

Abstract
Oxidative stress and inflammation are implicated in cardiovascular diseases such as atherosclerosis, reperfusion injury, hypertension, and heart failure. High levels of oxidative stress resulting from increased cardiac generation of reactive oxygen species (ROS) is thought to contribute to contractile and endothelial dysfunction, apoptosis and necrosis of myocytes, and extracellular matrix remodeling in the heart. ROS activate several transcription factors known as redox-regulated transcription factors, and these transcription factors play important roles in the pathophysiology of cardiovascular diseases. This review focuses on the pathological roles of environmental and redox stresses in cardiovascular diseases, especially severe cardiac dysfunction and the transition from compensated hypertrophy to heart failure. The aryl hydrocarbon receptor (AHR) and NF-E2 p45-related factor (Nrf2) are transcription factors involved in the regulation of drug-metabolizing enzymes. AHR has been studied as a receptor for environmental contaminants and as a mediator of chemical toxicity. However, other roles for AHR in cardiac and vascular development have recently been described. Moreover, Nrf2 protects against oxidative stress by increasing the transcription of genes, including those for several antioxidant enzymes. The roles of these transcription factors, AHR and Nrf2 in angiogenesis are also discussed in this review.
Keywords: Environmental cardiology, Oxidative stress, Redox-regulated transcription factors, Molecular mechanisms, Cardiovascular diseases
Introduction
Reactive oxygen species (ROS) are implicated in ailments of the cardiovascular system, such as atherosclerosis, reperfusion injury, hypertension, and heart failure [1, 2]. Increased oxidative stress resulting from the overproduction of ROS or low levels of available antioxidants is thought to contribute to contractile and endothelial dysfunction, apoptosis and necrosis of myocytes, and remodeling of the extracellular matrix in the myocardium [3]. Such oxidative stress has indeed been demonstrated in patients with various hypertensive disorders [4] as well as in animal models of heart failure [5, 6].
Roles of oxidative stress in cardiac dysfunction
Chronic heart failure is characterized by chronically and tonically enhanced sympathetic tone and hypercatecholaminemia [7], leading to left ventricular (LV) dysfunction. Downregulation and desensitization of β-adrenergic receptor signaling is a major contributing factor in the contractile dysfunction of the failing heart. Certain β-blockers are known to reduce the level of lipid peroxidation evident in the myocardium of patients with dilated cardiomyopathy (DCM) [8] and to inhibit the upregulation of the DNA binding activities of redox-regulated transcription factors in neonatal rat cardiac ventricular myocytes [9], suggesting that one of the beneficial effects of β-blockers in individuals with heart failure is attenuation of oxidative stress. In addition, superoxide production or biochemical markers of oxidative stress are increased in individuals with severe heart failure [10]. Oxidative stress is therefore an important susceptibility factor for cardiac dysfunction, and agents that reduce the level of such stress or interfere with the generation of intracellular ROS are potentially suitable for the treatment of chronic heart failure [11].
To determine the kinetics of the onset and progression of LV dysfunction resulting in heart failure in TO-2 hamsters (an appropriate model of DCM), we characterized the serial changes in LV function and structure by echocardiography and histological analysis, as well as by the extent of oxidative stress and β-adrenergic signaling in the LV myocardium [12, 13]. As shown in Fig. 1A, LV dysfunction first appeared at 8 weeks of age and deteriorated thereafter in TO-2 hamsters. Compared with the control, the glutathione redox ratio in the LV myocardium of TO-2 hamsters increased (indicating oxidative stress) at 4 weeks and became significantly higher after 8 weeks (Fig. 1B). The hearts of TO-2 hamsters had significantly lower levels of superoxide dismutase (SOD) activity from 8 weeks onward compared with the control hamsters (Fig. 1C). Moreover, adenylyl cyclase activity was significantly lower in the LV myocardium at 18 weeks (Fig. 1D). These observations indicated that myocardial oxidative stress was actually enhanced in the initial development of LV dysfunction. Both the activation of myocardial oxidative stress and impairment of β-adrenergic signaling became prominent at the stage of severe LV dysfunction, suggesting the involvement of myocardial oxidative stress in the development of β-adrenergic desensitization, leading to cardiac dysfunction.
Fig. 1.
