

Dosage compensation occurs by epigenetic regulation of sex chromosomes. Drosophila X-linked gene transcription is increased in males via MSL histone acetyltransferase complex recruitment, yet it is unknown how MSL specifically targets the X chromosome. Here, Kuroda and colleagues map genome-wide MSL-binding sites in newly evolving sex chromosome arms in Drosophila miranda. This study provides functional evidence for MSL binding to chromatin entry sites with sequence specificity followed by spreading and thus adds new insight into the evolution of dosage compensation mechanisms.
Keywords: dosage compensation, MSL complex, Drosophila, chromatin
Abstract
Dosage compensation has arisen in response to the evolution of distinct male (XY) and female (XX) karyotypes. In Drosophila melanogaster, the MSL complex increases male X transcription approximately twofold. X-specific targeting is thought to occur through sequence-dependent binding to chromatin entry sites (CESs), followed by spreading in cis to active genes. We tested this model by asking how newly evolving sex chromosome arms in Drosophila miranda acquired dosage compensation. We found evidence for the creation of new CESs, with the analogous sequence and spacing as in D. melanogaster, providing strong support for the spreading model in the establishment of dosage compensation.
One of the central questions in biology is how complex regulatory systems become established. Here, we study the evolution of dosage compensation, which was first described in Drosophila melanogaster. Dosage compensation occurs by increasing transcription of X-linked genes in males approximately twofold through recruitment of the MSL histone acetyltransferase complex (Hamada et al. 2005; Straub et al. 2005). How the MSL complex specifically targets the X chromosome is a major question, as its X-specific binding pattern lacks a consensus sequence at the majority of its binding sites (Alekseyenko et al. 2006; Gilfillan et al. 2006).
In our current model for targeting, the complex is thought to assemble on two noncoding RNAs (roX1 and roX2) expressed from the X chromosome and on 150–250 additional nucleation sites on X termed “chromatin entry sites” (CESs) (Kelley et al. 1999). CESs are spaced along the X at, on average, ∼50-kb intervals and contain a conserved sequence motif: the “MSL recognition element” (MRE) (Alekseyenko et al. 2008). Next, the MSL complex is thought to spread from CESs in cis, to bind most active genes on X in a sequence-independent manner. Spreading to active gene bodies occurs at least in part through recognition of a histone modification, H3K36me3, associated with active transcription (Larschan et al. 2007; Bell et al. 2008; Sural et al. 2008). The MSL complex then catalyzes site-specific acetylation of histone H4 (H4K16ac) on X-linked genes, which is associated with increased transcription (Turner et al. 1992; Bone et al. 1994; Hilfiker et al. 1997; Smith et al. 2001).
While a role for roX RNAs in dosage compensation is well established (Meller et al. 1997; Kelley et al. 1999; Meller and Rattner 2002), the role of cis-acting CESs is more difficult to define because of their large number and dispersed localization along the length of the X. About 150 CESs were initially mapped on the D. melanogaster X, from which the GA-rich MRE motif was identified (Alekseyenko et al. 2008; Straub et al. 2008). CESs moved from X to autosomes are sufficient to attract the MSL complex to an ectopic location, and the MRE sequence is necessary for this recognition (Alekseyenko et al. 2008). However, the degenerate 21-base-pair (bp) motif is only ∼1.7-fold enriched on X and is found on many additional locations on both X and autosomes, where it does not act as a CES. Additional specificity may come from coincidence with active chromatin—in particular, H3K36me3 associated with active transcription (Alekseyenko et al. 2012). Determining whether or not CESs with these same characteristics are acquired during the evolution of a new sex chromosome would be a strong test of the spreading model.
