

Abstract
Background
In recent years, findings in a number of animal and human models have ignited renewed interest in the type 3 deiodinase (D3), the main enzyme responsible for the inactivation of thyroid hormones. The induction of D3 in models of illness and injury has raised critical questions about the physiological significance of reduced thyroid hormone availability in those states. Phenotypes in transgenic mice lacking this enzyme also point to important developmental roles for D3. A critical determinant of D3 expression is genomic imprinting, an epigenetic phenomenon that regulates a small number of dosage-critical genes in the mammalian genome. The D3 gene (Dio3) is imprinted and preferentially expressed from one of the alleles in most tissues.
Scope of review
In the context of the physiological significance of D3 and the characteristics and purported origins of genomic imprinting, we review the current knowledge about the epigenetic mechanisms specifying gene dosage in the Dio3 locus.
Major conclusions
Altered Dio3 dosage is detrimental to development, suggesting that the level of thyroid hormone action needs to be exquisitely tailored in a timely fashion to the requirements of particular tissues. An appropriate Dio3 dosage is the result of the coordinated action of certain genomic elements and epigenetic marks in the Dlk1-Dio3 domain.
General significance
The imprinting of Dio3 prompts intriguing questions about why the level of thyroid hormone signaling should be regulated in this rare epigenetic manner, and to what extent altered Dio3 expression due to aberrant imprinting may be implicated in human conditions. This article is part of a Special Issue entitled Thyroid hormone signalling.
Keywords: Genomic imprinting, Type 3 deiodinase, Dio3, Thyroid hormone, Mouse chromosome 12, Uniparental disomy of chromosome 14
1. Introduction
Thyroid hormones exert broad biological effects in all mammalian species. Their critical roles in the regulation of growth, development and metabolism are particularly remarkable, but most tissues express some form of thyroid hormone receptor and, thus, are a target for thyroid hormones. As a result, alterations in the mechanisms regulating the availability of thyroid hormone or its signaling have the potential to affect many physiological systems and, in humans, be the cause of developmental abnormalities and adult patho-physiological states.
Multiple factors, as reviewed in this special issue, influence the degree of thyroid hormone action that occurs in a given cell. One of them is the type 3 deiodinase (D3), a selenoenzyme that converts the two main hormones produced by the thyroid gland, the pro-hormone thyroxine (T4) and the active hormone triiodothyronine (T3), into metabolites with little or no biological activity [1–3]. Given this enzymatic activity, D3 is positioned to decrease T3 availability in the tissues in which it is expressed and, therefore, to reduce the local level of thyroid hormone action. The ample tissue profile of D3 expression, especially during development, suggests roles for D3 in multiple systems. This notion is being increasingly confirmed in animal models, adding physiological significance to the mechanisms regulating D3 expression.
In this regard, a puzzling observation about the D3 gene (Dio3) is that it is subject to genomic imprinting [4–6], an epigenetic phenomenon affecting a small number of genes that results in the preferential expression of one of the alleles [7]. This characteristic sets the D3 apart from other determinants of thyroid hormone action, and raises the possibility that the epigenetic mechanisms governing the allelic expression of Dio3 are an important determinant of thyroid hormone levels in tissues, and are critical to ensure normal development and physiology.
Here we briefly summarize past and recent observations about the physiological significance of D3, review the current knowledge about the genomic imprinting of Dio3 and the potential implications for human disease. We also briefly discuss its potential role in developmental plasticity, evolution and in the epigenetic inheritance of biological traits.
2. Pathophysiological significance of D3
Although a detailed account of this topic is not the focus of the present review, it is important to fully understand the impact that alterations in Dio3 expression could have on development and physiology. This will provide a context in which to evaluate the significance of Dio3 imprinting in health and disease.
The existence of the D3 (also called type III, 5- or inner-ring deiodinase) has been known for more than 30 years. Initially, D3 garnered less attention than the other deiodinase enzymes of the family, the type 1 (D1) and the type 2 (D2), as investigators were more interested in the activation of thyroid hormones, a process of particular importance in preventing the devastating effects that the lack of thyroid hormones exert on the central nervous system. At the time, it was simply – and rightly – assumed that D3 played a role in protecting tissue under circumstances of thyroid hormone excess, i. e., as a result of hyperthyroidism due to pathology of the thyroid gland, or during fetal development, a time when thyroid hormone levels are much lower than those in the mother.
Partially consistent with this hypothesis was the expression profile of D3, which exhibits a marked developmental pattern (Fig. 1A). D3 activity can be found in most tissues during fetal and early neonatal life [8]. In contrast, during adult life, high D3 expression is limited to the central nervous system [9–11], with lower expression levels found in the skin [12] and certain endocrine organs including the uterus, ovary and adrenal gland [13]. Most other tissues feature very little or no D3 expression in adulthood. For most tissues, D3 activity decreases one to two orders of magnitude from fetal life to adulthood (Fig. 1A). These observations, together with the very high expression of D3 in the placenta and pregnant uterus [14,15], suggested an important role for this enzyme during development and in the regulation of neural and endocrine functions.
Fig. 1.
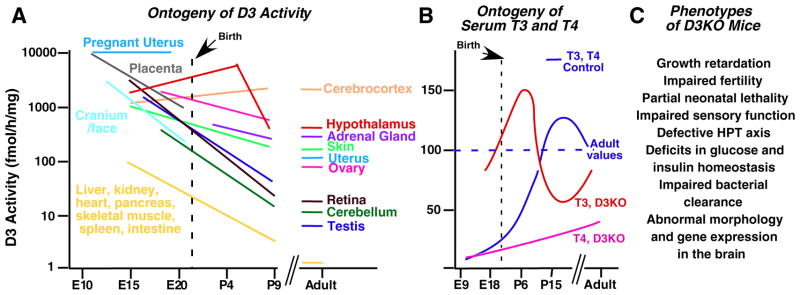
Physiological significance of D3. A, Ontogeny and relative level of D3 activity in rodent tissues (patterns are approximate, based on cited literature). Note that D3 activity is reduced by one or two orders of magnitude in most tissues from fetal life to adulthood. B, Ontogeny of serum thyroid hormone levels in normal animals and in D3KO mice. During development D3KO mice are exposed to excessive levels of T3. Values are expressed as a percentage of normal adult values (horizontal dotted line). C, Summary of reported phenotypes in D3KO mice.
As fetal development is a time of rapid cell proliferation and timely differentiation, it had been postulated that the high D3 expression at these stages was aimed at preventing tissues form undue exposure to thyroid hormones, a hypothesis consistent with the known roles of thyroid hormones in cell differentiation.
In this regard, a review on the role of D3 and other deiodinases in cell proliferation and differentiation and in signaling pathways related with these processes can be found in this issue [16].
2.1. D3 induction in fasting, illness and models of inflammation and injury
In the rodent, most adult tissues exhibit very low or undetectable levels of D3 activity. However, a number of physiological challenges, injuries and inflammatory processes can lead to remarkable inductions of D3 in certain tissues that would not express any D3 in a healthy state [17]. Thus, in humans and rodents, D3 is induced in the liver and skeletal muscle during critical illness [18–20] and after fasting [21,22]. These physiological states are typically associated with the “euthyroid sick syndrome”, which is characterized by low levels of serum T3 and T4 despite the absence of any pathology in the hypothalamic–pituitary–thyroid axis [23]. Although the mechanisms underlying this syndrome are not fully understood, it is possible that an induction of D3 in certain tissues may contribute to this phenomenon.
In certain models of injury and inflammation, D3 expression can also be induced in tissues and cell types which would not normally express D3 in healthy conditions [17]. This type of D3 induction has been found in the heart when subjected to physiological insults such as myocardial infarction and pulmonary hypertension [24,25]. Induction of local D3 has also been observed in infiltrating cells during lung infection [26], after sciatic nerve lesion [27], and during chronic hind limb inflammation [28].
The clinical significance of this re-induction of D3 in adult tissues after an injury or physiological insult is largely unknown. The return of D3 expression in some tissues to levels that resemble those present early in development may reflect the need for new cell proliferation and differentiation processes to supply the cell needs and required endocrine microenvironment of the healing tissue. The increased D3 expression and the subsequently low local levels of thyroid hormones may be an integral component of the response to certain physiological challenges or necessary for optimal recovery. However, D3 induction in some of these models could just be a by-product of inflammation and potentially detrimental for health outcomes.
More studies are needed to determine in each of these models whether the role of D3 induction is adaptive or maladaptive, but this phenomenon may have important implications for human conditions, as it raises the possibility of clinical manipulation of the local or systemic thyroid hormone environment to foster recovery from illness and injury. In addition, the mechanism/s by which these conditions can induce D3 are unknown, but the susceptibility of this gene to expression dosage alteration in particular patho-physiological states may relate to its regulation by epigenetic mechanisms (see below) and warrants further investigation.
2.2. Observations in D3KO mice
An important body of evidence about the physiological significance of D3 comes from studies in D3-deficient (D3KO) mice. As expected from the high D3 expression during fetal and neonatal life, D3KO mice are overexposed to T3 at those stages [29] (Fig. 1B), which are critical to the maturation of the hypothalamic–pituitary–thyroid (HPT) axis. As a result, D3KO animals present in adulthood with a moderate degree of hypothyroidism that varies slightly between genetic backgrounds, but that is characterized by a reduced serum T4, normal or slightly reduced serum T3 and slightly elevated or unaltered serum thyrotropin [29]. This hypothyroidism is the result of abnormal regulation and/or programming of the HPT axis, as physiological defects in hormonal responses are found in the hypothalamus, pituitary and thyroid gland [30].
D3KO mice manifest phenotypes affecting many other systems (Fig. 1C). Their immune response is deficient, as they show reduced bacterial clearance in a model of lung infection [31]. Their hearing and vision are impaired, as a result of abnormalities in cochlear and retinal development [32,33]. They are markedly growth retarded and their fertility and neonatal viability are reduced [29]. Insulin secretion and glucose homeostasis are impaired in D3KO mice [34]. The level of T3 action in most areas of the central nervous system is altered in developing, adult and aged animals, significantly altering gene expression patterns [35] and potentially affecting many aspects of brain development and function. D3KO mice also exhibit a complex phenotype affecting energy balance (A. Hernandez, unpublished observations). In addition, preliminary results from our laboratory suggest that defects in craniofacial structures and brain morphology, lifespan, adiposity and energy balance, behavior and heart physiology are associated with loss of D3 function.