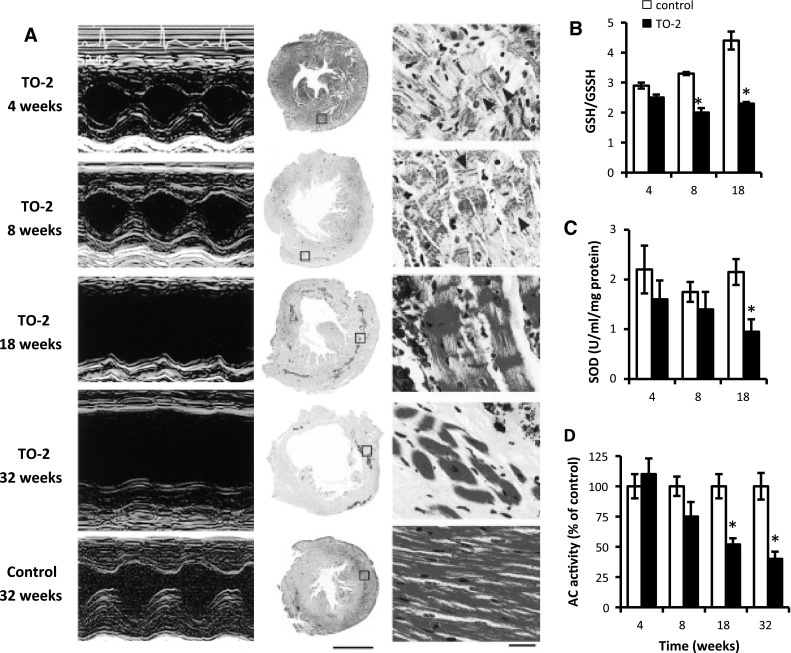
Serial changes in left ventricular (LV) function and extent of oxidative stress in TO-2 and control hamsters. A Representative echocardiograms and representative light photomicrographs of LV cross-sections stained with hematoxylin–eosin. B–D Glutathione redox ratio (GSH/GSSG; oxidized glutathione/reduced glutathione) (B), superoxide dismutase (SOD) enzyme activity (C), and adenylyl cyclase (AC) activity (D) in the LV myocardium. Data are mean ± standard error of the mean (SEM) of 8 animals in each experiment. *P < 0.05 versus age-matched controls
Doxorubicin (DOX) is one of the most widely used drugs in the treatment of a variety of human neoplasms [14]. However, long-term treatment with DOX can result in the development of cardiomyopathy and congestive heart failure in a process that involves multiple factors, including the generation of free radicals that damage cellular membranes [15, 16], disturbance of adrenergic function, alterations in intracellular Ca2+ homeostasis [17], myocardial cell apoptosis [18], and selective inhibition of the expression of cardiac muscle-specific proteins [19]. We investigated the mechanism of DOX-induced cardiotoxicity in mice as well as whether polyethylene glycol-conjugated SOD (PEG-SOD) protects against such toxicity. PEG-SOD is a membrane-permeable antioxidant with a long half-life in plasma [20], and pretreatment with this conjugate has been reported to protect against reperfusion-induced arrhythmias as well as myocardial ischemia–reperfusion injury in animal models [21, 22]. Our results demonstrated that DOX-induced cardiotoxicity was associated with increased oxidative stress, apoptosis, and impaired angiogenesis in the left ventricle of mice [23]. Furthermore, treatment with PEG-SOD ameliorated DOX-induced cardiac dysfunction, and this effect was mediated through inhibition of DOX-induced upregulation of nuclear factor-κB (NF-κB) signaling, lowering the levels of hexanoyl-lysine (HEL), a marker of free radical-induced lipid peroxidation, and suppression of the activation of Akt and Akt-regulated gene expression [23]. These results suggested that the antioxidant PEG-SOD prevented cardiac dysfunction induced by DOX through normalization of oxidative stress and redox-regulated NF-κB signaling.
Association between cardiac function and regulation of redox-regulated transcription factors
Reactive oxygen species activate several transcription factors, including NF-κB, activator protein-1 (AP-1), early growth response gene-1 (Egr-1), surfactant protein 1 (SP1), and E26 transformation specific-1 (Ets-1) [24]. The binding sites for NF-κB and AP-1 are present in the promoter regions of various genes that are important in the pathogenesis of cardiovascular diseases [25]. Expression of Egr-1 is the highest in the brain and heart and is increased in the intact heart or neonatal cardiomyocytes following stimulation with endothelin, angiotensin II, and DOX [26, 27]. SP1 and Ets-1 are expressed in a variety of cell types, including endothelial cells, vascular smooth muscle cells, and cardiomyocytes, and they regulate the transcription of several genes that function in angiogenesis or remodeling of the extracellular matrix [28, 29]. Peroxisome proliferator-activated receptor-α (PPAR-α) belongs to the nuclear receptor superfamily and contributes to the regulation of the expression of genes for transport proteins and enzymes that participate in fatty acid and triglyceride metabolism [30]. PPAR-α is expressed in cells of the cardiovascular system, including endothelial cells, vascular smooth muscle cells, monocytes–macrophages, and cardiomyocytes [31–33]. The results of several recent animal studies have suggested the substantial involvement of PPAR-α deactivation in the phenotypic changes that accompany cardiac growth in conditions of pressure overload, and that a compromised PPAR-α activity may participate in the deterioration of compensated LV hypertrophy to heart failure in hypertensive heart disease [34, 35]. Furthermore, Gomez-Garre et al. [36] have shown that PPAR-α and PPAR-γ are selectively activated in the atria of patients with end-stage heart failure and might be functionally important in the maintenance of atrial morphology. We investigated the effects of fenofibrate, a ligand and activator of PPAR-α, on the development of myocardial hypertrophy and progression from compensated LV hypertrophy to heart failure in Dahl salt-sensitive (DS) rats [37]. In the same study we also examined the effects of this substance on the inflammatory response and on the activities of redox-regulated transcription factors in the heart of these animals. DS rats fed a high-salt diet developed cardiac hypertrophy at 12 weeks of age, but this was significantly attenuated in rats treated with fenofibrate. At 18 weeks of age, the LV end-diastolic diameter was significantly greater in rats fed the high-salt diet than in rats fed the low-salt diet, but this was significantly attenuated in rats treated with fenofibrate. A marked increase in the extent of interstitial fibrosis in the left ventricle was noted in 18-week-old rats fed the high-salt diet compared to rats fed the low-salt diet (Fig. 2A). This increase in cardiac fibrosis was greatly reduced by treatment with both low- and high-dose fenofibrate. Infiltration of macrophages and T lymphocytes into the left ventricle was also pronounced in rats fed the high-salt diet compared with those fed the low-salt diet, and the accumulation of such cells was again suppressed by fenofibrate (Fig. 2A). Furthermore, the DNA binding activities of NF-κB and AP-1 were significantly higher in rats fed the high-salt diet compared with those fed the low-salt diet. Treatment with both low- and high-dose fenofibrate resulted in a significant reduction in the DNA binding activities of these transcription factors (Fig. 2B, C). The PPAR-α activator fenofibrate attenuated the progression of heart failure and improved the survival rate in this rat model. These effects were associated with inhibition of the inflammatory response and suppression of activation of redox-regulated transcription factors in the left ventricle. The findings suggested that PPAR-α seems to function as a modulator of inflammation in the heart and that its activation may have beneficial effects on cardiac metabolism and function in normal and diseased states.