In the genus Drosophila, autosomes have repeatedly fused to the ancestral sex chromosomes, creating neo-sex chromosomes (Charlesworth 1978; Lucchesi 1978). Depending on how long ago these fusions occurred, neo-sex chromosomes are at varying stages in the transition into a pair of fully differentiated, heteromorphic sex chromosomes. Two such independent fusions occurred in Drosophila miranda; therefore, this species harbors three X chromosome arms of different ages (Fig. 1A). Specifically, XL is >60 million years old and is the ancestral sex chromosome of the genus Drosophila (i.e., it is also the X in D. melanogaster). XR is shared by members of the Drosophila pseudoobscura subgroup and is >15 million years old, while the neo-X chromosome (1 million to 2 million years old) still largely resembles an autosome (Bachtrog and Charlesworth 2002) but is beginning to evolve partial dosage compensation (Bone and Kuroda 1996; Marin et al. 1996; Steinemann et al. 1996). Thus, D. miranda provides a unique opportunity for studying X chromosome arms of dramatically different ages within the same genome. In particular, we can address the following question: Would the selective pressure on a new X chromosome give rise to a novel strategy for acquiring MSL binding, or would the MSL targeting system display the same characteristics as seen in D. melanogaster with regard to the recognition sequence, the number and spacing of CESs, and the H3K36me3-dependent final targeting to active genes? De novo acquisition of these characteristics would be strong evidence for their functional significance.
Figure 1.

Karyotype evolution in D. pseudoobscura and D. miranda and the acquisition of dosage compensation on newly formed X chromosomes. (A) The fusion of an autosome (chromosome 3L) with the ancestral X chromosome ∼15 million years ago formed a large metacentric X chromosome (XL + XR) that is shared between D. pseudoobscura and D. miranda. In the latter species, a more recent fusion between another autosome (2R in D. melanogaster) with the Y chromosome formed a neo-Y chromosome 1 million to 2 million years ago, leaving the remaining homolog to evolve into the neo-X. The ancestral X chromosome is shown in dark red, XR is in red, neo-X is in pink, and Y and neo-Y are in green. (B) Polytene chromosomes of a D. miranda MSL3-TAP transgenic male larva immunostained for the TAP epitope (red) and anti-H3K9me2 (green). XL, XR, and the neo-X are targeted by the MSL complex. The neo-Y, which no longer synapses with the neo-X, shows a pronounced accumulation of heterochromatin. Bar, 5 μm.
Results and Discussion
To study the MSL-binding pattern in D. miranda, we constructed a transgene containing a genomic copy of the D. miranda wild-type msl3 gene, expressed from its endogenous promoter, with insertion of a C-terminal protein A epitope (TAP tag). The transgene expressed the tagged MSL3 protein that bound the single X chromosome and rescued the viability of msl3 mutant males in D. melanogaster (data not shown), demonstrating both its conservation and function. We obtained several transgenic D. miranda lines, including one with an insertion on the neo-Y chromosome, and immunostaining of polytene chromosomes demonstrated that the MSL3-TAP protein was appropriately expressed in males and correctly identified the X chromosomes. Consistent with previous findings (Bone and Kuroda 1996; Marin et al. 1996), the MSL complex is localized to chromosomes XL and XR and the neo-X (Fig. 1B). The neo-Y, which is highly similar in sequence to the neo-X but known to be degenerating in its coding capacity (Steinemann and Steinemann 1992; Zhou and Bachtrog 2012), does not attract the MSL complex, instead harboring a silent chromatin mark.
