comparison test **P<0.05, ***P<0.005. Alleles used in this analysis egl-13(ku194), ets-5(tm1734) and ahr-1(ia03). See materials and methods for neuronal scoring criteria used.
(EPS)
Expression of the O2- and CO2-sensing neuron terminal gene battery is affected in egl-13 mutant animals. (A) Effects in the expression of O2- and CO2-sensing neuron reporters in egl-13(ku194) mutant animals differ from gene to gene and from cell to cell. For example, flp-19 expression is strongly affected in both URX and BAG neurons, whereas gcy-33 expression is more strongly affected in the URX neurons than BAG neurons. Fluorescent reporters for the sister cells of the URX (CEPD) and BAG (SMDV) neurons are unaffected in egl-13(ku194) mutant animals. (B) The deletion in the egl-13 locus of rp23 leaves the BAG-element intact (see Figure 5). As a result, BAG terminal fate marker expression is not significantly affected in egl-13(rp23) mutant animals. In both tables, quantification indicates the percentage of animals in which fluorescence was observed (ON), not observed (OFF) or asymmetrically affected left expressed (L ON) or right expressed (R ON). “−” indicates that the reporter is not expressed in those neurons. Animals were scored at the young adult stage. See materials and methods for neuronal scoring criteria used.
(EPS)
Strains used in this study.
(EPS)
Acknowledgments
We thank members of the Pocock Laboratory and Oliver Hobert, Baris Tursun, and Iva Greenwald for comments on the manuscript; we thank the following people for strains and reagents: Oliver Hobert (Columbia University), Cornelia Bargmann (Rockefeller University), Mark Alkema (University of Massachusetts Medical School). Some strains were provided by the CGC and by Shohei Mitani at the National Bioresource Project (Japan). We also thank Thomas Burglin and Peter Swoboda at the Karolinska Institue for use of their COPAS Biosorter facility, under the C. elegans Nordforsk Researcher Network of Shared Technology Platforms.
Funding Statement
This work was supported by a grant from the Lundbeck Foundation (www.lundbeckfonden.dk) (grant number R93-A8391) and by an ERC Starting Grant (281869—elegansNeurocircuits). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Prahlad V, Cornelius T, Morimoto RI (2008) Regulation of the cellular heat shock response in Caenorhabditis elegans by thermosensory neurons. Science 320: 811–814 doi:10.1126/science.1156093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pocock R, Hobert O (2010) Hypoxia activates a latent circuit for processing gustatory information in C. elegans. Nat Neurosci 13: 610–614 doi:10.1038/nn.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chang AJ, Bargmann CI (2008) Hypoxia and the HIF-1 transcriptional pathway reorganize a neuronal circuit for oxygen-dependent behavior in Caenorhabditis elegans. Proc Natl Acad Sci USA 105: 7321–7326 doi:10.1073/pnas.0802164105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bretscher AJ, Busch KE, de Bono M (2008) A carbon dioxide avoidance behavior is integrated with responses to ambient oxygen and food in Caenorhabditis elegans. Proc Natl Acad Sci USA 105: 8044–8049 doi:10.1073/pnas.0707607105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Semenza GL (2012) Hypoxia-inducible factors in physiology and medicine. Cell 148: 399–408 doi:10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sharabi K, Lecuona E, Helenius IT, Beitel GJ, Sznajder JI, et al. (2009) Sensing, physiological effects and molecular response to elevated CO2 levels in eukaryotes. J Cell Mol Med 13: 4304–4318 doi:10.1111/j.1582-4934.2009.00952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kaelin WG, Ratcliffe PJ (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Molecular Cell 30: 393–402 doi:10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 8. Guais A, Brand G, Jacquot L, Karrer M, Dukan S, et al. (2011) Toxicity of carbon dioxide: a review. Chem Res Toxicol 24: 2061–2070 doi:10.1021/tx200220r. [DOI] [PubMed] [Google Scholar]
- 9. Suh GSB, Wong AM, Hergarden AC, Wang JW, Simon AF, et al. (2004) A single population of olfactory sensory neurons mediates an innate avoidance behaviour in Drosophila. Nature 431: 854–859 doi:10.1038/nature02980. [DOI] [PubMed] [Google Scholar]