Fig. 2.
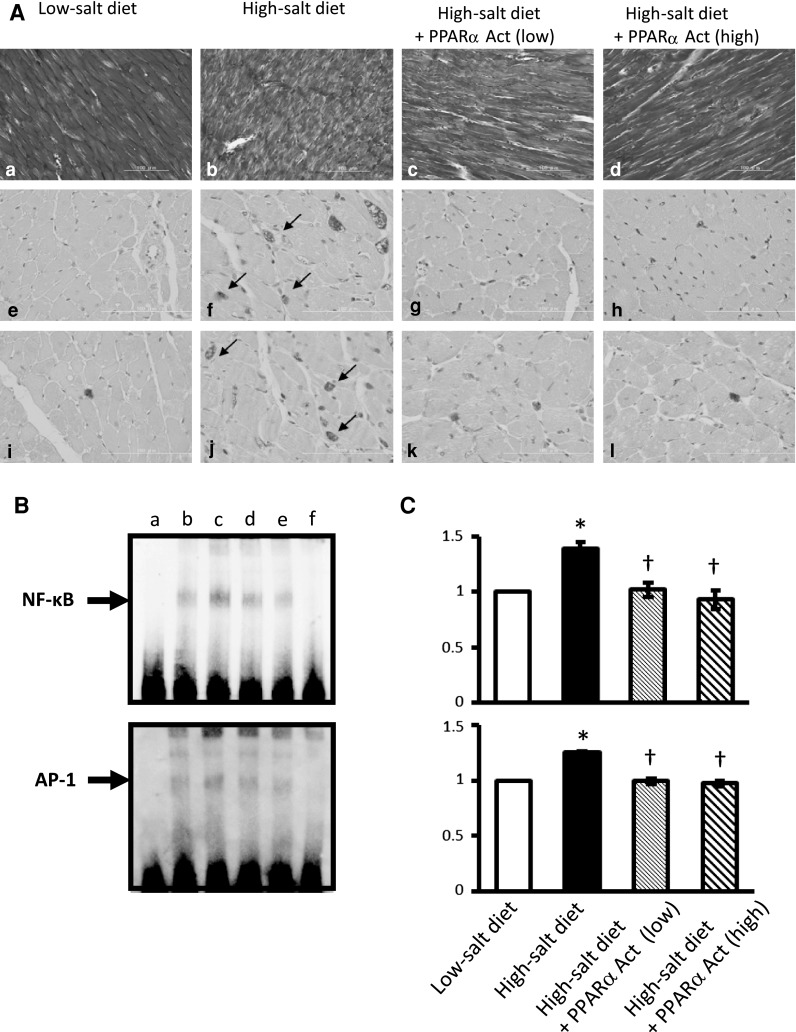
Cardiac inflammatory responses and DNA binding activities of redox-regulated transcription factors in the left ventricle of Dahl salt-sensitive (DS) rats. Aa–d Interstitial collagen deposition in the left ventricle as demonstrated by Azan-Mallory staining. e–l Immunostaining of LV tissue for macrophages (e–h) and T lymphocytes (i–l) of DS rats fed a low-salt diet (0.3 % NaCl) or a high-salt diet and treated either with vehicle (8 % NaCl) or with fenofibrate at doses of 30 mg/kg per day [8 % NaCl + peroxisome proliferator-activated receptor-α (PPARα) Act (low)] or 50 mg/kg per day [8 % NaCl + PPARα Act (high)]. Arrows Infiltrated immunoreactive cells. B Representative electrophoretic mobility-shift assays for the binding activities of nuclear factor-κB (NF-κB) and activator protein-1 (AP-1). Lanes: a Free extracts, b low-salt diet group, c high-salt diet group, d high-salt diet + PPARα Act (low) group, e high-salt diet + PPARα Act (high) group, f cold probe. C Quantitative data expressed relative to the corresponding value of the low-salt diet group. Results are presented as the mean ± SEM of 4 animals in each experiment. *P < 0.05 vs. rats fed the low-salt diet, †P < 0.05 vs. rats fed the high-salt diet and treated with vehicle
Environmental stress responses in cardiovascular diseases
Cigarette smoking is a major risk factor for ischemic heart disease, peripheral vascular disease, and chronic obstructive pulmonary disease [38, 39]. It has adverse effects on vascular biology, inducing endothelial dysfunction and arterial stiffness [40], and it inhibits angiogenesis by pulmonary artery endothelial cells in the setting of severe vascular obstruction and lung tissue ischemia [41, 42]. The effects of benzo[a]pyrene (B[a]P) in cigarette smoking and of other polycyclic and halogenated aromatic hydrocarbons in the environment are mediated by the aryl hydrocarbon receptor (AHR) [43]. The ligand-bound form of the AHR and AHR nuclear translocator (ARNT), which belongs to the Per–Arnt–Sim (PAS) family of basic helix–loop–helix transcription factors, form a heterodimeric transcription factor [44] which binds to the xenobiotic response elements (XREs) in the promoter regions of the target genes, such as that encoding cytochrome P450 1A1 (CYP1A1) [45]. Activation of vascular endothelial growth factor (VEGF) is mediated by the binding of another basic helix–loop–helix transcription factor, hypoxia-inducible factor-1α (HIF-1α) [46], which also dimerizes with ARNT [47]. To examine the effects of cigarette smoking on angiogenesis, we treated mice with B[a]P, one of the polycyclic and halogenated aromatic hydrocarbons found in tobacco smoke, in an animal model in which ischemia was induced surgically in a hindlimb [48]. Oral exposure to B[a]P resulted in significant inhibition of ischemia-induced increase in hindlimb blood flow in wild-type mice (Fig. 3A, B). AHR deficiency attenuated the inhibition of ischemia-induced angiogenesis by B[a]P, and the B[a]P-induced increase in the amount of CYP1A1 mRNA in wild-type mice was not apparent in AHR-null mice. Ischemia-induced angiogenesis was markedly enhanced in AHR-null mice compared with that in wild-type animals [49]. Ischemia-induced upregulation of the expression