We performed MSL3-TAP chromatin immunoprecipitation (ChIP) combined with deep sequencing (ChIP-seq) to map the D. miranda MSL-binding sites at high resolution. Input DNA and DNA purified from the MSL3-TAP pull-down from larval chromatin were sequenced using 100-bp paired-end reads on an Illumina Genome Analyzer. We also performed ChIP-seq on separate male and female wild-type (nontransgenic) larval chromatin for H3K36me3 to detect active genes and for H4K16ac, the mark associated with MSL binding in gene bodies. Reads were mapped against an improved D. miranda genome assembly (see the Materials and Methods). Consistent with the polytene chromosome immunostaining, positive clusters of MSL binding are highly enriched on the ancestral XL, on XR, and on the neo-X but not on the autosomes (Fig. 2A). Our ability to distinguish the neo-X from the neo-Y is currently incomplete due to the relatively high sequence identity retained in short reads of coding sequences (98.9% identical on average) and the inability to construct long contigs on the neo-Y due to the invasion of repetitive sequences and transposons throughout the chromosome. Therefore, our mapping to the neo-X, where ambiguous, was supported by the exclusive immunostaining of the MSL complex on neo-X and not neo-Y and by analysis using only ChIP-seq reads that have neo-X specific single-nucleotide polymorphisms (SNPs) (data not shown).
Figure 2.
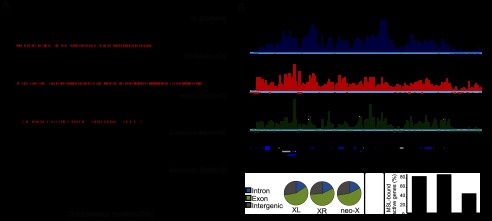
Genome-wide pattern of MSL complex binding in D. miranda. (A) Enrichment profile of MSL binding in the D. miranda genome. ChIP-seq enrichment ratios are plotted for MSL3-TAP binding (Y-axis) relative to chromosomal position (X-axis). The red marks along the X-axis indicate the position of inferred CESs. (B) GBrowse screen capture of an ∼130-kb region of chromosome XL showing enrichment ratios for male third instar larval MSL binding and histone marks. Gene models are shown below. (C) Preferential binding of the MSL complex to exons on all three X chromosome arms. (D) Fraction of H3K36me3-marked active genes bound by the MSL complex on the three X chromosome arms.
In D. melanogaster, the MSL complex primarily binds to active gene bodies marked by H3K36me3 (Alekseyenko et al. 2006; Larschan et al. 2007) and catalyzes the H4K16ac mark associated with increased transcription (Smith et al. 2001). Therefore, we compared the MSL3-TAP profiles with the H3K36me3 and H4K16ac profiles in males and found a significant correlation on XL, XR, and the neo-X (P < 10−3, Spearman's rank correlation) (Fig. 2B; Supplemental Fig. S1A), with MSL binding enriched on exons on all three chromosome arms (P < 0.0001) (Fig. 2C). Based on scoring H3K36me3-positive genes as active, the ChIP results demonstrate that the MSL complex in D. miranda exhibits a conserved localization on active gene bodies, as seen in D. melanogaster (Larschan et al. 2007; Bell et al. 2008). XL and XR are robustly targeted, while fewer active genes on the neo-X are recruiting the MSL complex (∼40% vs. >80% on XL/XR) (Fig. 2D), consistent with the partial acquisition of dosage compensation (Strobel et al. 1978). The binding of the MSL complex to active gene bodies is consistent with a conserved mechanism for increasing gene transcription, perhaps through facilitation of transcriptional elongation (Larschan et al. 2011).