- 10. Munger SD, Leinders-Zufall T, McDougall LM, Cockerham RE, Schmid A, et al. (2010) An olfactory subsystem that detects carbon disulfide and mediates food-related social learning. Curr Biol 20: 1438–1444 doi:10.1016/j.cub.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hallem EA, Sternberg PW (2008) Acute carbon dioxide avoidance in Caenorhabditis elegans. Proc Natl Acad Sci USA 105: 8038–8043 doi:10.1073/pnas.0707469105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wingrove JA, O'Farrell PH (1999) Nitric oxide contributes to behavioral, cellular, and developmental responses to low oxygen in Drosophila. Cell 98: 105–114 doi:10.1016/S0092-8674(00)80610-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jones WD, Cayirlioglu P, Kadow IG, Vosshall LB (2007) Two chemosensory receptors together mediate carbon dioxide detection in Drosophila. Nature 445: 86–90 doi:10.1038/nature05466. [DOI] [PubMed] [Google Scholar]
- 14. López-Barneo J, Ortega-Sáenz P, Pardal R, Pascual A, Piruat JI, et al. (2009) Oxygen sensing in the carotid body. Annals of the New York Academy of Sciences 1177: 119–131 doi:10.1111/j.1749-6632.2009.05033.x. [DOI] [PubMed] [Google Scholar]
- 15. Hu J, Zhong C, Ding C, Chi Q, Walz A, et al. (2007) Detection of near-atmospheric concentrations of CO2 by an olfactory subsystem in the mouse. Science 317: 953–957 doi:10.1126/science.1144233. [DOI] [PubMed] [Google Scholar]
- 16. Sim CK, Perry S, Tharadra SK, Lipsick JS, Ray A (2012) Epigenetic regulation of olfactory receptor gene expression by the Myb-MuvB/dREAM complex. Genes & Development 26: 2483–2498 doi:10.1101/gad.201665.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sylvia DM, Fuhrmann JJ, Hartel PG, Zuberer DA (2004) Principles and Applications of Soil Microbiology (2nd Edition). 2nd ed. Prentice Hall. 1 pp.
- 18. Gray JM, Karow DS, Lu H, Chang AJ, Chang JS, et al. (2004) Oxygen sensation and social feeding mediated by a C. elegans guanylate cyclase homologue. Nature 430: 317–322 doi:10.1038/nature02714. [DOI] [PubMed] [Google Scholar]
- 19. Cheung BHH, Cohen M, Rogers C, Albayram O, de Bono M (2005) Experience-Dependent Modulation of C. elegans Behavior by Ambient Oxygen. Current Biology 15: 905–917 doi:10.1016/j.cub.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 20. Zimmer M, Gray JM, Pokala N, Chang AJ, Karow DS, et al. (2009) Neurons detect increases and decreases in oxygen levels using distinct guanylate cyclases. Neuron 61: 865–879 doi:10.1016/j.neuron.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hallem EA, Spencer WC, McWhirter RD, Zeller G, Henz SR, et al. (2011) Receptor-type guanylate cyclase is required for carbon dioxide sensation by Caenorhabditis elegans. Proc Natl Acad Sci USA 108: 254–259 doi:10.1073/pnas.1017354108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bretscher AJ, Kodama-Namba E, Busch KE, Murphy RJ, Soltesz Z, et al. (2011) Temperature, oxygen, and salt-sensing neurons in C. elegans are carbon dioxide sensors that control avoidance behavior. Neuron 69: 1099–1113 doi:10.1016/j.neuron.2011.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ortiz CO (2006) Searching for Neuronal Left/Right Asymmetry: Genomewide Analysis of Nematode Receptor-Type Guanylyl Cyclases. Genetics 173: 131–149 doi:10.1534/genetics.106.055749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim K, Li C (2004) Expression and regulation of an FMRFamide-related neuropeptide gene family inCaenorhabditis elegans. J Comp Neurol 475: 540–550 doi:10.1002/cne.20189. [DOI] [PubMed] [Google Scholar]
- 25. Hanna-Rose W, Han M (1999) COG-2, a sox domain protein necessary for establishing a functional vulval-uterine connection in Caenorhabditis elegans. Development 126: 169–179. [DOI] [PubMed] [Google Scholar]
- 26. Brandt JP, Aziz-Zaman S, Juozaityte V, Martinez-Velazquez LA, Petersen JG, et al. (2012) A single gene target of an ETS-family transcription factor determines neuronal CO2-chemosensitivity. PLoS ONE 7: e34014 doi:10.1371/journal.pone.0034014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sulston JE, Schierenberg E, White JG, Thomson JN (1983) The embryonic cell lineage of the nematode Caenorhabditis elegans. Developmental Biology 100: 64–119. [DOI] [PubMed] [Google Scholar]
- 28. Lefebvre V, Li P, de Crombrugghe B (1998) A new long form of Sox5 (L-Sox5), Sox6 and Sox9 are coexpressed in chondrogenesis and cooperatively activate the type II collagen gene. EMBO J 17: 5718–5733 doi:10.1093/emboj/17.19.5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Satterlee JS, Sasakura H, Kuhara A, Berkeley M, Mori I, et al. (2001) Specification of thermosensory neuron fate in C. elegans requires ttx-1, a homolog of otd/Otx. Neuron 31: 943–956. [DOI] [PubMed] [Google Scholar]