of HIF-1α and ARNT, as well as that of target genes for these transcription factors, such as that for VEGF, were also enhanced in AHR-null mice (Fig. 3C). Furthermore, both the DNA binding activity of the HIF-1α–ARNT complex (Fig. 3D) and the association of HIF-1α and ARNT with the VEGF gene promoter were increased by ischemia to a greater extent in AHR-null mice than in wild-type mice, suggesting that AHR–ARNT signaling plays an important role in the regulation of ischemia-induced angiogenesis [50].
Fig. 3.

Changes in blood flow ratio after arterial ligation in the left hindlimb of aryl hydrocarbon receptor (AHR)-null or wild-type (WT) mice exposed to benzo[a]pyrene (B[a]P) or vehicle. A Laser Doppler perfusion imaging of blood flow immediately and 4 weeks after surgery and the commencement of weekly administration of B[a]P (125 mg/kg) or vehicle. B The ratio of blood flow in the ischemic (left) hindlimb to that in the normal (right) hindlimb measured immediately and each week after surgery and the commencement of weekly administration of B[a]P or vehicle. All quantitative data are given as the mean ± SEM of values from 8 mice per group. *P < 0.05 vs. corresponding value for vehicle-treated WT mice, †P < 0.05 vs. corresponding value for B[a]P-treated WT mice. C Representative immunoblot (IB) analysis of hypoxia-inducible factor-1α (HIF-1α), AHR nuclear translocator (ARNT), and lamin (loading control) in nuclear extracts, and vascular endothelial growth factor (VEGF) in ischemic or non-ischemic tissue at 1 week after surgery. D Quantitative data of the DNA binding activity of HIF-1α–ARNT in nuclear extracts of tissue isolated 1 week after surgery. All data are mean ± SEM of values from 6 mice per group. *P < 0.05 vs. non-ischemic hindlimb of WT mice; †P < 0.05 vs. non-ischemic hindlimb of AHR-null mice, ‡P < 0.05 vs. ischemic hindlimb of wild-type mice
The transcription factor Nrf2 [nuclear factor-erythroid 2 (NF-E2)-related factor 2; also known as NF-E2 like 2 (Nfe2l2)] is a member of the NF-E2 basic-leucine zipper family of proteins and interacts with the antioxidant response element (ARE) that is present in the promoter regions of genes for phase 2 detoxifying enzymes [51]. It also protects against oxidative stress through ARE-mediated transcriptional activation of the genes for several antioxidant enzymes [52]. Nrf2 signaling was reported to protect against renal oxidative damage induced by ferric nitrilotriacetate [53], mitochondrial complex II inhibitor-mediated neurotoxicity [54], and pulmonary hyperoxic injury [55]. Moreover, genetic ablation of Nrf2 was found to increase susceptibility to severe airway inflammation and to cigarette smoke-induced emphysema in mice [56, 57]. Our group investigated the potential role of Nrf2 in neovascularization with a murine surgical model of ischemia [58]. Nrf2-deficient animals (mice lacking the transcription factor Nrf2) showed markedly enhanced ischemia-induced neovascularization, underexpression of Nrf2 target genes encoding antioxidant enzymes, and potentiated inflammatory response [58]. The promotion of neovascularization by Nrf2 deficiency may thus be attributed to the enhanced inflammatory response resulting from impaired antioxidant defense and ROS accumulation in endothelial cells.
