

Abstract
Obesity is a leading risk factor for impaired glucose tolerance and type 2 diabetes (T2D). Although the cause of the obesity epidemic is multi-factorial and not entirely clear, the recent acceleration in incidence is too rapid to be accounted for only by genetics, the wide availability of calorie-rich foods, and increasingly sedentary lifestyles. Accumulating data suggest that the important causes of the obesity epidemic may be related to developmental and early life environmental conditions. The concept of the developmental origins of health and disease (DOHaD) suggests that adverse influences early in development, particularly during intrauterine life, may result in permanent changes in the physiology and metabolism of the infant, which in turn result in an increased risk of non-communicable diseases in adulthood. For example, undernutrition during pregnancy and rapid postnatal weight gain are associated with obesity and T2D in the adult offspring. Moreover, increasing evidence suggests that early-life exposure to a wide range of chemicals has a significant impact on the causes of metabolic disorders. Although the underlying molecular mechanisms remain to be determined, these factors can affect epigenetic processes, such as DNA methylation, allowing the developmental environment to modulate gene transcription. The objective of this review article was to summarize recent progress in the biomedical implications of the DOHaD concept, focusing on the pathogenesis of obesity and T2D, and to discuss a future direction for preventive strategies from a public health perspective.
Keywords: Chemicals, Developmental origins of health and disease, Epigenetics, Obesity, Type 2 diabetes
Introduction
Non-communicable diseases (NCDs), such as obesity, type 2 diabetes (T2D), and the metabolic syndrome, have long been believed to stem from factors typifying the way many people live today, which include an overly high-calorie diet and low level of physical activity. The obesity epidemic continues to pose one of the largest worldwide threats to the health of the population of almost every country in the world [1, 2]. The metabolic syndrome, a complex condition linked to obesity that is characterized by a cluster of risk factors, including hypertension, dyslipidemia, and glucose intolerance, is also becoming increasingly prevalent [3].
The reason for the rapid expansion of this epidemic remains incompletely understood. The recent acceleration in incidence is too rapid to be accounted for only by genetics, increases in the caloric density of food, and declines in physical activity. It is now assumed that environmental factors acting early in life, especially during fetal life, have profound effects on the susceptibility to disease later in life. Several studies, both in humans and experimental animals, suggest that altered risk of adult diseases may be linked to the maternal environmental and nutritional status around conception and implantation [4, 5]. The observation that early human development affects the risk of NCDs in later life has been confirmed by epidemiological studies [6–8]. Specifically, low birth weight (LBW) has been associated with increased blood pressure, dyslipidemia, and impaired glucose metabolism during adulthood [9–12].
In this review, the concept of the development origins of health and disease (DOHaD) and its possible underlying molecular mechanisms are summarized, followed by future directions for preventive strategies for obesity and T2D in the public health sector.
Developmental origins of health and disease
The concept of DOHaD is based on the assumption that environmental factors acting early in life (usually during fetal life) have profound effects on the predisposition to disease later in life. The idea that developmental factors may influence the susceptibility to disease much later in life was given impetus by a series of epidemiological studies by Barker et al. [13–18]. These authors reported that LBW babies who survived infancy and childhood were at increased risk of having risk factors for coronary heart disease in adulthood. LBW is considered to be a marker for an adverse fetal environment and fetal stress. Correction for known risk factors, such as diet, smoking, and exercise, did not have a major impact on the relationships between LBW and subsequent risk of NCDs [19–23]. Relationships have now been described between LBW and many risk factors for coronary heart disease, including hypertension, dyslipidemia, and T2D [24]. LBW has also been associated with increased insulin resistance later in life [10].
Barker et al. [5, 15] proposed the thrifty phenotype hypothesis in an attempt to explain the associations between poor fetal growth and increased risk of obesity and T2D in adult life. Thrifty genotype confers a survival advantage in a poor food environment by reducing glucose uptake and limiting body growth. However, when individuals of this genotype encounter an environment of plentiful food, they are at risk of developing obesity and T2D. This hypothesis can explain the molecular basis of DOHaD. Since this hypothesis was proposed, many studies worldwide have provided the epidemiological evidence for this concept [16, 19–24].
The concept of DOHaD was further supported by the ‘Dutch hunger winter’ studies. The Dutch hunger winter can be regarded as a unique ‘experiment of history’ and shows how maternal undernutrition during specific gestational time windows may affect later health outcome. The winter of 1944–45 in the Netherlands, which was occupied by the Germans in May 1940, is known as the ‘hunger winter.’ Despite the war, nutrition in the Netherlands had generally been adequate up to October 1944. In October 1944, the German authorities blocked all food supplies to the occupied west of the Netherlands. These were restored immediately after liberation on May 5, 1945. Therefore, children exposed to famine in utero during the hunger winter were well nourished in childhood and had accelerated weight gain [25]. The hunger winter cohort was used to examine how maternal undernutrition during specific gestational time windows affects the subsequent life course of offspring who experienced the famine in utero.
The results of the studies of the Dutch hunger winter have given important insights in the causes of DOHaD. A summary of the study outcomes is provided in Table 1. The offspring of women exposed to famine in early gestation, although of normal birth weight, had increased risk of obesity and a threefold increase in the risk of coronary heart disease as adults [26, 27]. The offspring of women exposed to famine in mid- and late gestation were born smaller than unexposed babies and had an increased risk of impaired glucose tolerance as adults [28, 29]. In addition to the studies on the Dutch hunger winter, many other epidemiological studies have indicated that obesity and T2D are highly prevalent in subjects born with LBW [30–34]. The Great Chinese Famine that affected the Chongqing population during 1959–1961 led to overweight females after 50 years [35]. Individuals exposed in utero and infancy to the Nigerian civil war famine were at increased risk of obesity and T2D about 40 years later [36]. Thus, the effects of LBW on the development of obesity and T2D in adult life have been proven in different studies.
Table 1.
Studies of the Dutch hunger winter families
| Subjects | Major findings | References |
|---|---|---|
| Obesity and type 2 diabetes | ||
| Age 19 years | Obesity | Ravelli et al. [6] |
| Age 50 years | 2-h glucose levels were elevated after a glucose load | Ravelli et al. [28] |
| Increase in body mass index and waist circumference in women | Ravelli et al. [26] | |
| Age 58 years | 2-h glucose levels were elevated after a glucose load | de Rooij SR et al. [127] |
| Impaired insulin secretion after a glucose load | de Rooij SR et al. [128] | |
| Glucose intolerance was differed by PPAR-gamma 2 genotype | de Rooij SR et al. [129] | |
| Increased weight and fat deposition in women | Stein et al. [130] | |
| No association between prenatal famine and metabolic syndrome | de Rooij et al. [131] | |
| Lipid profiles | ||
| Age 50 years | Increased ratio of low-density to high-density lipoproteins | Roseboom et al. [132] |
| Age 59 years | Increased total cholesterol and triglyceride levels in women | Lumey et al. [133] |
| Blood pressure | ||
| Age 50 years | No differences were found in systolic or diastolic pressure | Roseboom et al. [134] |
| No association with adult blood pressure | Roseboom et al. [135] | |
| Age 59 years | Moderate increase in systolic but not diastolic blood pressure | Stein et al. [136] |
| Greater blood pressure increase during stress | Painter et al. [137] | |
| Atherosclerosis and mortality | ||
| Age 50 years | Increased prevalence of coronary heart disease in those exposed to the famine in early gestation | Roseboom et al. [138] |
| Higher incidence of coronary artery disease | Painter et al. [139] | |
| Age 57 years | No effect on adult mortality | Painter et al. [140] |
| Reduced carotid artery intima media thickness | Painter et al. [141] | |
| Age between 18 and 64 years | Higher overall adult mortality risk in women | van Abeelen et al. [142] |
| Miscellaneous | ||
| Age 24–48 years | Increased risk for schizophrenia | Susser et al. [143] |
| Age 50 years | Decreased factor VII concentrations | Roseboom et al. [144] |
| Increased prevalence of obstructive airways disease | Lopuhaa et al. [145] | |
| Age 58 years | No difference in cortisol concentrations after dexamethasone suppression test | de Rooij et al. [146] |
PPAR Peroxisome proliferator-activated receptor
Intriguingly, adverse events during pregnancy not only affect the offspring of that pregnancy but also the next generation. Women who were severely undernourished during the first trimester of pregnancy gave birth to babies who were of normal birth weight, but those babies themselves then went on to give birth to smaller babies in the next generation [37]. In a cohort of North American Indians, poor maternal nutrition was reported to be associated with an increased risk of T2D over several generations [21].
Mechanisms of the developmental origins of metabolic diseases
Although our understanding of the molecular mechanisms underlying the effects of fetal undernutrition and LBW on the development of NCDs later in life is far from complete, possible mechanisms have been proposed. Generally, organisms possess an evolved ability to respond to external signals by adjusting their phenotype during development to match their environment. Thus, poor nutrition of the pregnant mother may signal to the fetus that nutrients are scarce in the postnatal environment. Therefore, a fetus that is exposed to signals it interprets as reflecting nutrient deficiency or maternal stress will adapt its metabolic trajectory to suit an environment of limited energy availability. When the postnatal environment then fails to match the experienced prenatal environment, maladaptation occurs, resulting in onset of obesity and T2D [38]. Indeed, it has been shown that those who were most likely to develop T2D in adult life had LBW and underwent rapid postnatal weight gain [39]. In humans, the combination of LBW and rapid childhood growth has been associated with later insulin resistance [38, 39]. Recent animal studies have confirmed that it is mainly the discrepancy between the pre- and postnatal environments that affects adult onset diseases, rather than gestational undernutrition itself [40, 41]. The process whereby a stimulus or insult during a sensitive or critical period has irreversible long-term effects on development is often referred to as ‘metabolic programming’ [42].