We next asked whether we could detect high-occupancy CESs for the MSL complex on XL, XR, and the neo-X. These were first detected in D. melanogaster msl3 mutants, where the initial MSL binding is intact, but the spreading step to active genes is impaired (Alekseyenko et al. 2008). Subsequent ChIP-seq analysis in D. melanogaster demonstrated that CESs could be identified by their high MSL occupancy, even in a wild-type context (Alekseyenko et al. 2008). Therefore, we examined the relative occupancy of MSL-bound regions in D. miranda and found a similar bimodal distribution of MSL enrichment on both XL and XR (Fig. 3A). Using a threshold of 9.19-fold enrichment of MSL3-IP versus Input, we identified 132 CESs on XL and 215 CESs on XR in D. miranda (Supplemental Table S1). The median distance between CESs is 51 kb on XL (similar to D. melanogaster, with a median distance of 49 kb) and 39 kb on XR. To determine whether D. miranda uses a sequence motif for MSL targeting similar to that of D. melanogaster, we performed motif searches on the 500-bp regions surrounding these top peaks and identified a 21-bp GA-rich motif that is virtually indistinguishable from the motif discovered in D. melanogaster (Fig. 3B). This motif is enriched at the center of the putative CESs on each chromosome (Supplemental Fig. S1B) and is ∼1.5-fold enriched on XL and XR compared with autosomes, similar to patterns of enrichment in D. melanogaster (Supplemental Fig. S1C). The enrichment of the MRE motif on XL and XR increases when only counting motifs that reside within genomic regions bearing the H3K36me3 histone modification (to more than fivefold for XL and more than threefold for XR) (Supplemental Fig. S1D). Therefore, MRE binding in D. miranda may be more likely to occur in an active chromatin environment, as in D. melanogaster (Alekseyenko et al. 2012).
Figure 3.

Evolution of the MRE motif on the D. miranda sex chromosome arms. (A) MSL enrichment at binding sites identified on XL, XR, and the neo-X. The distribution of total MSL binding is plotted on the Y axis versus enrichment level on the X axis for each X chromosome arm. Binding shows a bimodal distribution on XL and XR, with putative CESs defined as those with high MSL occupancy (to the right of the dotted line). (B) Motif searches revealed a conserved 21-bp GA-rich motif, the MRE, within CESs on XL (MEME E-value: 2.1 × 10−343) and XR (MEME E-value: 3.6 × 10−685) and an MRE-like consensus sequence, potentially still evolving, on the neo-X (MEME E-value: 1.1 × 10−152).
The D. miranda neo-X became a sex chromosome between 1 million and 2 million years ago, with several lines of evidence suggesting that there has not been sufficient time to fully evolve dosage compensation. Many neo-Y genes are still functional, and thus their homologs on the neo-X may not require compensation (Bachtrog et al. 2008; Zhou and Bachtrog 2012). Indeed, a smaller fraction of genes are bound by the MSL complex on the neo-X, and we identify fewer CESs on the neo-X compared with XL and XR (Fig. 3A). Using the same threshold as for XL and XR, we found 68 CESs on the neo-X, with a median distance of 68 kb between them (Supplemental Table S1). Interestingly, CESs on the neo-X are less identifiable by both occupancy level and sequence. The MSL enrichment plot does not show a clear bimodal distribution on the neo-X, displaying a shoulder rather than a discrete peak (Fig. 3A), and shows lower overall occupancy than CESs on XL and XR (Supplemental Fig. S1E). In addition, a motif search of the candidate 68 neo-X CESs yielded a GA-rich motif that exhibits similarity to the conserved MRE, but the individual peaks in the logo are less pronounced (Fig. 3B). This is consistent with a model in which many CESs on the neo-X are still evolving the optimal binding motif recognized by the MSL complex. H3K36me3-marked genes near these CESs are more likely to be MSL-bound than those more distant (Supplemental Fig. S1F), suggesting that these newly evolving CESs may facilitate MSL spreading to neighboring genes.
How might CESs arise on the neo-X? We selected one of the CESs from neo-X that exhibited the highest occupancy in our ChIP-seq data and performed PCR of male genomic DNA with flanking primers to search specifically for the differences between the two chromosomal copies. Interestingly, we discovered an expansion of the neo-X sequence compared with the corresponding neo-Y DNA fragment. This amplification was of a simple GA repeat sequence compatible with, but larger than, a typical MRE motif (Fig. 4A; Supplemental Fig. S2A). Such an expansion might have been created by errors during DNA replication or unequal crossing over. Therefore, this result suggests one potential mode for the creation of a CES on an evolving X chromosome.
Figure 4.