Overall, the picture emerging from all these studies suggests that Dio3 expression is critical to protect many tissues and cell types from excessive thyroid hormone action. This seems particularly true for tissues regulating neurological and endocrine functions, developing tissues, and those tissues that have been subjected to injury, inflammatory processes or specific physiological challenges. Moreover, the dynamic expression profile of D3 during embryonic and early postnatal development suggests that its appropriate expression dosage is critical for regulating local thyroid hormone action. Although the precise role of D3 and limited thyroid hormone action in all these scenarios is not fully understood, the evidence so far indicates that appropriate Dio3 expression is of importance for functional and developmental outcomes. It is in this context that the genomic imprinting and epigenetic regulation of Dio3 acquire its full relevance.
3. General characteristics of genomic imprinting
In contrast to the majority of genes that are expressed from both parental chromosomes, imprinted genes are expressed from a single allele based on their parental origin. The discovery of imprinted genes in the early 1980s [36,37] provided some of the first evidence of epigenetic regulation, since in order for two alleles of identical sequence to be differentially expressed in a heritable manner, they must be marked with further modifications to the gene that direct their expression. To date approximately 100 imprinted genes have been described in the mouse [38] and a similar number are thought to exist in other mammalian species including humans. Of these ~100 genes, approximately half are expressed from the maternally inherited chromosome, and the remainder are expressed from the paternally inherited chromosome. The majority of imprinted genes are located in genomic clusters rather than randomly scattered across the genome. Imprinting clusters can contain genes that are mono-allelically expressed from either parental chromosome, and genes within a given cluster are regulated in a coordinated manner by a common imprinting control region (ICR) (reviewed by Ferguson-Smith [39]).
3.1. Properties of ICRs
Control of gene expression by imprinting involves several stages; firstly, an epigenetic mark must be established in the gametes to distinguish the two parental alleles. Secondly, this mark must be maintained throughout development, specifically during global epigenetic reprogramming events that occur in early embryogenesis, but must be erased in the germ line to allow for the imprint to be reset according to the sex of the individual. Finally, in somatic cells the imprint must control gene expression in cis.
ICRs were initially identified as CG-rich DNA sequences that exhibited differences in the levels of 5-methyl cytosine according to their parental origin. ICRs are differentially methylated between sperm and oocyte, and they retain these methylation differences during fertilization and preimplantation development, and in the majority of somatic cells. During gametogenesis this methylation is erased and reset before gamete maturation (reviewed in Ferguson-Smith [39]).
In addition to DNA methylation, all ICRs are marked by signature modifications to histone proteins, specifically trimethylation of lysine 4 (H3K4me3) and lysine 9 (H3K9me3) of histone H3 and trimethylation of lysine 20 of histone H4 (H4K20me3) [40].
3.2. Imprinting establishment
In both male and female germ lines the establishment of methylation relies on the de-novo methyltransferase DNMT3A and its cofactor DNMT3L [41–43]. The mechanism by which methylation is targeted to ICRs is still unknown, but may involve recruitment of repressive chromatin by small RNAs [44], sequence periodicity [45], or other epigenetic modifications. In addition, ICR sequences may be selectively shielded from de-novo methylation in the germ line by transcription and histone modifications (reviewed by Smallwood [46]).
3.3. Post-fertilization maintenance of imprinting marks
Following fertilization the chromatin from both parents is subject to reprogramming by the oocyte cytoplasm. Both parental genomes are demethylated in the embryo before implantation by both active and replication-dependent mechanisms [47]. However, ICRs are not demethylated during this period and this protection from genome-wide epigenetic remodeling is dependent upon the Kruppel-associated box domain (KRAB)-containing zinc finger protein ZFP57 [48] and the developmental pluripotency-associated protein 3 (DPPA3/STELLA) [49]. ZFP57 binds to all known ICRs at a hexanucleotide motif when this sequence is methylated [50]. Binding of ZFP57 to the methylated ICR recruits binding in cis of the KAP1 complex and subsequent recruitment of DNA methyltransferases and histone H3K9 methyltransferases, and thus maintenance of the ICR [50,51]. Loss of Zfp57 in vitro or in vivo results in failure of ICRs to maintain their methylation and consequently misexpression of imprinted genes in both the mouse and human [48,50–52].
3.4. Control of gene expression
Two types of ICR have been described; those that are methylated in oocytes and unmethylated in sperm, which form the majority of ICRs, and those methylated in sperm but not oocytes. ICRs that are methylated in oocytes all overlap a CpG island promoter for either a protein coding gene or a non-protein coding macro RNA ([39]). In this case silencing is associated with promoter methylation. In loci containing macro RNAs there is another level of complexity since transcription of the macro RNA prevents cis expression of protein encoding genes by recruitment of repressive chromatin, transcriptional interference or by coating the chromosome in cis (reviewed by Koerner [53]). Methylation of the promoter on the maternally inherited allele prevents macro RNA expression and thus allows transcription of neighboring genes.
ICRs that are methylated in sperm are all located in intergenic regions that are not CpG island promoters. The mechanism by which these ICRs control the transcription of neighboring genes is probably not identical in each case, but in several clusters the ICR overlies a binding site for an insulator or enhancer-blocking element. When insulator proteins bind DNA they are able to prevent cis interactions between neighboring regions, such as between promoter and enhancer regions, and thus repress transcription. Methylation of the ICR prevents insulator binding, and thus parental allele-specific gene expression is achieved (reviewed by Ferguson-Smith [39]).
Some imprinted genes exhibit tissue-specific absence of imprinting. Biallelic expression is achieved by a variety of mechanisms such as the utilization of enhancer elements that spatially bypass the imprinting machinery [54], or by selective biallelic methylation of the ICR [55]. In addition some imprinted genes can employ alternative promoters that are methylated in a tissue-specific and parental specific manner to achieve opposite imprinting in different tissues [56,57].
3.5. Phenotypes affected by imprinted genes
Chromosomal domains in the mouse containing imprinted genes were initially identified in the 1980s by screening for phenotypes associated with aberrant chromosome segregation [58]. Many of these phenotypes were associated with embryonic growth, and subsequently it has become clear that imprinted genes encode proteins and non-coding RNAs (ncRNAs) that are critical for the control of placentation, embryonic growth and for perinatal phenotypes associated with growth, suckling and thermogenesis. In addition, modulation of imprinted gene dosage can affect postnatal behavior and metabolism (Reviewed in [59–61]).
In humans, genetic defects affecting imprinted genes lead to severe phenotypes. Conditions such as the Prader-Willi, Angelman, and Beckwith-Wiedemann syndromes are the result of an abnormal dosage of imprinted genes [62–66] and usually present with abnormalities in growth, development, metabolism, behavior and endocrine and neurological functions. Abnormal expression of the imprinted gene GNAS, which is critical for the signal transduction pathway of many hormones, may cause endocrine pathologies including pseudohypoparathyroidism and McCune-Albright syndrome [67–70].
4. The Dlk1-Dio3 imprinted cluster in chromosome 12
4.1. Genes in the cluster
The existence of imprinted genes on mouse chromosome 12 was predicted by the phenotypes of mice carrying Robertsonian or reciprocal translocation in chromosomes resulting in either two paternal chromosomes and no maternal chromosomes or the inverse [71,72]. Subsequently imprinted genes have been mapped to distal chromosome 12, comprising of a cluster that spans approximately 1.5 Mb (Fig. 2A, [73]). A highly conserved imprinting cluster exists on human chromosome 14 [74].
Fig. 2.

The mouse chromosome 12 imprinted cluster. A, The wild type locus. The Dlk1-Dio3 imprinted domain is shown (1.5 Mb, not to scale) on the two parental chromosomes. Following maternal transmission (top) there is expression of a long non-coding RNA (yellow) containing transcripts for Gtl2, Rtl1AS, snoRNAs and Mirg. This transcript arises from a non-methylated Gtl2 promoter (open lollipop), which is in cis to an unmethylated ICR (white circle). On the same chromosome Begain 1b, Dlk1, Rtl1 and Dio3 are not expressed (gray boxes). On the paternally-inherited chromosome (bottom) the ncRNAs are silent (gray boxes) in cis to a methylated Gtl2 promoter and ICR. The protein-encoding Begain 1b, Dlk1, Rtl1 and Dio3 genes are all expressed from this chromosome (green boxes). Dlk1 and Dio3 are driven by unmethylated CpG island promoters (open lollipops at start of genes) on both parental chromosomes, and there is an additional region of methylation in exon 5 of Dlk1 which is hypermethylated on the paternally-inherited chromosome. B, The maternally inherited ICR deletion. When a deletion of the ICR (red star) is transmitted through the female germline there is an epigenotype switch on this chromosome such that it resembles the paternally-inherited chromosome. The gtl2 promoter becomes methylated, the ncRNAs are silenced, and the protein encoding genes are reactivated in cis to the deletion. The paternally-inherited chromosome is unaffected by the deletion. C, Paternally-inherited transgene insertion. When a transgene insertion between the ICR and Gtl2 promoter (red box) is inherited of the paternal chromosome the Gtl2 promoter fails to become methylated in cis. This results in activation of the (normally silenced) ncRNAs and partial silencing of the protein-encoding genes.
The genes within the cluster have a variety of functions; Begain encodes a peptide specific to the nervous system, one variant of which is imprinted (Begain 1b, [75]), the delta-like homologue 1 (Dlk1) encodes a non-canonical Notch ligand which controls maturation in a number of stem cell populations [76]. Rtl1 is a retrotranposon-like gene that is essential for placental morphogenesis [77], and Dio3 marks the telomeric end of the domain (Fig. 2A). Several ncRNA genes are also present, though their functions are less well understood; the Gtl2 transcript has no assigned function to date, however its misexpression has been associated with tumorigenesis [78,79]; Rtl1 antisense (Rtl1AS) encodes microRNAs that target Rtl1 [80], though they may have additional targets, Mirg encodes a primary transcript for a large number of microRNAs [81], and the CD-box containing small nucleolar RNA products (snoRNAs) that are thought to catalyze RNA editing [82].