The stress response may be involved in the pathogenesis of DOHaD via the regulation of the hypothalamic–pituitary–adrenal (HPA) axis, which has potent effects on metabolism and vasculature [43, 44]. Changes in the HPA axis have been proposed as a mechanism behind the epidemiological link between LBW and later increased blood pressure [45]. Offspring of rat dams given dexamethasone during pregnancy have reduced birth weight and increased blood pressure and glucose intolerance in adulthood [46, 47]. Intrauterine stress is associated with insulin resistance and accompanying dysregulation of the HPA axis, with chronically excessive adrenal glucocorticoid secretion and increased stress responses [48]. Mechanistic analysis has shown that intrauterine glucocorticoid exposure leads to reduced numbers of glucocorticoid receptors in the hypothalamus, resulting in impaired negative feedback and hence long-term upregulation of the HPA axis after birth [49]. This, in turn, may contribute to increased blood pressure and glucose intolerance. In humans, exposure to antenatal betamethasone caused signs of insulin resistance in adult offspring at 30 years of age [50]. Thus, increased maternal corticosteroid levels as a result of stress induced by reduced nutrient availability induce hypertension in the offspring [51]. A recent study reported that maternal iron restriction, independent of maternal macronutrient or caloric intake, also works as a fetal stressor that programs metabolic and circulatory functions in the offspring [52].
Animal studies are used to clarify the underlying mechanisms of clinical observations in more detail. Fetal nutrition is a key regulator of fetal growth and thus an obvious candidate as an influence on programming [53]. In fact, rats living in a postnatal obesogenic environment have very different physiological responses depending on whether they were born to undernourished or well-fed mothers, with the former becoming more obese, more hypertensive, and more hyperphagic than the latter [54]. Imbalance of protein and carbohydrate intake during pregnancy has been associated with reduced birth weight and increased blood pressure in the offspring [55]. Many studies have shown that prenatal protein restriction results in LBW and programs hypertension in experimental animals [56–58]. Embryos collected from mothers fed a low-protein diet have reduced cell numbers [59]. In particular, a strong correlation has been found between nephron number and birth weight [60], and a reduction in nephron number is associated with increased blood pressure [61]. Exposure of rats to a low-protein diet in utero also decreases β-cell proliferation, islet size, and islet vascularization [62]. Subsequent accelerated growth leads to an excessive metabolic demand on this limited cell mass.
Transcriptome-wide analysis using the Affymetrix Mouse 430A_2.0 array (Affymetrix, Santa Clara, CA) showed that feeding mice a protein-restricted diet between gestational days 10.5 and 17.5 altered the expression of 235 genes in the placenta [63]. In the adult male offspring of dams fed a protein-restricted diet, 311 genes in the liver differed significantly from those in the offspring of control dams, as determined by the Agilent 014879 whole rat genome array (Agilent Technologies, Santa Clara, CA) [64]. Thus, an altered maternal diet during pregnancy induces persistent changes in the transcriptome. Whatever the mechanisms, these events may occur in humans if undernutrition in utero is followed by an abundant postnatal diet, such in small babies born into a food-rich Western society.
Role of epigenetics
The induction of altered phenotypes during development in response to environmental stimuli involves epigenetic changes. Epigenetic regulation has been defined as “heritable changes in gene function that occur without changes in the nucleotide sequence” [65]. Epigenetic factors include DNA methylations, histone modifications, and microRNAs. Epigenetic changes, in particular in DNA methylation, provide a ‘memory’ of developmental plastic responses to the early environment and are central to the generation of phenotypes and their stability throughout the life course. Methylation at the 5′ position of cytosine in DNA within a CpG dinucleotide (the p denotes the intervening phosphate group) is a common modification in mammalian genomes and constitutes a stable epigenetic mark that is transmitted through DNA replication and cell division [66]. CpG dinucleotides are not randomly distributed throughout the genome but are clustered at the 5′ ends of genes/promoters in regions known as CpG islands. Hypermethylation of these CpG islands is generally associated with transcriptional repression, while hypomethylation of CpG islands is generally associated with transcriptional activation.
Data from animal models suggest that epigenetic processes are an important link between the early life environment and altered metabolism and body composition in the adult offspring [67, 68]. A growing body of literature has reported a role for epigenetic factors in the complex interplay between genes and the environment [4, 69–71]. Epigenetic changes may explain how an altered maternal diet during pregnancy, such as a protein restricted diet, induces persistent changes in the transcriptome.
Environmental developmental influences, such as the maternal diet or chemical exposure, can affect the offspring phenotype via epigenetic effects [72, 73]. A diet that is poor or enriched in methyl donors and cofactors of DNA methylation, especially during fetal growth and development, may influence the epigenotype. For example, a methyl-rich maternal diet during gestation was found to alter the body composition of the offspring in agouti mice, which was accompanied by epigenetic changes in metabolic control genes [74]. Thus, environmentally induced epigenetic modifications alter gene expression and may increase the offspring’s susceptibility for later disease [75, 76].
It has been shown in rodent models that unbalanced maternal diets during pregnancy induce changes in DNA methylation and covalent histone modifications in the 5′ regulatory regions of specific non-imprinted genes, affecting the offspring’s later body composition and metabolic phenotype [77, 78]. For example, the offspring of rat dams fed a protein-restricted diet had lower levels of CpG methylation and greater expression of the peroxisome proliferators-activated receptor (PPAR) α and glucocorticoid receptor (GR) genes in the liver than control animals [77]. Changes in the epigenetic regulation of these genes may result in alterations in the activity of pathways controlled by their target genes, such as phosphoenolpyruvate carboxykinase and acyl-CoA oxidase, which subsequently affects lipid and carbohydrate metabolism. Hypomethylation of the hepatic PPARα and GR promoters has been reported in both F1 and F2 offspring of F0 rats fed a protein-restricted diet during pregnancy without further nutritional challenge to the F1 generation, indicating that changes in the epigenome that occur during development may be passed on to subsequent generations [68].
Intrauterine growth restriction has been associated with progressive epigenetic silencing of Pdx1, a pancreatic and duodenal homeobox 1 transcription factor critical for β-cell development, resulting in impaired β-cell function and T2D in the adult offspring in rats [79]. Lack of methylation of the retrotransposon with a methylation-sensitive promoter was associated with subsequent offspring obesity in mice [80]. In contrast, supplementation of the diets of pregnant animals with methyl donors, such as folic acid, vitamin B12, choline, or betaine, increased DNA methylation of specific genes in the offspring [74, 81–83]. Overall, these results show that dietary modifications can induce altered phenotypes through epigenetic changes in specific genes and that these changes in phenotype can be modulated by nutritional interventions during pregnancy.
As yet there are limited published human data linking maternal nutrition to epigenetic changes in the offspring. Adults who were exposed to famine in utero showed altered DNA methylation in the promoters of several imprinted and non-imprinted genes in white blood cells. Two studies on the Dutch hunger winter families reported the effects of prenatal undernutrition on promoter methylation [84, 85]. Individuals with periconceptual exposure to famine had reduced DNA methylation of the imprinted insulin-like growth factor-2 (IGF2) gene, a key factor in human growth and development, as compared with their unexposed, same-sex siblings at 59 years of age [84]. In addition, individuals from Dutch hunger winter families also had lower DNA methylation of the imprinted INS-IGF2 (INSIGF) gene, but increased DNA methylation of the guanine nucleotide-binding protein (GNASAS), maternally expressed 3 (MEG3), interleukin-10 (IL10), ATP-binding cassette A1 (ABCA1), and leptin (LEP) genes in parallel with impaired glucose tolerance compared with their unexposed same-sex siblings [85]. A recent study reported that higher methylation of retinoid X receptor-α chr9 was associated with lower maternal carbohydrate intake in early pregnancy, which is also associated with higher neonatal adiposity [53]. Thus, it is clear that fetal stress can affect the methylation status of several subsets of genes, supporting the hypothesis that associations between early developmental conditions and health outcomes later in life may be mediated by changes in the epigenetic information. However, the differences in methylation status of these genes between the exposed and unexposed individuals were relatively small, although statistically significant. Further studies with clear end points are needed to determine whether measurement of epigenetic marks in early life can be used as biomarkers to identify individuals who have experienced environmental perturbations in development and thus who are more likely to develop obesity and metabolic disease in later adulthood. It is crucial to identify such epigenetic marks that are predictive of a later phenotype so that they can be used as relevant biomarkers for disease prevention.
Role of chemicals
Because the obesity epidemic coincided with the rapid increase in the use of industrial chemicals, the hypothesis that exposure to chemicals, combined with genetic predisposition and consumption of a high-calorie diet, may be a major contributor to the obesity epidemic was proposed nearly 10 years ago [86, 87]. Since then, a number of studies have specifically addressed the effects of environmental chemical exposure on weight gain. Chemicals that affect human fetal development are generally called endocrine-disrupting chemicals (EDCs). EDCs are compounds that act upon the body’s hormonal systems and include industrial contaminants, plastics, pesticides, and other compounds [88]. EDC contamination is a global problem. Bisphenol A (BPA), the prototypical EDC, is produced in large quantities for use in the production of polycarbonate and epoxy resins. However, when used in food and drink containers, it can ‘leach’ into the contents, resulting in ingestion of BPA with food and drink [89]. As a result, human exposure to BPA is widespread, and BPA has been detected in urine in more than 90 % of all human samples tested [89, 90]. Some EDCs are highly resistant to degradation and remain persistent in the environment. For example, persistent organic pollutants, which include polychlorinated dibenzo-p-dioxins, polychlorinated dibenzofurans, and polychlorinated biphenyls (PCBs) can accumulate in the human body, especially in adipose tissue because of their lipophilic nature. Although the production of PCBs was banned by the USA in the 1970s, PCBs remain ubiquitous contaminants in the human population even today because of their stability [91]. Many studies have been conducted to study the link between chemical exposure and metabolic disorders. The findings of studies on the effects of environmental chemicals on obesity and T2D are summarized in Table 2.
Table 2.