Conclusion and future perspectives
Increased oxidative stress is implicated in the pathogenesis of heart failure, and inflammation is thought to play an important role in the progression of cardiovascular diseases. Myocardial oxidative stress is actually enhanced even at the initial stages of LV dysfunction. Treatments that reduce the level of inflammation promoted by activating the redox-regulated transcription factors are thus important to improve hemodynamic function in patients with advanced heart failure.
Heart diseases develop as a result of complex interactions between genes and environment. Although we demonstrated the significant associations of gene polymorphisms with cardiovascular diseases [59, 60], the odds ratio for the effect of the genotype of susceptibility genes to cardiovascular diseases was only moderate. This evidence suggests that large subsets of genes involved in many processes, such as blood pressure regulation, cholesterol metabolism, insulin sensitivity, redox signaling, antioxidant defense, and inflammation, contribute to the risk of complex traits, such as cardiovascular diseases. Since lifestyle choices, such as smoking, diet, and exercise, are considered to be key factors in cardiovascular diseases, exposure to pollutants and environmental chemicals could elevate the risk of cardiovascular diseases. In fact, exposure to fine particles, arsenic, lead, cadmium, solvents, and pesticides have been linked to increased incidence of cardiovascular diseases [61]. These effects might be attributed to changes in the synthesis or reactivity of nitric oxide that may be caused by environmental oxidants or increased endogenous production of ROS [62, 63]. To contribute to the field of environmental cardiology, we need to elucidate the underlying physiological and molecular mechanisms and estimate the relative susceptibility of individuals for cardiovascular diseases.
Acknowledgments
This study was supported in part by grants from the Japan Society for the Promotion of Science, from the Takeda Science Foundation, from the Astellas Foundation for Research on Metabolic Disorders, and from the Suzuken Memorial Foundation.
Conflict of interest
None.
References
- 1.Alexander RW. Hypertension and the pathogenesis of atherosclerosis. Oxidative stress and the mediation of arterial inflammatory response: a new perspective. Hypertension. 1995;25:155–161. doi: 10.1161/01.HYP.25.2.155. [DOI] [PubMed] [Google Scholar]
- 2.Ide T, Tsutsui H, Kinugawa S, Suematsu N, Hayashidani S, Ichikawa K, et al. Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ Res. 2000;86:152–157. doi: 10.1161/01.RES.86.2.152. [DOI] [PubMed] [Google Scholar]
- 3.Chien KR. Stress pathways and heart failure. Cell. 1999;98:555–558. doi: 10.1016/S0092-8674(00)80043-4. [DOI] [PubMed] [Google Scholar]
- 4.Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, et al. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol. 2003;41:2164–2171. doi: 10.1016/S0735-1097(03)00471-6. [DOI] [PubMed] [Google Scholar]
- 5.Khaper N, Singal PK. Modulation of oxidative stress by a selective inhibition of angiotensin II type 1 receptors in MI rats. J Am Coll Cardiol. 2001;37:1461–1466. doi: 10.1016/S0735-1097(01)01126-3. [DOI] [PubMed] [Google Scholar]
- 6.Ichihara S, Noda A, Nagata K, Obata K, Xu J, Ichihara G, et al. Pravastatin increases survival and suppresses an increase in myocardial metalloproteinase activity in a rat model of heart failure. Cardiovasc Res. 2006;69:726–735. doi: 10.1016/j.cardiores.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Cohn JN, Levine TB, Olivari MT, Garberg V, Lura D, Francis GS, et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med. 1984;311:819–823. doi: 10.1056/NEJM198409273111303. [DOI] [PubMed] [Google Scholar]
- 8.Kukin ML, Kalman J, Charney RH, Levy DK, Buchholz-Varley C, Ocampo ON, et al. Prospective, randomized comparison of effect of long-term treatment with metoprolol or carvedilol on symptoms, exercise, ejection fraction, and oxidative stress in heart failure. Circulation. 1999;99:2645–2651. doi: 10.1161/01.CIR.99.20.2645. [DOI] [PubMed] [Google Scholar]
- 9.Koitabashi N, Arai M, Tomaru K, Takizawa T, Watanabe A, Niwano K, et al. Carvedilol effectively blocks oxidative stress-mediated downregulation of sarcoplasmic reticulum Ca2+-ATPase 2 gene transcription through modification of Sp1 binding. Biochem Biophys Res Commun. 2005;328:116–124. doi: 10.1016/j.bbrc.2004.12.139. [DOI] [PubMed] [Google Scholar]