Four of these genes are expressed from the paternally inherited chromosome and encode proteins (Begain 1b, Dlk1, Rtl1 and Dio3). These genes are silenced on the maternally inherited chromosome, from which there is expression of several ncRNAs, which initiate at the promoter of the Gtl2 gene (Gtl2, Rtl1AS, Mirg and C/D snoRNAs) and form a long polycistronic transcript (Fig. 2A).
4.2. Control of imprinting in the Dlk1-Dio3 cluster
Imprinting of the cluster is controlled by an intergenic ICR (also known as the intergenic differentially methylated region or IG-DMR) which is methylated in sperm, unmethylated in oocytes, and retains paternally-derived methylation in the majority of somatic cells after fertilization (Fig. 2A, [73]). Further regions of differential methylation (DMRs) are found within the Dlk1 gene and at the promoter CpG island of Gtl2, both of which are hyper-methylated on the paternally-inherited chromosome (Fig. 2A) [73]. Deletion of the ICR on the maternally-inherited allele results in an imprinting switch occurring in cis to the deletion; the ncRNAs, which are expressed from the maternal allele in normal conditions, are now silenced, and the protein-encoding genes – normally repressed – are now expressed. This leads to bi-allelic expression of Dio3 (overexpression) and presumably to a decrease in thyroid hormone action. In contrast, paternal inheritance of the ICR deletion has no effect on gene expression, the ncRNAs remain silent and the protein-encoding genes retain expression (Fig. 2B) [83].
It is not currently known how the ICR acts to control gene expression across the entire 1.5 Mb domain, though several models have been proposed [84]. However, it is clear that one important action of the ICR is to direct the methylation status of the Gtl2 promoter. Loss of the ICR on the maternally inherited chromosome causes the Gtl2 promoter to gain methylation, which is associated with silencing of all of the downstream ncRNA transcripts (Fig. 2B, [83]). In addition, disrupting the action of the ICR by insertion of a transgene between it and the Gtl2 promoter results in loss of Gtl2 promoter methylation and expression of Gtl2 and associated transcripts (Fig. 2C, [85,86]. In both cases changes in the activity of the Gtl2 promoter have a reciprocal effect on the expression of the protein-coding genes, including Dio3, suggesting that transcription across this region has a regulatory function. However, it is unclear whether transcription is continuously required in all somatic cells, or if there is a critical developmental window in which transcription imposes a stable epigenetic state on the maternally inherited chromosome. To date there is no evidence that either parental chromosome acquires a domain-wide coating of epigenetic marks associated with repressed chromatin [87], but there is some evidence that the paternally-inherited chromosomal domain may occupy a different subnuclear localization than the maternally-inherited domain [88].
4.3. The Dio3 gene
The Dio3 gene is located at the distal end of the chromosome 12 imprinted region [89] and forms the telomeric boundary of the domain, since the neighboring gene Ppp2a5c is biallelically expressed [75]. The Dio3 mRNA is 2.1 kb in length and transcribed from a single 1.85 kb exon. This is the most abundant transcript in developing tissues and placenta. It includes an in-frame TGA triplet that codes for selenocysteine. However the insertion of this amino acid into the peptide sequence is dependent upon a sequence within the 3′UTR, the selenocysteine insertion sequence (SECIS, Fig. 3). Human DIO3 maps to the syntenic imprinted region 14q3, and is highly conserved. A high degree of nucleotide identity is also noted in the SECIS-containing portion of the 3′UTR and in the overall 3′UTR including the presence of a consensus polyadenylation sequence at the 3′end (Fig. 3) [90]. The promoter for both the human and mouse genes is located in a CpG island [90], which is unmethylated on both parental chromosomes (Fig. 3) [91].
Fig. 3.
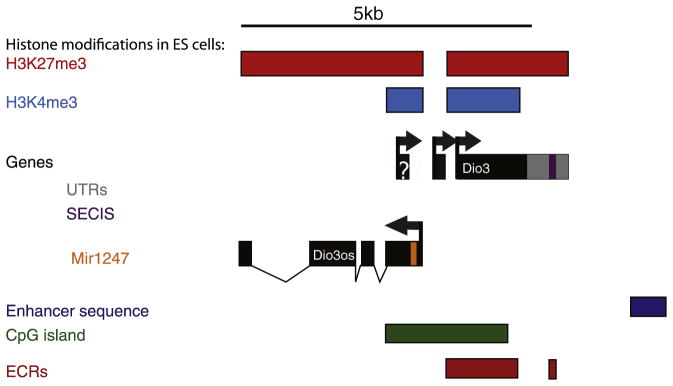
Features associated with the Dio3 gene. The major fetal and placental isoform of Dio3 is transcribed from a single 2.1 kb exon with a long 3′UTR (gray) containing the SECIS sequence (purple). The sequence containing the coding region of the gene and the 3′UTR is highly conserved in vertebrates (red boxes). Other transcripts have been described in the brain that may arise from alternative promoters (?), but have not yet been mapped. Dio3os is transcribed in the opposite direction to Dio3, and gives rise to at least 5 alternatively spliced transcripts. The 5′ end of these reported transcripts all originate within the same CpG island as the Dio3 promoter (green box). Exon 1 of Dio3os contains a conserved microRNA, mir1247 (orange box). Approximately 5 kb telomeric to the Dio3 promoter there is a conserved enhancer that can drive Dio3 and Dio3os expression in vitro (blue box). In ES cells the Dio3 promoter is decorated with modified histones, H3K27me3 (red bar) and H3K4me3 (blue bar), indicative of a ‘poised’ chromatin state.
However, additional, larger Dio3 transcripts (approximately 2.6 and approximately 3.2 kb) have been detected in the adult and hyperthyroid rat brain [92,93]. Furthermore, a similar 3.2-kb D3 transcript is present in various adult human tissues [94]. There is evidence that these larger Dio3 transcripts contain additional 5′-untranslated sequence [95], but their imprinting status is unknown and the promoter(s) responsible for their transcription has not been identified.
A second gene is present at the Dio3 locus, and encodes a spliced non-coding RNA that is transcribed in the opposite orientation, Dio3 opposite strand (Fig. 3, Dio3os). Based on the analysis of Dio3os cDNA clones, Dio3os encodes at least 5 transcripts, and their most 5′ end lies within the CpG island that overlaps the Dio3 promoter [94]. The Dio3os promoter has not been identified. Exon 1 of DIO3OS in humans includes the sequence for the pre-microRNA hsa-mir1247, and the homologous sequence in the mouse encodes mmir1247. This microRNA is expressed in human ES cells and induced ~13× upon their induction to embryoid bodies [96]. Dio3os is not imprinted in the mouse fetus [97] and it is not known whether it plays any mediatory role in regulating the expression or imprinting of Dio3. An enhancer that is highly conserved between mouse and human can activate the Dio3 and Dio3os promoters in vitro [94,98], but its specific role in vivo is unclear.
The Dio3 promoter is regulated by the polycomb group of proteins that mediate tissue-specific gene silencing during development [99,100]. Evidence for this is provided by high throughput assays that measure enrichment of DNA binding factors and histone modifications. In such a study the Dio3 CpG island promoter was found to be enriched for histone H3K27me3 in embryonic stem cells, which is catalyzed by the polycomb 2 complex [99]. The active modification H3K4me3 was also enriched at this region [100]. The juxtaposition of these two histone marks in ES cells is thought to mark promoters that are ‘poised’ for expression. Upon restriction of cell fate the status of the promoter is then resolved as active or repressed depending on cell type. Interestingly, the neighboring Dlk1 gene also has an unmethylated CpG island promoter that is ‘poised’ for transcription in ES cells [100].
In common with the other paternally-expressed protein encoding genes on chromosome 12, Dio3 expression is controlled by the ICR, since maternal inheritance of the ICR deletion results in biallelic expression of Dio3 (Fig. 2B, [83]). The ICR therefore acts over several hundred kilobases to affect the Dio3 promoter. Dio3 is frequently reported to have an imprinting bias since its expression from the maternally-inherited chromosome is not completely silenced [91]. Dio3 imprinting is somewhat relaxed in the placenta [101] and its expression can be biallelic or even maternally-expressed in certain areas of the brain [102]. It is possible that this observation represents a different imprinting status in different tissues or cell types, or they may be due to different allelic activity of alternative Dio3 promoters, given the presence of larger Dio3 transcripts in the brain. Recently, neighboring Dlk1 was shown to have a selective loss of imprinting in the neurogenic niche, and this was associated with biallelic methylation of the ICR [55]. Since Dio3 imprinting is also critically controlled by this element it would be interesting to determine if Dio3 follows the same imprinting pattern as Dlk1 in the context of neurogenesis. It will be also helpful to determine to what extent the methylation status of the ICR is consistent with Dio3 imprinting in those tissues with bi-allelic or maternal expression of Dio3.
5. The implications of Dio3 imprinting for development and disease
5.1. Developmental consequences of altered imprinting in the Dlk1-Dio3 domain
A consequence of genomic imprinting is that it renders its targets functionally haploid. Since only one parental copy is expressed in an individual, any mutations inherited at this locus may not be compensated for by a second allele. In addition, many functions performed by imprinted genes are exquisitely sensitive to gene dosage, such that doubling the level of mRNA expression by loss of imprinting can cause significant phenotypic consequences (reviewed in [59]).
Phenotypes arising from both loss of gene function, and over-expression of dosage-sensitive genes in the Dlk1-Dio3 domain have been observed when imprinting is perturbed as a result of uniparental disomy (UPD) of chromosome 12 [72]. In such models, both copies of chromosome 12 are inherited from the same parent. Mice with paternal UPD12 (associated with loss of ncRNA expression and double expression of Begain 1b, Dlk1, Rtl1 and Dio3) manifest placentomegaly and die late in gestation, while those with maternal UPD12 (which do not express the protein-encoding genes including Dio3, but overexpress the ncRNAs) are growth retarded and die perinatally. Skeletal defects are also noticed in mice with either type of parental disomy [72].