Studies on environmental chemicals on obesity and type 2 diabetes
| Subjects | Chemicalsa | Major findings | References |
|---|---|---|---|
| Human studies | |||
| Adults in southern Taiwan | Arsenic | High prevalence of T2D | Lai et al. [147] |
| Air Force veterans | TCDD | Increased prevalence of T2D | Henriksen et al. [148] |
| Air Force veterans | TCDD | Serum level was correlated with incidence of T2D | Longnecker et al. [149] |
| Pubertal boys | DDE | Increased body weight | Gladen et al. [150] |
| Adults | POPs | Correlated with the prevalence of T2D | Lee et al. [100] |
| Adult women | PCB | Increased incidence of T2D | Vasiliu et al. [102] |
| Mexican Americans | p, p′-DDT | Increased prevalence of T2D | Cox et al. [151] |
| Adult native Americans | HCB | Serum level was positively correlated with incidence of T2D | Codru et al. [152] |
| U.S. population | Organochlorine pesticides | Positively associated with metabolic syndrome | Lee et al. [153] |
| Children aged 6 years | HCB | Increased BMI (body mass index) and body weight | Smink et al. [97] |
| Yucheng poisoning women | PCBs | Increased prevalence of T2D | Wang et al. [95] |
| Residents in Cd-contaminated area | Cd | Correlated with diabetic nephropathy | Hanswell-Elkins et al. [154] |
| Air Force veterans | TCDD | Increased prevalence of T2D | Michalek et al. [155] |
| Adult female offspring | DDE | Increased weight and BMI | Karmaus et al. [98] |
| Women aged 50–59 years | p, p′-DDE | Increased prevalence of T2D | Rignell-Hydbom et al. [156] |
| Workers | PFOS | Increased prevalence of T2D | Lundin et al. [157] |
| Children aged 3 years | DDE and PCBs | Intrauterine exposure was associated with BMI | Verhulst et al. [103] |
| Great Lakes sport fish consumers | DDE | Increased incidence of T2D | Turyk et al. [158] |
| Women in southern Spain | Nonylphenol | Positively associated with BMI | Lopez-Espinosa et al. [159] |
| Adults of eastern Slovakia | PCBs | Increased prevalence of T2D | Ukropec et al. [160] |
| Koreans | Organochlorine pesticides | Increased prevalence of T2D | Son et al. [161] |
| Adults | PCBs | Increased prevalence of T2D | Lee et al. [162] |
| Koreans | Heptachlor epoxide | Positively associated with metabolic syndrome | Park et al. [163] |
| Children at 14 months | DDE | Elevated BMI | Mendez et al. [164] |
| Adults aged 18–74 | BPA | Higher exposure was associated with general and central obesity | Carwile et al. [165] |
| Women aged 20 years | PFOA | Maternal PFOA concentrations were positively associated with BMI | Halldorsson et al. [104] |
| Adult populations of Catalonia | PCBs and HCBs | Positively associated with diabetes and prediabetes | Gasull et al. [166] |
| Animal studies | |||
| Rats | Cd | Neonatal exposure increased diabetic prevalence | Merali et al. [167] |
| Female mice | BPA | Perinatal exposure led to obesity | Howdeshell et al. [107] |
| Female rats | BPA | Perinatal exposure led to obesity | Rubin et al. [168] |
| Rats | BPA | Perinatal exposure led to impaired glucose tolerance | Wei et al. [109] |
| Female ewe lambs | Octylphenols | Gestational exposure led to obesity | Wright et al. [112] |
| Mice | DES | Perinatal exposure led to obesity | Newbold et al. [106] |
| Mice | TBT | In utero exposure led to obesity | Grun et al. [117] |
| Female mice | BPA | Perinatal and postnatal exposure led to obesity | Miyawaki et al. [169] |
| Female mice | PFOS | Work as developmental obesogen | Hines et al. [170] |
| Female rats | BPA | Gestational exposure led to obesity | Somm et al. [108] |
| Mice | TBT | In utero exposure led to multipotent stem cells to become adipocytes | Kirchner et al. [118] |
TD2 Type 2 diabetes, BMI body mass index
a TCDD 2, 3, 7, 8-Tetrachlorodibenzo-p-dioxin, DDE dichlorodiphenyl-dichloroethylene, POPs persistent organic pollutants, PCB polychlorinated biphenyl, p, p′-DDT dichlorodiphenyltrichloroethane, HCB hexachlorobenzene, Cd cadmium, PFOS perfluorooctanesulfonic acid, BPA bisphenol A, PFOA perfluorooctanoate, DES diethylstilbestrol, TBT tributyltin, PFOS perfluorooctanoic acid
Epidemiological studies on maternal smoking have indicated that the adjusted odds ratio for obesity is between 1.5- and 2.0-fold greater if children were exposed during, but not before or after, pregnancy [92–94], suggesting that there are critical periods during embryogenesis when the embryo is the most sensitive to exposure to xenobiotics. These studies also indicated that chemical toxicants, such as nicotine, can contribute to the etiology of later obesity. In humans, there is epidemiological evidence for the association between developmental exposure to chemicals and metabolic disorders later in life [95]. It has been demonstrated that chemicals cross the placenta and directly affect the fetus [96]. Maternal exposure to chemical substances during pregnancy has been associated with an increased body mass index (BMI) in the offspring [97–99]. Several studies have indicated that organochlorine exposure may be associated with the development of T2D [97, 98, 100–103]. For example, the results of a Spanish cohort study showed that prenatal exposure to the organochlorine hexachlorobenzene was associated with increased BMI at age 6 years [97]. Prenatal exposure to dichlorodiphenyl-dichloroethylene (DDE) was significantly associated with increased weight and BMI in adult female offspring [98]. DDE in cord blood was associated with increased BMI in young children, and this effect was exacerbated by maternal smoking [103]. A recent prospective study reported that in utero exposure to perfluorooctanoate was positively associated with risk of overweight at age 20 years in female but not in male offspring [104]. Collectively, these results support the hypothesis that the fetus is vulnerable to exposure to environmental chemicals, resulting in an increased risk of excessive body weight gain and susceptibility to T2D later in life. Moreover, gender and the postnatal environment in combination with prenatal chemical exposure can modify the onset, as well as the progression and outcome of the disease.
In experimental cell cultures and animal studies, a variety of chemicals have been shown to act as adipogenesis or obesity-inducing agents. A number of chemicals are known to promote obesity by increasing the number of adipocytes or the storage of fat into existing adipocytes. The organochlorine compound 1, 1, 1-trichloro-2, 2-bis(p-chlorophenyl)-ethane (p, p′-DDT) induces a concentration-dependent increase in in vitro 3T3-L1 adipocyte differentiation [105]. In mouse studies, treatment with the synthetic estrogen diethylstilbestrol (DES) on days 1–5 of neonatal life at 0.001 mg/day increased body weight and the percentage of body fat [106]. Treatment of pregnant mice with xenoestrogen BPA resulted in increased body weight in the female offspring on postnatal day 22 compared with unexposed controls [107]. Recent studies have indicated that perinatal exposure to approximately 70 μg/kg/day BPA via drinking water alters early adipogenesis and increases body weight in rodent models [108, 109]. Mice perinatally exposed to DES or the phytoestrogen genistein increased weight after puberty [106, 110, 111]. Wright et al. [112] reported that exposure to octylphenol, another xenoestrogen, during fetal and postnatal life in female lambs led to increased weight at puberty. During development, estrogen induces an increase in adipocyte numbers and affects adipocyte function [113]. Overall, these results show that exposure to organochlorine compounds and/or xenoestrogens during sensitive windows of development may have obesogenic effects, and may lead to permanent changes in the metabolic pathways that regulate body weight.
Other chemicals that can work as potential obesogens are the organotins. Organotins represent a class of persistent organic pollutants that may reach harmful levels in exposed populations [114]. In culture systems, organotins induce adipocyte differentiation [115, 116]. Tributyltin is also known to induce adipogenesis in vivo. Mice treated prenatally with tributyltin are born with more stored fat than controls [117]. Multipotent stromal cells harvested from white adipose tissue at 8 weeks of age in mice prenatally exposed to tributyltin had high numbers of preadipocytes and cells preprogrammed to prefer the adipogenic fate, an effect that will likely lead to an increase in adipose mass over time [118]. Organotins work via direct activation of the PPARγ–retinoid X receptor (RXR) heterodimer [116]. Activation of the PPARγ–RXR heterodimer has been found to favor lipid biosynthesis and storage, and induces adipocyte differentiation. Interestingly, a recent report indicated that tributyltin can work as a xenoestrogen via estrogen receptors in vivo [119]. Among commonly used phthalates as plasticizers, monoethyl-hexyl-phthalate can work as an activator for PPARγ [120].
The mechanisms of action of these chemicals are diverse and probably involve epigenetic molecular changes, including DNA methylation and histone modifications [121]. Environmental compounds may induce the establishment of specific epigenetic patterns during key developmental periods that influence phenotypic variation, which in some cases lead to disease states [122, 123]. Indeed, maternal exposure to BPA decreased DNA methylation in the retrotransposon upstream of the agouti gene in mice and shifted coat color distribution in the offspring by stably altering the epigenome [74]. Whatever the mechanism, complex events, including exposure to obesogenic or diabetogenic chemicals during development, may be contributing to the obesity and T2D epidemics. Clarifying the mechanisms involved in weight homeostasis is a novel target in the study of abnormal programming induced by environmental chemicals, which should be re-named ‘metabolic disrupting chemicals’ [124].
Future perspectives
We are in the midst of a global epidemic of obesity with substantial negative health and socio-economic consequences. There is now growing evidence that developmental influences have lifelong effects on metabolic function (Fig. 1). Perturbations of the developmental milieu can have a profound impact on the onset and incidence of obesity and T2D. Phenotypic outcomes with long-term consequences involve the interplay between environmental, developmental, and genetic influences. The pathogenesis of obesity and T2D resides in a mixture of genetic and environmental factors. It is therefore probable that not only maternal nutrition and stress, but also maternal size, parity, and maternal age can affect the offspring phenotype and/or epigenetic states. Moreover, possible interplay among prenatal exposure and the postnatal environmental factors, such as nutrition, stress, chemical exposure, and aging, can affect disease outcome. Further studies are merited to clarify the interactions that mediate metabolic outcomes.