- 10.Tsutamoto T, Wada A, Matsumoto T, Maeda K, Mabuchi N, Hayashi M, et al. Relationship between tumor necrosis factor-alpha production and oxidative stress in the failing hearts of patients with dilated cardiomyopathy. J Am Coll Cardiol. 2001;37:2086–2092. doi: 10.1016/S0735-1097(01)01299-2. [DOI] [PubMed] [Google Scholar]
- 11.Nojiri S, Daida H, Inaba Y. Antioxidants and cardiovascular disease: still a topic of interest. Environ Health Prev Med. 2004;9:200–213. doi: 10.1007/BF02898101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishizawa T, Iwase M, Kanazawa H, Ichihara S, Ichihara G, Nagata K, et al. Serial alterations of β-adrenergic signaling in dilated cardiomyopathic hamsters—possible role of myocardial oxidative stress. Circ J. 2004;68:1051–1060. doi: 10.1253/circj.68.1051. [DOI] [PubMed] [Google Scholar]
- 13.Ichihara S, Yamada Y, Ichihara G, Kanazawa H, Hashimoto K, Kato Y, et al. Attenuation of oxidative stress and cardiac dysfunction by bisoprolol in an animal model of dilated cardiomyopathy. Biochem Biophys Res Commun. 2006;350:105–113. doi: 10.1016/j.bbrc.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 14.Blum RH, Carter SK. Adriamycin. A new anticancer drug with significant clinical activity. Ann Intern Med. 1974;80:249–259. doi: 10.7326/0003-4819-80-2-249. [DOI] [PubMed] [Google Scholar]
- 15.Keizer HG, Pinedo HM, Schuurhuis GJ, Joenje H. Doxorubicin (adriamycin): a critical review of free radical-dependent mechanisms of cytotoxicity. Pharmacol Ther. 1990;47:219–231. doi: 10.1016/0163-7258(90)90088-J. [DOI] [PubMed] [Google Scholar]
- 16.Rajagopalan S, Politi PM, Sinha BK, Myers CE. Adriamycin-induced free radical formation in the perfused rat heart: implications for cardiotoxicity. Cancer Res. 1988;48:4766–4769. [PubMed] [Google Scholar]
- 17.Kim DH, Landry 3rd AB, Lee YS, Katz AM. Doxorubicin-induced calcium release from cardiac sarcoplasmic reticulum vesicles. J Mol Cell Cardiol. 1989;21:433–6. [DOI] [PubMed]
- 18.Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M, Voipio-Pulkki LM. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000;60:1789–1792. [PubMed] [Google Scholar]
- 19.Jeyaseelan R, Poizat C, Baker RK, Abdishoo S, Isterabadi LB, Lyons GE, et al. A novel cardiac-restricted target for doxorubicin. CARP, a nuclear modulator of gene expression in cardiac progenitor cells and cardiomyocytes. J Biol Chem. 1997;272:22800–22808. doi: 10.1074/jbc.272.36.22800. [DOI] [PubMed] [Google Scholar]
- 20.Veronese FM, Caliceti P, Schiavon O, Sergi M. Polyethylene glycol-superoxide dismutase, a conjugate in search of exploitation. Adv Drug Deliv Rev. 2002;54:587–606. doi: 10.1016/S0169-409X(02)00029-7. [DOI] [PubMed] [Google Scholar]
- 21.Tamura Y, Chi LG, Driscoll EM, Jr, Hoff PT, Freeman BA, Gallagher KP, et al. Superoxide dismutase conjugated to polyethylene glycol provides sustained protection against myocardial ischemia/reperfusion injury in canine heart. Circ Res. 1988;63:944–959. doi: 10.1161/01.RES.63.5.944. [DOI] [PubMed] [Google Scholar]
- 22.Yamakawa T, Kadowaki Y, Garcia-Alves M, Yokoyama M, Iwashita Y, Nishi K. Effects of polyoxyethylene-modified superoxide dismutase on reperfusion induced arrhythmia in isolated rat and guinea-pig hearts. J Mol Cell Cardiol. 1989;21:441–452. doi: 10.1016/0022-2828(89)90784-0. [DOI] [PubMed] [Google Scholar]
- 23.Ichihara S, Yamada Y, Kawai Y, Osawa T, Furuhashi K, Duan Z, et al. Roles of oxidative stress and Akt signaling in doxorubicin cardiotoxicity. Biochem Biophys Res Commun. 2007;359:27–33. doi: 10.1016/j.bbrc.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 24.Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996;10:709–720. doi: 10.1096/fasebj.10.7.8635688. [DOI] [PubMed] [Google Scholar]
- 25.Frantz S, Fraccarollo D, Wagner H, Behr TM, Jung P, Angermann CE, et al. Sustained activation of nuclear factor kappa B and activator protein 1 in chronic heart failure. Cardiovasc Res. 2003;57:749–756. doi: 10.1016/S0008-6363(02)00723-X. [DOI] [PubMed] [Google Scholar]
- 26.Khachigian LM, Lindner V, Williams AJ, Collins T. Egr-1-induced endothelial gene expression: a common theme in vascular injury. Science. 1996;271:1427–1431. doi: 10.1126/science.271.5254.1427. [DOI] [PubMed] [Google Scholar]