Mice with deletion of the ICR in the maternal allele also exhibit perinatal lethality as a consequence of overexpression of the paternally-expressed genes [83]. Although it is not possible to discern the contribution of particular genes to those phenotypes, the results indicate that appropriate genomic imprinting in the Dlk1-Dio3 domain is necessary for normal viability, growth and development.
5.2. Pathological implications for humans
To date, no direct mutations on the D3 gene with functional consequences have been reported in humans. Relevant evidence for the potential role of DIO3 and the DLK1-DIO3 imprinted domain in humans comes from a number of patients with UPD of chromosome 14 [103–105]. As the human homolog of the Dlk1-Dio3 domain is located in the distal arm of chromosome 14 [89], these patients’ symptoms are presumably the consequence of misexpression of the imprinted genes in the domain. Chromosomal UPD in humans is relatively rare, and usually the result of aberrant chromosomal translocations, or the resolution of chromosomal trisomies very early in development, as evidenced in some cases by placental mosaicism of cells with chromosomal trisomy [106]. A number of common features have been shown in these patients. Maternal UPD14 is associated with growth retardation, hypotonia, small hands and feet, neonatal feeding problems, facial dysmorphisms, hydrocephalus, early puberty, mild mental retardation and slightly increased adiposity [103,107,108]. Patients with paternal UPD14 exhibit severe mental retardation, skeletal abnormalities (“coat-hanger” ribs) and facial dysmorphisms [105,109–112]. As no other imprinted genes have been described on this human chromosome, these observations indicate that aberrant expression of imprinted genes in the DLK1-DIO3 domain is likely responsible for the abnormalities associated with these conditions. This is further supported by reports of deletions and epimutations in the domain in association with characteristics typical of UPD14 [113–115], and by the recapitulation of UPD14 phenotypes in patients in which the UPD affects only a chromosomal segment in which the imprinted domain is located [116].
Aberrant expression of DLK1, DIO3 and other genes in the imprinted domain has been associated with some cellular malignancies in humans, suggesting that the cluster may also play a role in cellular transformation [95,117–119].
5.3. The contribution of Dio3 to phenotypes of abnormal imprinting
The phenotype of the D3KO mouse is very rich and surprisingly insightful about the developmental roles of Dio3, but it is the consequence of a constitutive, extremely low Dio3 dosage. However, the degree of Dio3 expression from the paternal allele may vary in some tissues, like placenta and brain. Thus, although in general Dio3 is preferentially expressed from the paternal allele, most tissues still exhibit significant residual expression from the maternal copy of the gene. In this context, it is likely that certain tissues, and the phenotypes associated with them, will be more sensitive than others to abnormal imprinting of Dio3.
5.4. Similarities between D3KO and maternal UPD phenotypes
All imprinted genes in the Dlk1-Dio3 domain are likely contributing to the UPD syndromes. However, we can estimate the individual contribution of Dio3 by comparing the abnormalities of the D3KO mouse to those observed in mouse and human maternal UPD12 and 14, respectively. The perinatal lethality and growth retardation of maternal UPD12 mice are also observed in D3KO mice. In the latter, neonatal lethality is partial (30–70%) in most genetic backgrounds [29], but it is close to 100% in commonly used strains such as C57Bl/6, suggesting that abnormal Dio3 expression is partly responsible for this outcome. Mice lacking the ICR on the maternal allele also exhibit perinatal lethality [83]. This phenotype is not rescued by normalization of the Dlk1 or Rtl1 expression [120], suggesting that aberrant expression of other imprinted genes in the domain, including Dio3, are contributing to this phenotype.
In humans, several genes of the DLK1-DIO3 domain are imprinted and exhibit similar parent-of-origin allelic expression as the mouse homologs [74], but no information about the imprinting status of the human DIO3 has been reported. A preliminary observation in tissue from adult skin indicates that human DIO3 is imprinted [121]. If this is the case and human DIO3 exhibits a similar tissue and developmental imprinting pattern as that in the mouse, one would expect that patients with maternal UPD14 are D3-deficient and should exhibit characteristics similar to that in D3KO mice. Published and unpublished observations reveal that some maternal UPD14 phenotypic features are also present in D3KO mice (Fig. 1C), including growth retardation [29], hydrocephalus, facial dysmorphisms and age of puberty onset. These similarities suggest that deficient D3 expression could be partly responsible for the phenotypes associated with abnormal imprinting of the Dlk1-Dio3 domain in mouse and humans.
To our knowledge, no data on the thyroid status of UPD14 patients have been published. In three case reports of maternal UPD14 [122–124], the authors state that endocrine and thyroid function tests were performed in the patients with normal results, although no specifics about the exact parameters determined are mentioned. If thyroid hormone abnormalities exist in these individuals and are similar to those in D3KO mice, they are unlikely to be revealed by normal tests, as thyroid hormone parameters in humans that correspond to those in D3KO mice will mostly be within the range of normal clinical values. The identification of a potential HPT axis phenotype in maternal UPD14 subjects may require a physiological challenge.
5.5. Dio3 imprinting from an evolutionary perspective
The reason why genomic imprinting occurs in the first place is still controversial. At first glance, genomic imprinting does not make sense from an evolutionary perspective, since the repression of one copy of a given gene leaves an organism more susceptible to the deleterious effects of genetic mutations in the copy that is normally expressed.
Based on available data, a number of theories have been postulated on the subject [125–127], and they are continually tested and modified by new research. Genomic imprinting evolved in a period of rapid speciation during mammalian divergence and, among the animal kingdom, it is essentially limited to mammals [128]. Interestingly, imprinted gene functions appear to converge on mammalian-specific traits such as invasive placentation, suckling and postnatal adaptations to independent life [59,61,129]. Since these processes are also concerned with the allocation of resources from the mother to offspring, it has been postulated that imprinting may have evolved as a result of a conflict between maternal and paternal genomes [126]. This ‘parental conflict’ theory postulates that in species with multiple paternities, offspring are unequally related to one another. Thus, genes from the father should act to maximize their resource usage, whereas genes from the mother would seek the survival of all her offspring, regardless of paternity, without compromising her own future reproductive function. Consequently paternally expressed imprinted genes are predicted to promote growth and resource allocation, and maternally-expressed genes to restrain it. While this theory fits well for some imprinted genes [126], the phenotypes of some imprinted gene deletions are consistent with a role for imprinting in the co-adaptation of mother and fetus [130].
Environmental factors, potentially driving natural selection and adaptation, are also speculated to influence genomic imprinting. Given the increasing evidence of epigenetic effects exerted by environmental factors, the unique regulatory mechanisms governing the expression of imprinted genes could be particularly susceptible to the environment [131]. In this regard, as iodine is a critical component of thyroid hormones, there is the intriguing possibility that, as mammals colonized other iodine-poor, land-based ecosystems, a deficiency in this element could have played a role in the genomic imprinting of Dio3. This would have reduced Dio3 expression and increased the efficiency of thyroid hormones. Advances in our understanding of the evolutionary reasons of genomic imprinting may provide new insights into the physiological roles of Dio3, and vice versa.
Comparative genomic analysis indicates that Dlk1 and Dio3 are the ancestral genes in the chromosome 12 cluster, and they display tight linkage and strong conservation in all vertebrates. The two genes are approximately 10 kb apart in the fish Takifugu rubripes and 370 kb apart in chicken, compared to ~800 kb apart in the human and mouse [132]. The linkage of the genes precedes the evolution of imprinting of the locus, which occurred after the divergence of the marsupial and eutherian mammals, and suggests that both genes may share important physiological roles. The acquisition of imprinting at the cluster is coincident with transcription of the ncRNAs and maintenance of an active Rtl1 gene [132].
5.6. Transgenerational epigenetic effects of thyroid hormones
More than forty years ago, several investigators used a rat model to analyze the effects of excessive exposure to thyroid hormones during development [133,134]. Rat neonates were rendered thyrotoxic by the injection of pharmacological doses of T4 for a few days after birth. As adults, these animals showed growth and HPT axis phenotypes that, although less severe, strikingly resemble those in D3KO mice [135]. Additional studies showed that the untreated offspring and descendants of those animals, as well as that of animals subjected to other thyroid hormone manipulations, exhibited modest but significant abnormalities [136,137]. As these effects could be transmitted for two more generations through the paternal line, they indicate that the degree of exposure to thyroid hormones can produce transgenerational epigenetic effects in the descendants. This raises the possibility that alterations in the genomic imprinting of Dio3, and thus in the level of exposure to thyroid hormone, may do likewise.
6. Summary
Many questions remain and much knowledge is yet to be gained on the topics covered here, but the data available is sufficiently indicative of the importance of the mechanisms regulating the genomic imprinting of Dio3 and associated genes may have for proper development, normal biological functions in adulthood and adequate responses to physiological challenges. It is possible that the epigenetic dysregulation of Dio3 may not be of appreciable consequence for certain tissues or biological processes. However, given the broad physiological influence of D3, it is reasonable to expect that important aspects of development and physiology will be markedly affected by an altered dosage of Dio3. It is also likely that epigenetic abnormalities in the regulation of DIO3 will be relevant to human conditions.
Acknowledgments
FP7 Epigenesis (M.C.) and the National Institute of Mental Health (MH083220, A.H.) supported this work.
Footnotes
This article is part of a Special Issue entitled Thyroid hormone signalling.