- 30. Flames N, Hobert O (2009) Gene regulatory logic of dopamine neuron differentiation. Nature 458: 885–889 doi:10.1038/nature07929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tursun B, Patel T, Kratsios P, Hobert O (2011) Direct conversion of C. elegans germ cells into specific neuron types. Science 331: 304–308 doi:10.1126/science.1199082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qin H, Zhai Z, Powell-Coffman JA (2006) The Caenorhabditis elegans AHR-1 transcription complex controls expression of soluble guanylate cyclase genes in the URX neurons and regulates aggregation behavior. Developmental Biology 298: 606–615 doi:10.1016/j.ydbio.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 33. Guillermin ML, Castelletto ML, Hallem EA (2011) Differentiation of carbon dioxide-sensing neurons in Caenorhabditis elegans requires the ETS-5 transcription factor. Genetics 189: 1327–1339 doi:10.1534/genetics.111.133835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Azim E, Jabaudon D, Fame RM, Macklis JD (2009) SOX6 controls dorsal progenitor identity and interneuron diversity during neocortical development. Nat Neurosci 12: 1238–1247 doi:10.1038/nn.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lai T, Jabaudon D, Molyneaux BJ, Azim E, Arlotta P, et al. (2008) SOX5 controls the sequential generation of distinct corticofugal neuron subtypes. Neuron 57: 232–247 doi:10.1016/j.neuron.2007.12.023. [DOI] [PubMed] [Google Scholar]
- 36. Scott CE, Wynn SL, Sesay A, Cruz C, Cheung M, et al. (2010) SOX9 induces and maintains neural stem cells. Nat Neurosci 13: 1181–1189 doi:10.1038/nn.2646. [DOI] [PubMed] [Google Scholar]
- 37. Pevny L, Placzek M (2005) SOX genes and neural progenitor identity. Current Opinion in Neurobiology 15: 7–13 doi:10.1016/j.conb.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 38. Bergsland M, Ramsköld D, Zaouter C, Klum S, Sandberg R, et al. (2011) Sequentially acting Sox transcription factors in neural lineage development. Genes & Development 25: 2453–2464 doi:10.1101/gad.176008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morales AV, Perez-Alcala S, Barbas JA (2007) Dynamic Sox5 protein expression during cranial ganglia development. Dev Dyn 236: 2702–2707 doi:10.1002/dvdy.21282. [DOI] [PubMed] [Google Scholar]
- 40. Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mello CC, Kramer JM, Stinchcomb D, Ambros V (1991) Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10: 3959–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Flibotte S, Edgley ML, Chaudhry I, Taylor J, Neil SE, et al. (2010) Whole-genome profiling of mutagenesis in Caenorhabditis elegans. Genetics 185: 431–441 doi:10.1534/genetics.110.116616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ramot D, Johnson BE, Berry TL, Carnell L, Goodman MB (2008) The Parallel Worm Tracker: a platform for measuring average speed and drug-induced paralysis in nematodes. PLoS ONE 3: e2208 doi:10.1371/journal.pone.0002208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tsunozaki M, Chalasani SH, Bargmann CI (2008) A behavioral switch: cGMP and PKC signaling in olfactory neurons reverses odor preference in C. elegans. Neuron 59: 959–971 doi:10.1016/j.neuron.2008.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
egl-13 mutants have anchor cell fusion defects. (A) egl-13(ku194) mutant hermaphrodites are severely egg-laying defective due to a defect in anchor cell fusion (right, compared to the wild-type adult on the left). (B) egl-13 mutant alleles isolated from our forward genetic screens (rp14, 22, 23 and 26) all have anchor cell fusion defects comparable to the ku194 allele. In the wild-type vulva, the anchor cell fuses to the utse cells to form the mature uterine-vulval connection (green arrowhead, top left). In egl-13 mutant animals, the anchor cell fails to fuse to the utse (red arrowheads) and blocks egg-laying. (C) Scoring of neuronal phenotypes in egl-13 mutants. rp13 was isolated with the BAG marker, gcy-33prom::gfp. rp22 and rp23 were isolated with the URX marker, flp-8prom::GFP and rp26 was isolated with the BAG, URX, AQR and PQR marker, gcy-33prom::GCY-33::gfp. Expression of each marker in wild-type animals is stated with a +. Scorings were conducted as stated in the materials and methods section.