- 27.Saadane N, Alpert L, Chalifour LE. TAFII250, Egr-1, and D-type cyclin expression in mice and neonatal rat cardiomyocytes treated with doxorubicin. Am J Physiol Heart Circ Physiol. 1999;276:H803–H814. doi: 10.1152/ajpheart.1999.276.3.H803. [DOI] [PubMed] [Google Scholar]
- 28.Kadonaga JT, Carner KR, Masiarz FR, Tjian R. Isolation of cDNA encoding transcription factor SP1 and functional analysis of the DNA binding domain. Cell. 1987;51:1079–1090. doi: 10.1016/0092-8674(87)90594-0. [DOI] [PubMed] [Google Scholar]
- 29.Hultgardh-Nilsson A, Cercek B, Wang JW, Naito S, Lovdahl C, Sharifi B, et al. Regulated expression of the ets-1 transcription factor in vascular smooth muscle cells in vivo and in vitro. Circ Res. 1996;78:589–595. doi: 10.1161/01.RES.78.4.589. [DOI] [PubMed] [Google Scholar]
- 30.Motojima K, Passilly P, Peters JM, Gonzalez FJ, Latruffe N. Expression of putative fatty acid transporter genes are regulated by peroxisome proliferator-activated receptor α and γ activators in a tissue- and inducer-specific manner. J Biol Chem. 1998;273:16710–16714. doi: 10.1074/jbc.273.27.16710. [DOI] [PubMed] [Google Scholar]
- 31.Inoue I, Shino K, Noji S, Awata T, Katayama S. Expression of peroxisome proliferator-activated receptor α (PPARα) in primary cultures of human vascular endothelial cells. Biochem Biophys Res Commun. 1998;246:370–374. doi: 10.1006/bbrc.1998.8622. [DOI] [PubMed] [Google Scholar]
- 32.Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, et al. Activation of human aortic smooth-muscle cells is inhibited by PPARα but not by PPARγ activators. Nature. 1998;393:790–793. doi: 10.1038/31701. [DOI] [PubMed] [Google Scholar]
- 33.Gilde AJ, van der Lee KA, Willemsen PH, Chinetti G, van der Leij FR, van der Vusse GJ, et al. Peroxisome proliferator-activated receptor (PPAR) alpha and PPAR beta/delta, but not PPAR gamma, modulate the expression of genes involved in cardiac lipid metabolism. Circ Res. 2003;92:518–524. doi: 10.1161/01.RES.0000060700.55247.7C. [DOI] [PubMed] [Google Scholar]
- 34.Goikoetxea MJ, Beaumont J, Diez J. Peroxisome proliferators-activated receptor alpha and hypertensive heart failure. Drugs. 2004;64:9–18. doi: 10.2165/00003495-200464002-00003. [DOI] [PubMed] [Google Scholar]
- 35.Iglarz M, Touyz RM, Amiri F, Lavoie M-F, Diep QN, Schiffrin EL. Effect of peroxisome proliferators-activated receptor-α and –γ activators on vascular remodeling in endothelin-dependent hypertension. Arterioscler Thromb Vasc Biol. 2003;23:45–51. doi: 10.1161/01.ATV.0000047447.67827.CD. [DOI] [PubMed] [Google Scholar]
- 36.Gomez-Garre D, Herraiz M, Gonzalez-Rubio ML, Bernal R, Aragoncillo P, Carbonell A, et al. Activation of peroxisome proliferator-activated receptor-alpha and -gamma in auricular tissue from heart failure patients. Eur J Heart Fail. 2005;12:422–428. doi: 10.1016/j.ejheart.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 37.Ichihara S, Obata K, Yamada Y, Nagata K, Noda A, Ichihara G, et al. Attenuation of cardiac dysfunction by a PPAR-alpha agonist is associated with down-regulation of redox-regulated transcription factors. J Mol Cell Cardiol. 2006;41:318–329. doi: 10.1016/j.yjmcc.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 38.Celermajer D, Sorensen K, Georgakopoulos D, Bull C, Thomas O, Robinson J, et al. Cigarette smoking is associated with dose-related and potentially reversible impairment of endothelium-dependent dilation in healthy young adults. Circulation. 1993;88:2149–2155. doi: 10.1161/01.CIR.88.5.2149. [DOI] [PubMed] [Google Scholar]
- 39.Sethi JM, Rochester CL. Smoking and chronic obstructive pulmonary disease. Clin Chest Med. 2000;21:67–86. doi: 10.1016/S0272-5231(05)70008-3. [DOI] [PubMed] [Google Scholar]
- 40.Celermajer DS, Adams MR, Clarkson P, Robinson J, McCredie R, Donald A, et al. Passive smoking and impaired endothelium-dependent arterial dilatation in healthy young adults. N Engl J Med. 1996;334:150–154. doi: 10.1056/NEJM199601183340303. [DOI] [PubMed] [Google Scholar]
- 41.Su Y, Cao W, Han Z, Block ER. Cigarette smoke extract inhibits angiogenesis of pulmonary artery endothelial cells: the role of calpain. Am J Physiol Lung Cell Mol Physiol. 2004;287:L794–L800. doi: 10.1152/ajplung.00079.2004. [DOI] [PubMed] [Google Scholar]
- 42.Haj Mouhamed D, Ezzaher A, Neffati F, Douki W, Gaha L, Najjar MF. Effect of cigarette smoking on plasma uric acid concentrations. Environ Health Prev Med 2011;16:307–12. [DOI] [PMC free article] [PubMed]