References
- 1.Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev. 2002;23:38–89. doi: 10.1210/edrv.23.1.0455. [DOI] [PubMed] [Google Scholar]
- 2.Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, Zeold A, Bianco AC. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling [Review] [446 refs] Endocr Rev. 2008;29:898–938. doi: 10.1210/er.2008-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hernandez A. Structure and function of the type 3 deiodinase gene. Thyroid. 2005;15:865–874. doi: 10.1089/thy.2005.15.865. [DOI] [PubMed] [Google Scholar]
- 4.Tsai CE, Lin SP, Ito M, Takagi N, TS, Ferguson-Smith AC. Genomic imprinting contributes to thyroid hormone metabolism in the mouse embryo. Curr Biol. 2002;12:1221–1226. doi: 10.1016/s0960-9822(02)00951-x. [DOI] [PubMed] [Google Scholar]
- 5.Yevtodiyenko A, Carr MS, Patel N, Schmidt JV. Analysis of candidate imprinted genes linked to Dlk1-Gtl2 using a congenic mouse line. Mamm Genome. 2002 Nov;13(11):633–638. doi: 10.1007/s00335-002-2208-1. [DOI] [PubMed] [Google Scholar]
- 6.Hernandez A, Fiering S, Martinez E, Galton VA, StGermain DL. The gene locus encoding the iodothyronine deiodinase type 3 (Dio3) is imprinted in the fetus and expresses antisense transcripts. Endocrinology. 2002;143:4483–4486. doi: 10.1210/en.2002-220800. [DOI] [PubMed] [Google Scholar]
- 7.Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- 8.Huang T, Chopra IJ, Boado R, Solomon DH, Chua Teco GN. Thyroxine inner ring monodeiodinating activity in fetal tissues of the rat. Pediatr Res. 1988;23:196–199. doi: 10.1203/00006450-198802000-00014. [DOI] [PubMed] [Google Scholar]
- 9.Huang T, Beredo A, Solomon DH, Chopra IJ. The inner ring (5-) mono-deiodination of thyroxine (T4) in cerebral cortex during fetal, neonatal, and adult life. Metabolism. 1986;35:272–277. doi: 10.1016/0026-0495(86)90213-1. [DOI] [PubMed] [Google Scholar]
- 10.Escobar-Morreale HF, Obregon MJ, Hernandez A, Escobar del Rey F, Morreale de Escobar G. Regulation of iodothyronine deiodinase activity as studied in thyroidectomized rats infused with thyroxine or triiodothyronine. Endocrinology. 1997;138:2559–2568. doi: 10.1210/endo.138.6.5212. [DOI] [PubMed] [Google Scholar]
- 11.Kaplan MM, Yaskoski KA. Phenolic and tyrosyl ring deiodination of iodothyronines in rat brain homogenates. J Clin Invest. 1980;66:551–562. doi: 10.1172/JCI109887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang T, Chopra IJ, Beredo A, Solomon DH, Chua Teco GN. Skin is an active site of inner ring monodeiodination of thyroxine to 3,3′,5′-triiodothyronine. Endocrinology. 1985;117:2106–2113. doi: 10.1210/endo-117-5-2106. [DOI] [PubMed] [Google Scholar]
- 13.Bates JM, StGermain DL, Galton VA. Expression profiles of the three iodothyronine deiodinases, D1, D2 and D3, in the developing rat. Endocrinology. 1999;140:844–851. doi: 10.1210/endo.140.2.6537. [DOI] [PubMed] [Google Scholar]
- 14.Galton VA, Martinez E, Hernandez A, StGermain EA, Bates JM, StGermain DL. Pregnant rat uterus expresses high levels of the type 3 iodothyronine deiodinase. J Clin Invest. 1999;103:979–987. doi: 10.1172/JCI6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang SA, Dorfman DM, Genest DR, Salvatore D, Larsen PR. Type 3 iodothyronine deiodinase is highly expressed in the human uteroplacental unit and in fetal epithelium. J Clin Endocrinol Metab. 2003;88:1384–1388. doi: 10.1210/jc.2002-021291. [DOI] [PubMed] [Google Scholar]
- 16.Salvatore D, Larsen PR. Role of deiodinases in tissue (de)differentiation. in (this issue) [Google Scholar]
- 17.Huang SA, Bianco AC. Reawakened interest in type III iodothyronine deiodinase in critical illness and injury [Review] [67 refs] Nat Clin Pract Endocrinol Metab. 2008;4:148–155. doi: 10.1038/ncpendmet0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peeters RP, Wouters PJ, Kaptein E, van Toor H, Visser TJ, Van den Berghe G. Reduced activation and increased inactivation of thyroid hormone in tissues of critically ill patients. J Clin Endocrinol Metab. 2003;88:3202–3211. doi: 10.1210/jc.2002-022013. [DOI] [PubMed] [Google Scholar]
- 19.Peeters RP, Wouters PJ, van Toor H, Kaptein E, Visser TJ, Van den Berghe G. Serum 3,3′,5′-triiodothyronine (rT3) and 3,5,3′-triiodothyronine/rT3 are prognostic markers in critically ill patients and are associated with postmortem tissue deiodinase activities. J Clin Endocrinol Metab. 2005;90:4559–4565. doi: 10.1210/jc.2005-0535. [DOI] [PubMed] [Google Scholar]
- 20.Debaveye Y, Ellger B, Mebis L, Visser TJ, Darras VM, Van den Berghe G. Effects of substitution and high-dose thyroid hormone therapy on deiodination, sulfoconjugation, and tissue thyroid hormone levels in prolonged critically ill rabbits. Endocrinology. 2008 Aug;149(8):4218–4228. doi: 10.1210/en.2007-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Darras VM, Cokelaere M, Dewil E, Arnouts S, Decuypere E, Kuhn ER. Partial food restriction increases hepatic inner ring deiodinating activity in the chicken and the rat. Gen Comp Endocrinol. 1995;100:334–338. doi: 10.1006/gcen.1995.1164. [DOI] [PubMed] [Google Scholar]
- 22.Van der Geyten S, Van Rompaey E, Sanders JP, Visser TJ, Kuhn ER, Darras VM. Regulation of thyroid hormone metabolism during fasting and refeeding in chicken. Gen Comp Endocrinol. 1999;116:272–280. doi: 10.1006/gcen.1999.7368. [DOI] [PubMed] [Google Scholar]
- 23.Chopra IJIe. Mechanisms of altered serum thyroid hormones in nonthyroidal illness. In: Wu SY, editor. Thyroid Hormone Metabolism. Blackwell Scientific Publications; Oxford: 1991. pp. 195–210. [Google Scholar]
- 24.Wassen FW, Schiel AE, Kuiper GG, Kaptein E, Bakker O, Visser TJ, Simonides WS. Induction of thyroid hormone-degrading deiodinase in cardiac hypertrophy and failure. Endocrinology. 2002;143:2812–2815. doi: 10.1210/endo.143.7.8985. [DOI] [PubMed] [Google Scholar]
- 25.Olivares EL, Marassi MP, Fortunato RS, da Silva AC, Costa-e-Sousa RH, Araujo IG, Mattos EC, Masuda MO, Mulcahey MA, Huang SA, Bianco AC, Carvalho DP. Thyroid function disturbance and type 3 iodothyronine deiodinase induction after myocardial infarction in rats a time course study. Endocrinology. 2007 Oct;148(10):4786–4792. doi: 10.1210/en.2007-0043. [DOI] [PubMed] [Google Scholar]
- 26.Boelen A, Boorsma J, Kwakkel J, Wieland CW, Renckens R, Visser TJ, Fliers E, Wiersinga WM. Type 3 deiodinase is highly expressed in infiltrating neutrophilic granulocytes in response to acute bacterial infection. Thyroid. 2008;18:1095–1103. doi: 10.1089/thy.2008.0090. [DOI] [PubMed] [Google Scholar]
- 27.Li WW, Le Goascogne C, Ramauge M, Schumacher M, Pierre M, Courtin F. Induction of type 3 iodothyronine deiodinase by nerve injury in the rat peripheral nervous system. Endocrinology. 2001;142:5190–5197. doi: 10.1210/endo.142.12.8532. [DOI] [PubMed] [Google Scholar]
- 28.Boelen A, Kwakkel J, Alkemade A, Renckens R, Kaptein E, Kuiper G, Wiersinga WM, Visser TJ. Induction of type 3 deiodinase activity in inflammatory cells of mice with chronic local inflammation. Endocrinology. 2005;146:5128–5134. doi: 10.1210/en.2005-0608. [DOI] [PubMed] [Google Scholar]
- 29.Hernandez A, Martinez ME, Fiering S, Galton VA, St Germain D. Type 3 deiodinase is critical for the maturation and function of the thyroid axis. J Clin Invest. 2006;116:476–484. doi: 10.1172/JCI26240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hernandez A, Martinez E, Liao X, Van Sande J, Refetoff S, Galton VA, St Germain D. Type 3 deiodinase deficiency results in functional abnormalities at multiple levels of the thyroid axis. Endocrinology. 2007;148:5680–5687. doi: 10.1210/en.2007-0652. [DOI] [PubMed] [Google Scholar]
- 31.Boelen A, Kwakkel J, Wieland CW, St Germain D, Fliers E, Hernandez A. Impaired bacterial clearance in type 3 deiodinase deficient mice infected with Streptococcus pneumoniae. Endocrinology. 2009;150:1984–1990. doi: 10.1210/en.2008-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ng L, Hernandez A, He W, Ren T, Srinivas M, Ma M, Galton VA, St Germain DL, Forrest D. A protective role for type 3 deiodinase, a thyroid hormone-inactivating enzyme, in cochlear development and auditory function. Endocrinology. 2009;150:1952–1960. doi: 10.1210/en.2008-1419. [Erratum appears in Endocrinology. 2009 May;150(5):2499] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ng L, Lyubarsky A, Nikonov SS, Ma M, Srinivas M, Kefas B, St Germain DL, Hernandez A, Pugh EN, Jr, Forrest D. Type 3 deiodinase, a thyroid-hormone-inactivating enzyme, controls survival and maturation of cone photoreceptors. J Neurosci. 2010;30:3347–3357. doi: 10.1523/JNEUROSCI.5267-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Medina MC, Molina J, Gadea Y, Fachado A, Murillo M, Simovic G, Pileggi A, Hernandez A, Edlund H, Bianco AC. The thyroid hormone-inactivating type III deiodinase is expressed in mouse and human beta-cells and its targeted inactivation impairs insulin secretion. Endocrinology. 2011;152:3717–3727. doi: 10.1210/en.2011-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hernandez A, Quignodon L, Martinez ME, Flamant F, StGermain DL. Type 3 deiodinase deficiency causes spatial and temporal alterations in brain T3 signaling that are dissociated from serum thyroid hormone levels. Endocrinology. 2010;151:5550–5558. doi: 10.1210/en.2010-0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Surani MA, Barton SC, Norris ML. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature. 1984;308:548–550. doi: 10.1038/308548a0. [DOI] [PubMed] [Google Scholar]