(EPS)
BAG and URX lineages. Lineage diagrams of the BAG and URX neurons. Neurons whose fate are affected in egl-13(ku194) mutants are indicated in red, unaffected in green and untested in black. See Table S2 for fate markers used.
(EPS)
egl-13 expression pattern. Expression pattern of egl-13 at different stages (330–360 mins, 550 mins and L1 larva), using the egl-13prom1::gfp reporter transgene. Fluorescence micrographs (right) and differential interference contrast (DIC) microscopy images (left). Scale bar, 10 µm.
(EPS)
egl-13 acts cell autonomously to control O2- and CO2-sensing neuron fate. (A) Protein domain structure of EGL-13 isoforms A+D. (B) Extrachromosomal transgenic rescue of egl-13(ku194) mutant phenotypes, in the gcy-33prom::gcy-33::gfp strain, using egl-13prom1-driven egl-13isoformAcDNA (left) and egl-13isoformDcDNA (right). Both isoforms equally rescue the egl-13(ku194) mutant phenotype. n = 46–61. *P<0.05, ***P<0.001 indicates significant difference from non-transgenic egl-13 mutant animals. (C) Transgenic lines expressing egl-13 cDNA under the control of egl-13prom1 rescues all the egl-13 alleles retrieved from the genetic screens. Rescue of rp14 in BAG (gcy-33prom::gfp), rp22 in URX (flp-8prom::gfp), rp23 in URX (flp-8prom::gfp) and rp26 in BAG, URX, AQR and PQR (gcy-33prom::GCY-33::gfp). n = 47–87. **P<0.01, ***P<0.001 indicates significant difference from non-transgenic egl-13 mutant animals. See materials and methods for neuronal scoring criteria used.
(EPS)
Differential regulation of O2- and CO2-sensing neuron terminal differentiation markers by egl-13, ets-5, and ahr-1. (A) Requirement of egl-13 and ets-5 for the expression of BAG terminal differentiation markers: flp-13, flp-17, flp-19, gcy-9, gcy-31 and gcy-33. egl-13 and ets-5 are both absolutely required for the expression of flp-13 and flp-19 suggesting that they act in a common pathway. gcy-9 is predominantly regulated by ets-5 with egl-13 playing a lesser role. flp-17 is regulated by both egl-13 and ets-5 acting in parallel pathways. Finally, gcy-31 and gcy-33 expression is predominantly regulated by egl-13. + = wt, red bars = mutant backgrounds. (B) Requirement of egl-13 and ahr-1 for the expression of URX terminal differentiation markers: flp-8, flp-19 and gcy-33. egl-13 and ahr-1 act in parallel pathways to regulate flp-8 expression. egl-13 is absolutely required for the expression of flp-19 whereas ahr-1 is less important. egl-13 and ahr-1 are both required for gcy-33 expression, probably via the same pathway. + = wt, blue bars = mutant backgrounds. Statistical model applied is a one-way ANOVA with Newman-Keuls multiple comparison test **P<0.05, ***P<0.005. Statistical model applied is a one-way ANOVA with Newman-Keuls multiple comparison test **P<0.05, ***P<0.005. Alleles used in this analysis egl-13(ku194), ets-5(tm1734) and ahr-1(ia03). See materials and methods for neuronal scoring criteria used.
(EPS)
Expression of the O2- and CO2-sensing neuron terminal gene battery is affected in egl-13 mutant animals. (A) Effects in the expression of O2- and CO2-sensing neuron reporters in egl-13(ku194) mutant animals differ from gene to gene and from cell to cell. For example, flp-19 expression is strongly affected in both URX and BAG neurons, whereas gcy-33 expression is more strongly affected in the URX neurons than BAG neurons. Fluorescent reporters for the sister cells of the URX (CEPD) and BAG (SMDV) neurons are unaffected in egl-13(ku194) mutant animals. (B) The deletion in the egl-13 locus of rp23 leaves the BAG-element intact (see Figure 5). As a result, BAG terminal fate marker expression is not significantly affected in egl-13(rp23) mutant animals. In both tables, quantification indicates the percentage of animals in which fluorescence was observed (ON), not observed (OFF) or asymmetrically affected left expressed (L ON) or right expressed (R ON). “−” indicates that the reporter is not expressed in those neurons. Animals were scored at the young adult stage. See materials and methods for neuronal scoring criteria used.
(EPS)
Strains used in this study.
(EPS)