Fig. 1.
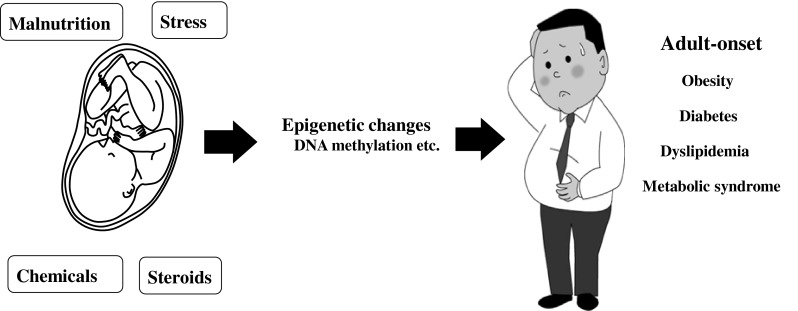
Influences during critical fetal periods may cause the adult onset of non-communicable diseases, such as obesity and type 2 diabetes
Transient environmental conditions during human gestation can be recorded as persistent changes in the epigenome. Epigenetic changes, in particular those in DNA methylation, are central to the generation of novel phenotypes and their stability throughout the life course. In the future, epigenetic research may have substantial benefits for public health because specific components of the epigenetic state at birth may predict later obesity and T2D. Researchers need to identify such effective epigenetic biomarkers which can be measured at early stage of life. These epigenetic marks may be important biomarkers, which not only indicate responses to prenatal challenges, but also predict later risk for obesity and T2D. Current studies are limited to analyzing specific candidate loci, and the complete epigenome has yet to be explored. New technologies, such as methods for rapid sequencing for differentially methylated regions of the genome, need to be developed before large-scale epidemiological studies can be conducted. It is likely that comprehensive studies of the epigenome will be helpful to shed light on gene–environment interactions in the pathogenesis of obesity and T2D.
Accumulating data indicate that low doses of environmental chemicals have adverse effects on human health [125]. These days, individuals are exposed to a mixture of EDCs. Indeed, dozens of environmental chemicals are detected in human tissues and fluids [126]. However, very little is known about how these chemicals act in combination. So far, almost all epidemiological studies have looked for the associations between metabolic disorders and the exposure to a single EDC at a single time, rather than to the whole mixture of toxicants to which human are exposed. These mixtures are likely to have unexpected and unpredictable effects. Further prospective studies with clear end points are required to determine whether exposure to mixtures of EDCs in early life can influence the development of obesity and T2D.
NCDs are preventable, but new initiatives are needed to institute prevention. If the risk of many common diseases of adulthood in our communities is largely determined before birth, adult lifestyle interventions will only reduce the risk transiently or to a small degree because they occur too late. Thus, from a public health perspective, it is important to determine whether interventions after the neonatal period can reverse the adverse effects of unbalanced prenatal nutrition. If the effects of adult lifestyle interventions are limited, maximum effect will be gained from timely interventions in early life. Improved nutrition will not only benefit the present population but may also reduce disease in future generations. If so, more care should be given to the consumption of a healthy diet during pregnancy and improved fetal nutrient availability, which may lead to a more normal birth weight and early life growth, thereby reducing the risk for programmed metabolic disease. Clear evidence and good communication of this evidence may lead to policy responses that will open the possibility of nutritional or pharmacological interventions to combat the rapid rise in obesity and T2D.
Acknowledgments
The author thanks two anonymous reviewers whose comments and suggestions greatly improved this review.
Conflict of interest
None.
Abbreviations
- BMI
Body mass index
- BPA
Bisphenol A
- DDE
Dichlorodiphenyl-dichloroethylene
- p, p′-DDT
1,1,1-trichloro-2, 2-bis(p-chlorophenyl)-ethane
- DES
Diethylstilbestrol
- DOHaD
Developmental origins of health and disease
- EDC
Endocrine-disrupting chemical
- GR
Glucocorticoid receptor
- HPA
Hypothalamic–pituitary–adrenal
- LBW
Low birth weight
- NCDs
Non-communicable diseases
- PCB
Polychlorinated biphenyls
- PPAR
Peroxisome proliferator-activated receptor
- T2D
Type 2 diabetes
References
- 1.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 2.Finucane MM, Stevens GA, Cowan MJ, Danaei G, Lin JK, Paciorek CJ, et al. National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet. 2011;377:557–567. doi: 10.1016/S0140-6736(10)62037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alexander CM, Landsman PB, Teutsch SM, Haffner SM. NCEP-defined metabolic syndrome, diabetes, and prevalence of coronary heart disease among NHANES III participants age 50 years and older. Diabetes. 2003;52:1210–1214. doi: 10.2337/diabetes.52.5.1210. [DOI] [PubMed] [Google Scholar]
- 4.Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359:61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Godfrey KM, Barker DJ. Fetal programming and adult health. Public Health Nutr. 2001;4:611–624. doi: 10.1079/PHN2001145. [DOI] [PubMed] [Google Scholar]
- 6.Ravelli GP, Stein ZA, Susser MW. Obesity in young men after famine exposure in utero and early infancy. N Engl J Med. 1976;295:349–353. doi: 10.1056/NEJM197608122950701. [DOI] [PubMed] [Google Scholar]
- 7.Hofman PL, Regan F, Jackson WE, Jefferies C, Knight DB, Robinson EM, et al. Premature birth and later insulin resistance. N Engl J Med. 2004;351:2179–2186. doi: 10.1056/NEJMoa042275. [DOI] [PubMed] [Google Scholar]
- 8.Gluckman PD, Hanson MA, editors. Developmental origins of health and disease. Cambridge: Cambridge University Press; 2006. [Google Scholar]
- 9.Huxley R, Neil A, Collins R. Unravelling the fetal origins hypothesis: is there really an inverse association between birthweight and subsequent blood pressure? Lancet. 2002;360:659–665. doi: 10.1016/S0140-6736(02)09834-3. [DOI] [PubMed] [Google Scholar]
- 10.Newsome CA, Shiell AW, Fall CH, Phillips DI, Shier R, Law CM. Is birth weight related to later glucose and insulin metabolism?—a systematic review. Diabet Med. 2003;20:339–348. doi: 10.1046/j.1464-5491.2003.00871.x. [DOI] [PubMed] [Google Scholar]
- 11.Huxley R, Owen CG, Whincup PH, Cook DG, Colman S, Collins R. Birth weight and subsequent cholesterol levels: exploration of the “fetal origins” hypothesis. JAMA. 2004;292:2755–2764. doi: 10.1001/jama.292.22.2755. [DOI] [PubMed] [Google Scholar]
- 12.Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science. 2004;305:1733–1736. doi: 10.1126/science.1095292. [DOI] [PubMed] [Google Scholar]
- 13.Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischemic heart disease in England and Wales. Lancet. 1986;1:1077–1081. doi: 10.1016/S0140-6736(86)91340-1. [DOI] [PubMed] [Google Scholar]
- 14.Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischemic heart disease. Lancet. 1989;2:577–580. doi: 10.1016/S0140-6736(89)90710-1. [DOI] [PubMed] [Google Scholar]
- 15.Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia. 1992;35:595–601. doi: 10.1007/BF00400248. [DOI] [PubMed] [Google Scholar]
- 16.Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. Lancet. 1993;341:938–941. doi: 10.1016/0140-6736(93)91224-A. [DOI] [PubMed] [Google Scholar]
- 17.Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia. 1993;36:62–67. doi: 10.1007/BF00399095. [DOI] [PubMed] [Google Scholar]
- 18.Phillips DI, Barker DJ, Hales CN, Hirst S, Osmond C. Thinness at birth and insulin resistance in adult life. Diabetologia. 1994;37:150–154. doi: 10.1007/s001250050086. [DOI] [PubMed] [Google Scholar]
- 19.Frankel S, Elwood P, Sweetnam P, Yarnell J, Smith GD. Birthweight, body-mass index in middle age, and incident coronary heart disease. Lancet. 1996;348:1478–1480. doi: 10.1016/S0140-6736(96)03482-4. [DOI] [PubMed] [Google Scholar]
- 20.Stein CE, Fall CH, Kumaran K, Osmond C, Cox V, Barker DJ. Fetal growth and coronary heart disease in south India. Lancet. 1996;48:1269–1273. doi: 10.1016/S0140-6736(96)04547-3. [DOI] [PubMed] [Google Scholar]
- 21.Benyshek DC, Martin JF, Johnston CS. A reconsideration of the origins of the type 2 diabetes epidemic among Native Americans and the implications for intervention policy. Med Anthropol. 2001;20:25–64. doi: 10.1080/01459740.2001.9966186. [DOI] [PubMed] [Google Scholar]
- 22.Kajantie E, Osmond C, Barker DJ, Forsen T, Phillips DI, Eriksson JG. Size at birth as a predictor of mortality in adulthood: a follow-up of 350000 person-years. Int J Epidemiol. 2005;34:655–663. doi: 10.1093/ije/dyi048. [DOI] [PubMed] [Google Scholar]
- 23.Leon DA, Lithell HO, Vagero D, Koupilova I, Mohsen R, Berglund L, et al. Reduced fetal growth rate and increased risk of death from ischemic heart disease: cohort study of 15000 Swedish men and women born 1915–29. Br Med J. 1998;317:241–245. doi: 10.1136/bmj.317.7153.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slomko H, Heo HJ, Einstein FH. Epigenetics of obesity and diabetes in humans. Endocrinology. 2012;153:1025–1030. doi: 10.1210/en.2011-1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lumey LH, Stein AD, Kahn HS, van der Pal-de Bruin KM, Blauw GJ, Zybert PA, et al. Cohort profile: the Dutch Hunger Winter families study. Int J Epidemiol. 2007;36:1196–204. [DOI] [PubMed]