- 37.McGrath J, Solter D. Completion of mouse embryogenesis requires both the maternal and the paternal genome. Cell. 1984;137:179–183. doi: 10.1016/0092-8674(84)90313-1. [DOI] [PubMed] [Google Scholar]
- 38.Williamson CM, Blake A, Beechey CV, Hancock J, Cattanach BM, Peters J. Mouse Imprinting Data and References. MRC Harwell; Oxforshire: 2012. http://www.har.mrc.ac.uk/research/genomic_imprinting/ [Google Scholar]
- 39.Ferguson-Smith AC. Genomic imprinting: the emergence of an epigentic paradigm. Nat Rev Genet. 2011:8. doi: 10.1038/nrg3032. [DOI] [PubMed] [Google Scholar]
- 40.McEwen KR, Ferguson-Smith AC. Distinguishing epigenetic marks of developmental and imprinting regulation. Epigenetics Chromatin. 2010;3:2. doi: 10.1186/1756-8935-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- 42.Bourchis D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–2539. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- 43.Bourchis D, Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431:96–99. doi: 10.1038/nature02886. [DOI] [PubMed] [Google Scholar]
- 44.Watanabe T, Tomizawa S, Mitsuya K, Totoki Y, Yamamoto Y, Kuramochi-Miyagawa S, Iida N, Hoki Y, Murphy PJ, Toyoda A, Gotoh K, Hiura H, Arima T, Fujiyama A, Sado T, Shibata T, Nakano T, Lin H, Ichiyanagi K, Soloway PD, Sasaki H. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science. 2011;332:848–852. doi: 10.1126/science.1203919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glass JL, Fazzari MJ, Ferguson-Smith AC, Greally JM. CG dinucleotide periodicities recognized by the Dnmt3a-Dnmt3L complex are distinctive at retroelements and imprinted domains. Mamm Genome. 2009;20:633–643. doi: 10.1007/s00335-009-9232-3. [DOI] [PubMed] [Google Scholar]
- 46.Smallwood SA, Kelsey G. De novo DNA methylation: a germ cell perspective. Trends Genet. 2012;28:33–42. doi: 10.1016/j.tig.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 47.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 48.Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, Ferguson-Smith AC. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell. 2008;15:547–557. doi: 10.1016/j.devcel.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamura T, Arai Y, Umehara H, Matsuhara M, Kimura T, Taniguchi H, Sekimoto T, Ikawa M, Yoneda Y, Okabe M, Tanaka S, Shiota K, Nakano T. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat Cell Biol. 2007;9:64–71. doi: 10.1038/ncb1519. [DOI] [PubMed] [Google Scholar]
- 50.Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, Baglivo I, Pedone PV, Grimaldi G, Riccio A, Trono D. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell. 2011;44:361–372. doi: 10.1016/j.molcel.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zuo X, Sheng J, Lau HT, McDonald CM, Andrade M, Cullen DE, Bell FT, Iacovino M, Kyba M, Xu G, Li X. The zinc finger protein ZFP57 requires its cofactor to recruit DNA methyltransferases and maintains the DNA methylation imprint in embryonic stem cells via its transcriptional repression domain. J Biol Chem. 2011 doi: 10.1074/jbc.M111.322644. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, Dayanikli P, Firth HV, Goodship JA, Haemers AP, Hahnemann JM, Kordonouri O, Masoud AF, Oestergaard E, Storr J, Ellard S, Hattersley AT, Robinson DO, Temple IK. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40:949–951. doi: 10.1038/ng.187. [DOI] [PubMed] [Google Scholar]
- 53.Koerner MW, Pauler FM, Huang R, Barlow DP. The function of non-coding RNAs in genomic imprinting. Development. 2009;136:1771–1783. doi: 10.1242/dev.030403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Charalambous M, Menheniott TR, Bennett WR, Kelly SM, Dell G, Dandolo L, Ward A. An enhancer element at the Igf2/H19 locus drives gene expression in both imprinted and non-imprinted tissues. Dev Biol. 2004;271:488–497. doi: 10.1016/j.ydbio.2004.04.022. [DOI] [PubMed] [Google Scholar]
- 55.Ferron SR, Charalambous M, Radford E, McEwen K, Wildner H, Hind E, Morante-Redolat JM, Laborda J, Guillemot F, Bauer SR, Farinas I, ferguson-Smith AC. Postnatal loss of Dlk1 imprinting in stem cells and niche astrocytes regulates neurogenesis. Nature. 2011;475:381–385. doi: 10.1038/nature10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arnaud P, Monk D, Hitchins M, Gordon E, Dean W, Beechey CV, Peters J, Craigen W, Preece M, Stanier P, Moore GE, Kelsey G. Conserved methylation imprints in the human and mouse GRB10 genes with divergent allelic expression suggests differential reading of the same mark. Hum Mol Genet. 2003;12:1005–1019. doi: 10.1093/hmg/ddg110. [DOI] [PubMed] [Google Scholar]
- 57.Peters J, Williamson CM. Control of imprinting at the Gnas cluster. Epigenetics. 2007;2:207–213. doi: 10.4161/epi.2.4.5380. [DOI] [PubMed] [Google Scholar]
- 58.Cattanach BM, Beechey CV. Autosomal and X-chromosome imprinting [Review] [60 refs] Dev Suppl. 1990:63–72. [PubMed] [Google Scholar]
- 59.Charalambous M, da Rocha ST, Ferguson-Smith AC. Genomic imprinting, growth control and the allocation of nutritional resources: consequences for postnatal life. Curr Opin Endocrinol Diabetes Obes. 2007;14:3–12. doi: 10.1097/MED.0b013e328013daa2. [DOI] [PubMed] [Google Scholar]
- 60.Curley JP. Is there a genomically imprinted social brain? Bioessays. 2011;33:662–668. doi: 10.1002/bies.201100060. [DOI] [PubMed] [Google Scholar]
- 61.Frontera M, Dickins B, Plagge A, Kelsey G. Imprinted genes, postnatal adaptations and enduring effects on energy homeostasis [Review] [138 refs] Adv Exp Med Biol. 2008;626:41–61. doi: 10.1007/978-0-387-77576-0_4. [DOI] [PubMed] [Google Scholar]
- 62.Knoll JH, Nicholls RD, Magenis RE, Graham JMJ, Lalande M, Latt SA. Angelman and Prader-Willi syndromes share a common chromosome 15 deletion but differ in the parental origin of the deletion. Am J Med Genet. 1989;32:285–290. doi: 10.1002/ajmg.1320320235. [DOI] [PubMed] [Google Scholar]
- 63.Shemer R, Hershko AY, Perk J, Mostoslavsky R, Tsuberi B, Cedar H, Buiting K, Razin A. The imprinting box of the Prader-Willi/Angelman syndrome domain. Nat Genet. 2000;26:440–443. doi: 10.1038/82571. [DOI] [PubMed] [Google Scholar]
- 64.Hatada I, Ohashi H, Fukushima Y, Kaneko Y, Inoue M, Komoto Y, Okada A, Ohishi S, Nabetani A, Morisaki H, Nakayama M, Niikawa N, Mukai T. An imprinted gene p57KIP2 is mutated in Beckwith–Wiedemann syndrome. Nat Genet. 1996;14:171–173. doi: 10.1038/ng1096-171. [DOI] [PubMed] [Google Scholar]
- 65.Lee MP, Hu RJ, Johnson LA, Feinberg AP. Human KVLQT1 gene shows tissue-specific imprinting and encompasses Beckwith–Wiedemann syndrome chromosomal rearrangements. Nat Genet. 1997;15:181–185. doi: 10.1038/ng0297-181. [DOI] [PubMed] [Google Scholar]
- 66.Maher ER, Reik W. Beckwith–Wiedemann syndrome: imprinting in clusters revisited. J Clin Invest. 2000;105:247–252. doi: 10.1172/JCI9340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rao VV, Schnittger S, Hansmann I. G protein Gs alpha (GNAS 1), the probable candidate gene for Albright hereditary osteodystrophy, is assigned to human chromosome 20q12-q13.2. Genomics. 1991;10:257–261. doi: 10.1016/0888-7543(91)90508-c. [DOI] [PubMed] [Google Scholar]
- 68.Mantovani G, Bondioni S, Lania AG, Corbetta S, de Sanctis L, Cappa M, Di Battista E, Chanson P, Beck-Peccoz P, Spada A. Parental origin of Gsalpha mutations in the McCune–Albright syndrome and in isolated endocrine tumors. J Clin Endocrinol Metab. 2004;89:3007–3009. doi: 10.1210/jc.2004-0194. [DOI] [PubMed] [Google Scholar]
- 69.Juppner H, Schipani E, Bastepe M, Cole DE, Lawson ML, Mannstadt M, Hendy GN, Plotkin H, Koshiyama H, Koh T, Crawford JD, Olsen BR, Vikkula M. The gene responsible for pseudohypoparathyroidism type Ib is paternally imprinted and maps in four unrelated kindreds to chromosome 20q13.3. Proc Natl Acad Sci U S A. 1998;95:11798–11803. doi: 10.1073/pnas.95.20.11798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu J, Litman D, Rosenberg MJ, Yu S, Biesecker LG, Weinstein LS. A GNAS1 imprinting defect in pseudohypoparathyroidism type IB. J Clin Invest. 2000;106:1167–1174. doi: 10.1172/JCI10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cattanach BM, Rasberry C. Evidence of imprinting involving the distal region of Ch12. Mouse Genome. 1993;91:858. [Google Scholar]