- 43.Schmidt JV, Bradfield CA. Ah receptor signaling pathways. Annu Rev Cell Dev Biol. 1996;12:55–89. doi: 10.1146/annurev.cellbio.12.1.55. [DOI] [PubMed] [Google Scholar]
- 44.Reyes H, Reisz-Porszansz S, Hankinson O. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science. 1992;256:1193–1195. doi: 10.1126/science.256.5060.1193. [DOI] [PubMed] [Google Scholar]
- 45.Dong L, Ma Q, Whitlock JP., Jr DNA binding by the heterodimeric Ah receptor: relationship to dioxin-induced CYP1A1 transcription in vivo. J Biol Chem. 1996;271:7942–7948. doi: 10.1074/jbc.271.14.7942. [DOI] [PubMed] [Google Scholar]
- 46.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wiesener MS, Turley H, Allen WE, Willam C, Eckardt KU, Talks KL, et al. Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1a. Blood. 1998;92:2260–2268. [PubMed] [Google Scholar]
- 48.Ichihara S, Yamada Y, Gonzalez FJ, Nakajima T, Murohara T, Ichihara G. Inhibition of ischemia-induced angiogenesis by benzo[a]pyrene in a manner dependent on the aryl hydrocarbon receptor. Biochem Biophys Res Commun. 2009;381:44–49. doi: 10.1016/j.bbrc.2009.01.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ichihara S, Yamada Y, Ichihara G, Nakajima T, Li P, Kondo T, et al. A role for the aryl hydrocarbon receptor in regulation of ischemia-induced angiogenesis. Arterioscler Thromb Vasc Biol. 2007;2:1297–1304. doi: 10.1161/ATVBAHA.106.138701. [DOI] [PubMed] [Google Scholar]
- 50.Ichihara S. Role of AHR in the development of the liver and blood vessels. In: Pohjanvirta R, editor. The AH receptor in biology and toxicology. Hoboken: Wiley; 2010. p. 413–21.
- 51.Kwak M-K, Wakabayashi N, Kensler TW. Chemoprevention through the Keap1-Nrf2 signaling pathway by phase 2 enzyme inducers. Mutat Res. 2004;555:133–148. doi: 10.1016/j.mrfmmm.2004.06.041. [DOI] [PubMed] [Google Scholar]
- 52.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 2004;36:1199–1207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 53.Kanki K, Umemura T, Kitamura Y, Ishii Y, Kuroiwa Y, Kodama Y, et al. A possible role of Nrf2 in prevention of renal oxidative damage by ferric nitrilotriacetate. Toxicol Pathol. 2008;36:353–361. doi: 10.1177/0192623307311401. [DOI] [PubMed] [Google Scholar]
- 54.Calkins MJ, Jakel RJ, Johnson DA, Chan K, Kan YW, Johnson JA. Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc Natl Acad Sci USA. 2005;102:244–249. doi: 10.1073/pnas.0408487101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cho H-Y, Jedlicka AE, Reddy SPM, Kensler TW, Yamamoto M, Zhang L-Y, et al. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 2002;26:175–182. doi: 10.1165/ajrcmb.26.2.4501. [DOI] [PubMed] [Google Scholar]
- 56.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ichihara S, Yamada Y, Liu F, Murohara T, Itoh K, Yamamoto M, et al. Ablation of the transcription factor Nrf2 promotes ischemia-induced neovascularization by enhancing the inflammatory response. Arterioscler Thromb Vasc Biol. 2010;30:1553–1561. doi: 10.1161/ATVBAHA.110.204123. [DOI] [PubMed] [Google Scholar]
- 59.Ichihara S, Yamada Y, Yokota M. Association of a G994 → T missense mutation in the plasma platelet-activation factor acetylhydrolase gene with genetic susceptibility to non-familial dilated cardiomyopathy in Japanese. Circulation. 1998;98:1881–1885. doi: 10.1161/01.CIR.98.18.1881. [DOI] [PubMed] [Google Scholar]
- 60.Yamada Y, Nishida T, Ichihara S, Sawabe M, Fuku N, Nishigaki Y, et al. Association of a polymorphism of BTN2A1 with myocardial infarction in East Asian populations. Atherosclerosis. 2011;215:145–152. doi: 10.1016/j.atherosclerosis.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 61.Bhatnagar A. Environmental cardiology: studying mechanistic links between pollution and heart disease. Cir Res. 2006;99:692–705. doi: 10.1161/01.RES.0000243586.99701.cf. [DOI] [PubMed] [Google Scholar]
- 62.Kumagai Y, Pi J. Molecular basis for arsenic-induced alteration in nitric oxide production and oxidative stress: implication of endothelial dysfunction. Toxicol Appl Pharmacol. 2004;198:450–457. doi: 10.1016/j.taap.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 63.Nel A, Xia T, Madler L, Li N. Toxic potential of materials at the nanolevel. Science. 2006;311:622–627. doi: 10.1126/science.1114397. [DOI] [PubMed] [Google Scholar]