- 26.Ravelli AC, van der Meulen JH, Osmond C, Barker DJ, Bleker OP. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am J Clin Nutr. 1999;70:811–816. doi: 10.1093/ajcn/70.5.811. [DOI] [PubMed] [Google Scholar]
- 27.Roseboom TJ, van der Meulen JH, Ravelli AC, Osmond C, Barker DJ, Bleker OP. Perceived health of adults after prenatal exposure the Dutch famine. Paediatr Perinat Epidemiol. 2003;17:391–397. doi: 10.1046/j.1365-3016.2003.00516.x. [DOI] [PubMed] [Google Scholar]
- 28.Ravelli AC, van der Meulen JH, Michels RP, Osmond C, Barker DJ, Hales CN, et al. Glucose tolerance in adults after prenatal exposure to famine. Lancet. 1998;351:173–177. doi: 10.1016/S0140-6736(97)07244-9. [DOI] [PubMed] [Google Scholar]
- 29.Painter RC, Roseboom TJ, Bleker OP. Prenatal exposure to the Dutch famine and disease in later life: an overview. Reprod Toxicol. 2005;20:345–352. doi: 10.1016/j.reprotox.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Phillips DI. Insulin resistance as a programmed response to fetal undernutrition. Diabetologia. 1996;39:1119–1122. doi: 10.1007/BF00400663. [DOI] [PubMed] [Google Scholar]
- 31.Curhan GC, Willett WC, Rimm EB, Spiegelman D, Ascherio AL, Stampfer MJ. Birth weight and adult hypertension, diabetes mellitus, and obesity in US men. Circulation. 1996;94:3246–3250. doi: 10.1161/01.CIR.94.12.3246. [DOI] [PubMed] [Google Scholar]
- 32.Nobili V, Alisi A, Panera N, Agostoni C. Low birth weight and catch-up-growth associated with metabolic syndrome: a ten year systematic review. Pediatr Endocrinol Rev. 2008;6:241–247. [PubMed] [Google Scholar]
- 33.Dufour S, Petersen KF. Disassociation of liver and muscle insulin resistance from ectopic lipid accumulation in low-birth-weight individuals. J Clin Endocrinol Metab. 2011;96:3873–3880. doi: 10.1210/jc.2011-1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barouki R, Gluckman PD, Grandjean P, Hanson M, Heindel JJ. Developmental origins of non-communicable disease: implications for research and public health. Environ Health. 2012;11:42. doi: 10.1186/1476-069X-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Wang X, Kong Y, Zhang JH, Zeng Q. The Great Chinese Famine leads to shorter and overweight females in Chongqing Chinese population after 50 years. Obesity. 2010;18:588–592. doi: 10.1038/oby.2009.296. [DOI] [PubMed] [Google Scholar]
- 36.Hult M, Tornhammar P, Ueda P, Chima C, Bonamy AK, Ozumba B, et al. Hypertension, diabetes and overweight: looming legacies of the Biafran famine. PLoS One. 2010;5:e13582. doi: 10.1371/journal.pone.0013582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stein AD, Lumey LH. The relationship between maternal and offspring birth weights after maternal prenatal famine exposure: the Dutch famine Birth Cohort Study. Hum Biol. 2000;72:641–654. [PubMed] [Google Scholar]
- 38.Eriksson JG, Forsen TJ, Osmond C, Barker DJ. Pathways of infant and childhood growth that lead to type 2 diabetes. Diabetes Care. 2003;26:3006–3010. doi: 10.2337/diacare.26.11.3006. [DOI] [PubMed] [Google Scholar]
- 39.Eriksson JG, Osmond C, Kajantie E, Forsen TJ, Barker DJ. Patterns of growth among children who later develop type 2 diabetes or its risk factors. Diabetologia. 2006;49:2853–2858. doi: 10.1007/s00125-006-0459-1. [DOI] [PubMed] [Google Scholar]
- 40.Dai Y, Thamotharan S, Garg M, Shin BC, Devakar SU. Superimposition of postnatal calorie restriction protects the aging male intrauterine growth-restricted offspring from metabolic maladaptations. Endocrinology. 2012;153:4216–4226. doi: 10.1210/en.2012-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garg M, Thamotharan M, Dai Y, Thamotharan S, Shin BC, Stout D, et al. Early postnatal caloric restriction protects adult male intrauterine growth-restricted offspring from obesity. Diabetes. 2012;61:1391–1398. doi: 10.2337/db11-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pinney SE, Simmons RA. Metabolic programming, epigenetics, and gestational diabetes mellitus. Curr Diab Rep. 2012;12:67–74. doi: 10.1007/s11892-011-0248-1. [DOI] [PubMed] [Google Scholar]
- 43.Barker DJ, Osmond C, Simmonds SJ, Wield GA. The relation of small head circumference and thinness at birth to death from cardiovascular disease in adult life. Br Med J. 1993;306:422–426. doi: 10.1136/bmj.306.6875.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phillips DI. Programming of the stress response: a fundamental mechanism underlying the long-term effects of the fetal environment? J Intern Med. 2007;261:453–460. doi: 10.1111/j.1365-2796.2007.01801.x. [DOI] [PubMed] [Google Scholar]
- 45.Benediktsson R, Lindsay RS, Noble J, Seckl JR, Edwards CR. Glucocorticoid exposure in utero: new model for adult hypertension. Lancet. 1993;341:339–341. doi: 10.1016/0140-6736(93)90138-7. [DOI] [PubMed] [Google Scholar]
- 46.Levitt NS, Lindsay RS, Holmes MC, Seckl JR. Dexamethasone in the last week of pregnancy attenuates hippocampal glucocorticoid receptor gene expression and elevates blood pressure in the adult offspring in the rat. Neuroendocrinology. 1996;64:412–418. doi: 10.1159/000127146. [DOI] [PubMed] [Google Scholar]
- 47.Nyirenda MJ, Lindsay RS, Kenyon CJ, Burchell A, Seckl JR. Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. J Clin Invest. 1998;101:2174–2181. doi: 10.1172/JCI1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buhl ES, Neschen S, Yonemitsu S, Rossbacher J, Zhang D, Morino K, et al. Increased hypothalamic–pituitary–adrenal axis activity and hepatic insulin resistance in low-birth weight rats. Am J Physiol Endocrinol Metab. 2007;293:E1451–E1458. doi: 10.1152/ajpendo.00356.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gluckman PD, Hanson MA, Beedle AS. Early life events and their consequences for later disease: a life history and evolutionary perspective. Am J Hum Biol. 2007;19:1–19. doi: 10.1002/ajhb.20590. [DOI] [PubMed] [Google Scholar]
- 50.Dalziel SR, Walker NK, Parag V, Mantell C, Rea HH, Rodgers A, et al. Cardiovascular risk factors after antenatal exposure to betamethasone: 30-year follow-up of a randomised controlled trial. Lancet. 2005;365:1856–1862. doi: 10.1016/S0140-6736(05)66617-2. [DOI] [PubMed] [Google Scholar]
- 51.Langley-Evans SC. Hypertension induced by foetal exposure to a maternal low-protein diet, in the rat, is prevented by pharmacological blockade of maternal glucocorticoid synthesis. J Hypertens. 1997;15:537–544. doi: 10.1097/00004872-199715050-00010. [DOI] [PubMed] [Google Scholar]
- 52.Bourque SL, Komolova M, McCabe K, Adams MA, Nakatsu K. Perinatal iron deficiency combined with a high-fat diet causes obesity and cardiovascular dysregulation. Endocrinology. 2012;153:1174–1182. doi: 10.1210/en.2011-1700. [DOI] [PubMed] [Google Scholar]
- 53.Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes. 2011;60:1528–1534. doi: 10.2337/db10-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vickers MH, Breier BH, Cutfield WS, Hofman PL, Gluckman PD. Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am J Physiol Endocrinol Metab. 2000;279:E83–E87. doi: 10.1152/ajpendo.2000.279.1.E83. [DOI] [PubMed] [Google Scholar]
- 55.Shiell AW, Campbell-Brown M, Haselden S, Robinson S, Godfrey KM, Barker DJ. High-meat, low-carbohydrate diet in pregnancy: relation to adult blood pressure in the offspring. Hypertension. 2001;38:1282–1288. doi: 10.1161/hy1101.095332. [DOI] [PubMed] [Google Scholar]
- 56.Langley-Evans SC, Phillips GJ, Jackson AA. In utero exposure to maternal low protein diets induces hypertension in weanling rats, independently of maternal blood pressure changes. Clin Nutr. 1994;13:319–324. doi: 10.1016/0261-5614(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 57.Maloney CA, Gosby AK, Phuyal JL, Denyer GS, Bryson JM, Caterson ID. Site-specific changes in the expression of fat-partitioning genes in weanling rats exposed to a low-protein diet in utero. Obes Res. 2003;11:461–468. doi: 10.1038/oby.2003.63. [DOI] [PubMed] [Google Scholar]
- 58.Langley-Evans SC. Critical differences between two low protein diet protocols in the programming of hypertension in the rat. Int J Food Sci Nutr. 2005;51:11–17. doi: 10.1080/096374800100859. [DOI] [PubMed] [Google Scholar]
- 59.Kwong WY, Wild AE, Roberts P, Willis AC, Fleming TP. Maternal undernutrition during the preimplantation period of rat development causes blastocyst abnormalities and programming of postnatal hypertension. Development. 2000;127:4195–4202. doi: 10.1242/dev.127.19.4195. [DOI] [PubMed] [Google Scholar]
- 60.McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev. 2005;85:571–633. doi: 10.1152/physrev.00053.2003. [DOI] [PubMed] [Google Scholar]
- 61.Alexander BT. Fetal programming of hypertension. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1–R10. doi: 10.1152/ajpregu.00417.2005. [DOI] [PubMed] [Google Scholar]