- 72.Georgiades P, Wadkins M, Surani MA, Ferguson-Smith AC. Parental origin-specific developmental defects in mice with uniparental disome of chromosome 12. Development. 2000;127:4719–4728. doi: 10.1242/dev.127.21.4719. [DOI] [PubMed] [Google Scholar]
- 73.Takada S, Tevendale M, Baker J, Georgiades P, Campbell E, Freeman T, Johnson MH, Paulsen M, Ferguson-Smith AC. Delta-like and gtl2 are reciprocally expressed, differentially methylated linked imprinted genes on mouse chromosome 12. Curr Biol. 2000;10:1135–1138. doi: 10.1016/s0960-9822(00)00704-1. [DOI] [PubMed] [Google Scholar]
- 74.Wylie AA, Murphy SK, Orton TC, Jirtle RL. Novel imprinted DLK1/GTL2 domain on human chromosome 14 contains motifs that mimic those implicated in IGF2/H19 regulation. Genome Res. 2000;10:1711–1718. doi: 10.1101/gr.161600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tierling S, Gasparoni G, Youngson N, Paulsen M. The Begain gene marks the centromeric boundary of the imprinted region on mouse chromosome 12. Mamm Genome. 2009;20:699–710. doi: 10.1007/s00335-009-9205-6. [DOI] [PubMed] [Google Scholar]
- 76.Sul HK. Pref1: role in adipogenesis and mesenchymal cell fate. Mol Endocrinol. 2009;23:1717–1725. doi: 10.1210/me.2009-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sekita Y, Wagatsuma H, Nakamura K, Ono R, Kagami M, Wakisaka N, Hino T, Suzuki-Migishima R, Ogura A, Ogata T, Yokoyama M, Kaneko-Ishino T, Ishino F. Role of retrotransposon-derived imprinted gene, Rtl1, in the feto-maternal interface of mouse placenta. Nat Genet. 2008;40:243–248. doi: 10.1038/ng.2007.51. [DOI] [PubMed] [Google Scholar]
- 78.Braconi C, Kogure T, Valeri N, Huang N, Nuovo G, Costinean S, Negrini M, Miotto E, Croce CM, Patel T. MicroRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Oncogene. 2011;30:4750–4756. doi: 10.1038/onc.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Benetatos L, Vartholomatos G, Hatzimichael E. MEG3 imprinted gene contribution in tumorigenesis. Int J Cancer. 2011;129:773–779. doi: 10.1002/ijc.26052. [DOI] [PubMed] [Google Scholar]
- 80.Seitz H, Youngson N, Lin SP, Dalbert S, Paulsen M, Bachellerie JP, Ferguson-Smith AC, Cavaille J. Imprinted microRNA genes transcribed antisense to a reciprocally imprinted retrotransposon-like gene. Nat Genet. 2003;34:261–262. doi: 10.1038/ng1171. [DOI] [PubMed] [Google Scholar]
- 81.Seitz H, Royo H, Bortolin ML, Lin SP, Ferguson-Smith AC, Cavaille J. A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 2004;14:1741–1748. doi: 10.1101/gr.2743304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cavaille J, Seitz H, Paulsen M, Ferguson-Smith AC, Bachellerie JP. Identification of tandemly-repeated C/D snoRNA genes at the imprinted human 14q32 domain reminiscent of those at the Prader-Willi/Angelman syndrome region. Hum Mol Genet. 2002;11:1527–1538. doi: 10.1093/hmg/11.13.1527. [DOI] [PubMed] [Google Scholar]
- 83.Lin SP, Youngson N, Takada S, Seitz H, Reik W, Paulsen M, Cavaille J, Ferguson-Smith AC. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12 [See comment] Nat Genet. 2003;35:97–102. doi: 10.1038/ng1233. [DOI] [PubMed] [Google Scholar]
- 84.da Rocha ST, Edwards CA, Ito M, Ogata T, Ferguson-Smith AC. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008;24:306–316. doi: 10.1016/j.tig.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 85.Steshina EY, Carr MC, Glick EA, Yevtodiyenko A, Appelbe OK, Schmidt JV. Loss of imprinting at the Dlk1-Gtl2 locus caused by insertional mutagenesis in the Gtl2 5′region. BMC Genet. 2006;7:44. doi: 10.1186/1471-2156-7-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sekita Y, Wagatsuma H, Irie M, Kobayashi S, Kohda T, Matsuda J, Yokoyama M, Ogura A, Schuster-Gossler K, Gossler A, Ishino F, Kaneko-Ishino T. Aberrant regulation of imprinted gene expression in Gtl2lacZ mice. Cytogenet Genome Res. 2006;113:223–229. doi: 10.1159/000090836. [DOI] [PubMed] [Google Scholar]
- 87.MIkkelsen TS, Ku M, Jaffe DB, Isaac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Menderhall E, O’Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. Genome-wide maps of chormatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Braem C, Recolin B, Rancourt RC, Angiolini C, Barthes P, Branchu P, Court F, Cathala G, Ferguson-Smith AC, Forne T. Genomic matrix attachment region and chromosome conformation capture quantitative real time PCR assays identify novel putative regulatory elements at the imprinted Dlk1/Gtl2 locus. J Biol Chem. 2008;283:18612–18620. doi: 10.1074/jbc.M801883200. [DOI] [PubMed] [Google Scholar]
- 89.Hernández A, Park J, Lyon GJ, Mohandas TK, St DL. Germain, localization of the type 3 iodothyronine deiodinase (DIO3) gene to human chromosome 14q32 and mouse chromosome 12F1. Genomics. 1998;53:119–121. doi: 10.1006/geno.1998.5505. [DOI] [PubMed] [Google Scholar]
- 90.Hernández A, Lyon GJ, Schneider MJ, StGermain DL. Isolation and characterization of the mouse gene for the type 3 iodothyronine deiodinase. Endocrinology. 1999;140:124–130. doi: 10.1210/endo.140.1.6423. [DOI] [PubMed] [Google Scholar]
- 91.Tsai CE, Lin SP, Ito M, Takagi N, Takada S, Ferguson-Smith AC. Genomic imprinting contributes to thyroid hormone metabolism in the mouse embryo. Curr Biol. 2002;12:1221–1226. doi: 10.1016/s0960-9822(02)00951-x. [DOI] [PubMed] [Google Scholar]
- 92.Croteau W, Whittemore SL, Schneider MJ, St Germain DL. Cloning and expression of a cDNA for a mammalian type III iodothyronine deiodinase. J Biol Chem. 1995;270:16569–16575. doi: 10.1074/jbc.270.28.16569. [DOI] [PubMed] [Google Scholar]
- 93.Tu HM, Legradi G, Bartha T, Salvatore D, Lechan R, Larsen PR. Regional expression of the type 3 iodothyronine deiodinase messenger ribonucleic acid in the rat central nervous system and its regulation by thyroid hormone. Endocrinology. 1999;140:784–790. doi: 10.1210/endo.140.2.6486. [DOI] [PubMed] [Google Scholar]
- 94.Hernandez A, Martinez E, Croteau W, St Germain D. Complex organization and structure of sense and antisense transcripts expressed from the DIO3 gene imprinted locus. Genomics. 2004;83:413–424. doi: 10.1016/j.ygeno.2003.08.024. [DOI] [PubMed] [Google Scholar]
- 95.Dentice M, Luongo C, Huang S, Ambrosio R, Elefante A, Mirebeau-Prunier D, Zavacki AM, Fenzi G, Grachtchouk M, Hutchin M, Dlugosz AA, Bianco AC, Missero C, Larsen PR, Salvatore D. Sonic hedgehog-induced type 3 deiodinase blocks thyroid hormone action enhancing proliferation of normal and malignant keratinocytes. Proc Natl Acad Sci U S A. 2007;104:14466–14471. doi: 10.1073/pnas.0706754104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Morin RD, O’Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu AL, Zhao Y, McDonald H, Zeng TB, Hirst M, Eaves CJ, Marra MA. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tierling S, Dalbert S, Schoppenhorst S, Tsai C, Oliger S, Ferguson-Smith AC, Paulsen M, Walter J. High-resolution map and imprinting analysis of the Gtl2-Dnchc1 domain on mouse chromosome 12. Genomics. 2006;87:225–235. doi: 10.1016/j.ygeno.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 98.Hernandez A, St Germain DL. Activity and response to serum of the mammalian thyroid hormone deiodinase 3 gene promoter: identification of a conserved enhancer. Mol Cell Endocrinol. 2003;206:23–32. doi: 10.1016/s0303-7207(03)00239-9. [DOI] [PubMed] [Google Scholar]
- 99.Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R. Polycombcomplexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 100.Bernstein BE, MIkkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 101.Lin SP, Coan P, da Rocha ST, Seitz H, Cavaille J, Teng PW, Takada S, Ferguson-Smith AC. Differential regulation of imprinting in the murine embryo and placenta by the Dlk1-Dio3 imprinting control region. Development. 2007;134:417–426. doi: 10.1242/dev.02726. [DOI] [PubMed] [Google Scholar]
- 102.Sittig LJ, Shukla PK, Herzing LB, Redei EE. Strain-specific vulnerability to alcohol exposure in utero via hippocampal parent-of-origin expression of deiodinase-III. FASEB J. 2011;25:2313–2324. doi: 10.1096/fj.10-179234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kagami M, Sekita Y, Nishimura G, Irie M, Kato F, Okada M, Yamamori S, Kishimoto H, Nakayama M, Tanaka Y, Matsuoka K, Takahashi T, Noguchi M, Tanaka Y, Masumoto K, Utsunomiya T, Kouzan H, Komatsu Y, Ohashi H, Kurosawa K, Kosaki K, Ferguson-Smith AC, Ishino F, Ogata T. Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat Genet. 2008;40:237–242. doi: 10.1038/ng.2007.56. [DOI] [PubMed] [Google Scholar]
- 104.Falk MJ, Curtis CA, Bass NE, Zinn AB, Schwartz S. Maternal uniparental disomy chromosome 14: case report and literature review [Review] [16 refs] Pediatr Neurol. 2005;32:116–120. doi: 10.1016/j.pediatrneurol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 105.Coveler KJ, Yang SP, Sutton R, Milstein JM, Wu YQ, Bois KD, Beischel LS, Johnson JP, Shaffer LG. A case of segmental paternal isodisomy of chromosome 14. [Review] [25 refs] Hum Genet. 2002;110:251–256. doi: 10.1007/s00439-002-0688-4. [DOI] [PubMed] [Google Scholar]
- 106.Towner DR, Shaffer LG, Yang SP, Walgenbach DD. Confined placental mosaicism for trisomy 14 and maternal uniparental disomy in association with elevated second trimester maternal serum human chorionic gonadotrophin and third trimester fetal growth restriction. Prenat Diagn. 2001;21:395–398. doi: 10.1002/pd.75. [DOI] [PubMed] [Google Scholar]
- 107.Kotzot D. Maternal uniparental disomy 14 dissection of the phenotype with respect to rare autosomal recessively inherited traits, trisomy mosaicism, and genomic imprinting. Ann Genet. 2004;47:251–260. doi: 10.1016/j.anngen.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 108.Cox H, Bullman H, Temple IK. Maternal UPD(14) in the patient with a normal karyotype: clinical report and a systematic search for cases in samples sent for testing for Prader-Willi syndrome. Am J Med Genet. 2004;127A:21–25. doi: 10.1002/ajmg.a.20611. [DOI] [PubMed] [Google Scholar]
- 109.Offiah AC, Cornette L, Hall CM. Paternal uniparental disomy 14: introducing the “coat-hanger” sign. Pediatr Radiol. 2003;33:509–512. doi: 10.1007/s00247-003-0931-8. [DOI] [PubMed] [Google Scholar]
- 110.Sutton VR, McAlister WH, Bertin TK, Kaffe S, Wang JC, Yano S, Shaffer LG, Lee B, Epstein CJ, Villar AJ. Skeletal defects in paternal uniparental disomy for chromosome 14 are re-capitulated in the mouse model (paternal uniparental disomy 12) Hum Genet. 2003;113:447–451. doi: 10.1007/s00439-003-0981-x. [DOI] [PubMed] [Google Scholar]
- 111.Kurosawa K, Sasaki H, Sato Y, Yamanaka M, Shimizu M, Ito Y, Okuyama T, Matsuo M, Imaizumi K, Kuroki Y, Nishimura G. Paternal UPD14 is responsible for a distinctive malformation complex. Am J Med Genet. 2002;110:268–272. doi: 10.1002/ajmg.10404. [DOI] [PubMed] [Google Scholar]
- 112.Naik S, Temple IK. Coat hanger appearances of the ribs: a useful diagnostic marker of paternal uniparental disomy of chromosome 14. Arch Dis Child. 2010;95:909. doi: 10.1136/adc.2010.185736. [DOI] [PubMed] [Google Scholar]
- 113.Zechner U, Kohlschmidt N, Rittner G, Damatova N, Beyer V, Haaf T, Bartsch O. Epimutation at human chromosome 14q32.2 in a boy with a upd(14)mat-like clinical phenotype. Clin Genet. 2009;75:251–258. doi: 10.1111/j.1399-0004.2008.01116.x. [DOI] [PubMed] [Google Scholar]
- 114.Hosoki K, Ogata T, Kagami M, Tanaka T, Saitoh S. Epimutation (hypomethylation) affecting the chromosome 14q32.2 imprinted region in a girl with upd(14) mat-like phenotype. Eur J Hum Genet. 2008 Aug;16(8):1019–1023. doi: 10.1038/ejhg.2008.90. [DOI] [PubMed] [Google Scholar]
- 115.Temple IK, Shrubb V, Lever M, Bullman H, Mackay DJ. Isolated imprinting mutation of the DLK1/GTL2 locus associated with a clinical presentation of maternal uniparental disomy of chromosome 14. J Med Genet. 2007;44:637–640. doi: 10.1136/jmg.2007.050807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Irving MD, Buiting K, Kanber D, Donaghue C, Schulz R, Offiah A, Mohammed SN, Oakey RJ. Segmental paternal uniparental disomy (patUPD) of 14q32 with abnormal methylation elicits the characteristic features of complete patUPD14. Am J Med Genet. 2010:1942. doi: 10.1002/ajmg.a.33449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Martin-Subero JI, Ammerpohl O, Bibikova M, Wickham-Garcia E, Agirre X, Alvarez S, Bruggemann M, Bug S, Calasanz MJ, Deckert M, Dreyling M, Du MQ, Durig J, Dyer MJ, Fan JB, Gesk S, Hansmann ML, Harder L, Hartmann S, Klapper W, Kuppers R, Montesinos-Rongen M, Nagel I, Pott C, Richter J, Roman-Gomez J, Seifert M, Stein H, Suela J, Trumper L, Vater I, Prosper F, Haferlach C, Cruz Cigudosa J, Siebert R. A comprehensive microarray-based DNA methylation study of 367 hematological neoplasms. PLoS One. 2009;4:e6986. doi: 10.1371/journal.pone.0006986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Luk JM, Burchard J, Zhang C, Liu AM, Wong KF, Shek FH, Lee NP, Fan ST, Poon RT, Ivanovska I, Philippar U, Cleary MA, Buser CA, Shaw PM, Lee CN, Tenen DG, Dai H, Mao M. DLK1-DIO3 genomic imprinted microRNA cluster at 14q32.2 defines a stemlike subtype of hepatocellular carcinoma associated with poor survival. J Biol Chem. 2011;286:30706–30713. doi: 10.1074/jbc.M111.229831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Astuti D, Latif F, Wagner K, Gentle D, Cooper WN, Catchpoole D, Grundy R, Ferguson-Smith AC, Maher ER. Epigenetic alteration at the DLK1-GTL2 imprinted domain in human neoplasia: analysis of neuroblastoma, phaeochromocytoma and Wilms’ tumour. Br J Cancer. 2005;92:1574–1580. doi: 10.1038/sj.bjc.6602478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Takahashi N, Kobayashi R, Kono T. Restoration of Dlk1 and Rtl1 is necessary but insufficient to rescue lethality in intergenic differentially methylated region (IG-DMR)-deficient mice. J Biol Chem. 2010;285:26121–26125. doi: 10.1074/jbc.M109.075325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hernandez A, Martinez E, Collison D, St Germain D. Preferential monoallelic expression of the human DIO3, a candidate gene for the maternal uniparental disomy of chromosome 14 phenotype, abstract. 85th annual Meeting of the American Endocrine Society; Philadelphia, PA. 2003. [Google Scholar]
- 122.Antonarakis SE, Blouin JL, Maher J, Avramopoulos D, Thomas G, Talbot CC., Jr Maternal uniparental disomy for human chromosome 14, due to loss of a chromosome 14 from somatic cells with t(13;14) trisomy 14. Am J Hum Genet. 1993;52:1145–1152. [PMC free article] [PubMed] [Google Scholar]
- 123.Hordijk R, Wierenga H, Scheffer H, Leegte B, Hofstra RM, Stolte-Dijkstra I. Maternal uniparental disomy for chromosome 14 in a boy with a normal karyotype. J Med Genet. 1999;36:782–785. doi: 10.1136/jmg.36.10.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Berends MJ, Hordijk R, Scheffer H, Oosterwijk JC, Halley DJ, Sorgedrager N. Two cases of maternal uniparental disomy 14 with a phenotype overlapping with the Prader-Willi phenotype. Am J Med Genet. 1999;84:76–79. doi: 10.1002/(sici)1096-8628(19990507)84:1<76::aid-ajmg16>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 125.Wood AJ, Oakey RJ. Genomic imprinting in mammals: emerging themes and established theories. PLoS Genet. 2006;2:e147. doi: 10.1371/journal.pgen.0020147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Haig D. Genomic imprinting and kinship: how good is the evidence? Annu Rev Genet. 2004;38:553–585. doi: 10.1146/annurev.genet.37.110801.142741. [DOI] [PubMed] [Google Scholar]
- 127.Reik W, Walter J. Evolution of imprinting mechanisms: the battle of the sexes begins in the zygote. Nat Genet. 2001;27:255–256. doi: 10.1038/85804. [DOI] [PubMed] [Google Scholar]
- 128.Killian JK, Buckley TR, Stewart N, Munday BL, Jirtle RL. Marsupials and Eutherians reunited: genetic evidence for the Theria hypothesis of mammalian evolution. Mamm Genome. 2001;12:513–517. doi: 10.1007/s003350020026. [DOI] [PubMed] [Google Scholar]
- 129.Charalambous M, Ferron SR, da Rocha ST, Murray AJ, Rowland T, Ito M, Schuster-Gossler K, Hernandez A, Ferguson-Smith AC. Imprinted gene dosage is critical for the transition to independent life. Cell Metabolism. 2012;15:209–221. doi: 10.1016/j.cmet.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Curley JP, Barton SC, Surani A, Keverne EB. Coadaptation in mother and infant regulated by a paternally expressed imprinted gene. Proc Biol Sci. 2004;271:1303–1309. doi: 10.1098/rspb.2004.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Edwards CA, Mungall AJ, Matthews L, Ryder E, Gray DJ, Pask AJ, Shaw G, Graves JAM, Rogers J, consortium S, Dunham I, Renfree MB, Ferguson-Smith AC. The evolution of the DLK1-DIO3 imprinted domain in mammals. PLoS Biol. 2008;6:e135. doi: 10.1371/journal.pbio.0060135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bakke JL, Lawrence N. Persistent thyrotropin insufficiency following neonatal thyroxine administration. J Lab Clin Med. 1966;67:477–482. [PubMed] [Google Scholar]
- 134.Bakke JL, Lawrence N, Wilber JF. The late effects of neonatal hyperthyoridism upon the hypothalamic–pituitary–thyroid axis in the rat. Endocrinology. 1974;95:406–411. doi: 10.1210/endo-95-2-406. [DOI] [PubMed] [Google Scholar]
- 135.Bakke JL, Lawrence N, Robinson S. Late effects of thyroxine injected into the hypothalamus of the neonatal rat. Neuroendocrinology. 1972;10:183–195. doi: 10.1159/000122088. [DOI] [PubMed] [Google Scholar]
- 136.Bakke JL, Lawrence NL, Robinson S, Bennett J. Endocrine studies in the untreated F1 and F2 progeny of rats treated neonatally with thyroxine Biol. Neonate. 1977;31:71–83. doi: 10.1159/000240946. [DOI] [PubMed] [Google Scholar]
- 137.Bakke JL, Lawrence NL, Robinson S, Bennett J. Observations on the untreated progeny of hypothyroid male rats. Metab Clin Exp. 1976;25:437–444. doi: 10.1016/0026-0495(76)90076-7. [DOI] [PubMed] [Google Scholar]