- 62.Snoeck A, Remacle C, Reusens B, Hoet JJ. Effect of a low protein diet during pregnancy on the fetal rat endocrine pancreas. Biol Neonate. 1990;57:107–118. doi: 10.1159/000243170. [DOI] [PubMed] [Google Scholar]
- 63.Gheorghe CP, Goyal R, Holweger JD, Longo LD. Placental gene expression responses to maternal protein restriction in the mouse. Placenta. 2009;30:411–417. doi: 10.1016/j.placenta.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lillycrop KA, Rodford J, Garratt ES, Slater-Jefferies JL, Godfrey KM, Gluckman PD, et al. Maternal protein restriction with or without folic acid supplementation during pregnancy alters the hepatic transcriptome in adult male rats. Br J Nutr. 2010;103:1711–1719. doi: 10.1017/S0007114509993795. [DOI] [PubMed] [Google Scholar]
- 65.Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293:1068–1070. doi: 10.1126/science.1063852. [DOI] [PubMed] [Google Scholar]
- 66.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 67.Drake AJ, Walker BR, Seckl JR. Intergenerational consequences of fetal programming by in utero exposure to glucocorticoids in rats. Am J Physiol Regul Integr Comp Physiol. 2005;288:R34–R38. doi: 10.1152/ajpregu.00106.2004. [DOI] [PubMed] [Google Scholar]
- 68.Burdge GC, Slater-Jefferies J, Torrens C, Phillips ES, Hanson MA, Lillycrop KA. Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. Br J Nutr. 2007;97:435–439. doi: 10.1017/S0007114507352392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burdge GC, Lillycrop KA. Nutrition, epigenetics, and developmental plasticity: implications for understanding human disease. Annu Rev Nutr. 2010;30:315–339. doi: 10.1146/annurev.nutr.012809.104751. [DOI] [PubMed] [Google Scholar]
- 70.Ooki S. Life course genetic epidemiologic study based on longitudinal twin-family data: a new perspective. Nihon Eiseigaku Zasshi. 2011;66:31–38. doi: 10.1265/jjh.66.31. [DOI] [PubMed] [Google Scholar]
- 71.Drong AW, Lindgren CM, McCarthy MI. The genetic and epigenetic basis of type 2 diabetes and obesity. Clin Pharmacol Ther. 2012;92:707–715. doi: 10.1038/clpt.2012.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Waterland RA, Jirtle RL. Early nutrition, epigenetic changes at transposons and imprinted genes, and enhanced susceptibility to adult chronic diseases. Nutrition. 2004;20:63–68. doi: 10.1016/j.nut.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 73.Godfrey KM, Lillycrop KA, Burdge GC, Gluckman PD, Hanson MA. Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr Res. 2007;61:5R–10R. doi: 10.1203/pdr.0b013e318045bedb. [DOI] [PubMed] [Google Scholar]
- 74.Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci USA. 2007;104:13056–13061. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes. 2009;58:2718–2725. doi: 10.2337/db09-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heindel JJ, vom Saal FS. Role of nutrition and environmental endocrine disrupting chemicals during the perinatal period on the aetiology of obesity. Mol Cell Endocrinol. 2009;304:90–96. doi: 10.1016/j.mce.2009.02.025. [DOI] [PubMed] [Google Scholar]
- 77.Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J Nutr. 2005;135:1382–1386. doi: 10.1093/jn/135.6.1382. [DOI] [PubMed] [Google Scholar]
- 78.Bogdarina I, Welham S, King PJ, Burns SP, Clark AJ. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circ Res. 2007;100:520–526. doi: 10.1161/01.RES.0000258855.60637.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing Pdx 1. J Clin Invest. 2008;118:2316–2324. doi: 10.1172/JCI32011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sinclair KD, Allegrucci C, Singh R, Gardner DS, Sebastian S, Bispham J, et al. DNA methylation, insulin resistance, and blood pressure in offspring determined by maternal periconceptional B vitamin and methionine status. Proc Natl Acad Sci USA. 2007;104:19351–19356. doi: 10.1073/pnas.0707258104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lillycrop KA, Phillips ES, Torrens C, Hanson MA, Jackson AA, Burdge GC. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPAR alpha promoter of the offspring. Br J Nutr. 2008;100:278–282. doi: 10.1017/S0007114507894438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Burdge GC, Lillycrop KA, Jackson AA, Gluckman PD, Hanson MA. The nature of the growth pattern and of the metabolic response to fasting in the rat are dependent upon the dietary protein and folic acid intakes of their pregnant dams and post-weaning fat consumption. Br J Nutr. 2008;99:540–549. doi: 10.1017/S0007114507815819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA. 2008;105:17046–17049. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, et al. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18:4046–4053. doi: 10.1093/hmg/ddp353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Baillie-Hamilton PF. Chemical toxins: a hypothesis to explain the global obesity epidemic. J Altern Complement Med. 2002;8:185–192. doi: 10.1089/107555302317371479. [DOI] [PubMed] [Google Scholar]
- 87.Heindel JJ. Endocrine disruptors and the obesity epidemic. Toxicol Sci. 2003;76:247–249. doi: 10.1093/toxsci/kfg255. [DOI] [PubMed] [Google Scholar]
- 88.Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, et al. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. 2009;30:293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Calafat AM, Ye X, Wong LY, Reidy JA, Needham LL. Exposure of the U.S. population to bisphenol A and 4-tertiary-octylphenol: 2003–2004. Environ Health Perspect. 2008;116:39–44. doi: 10.1289/ehp.10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV. Human exposure to bisphenol A. Reprod Toxicol. 2007;24:139–177. doi: 10.1016/j.reprotox.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 91.Mullerova D, Kopecky J. White adipose tissue: storage and effector site for environmental pollutants. Physiol Res. 2007;56:375–381. doi: 10.33549/physiolres.931022. [DOI] [PubMed] [Google Scholar]
- 92.Toschke AM, Koletzko B, Slikker W, Jr, Hermann M, von Kries R. Childhood obesity is associated with maternal smoking in pregnancy. Eur J Pediatr. 2002;161:445–448. doi: 10.1007/s00431-002-0983-z. [DOI] [PubMed] [Google Scholar]
- 93.Power C, Jefferis BJ. Fetal environment and subsequent obesity: a study of maternal smoking. Int J Epidemiol. 2002;31:413–419. doi: 10.1093/ije/31.2.413. [DOI] [PubMed] [Google Scholar]
- 94.Al Mamun A, Lawlor DA, Alati R, O’Callaghan MJ, Williams GM, Najman JM. Does maternal smoking during pregnancy have a direct effect on future offspring obesity? Evidence from a prospective birth cohort study. Am J Epidemiol. 2006;164:317–325. doi: 10.1093/aje/kwj209. [DOI] [PubMed] [Google Scholar]
- 95.Wang SL, Tsai PC, Yang CY, Leon Guo Y. Increased risk of diabetes and polychlorinated biphenyls and dioxins: a 24-year follow-up study of the Yucheng cohort. Diabetes Care. 2008;31:1574–1579. doi: 10.2337/dc07-2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Park JS, Bergman A, Linderholm L, Athanasiadou M, Kocan A, Petrik J, et al. Placental transfer of polychlorinated biphenyls, their hydroxylated metabolites and pentachlorophenol in pregnant women from eastern Slovakia. Chemosphere. 2008;70:1676–1684. doi: 10.1016/j.chemosphere.2007.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Smink A, Ribas-Fito N, Garcia R, Torrent M, Mendez MA, Grimalt JO, et al. Exposure to hexachlorobenzene during pregnancy increases the risk of overweight in children aged 6 years. Acta Paediatr. 2008;97:1465–1469. doi: 10.1111/j.1651-2227.2008.00937.x. [DOI] [PubMed] [Google Scholar]
- 98.Karmaus W, Osuch JR, Eneli I, Mudd LM, Zhang J, Mikucki D, et al. Maternal level of dichlorodiphenyl-dichloroethylene (DDE) may increase weight and body mass index in adult female offspring. Occup Environ Med. 2009;66:143–149. doi: 10.1136/oem.2008.041921. [DOI] [PubMed] [Google Scholar]
- 99.Janesick A, Blumberg B. Endocrine disrupting chemicals and the developmental programming of adipogenesis and obesity. Birth Defects Res C Embryo Today. 2011;93:34–50. doi: 10.1002/bdrc.20197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee DH, Lee IK, Song K, Steffes M, Toscano W, Baker BA, et al. A strong dose-response relation between serum concentration of persistent organic pollutants and diabetes: results from the National Health and Examination Survey 1999–2002. Diabetes Care. 2006;29:1638–1644. doi: 10.2337/dc06-0543. [DOI] [PubMed] [Google Scholar]
- 101.Porta M. Persistent organic pollutants and the burden of diabetes. Lancet. 2006;368:558–559. doi: 10.1016/S0140-6736(06)69174-5. [DOI] [PubMed] [Google Scholar]
- 102.Vasiliu O, Cameron L, Gardiner J, Deguire P, Karmaus W. Polybrominated biphenyls, polychlorinated biphenyls, body weight, and incidence of adult-onset diabetes mellitus. Epidemiology. 2006;17:352–359. doi: 10.1097/01.ede.0000220553.84350.c5. [DOI] [PubMed] [Google Scholar]
- 103.Verhulst SL, Nelen V, Hond ED, Deguire P, Karmaus W. Intrauterine exposure to environmental pollutants and body mass index during the first 3 years of life. Environ Health Perspect. 2009;117:122–126. doi: 10.1289/ehp.0800003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Halldorsson TI, Rytter D, Haug LS, Bech BH, Danielsen I, Becher G, et al. Prenatal exposure to perfluorooctanoate and risk of overweight at 20 years of age: a prospective cohort study. Environ Health Perspect. 2012;120:668–673. doi: 10.1289/ehp.1104034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moreno-Aliaga MJ, Matsumura F. Effects of 1,1,1-trichloro-2,2-bis(p-chlorophenyl)-ethane (p, p′-DDT) on 3T3-L1 and 3T3 F442A adipocyte differentiation. Biochem Pharmacol. 2002;63:997–1007. doi: 10.1016/S0006-2952(01)00933-9. [DOI] [PubMed] [Google Scholar]
- 106.Newbold RR, Padilla-Banks E, Snyder RJ, Jefferson WN. Developmental exposure to estrogenic compounds and obesity. Birth Defects Res A Clin Mol Teratol. 2005;73:478–480. doi: 10.1002/bdra.20147. [DOI] [PubMed] [Google Scholar]
- 107.Howdeshell KL, Hotchkiss AK, Thayer KA, Vandenberg JG, vom Saal FS. Exposure to bisphenol A advances puberty. Nature. 1999;401:763–764. doi: 10.1038/44517. [DOI] [PubMed] [Google Scholar]
- 108.Somm E, Schwitzgebel VM, Toulotte A, Cederroth CR, Combescure C, Nef S, et al. Perinatal exposure to bisphenol a alters early adipogenesis in the rat. Environ Health Perspect. 2009;117:1549–1555. doi: 10.1289/ehp.11342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wei J, Lin Y, Li Y, Ying C, Chen J, Song L, et al. Perinatal exposure to bisphenol A at reference dose predisposes offspring to metabolic syndrome in adult rats on a high-fat diet. Endocrinology. 2011;152:3049–3061. doi: 10.1210/en.2011-0045. [DOI] [PubMed] [Google Scholar]
- 110.Newbold RR, Padilla-Banks E, Jefferson WN. Perinatal exposure to environmental estrogens and the development of obesity. Mol Nutr Food Res. 2007;51:912–917. doi: 10.1002/mnfr.200600259. [DOI] [PubMed] [Google Scholar]
- 111.Newbold RR, Padilla-Banks E, Jefferson WN. Environmental estrogens and obesity. Mol Cell Endocrinol. 2009;304:84–89. doi: 10.1016/j.mce.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wright C, Evans AC, Evans NP, Duffy P, Fox J, Boland MP, et al. Effect of maternal exposure to the environmental estrogen, octylphenol, during fetal and/or postnatal life on onset of puberty, endocrine status, and ovarian follicular dynamics in ewe lambs. Biol Reprod. 2002;67:1734–1740. doi: 10.1095/biolreprod.101.002006. [DOI] [PubMed] [Google Scholar]
- 113.Cook PS, Naaz A. Role of estrogens in adipocyte development and function. Exp Biol Med. 2004;229:1127–1135. doi: 10.1177/153537020422901107. [DOI] [PubMed] [Google Scholar]
- 114.Rantakokko P, Turunen A, Verkasalo PK, Kiviranta H, Mannisto S, Vartiainen T. Blood levels of organotin compounds and their relation to fish consumption in Finland. Sci Total Environ. 2008;399:90–95. doi: 10.1016/j.scitotenv.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 115.Inadera H, Shimomura A. Environmental chemical tributyltin augments adipocyte differentiation. Toxicol Lett. 2005;74:226–234. doi: 10.1016/j.toxlet.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 116.Kanayama T, Kobayashi N, Mamiya T, Nakanishi T, Nishikawa J. Organotin compounds promote adipocyte differentiation as agonists of the peroxisome proliferators-activated receptor gamma/retinoid X receptor pathway. Mol Pharmacol. 2005;67:766–774. doi: 10.1124/mol.104.008409. [DOI] [PubMed] [Google Scholar]
- 117.Grun F, Watanabe H, Zamanian Z, Maeda L, Arima K, Cubacha R, et al. Endocrine-disrupting organotin compounds are potent inducers of adipogenesis in vertebrates. Mol Endocrinol. 2006;20:2141–2155. doi: 10.1210/me.2005-0367. [DOI] [PubMed] [Google Scholar]
- 118.Kirchner S, Kieu T, Chow C, Casey S, Blumberg B. Prenatal exposure to the environmental obesogen tributyltin predisposes multipotent stem cells to become adipocytes. Mol Endocrinol. 2010;24:526–539. doi: 10.1210/me.2009-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Penza M, Jeremic M, Marrazzo E, Maggi A, Ciana P, Rando G, et al. The environmental chemical tributyltin chloride (TBT) shows both estrogenic and adipogenic activities in mice which might depend on the exposure dose. Toxicol Appl Pharmacol. 2011;255:65–75. doi: 10.1016/j.taap.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 120.Feige JN, Gelman L, Rossi D, Zoete V, Metivier R, Tudor C, et al. The endocrine disruptor monoethyl-hexyl-phthalate is a selective peroxisome proliferator-activated receptor gamma modulator that promotes adipogenesis. J Biol Chem. 2007;282:19152–19166. doi: 10.1074/jbc.M702724200. [DOI] [PubMed] [Google Scholar]
- 121.Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308:1466–1469. doi: 10.1126/science.1108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Skinner MK, Anway MD, Savenkova MI, Gore AC, Crews D. Transgenerational epigenetic programming of the brain transcriptome and anxiety behavior. PLoS One. 2008;3:e3745. doi: 10.1371/journal.pone.0003745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Casals-Casas C, Desvergne B. Endocrine disruptors: from endocrine to metabolic disruption. Annu Rev Physiol. 2011;73:135–162. doi: 10.1146/annurev-physiol-012110-142200. [DOI] [PubMed] [Google Scholar]
- 125.Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR, Jr, Lee DH, et al. Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev. 2012;33:378–455. doi: 10.1210/er.2011-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Woodruff TJ, Zota AR, Schwartz JM. Environmental chemicals in pregnant women in the US: NHANES 2003–2004. Environ Health Perspect. 2011;119:878–885. doi: 10.1289/ehp.1002727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.de Rooij SR, Painter RC, Roseboom TJ, Phillips DI, Osmond C, Barker DJ, et al. Glucose tolerance at age 58 and the decline of glucose tolerance in comparison with age 50 in people prenatally exposed to the Dutch famine. Diabetologia. 2006;49:637–643. doi: 10.1007/s00125-005-0136-9. [DOI] [PubMed] [Google Scholar]
- 128.de Rooij SR, Painter RC, Phillips DI, Osmond C, Michels RP, Godsland IF, et al. Impaired insulin secretion after prenatal exposure to the Dutch famine. Diabetes Care. 2006;29:1897–1901. doi: 10.2337/dc06-0460. [DOI] [PubMed] [Google Scholar]
- 129.de Rooij SR, Painter RC, Phillips DI, Osmond C, Tanck NW, Defesche JC, et al. The effects of the Pro12Ala polymorphism of the peroxisome proliferators-activated receptor-gamma2 gene on glucose/insulin metabolism interact with prenatal exposure to famine. Diabetes Care. 2006;29:1052–1057. doi: 10.2337/dc05-1993. [DOI] [PubMed] [Google Scholar]
- 130.Stein AD, Kahn HS, Rundle A, Zybert PA, van der Pal-de Bruin K, Lumey LH. Anthropometric measures in middle age after exposure to famine during gestation: evidence from the Dutch Famine. Am J Clin Nutr. 2007;85:869–76. [DOI] [PubMed]
- 131.de Rooij SR, Painter RC, Holleman F, Bossuyt PM, Roseboom TJ. The metabolic syndrome in adults prenatally exposed to the Dutch famine. Am J Clin Nutr. 2007;86:1219–1224. doi: 10.1093/ajcn/86.4.1219. [DOI] [PubMed] [Google Scholar]
- 132.Roseboom TJ, van der Meulen JH, Osmond C, Barker DJ, Ravelli AC, Bleker OP. Plasma lipid profiles in adults after prenatal exposure to the Dutch famine. Am J Clin Nutr. 2000;72:1101–1106. doi: 10.1093/ajcn/72.5.1101. [DOI] [PubMed] [Google Scholar]
- 133.Lumey LH, Stein AD, Kahn HS, Romijn JA. Lipid profiles in middle-aged men and women after famine exposure during gestation: the Dutch Hunger Winter Families Study. Am J Clin Nutr. 2009;89:1737–1743. doi: 10.3945/ajcn.2008.27038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Roseboom TJ, van der Meulen JH, Ravelli AC, van Montfrans GA, Osmond C, Barker DJ, et al. Blood pressure in adults after prenatal exposure to famine. J Hypertens. 1999;17:325–330. doi: 10.1097/00004872-199917030-00004. [DOI] [PubMed] [Google Scholar]
- 135.Roseboom TJ, van der Meulen JH, van Montfrans GA, Ravelli AC, Osmond C, Barker DJ, et al. Maternal nutrition during gestation and blood pressure in later life. J Hypertens. 2001;19:29–34. doi: 10.1097/00004872-200101000-00004. [DOI] [PubMed] [Google Scholar]
- 136.Stein AD, Zybert PA, van der Pal-de Bruin K, Lumey LH. Exposure to famine during gestation, size at birth, and blood pressure at age 59 years: evidence from the Dutch Famine. Eur J Epidemiol. 2006;21:759–65. [DOI] [PubMed]
- 137.Painter RC, de Rooij SR, Bossuyt PM, Phillips DI, Osmond C, Barker DJ, et al. Blood pressure response to psychological stressors in adults after prenatal exposure to the Dutch famine. J Hypertens. 2006;24:1771–1778. doi: 10.1097/01.hjh.0000242401.45591.e7. [DOI] [PubMed] [Google Scholar]
- 138.Roseboom TJ, van der Meulen JH, Osmond C, Barker DJ, Ravelli AC, Schroeder-Tanka JM, et al. Coronary heart disease after prenatal exposure to the Dutch famine, 1944–45. Heart. 2000;84:595–598. doi: 10.1136/heart.84.6.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Painter RC, de Rooij SR, Bossuyt PM, Simmers TA, Osmond C, Barker DJ, et al. Early onset of coronary artery disease after prenatal exposure to the Dutch famine. Am J Clin Nutr. 2006;84:322–327. doi: 10.1093/ajcn/84.1.322. [DOI] [PubMed] [Google Scholar]
- 140.Painter RC, Roseboom TJ, Bossuyt PM, Osmond C, Barker DJ, Bleker OP. Adult mortality at age 57 after prenatal exposure to the Dutch famine. Eur J Epidemiol. 2005;20:673–676. doi: 10.1007/s10654-005-7921-0. [DOI] [PubMed] [Google Scholar]
- 141.Painter RC, de Rooij SR, Hutten BA, Bossuyt PM, de Groot E, Osmond C, et al. Reduced intima media thickness in adults after prenatal exposure to the Dutch famine. Atherosclerosis. 2007;193:421–427. doi: 10.1016/j.atherosclerosis.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 142.van Abeelen AF, Veenendaal MV, Painter RC, de Rooij SR, Dijkgraaf MG, Bossuyt PM, et al. Survival effects of prenatal famine exposure. Am J Clin Nutr. 2012;95:179–183. doi: 10.3945/ajcn.111.022038. [DOI] [PubMed] [Google Scholar]
- 143.Susser E, Neugebauer R, Hoek HW, Brown AS, Lin S, Labovitz D, et al. Schizophrenia after prenatal famine. Further evidence. Arch Gen Psychiatry. 1996;53:25–31. doi: 10.1001/archpsyc.1996.01830010027005. [DOI] [PubMed] [Google Scholar]
- 144.Roseboom TJ, van der Meulen JH, Ravelli AC, Osmond C, Barker DJ, Bleker OP, et al. Plasma fibrinogen and factor VII concentrations in adults after prenatal exposure to famine. Br J Haematol. 2000;111:112–117. doi: 10.1046/j.1365-2141.2000.02268.x. [DOI] [PubMed] [Google Scholar]
- 145.Lopuhaa CE, Roseboom TJ, Osmond C, Barker DJ, Ravelli AC, Bleker OP, et al. Atopy, lung function, and obstructive airways disease after prenatal exposure to famine. Thorax. 2000;55:555–561. doi: 10.1136/thorax.55.7.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.de Rooij SR, Painter RC, Phillips DI, Osmond C, Michels RP, Bossuyt PM, et al. Hypothalamic–pituitary–adrenal axis activity in adults who were prenatally exposed to the Dutch famine. Eur J Endocrinol. 2006;155:153–160. doi: 10.1530/eje.1.02193. [DOI] [PubMed] [Google Scholar]
- 147.Lai MS, Hsueh YM, Chen CJ, Shyu MP, Chen SY, Kuo TL, et al. Ingested inorganic arsenic and prevalence of diabetes mellitus. Am J Epidemiol. 1994;139:484–492. doi: 10.1093/oxfordjournals.aje.a117031. [DOI] [PubMed] [Google Scholar]
- 148.Henriksen GL, Ketchum NS, Michalek JE, Swaby JA. Serum dioxin and diabetes mellitus in veterans of Operation Ranch Hand. Epidemiology. 1997;8:252–258. doi: 10.1097/00001648-199705000-00005. [DOI] [PubMed] [Google Scholar]
- 149.Longnecker MP, Michalek JE. Serum dioxin level in relation to diabetes mellitus among Air Force veterans with background levels of exposure. Epidemiology. 2000;11:44–48. doi: 10.1097/00001648-200001000-00010. [DOI] [PubMed] [Google Scholar]
- 150.Gladen BC, Ragan NB, Rogan WJ. Pubertal growth and development and prenatal and lactational exposure to polychlorinated biphenyls and dichlorodiphenyl dichloroethene. J Pediatr. 2000;136:490–496. doi: 10.1016/S0022-3476(00)90012-X. [DOI] [PubMed] [Google Scholar]
- 151.Cox S, Niskar AS, Narayan KM, Marcus M. Prevalence of self-reported diabetes and exposure to organochlorine pesticides among Mexican Americans: Hispanic health and nutrition examination survey, 1982–1984. Environ Health Perspect. 2007;115:1747–1752. doi: 10.1289/ehp.10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Codru N, Schymura MJ, Negoita S, Akwesasne Task Force on Environment, Rei R, Carpenter DO. Diabetes in relation to serum levels of polychlorinated biphenyls and chlorinated pesticides in adult Native Americans. Environ Health Perspect. 2007;115:1442–7. [DOI] [PMC free article] [PubMed]
- 153.Lee DH, Lee IK, Porta M, Steffes M, Jacobs DR., Jr Relationship between serum concentrations of persistent organic pollutants and the prevalence of metabolic syndrome among non-diabetic adults: results from the National Health and Nutrition Examination Survey 1999–2002. Diabetologia. 2007;50:1841–1851. doi: 10.1007/s00125-007-0755-4. [DOI] [PubMed] [Google Scholar]
- 154.Hanswell-Elkins M, Satarug S, O’Rourke P, Moore M, Ng J, McGrath V, et al. Striking association between urinary cadmium level and albuminuria among Torres Strait Islander people with diabetes. Environ Res. 2008;106:379–383. doi: 10.1016/j.envres.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 155.Michalek JE, Pavuk M. Diabetes and cancer in veterans of Operation Ranch Hand after adjustment for calendar period, days of spraying, and time spent in Southeast Asia. J Occup Environ Med. 2008;50:330–340. doi: 10.1097/JOM.0b013e31815f889b. [DOI] [PubMed] [Google Scholar]
- 156.Rignell-Hydbom A, Lidfeldt J, Kiviranta H, Rantakokko P, Samsioe G, Agardh CD, et al. Exposure to p, p′-DDE: a risk factor for type 2 diabetes. PLoS One. 2009;4:e7503. doi: 10.1371/journal.pone.0007503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lundin JI, Alexander BH, Olsen GW, Church TR. Ammonium perfluorooctanoate production and occupational mortality. Epidemiology. 2009;20:921–928. doi: 10.1097/EDE.0b013e3181b5f395. [DOI] [PubMed] [Google Scholar]
- 158.Turyk M, Anderson H, Knobeloch L, Imm P, Persky V. Organochlorine exposure and incidence of diabetes in a cohort of Great Lakes sport fish consumers. Environ Health Perspect. 2009;117:1076–1082. doi: 10.1289/ehp.0800281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lopez-Espinosa MJ, Freire C, Arrebola JP, Navea N, Taoufiki J, Fernandez MF, et al. Nonylphenol and octylphenol in adipose tissue of women in Southern Spain. Chemosphere. 2009;76:847–852. doi: 10.1016/j.chemosphere.2009.03.063. [DOI] [PubMed] [Google Scholar]
- 160.Ukropec J, Radikova Z, Huckova M, Koska J, Kocan A, Sebokova E, et al. High prevalence of prediabetes and diabetes in a population exposed to high levels of an organochlorine cocktail. Diabetologia. 2010;53:899–906. doi: 10.1007/s00125-010-1683-2. [DOI] [PubMed] [Google Scholar]
- 161.Son HK, Kim SA, Kang JH, Chang YS, Park SK, Lee SK, et al. Strong associations between low-dose organochlorine pesticides and type 2 diabetes in Korea. Environ Int. 2010;36:410–414. doi: 10.1016/j.envint.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 162.Lee DH, Steffes MW, Sjodin A, Jones RS, Needham LL, Jacobs DR., Jr Low dose of some persistent organic pollutants predicts type 2 diabetes: a nested case-control study. Environ Health Perspect. 2010;118:1235–1242. doi: 10.1289/ehp.0901480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Park SK, Son HK, Lee SK, Kang JH, Chang YS, Jacobs DR, et al. Relationship between serum concentrations of organochlorine pesticides and metabolic syndrome among non-diabetic adults. J Prev Med Public Health. 2010;43:1–8. doi: 10.3961/jpmph.2010.43.1.1. [DOI] [PubMed] [Google Scholar]
- 164.Mendez MA, Garcia-Esteban R, Guxens M, Vrijheid M, Kogevinas M, Goni F, et al. Prenatal organochlorine compound exposure, rapid weight gain, and overweight in infancy. Environ Health Perspect. 2011;119:272–278. doi: 10.1289/ehp.1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Carwile JL, Michels KB. Urinary bisphenol A and obesity: NHANES 2003–2006. Environ Res. 2011;111:825–830. doi: 10.1016/j.envres.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gasull M, Pumarega J, Tellez-Plaza M, Castell C, Tresserras R, Lee DH, et al. Blood concentrations of persistent organic pollutants and prediabetes and diabetes in the general population of Catalonia. Environ Sci Technol. 2012;46:7799–7810. doi: 10.1021/es300712g. [DOI] [PubMed] [Google Scholar]
- 167.Merali Z, Singhal RL. Diabetogenic effects of chronic oral cadmium administration to neonatal rats. Br J Pharmacol. 1980;69:151–157. doi: 10.1111/j.1476-5381.1980.tb10895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rubin BS, Murray MK, Damassa DA, King JC, Soto AM. Perinatal exposure to low doses of bisphenol A affects body weight, patterns of estrous cyclicity, and plasma LH levels. Environ Health Perspect. 2001;109:657–680. doi: 10.1289/ehp.01109675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Miyawaki J, Sakayama K, Kato H, Yamamoto H, Masuno H. Perinatal and postnatal exposure to bisphenol a increases adipose tissue mass and serum cholesterol level in mice. J Atheroscler Thromb. 2007;14:245–252. doi: 10.5551/jat.E486. [DOI] [PubMed] [Google Scholar]
- 170.Hines EP, White SS, Stanko JP, Gibbs-Flournoy EA, Lau C, Fenton SE. Phenotypic dichotomy following developmental exposure to perfluorooctanoic acid (PFOA) in female CD-1 mice: low doses induce elevated serum leptin and insulin, and overweight in mid-life. Mol Cell Endocrinol. 2009;304:97–105. doi: 10.1016/j.mce.2009.02.021. [DOI] [PubMed] [Google Scholar]