Ectopic MSL targeting by a putative CES from the D. miranda neo-X and lack of activity from the corresponding region from the neo-Y. (A) Schematic of the GA expansion found in the CES identified on the neo-X compared with its putative progenitor sequence on the neo-Y. Red boxes indicate MRE motifs. (B,C) Transgenic msl3 mutant polytene chromosomes stained with anti-MSL2 (red) to identify all CESs and DAPI to identify all chromosome arms (blue). (B, green arrow) Lack of MSL immunostaining at the 37B7 cytological location of the neo-Y transgene. (C, white arrow) Robust MSL immunostaining at the same location when the insertion contains the neo-X CES.
Since the MRE is a degenerate sequence motif found at many locations on all chromosomes, we wondered whether the candidate CESs that we identified in D. miranda were functional and truly analogous to CESs discovered in D. melanogaster. Therefore, we used a transgenic assay in D. melanogaster as a stringent test for conservation. We selected one representative CES each from XL (from the roX2 gene) and XR (a mapped site with high MSL occupancy) as well as the GA expansion from the neo-X described above and its unexpanded counterpart from the neo-Y. We cloned these as 1.0 to 1.5-kb DNA segments into a transformation vector used previously to insert ectopic CESs into an autosomal location at cytological position 37B7 in D. melanogaster (Alekseyenko et al. 2008). We tested MSL binding in an msl3 mutant background in which the MSL pattern is reduced to CESs only and found that XL and XR constructs strongly recruited the MSL complex of D. melanogaster to the ectopic location on the autosome (Supplemental Fig. S2). In contrast, the neo-Y insertion failed to attract the MSL complex to the identical location (Fig. 4B), but its neo-X counterpart demonstrated robust MSL targeting (Fig. 4C). This result verifies the function of representative CESs identified in D. miranda and also confirms that the dosage compensation machinery is functionally interchangeable between species. Our results support a model in which the neo-X has evolved at least one CES by expanding a GA-rich sequence to create a potent new MSL-binding site.
In summary, our results are highly compatible with existing models for sex chromosome evolution and the resulting selective pressure to acquire dosage compensation (Charlesworth 1978; Lucchesi 1978). We found full acquisition of MSL targeting to XR over an estimated time span of 15 million years. The MRE targeting sequence is conserved on XL and XR, including its average spacing along the chromosome. Thus, our results do not support a model in which rapid evolution of MSL proteins was proposed to lead to distinct MRE recognition sequences in divergent Drosophila species (Levine et al. 2007; Rodriguez et al. 2007; Bachtrog 2008). Instead, both the protein components and the recognition sequences are functionally conserved over long evolutionary distances. The partial acquisition of MSL targeting over 1 million to 2 million years on the neo-X, while the MSL complex remains excluded from the neo-Y, has occurred in concert with the differentiation of these formerly homologous chromosome arms. The neo-X is acquiring new MSL entry sites, not yet fully optimized, while the neo-Y has acquired repetitive elements and gene silencing. The evolution of MSL binding on the neo-X chromosome provides strong support for both steps of the MSL-spreading model: initiation from sequence-dependent CESs and sequence-independent spreading to active genes.
Recently, it has become evident that strikingly different modes of dosage compensation in Caenorhabditis elegans and mammals may use strategies remarkably similar to the mechanism discovered in Drosophila for whole-chromosome targeting (Csankovszki et al. 2004; Pinter et al. 2012). In each case, the proposed strategies involve initial recognition of ∼150 nucleation sites along the length of the selected chromosome, followed by spreading in cis to control the chromosome as a whole. Thus, it is tempting to speculate that dosage compensation is not unique in using such a strategy but instead reveals that spreading may be an ancient mechanism for genome organization and function.
Materials and methods
Genome assembly and annotation
We produced an updated assembly of the D. miranda genome using a combination of Illumina reads (90-fold genomic coverage; various libraries with insert sizes from 170 bp up to 10 kb; 100-bp-long paired end reads) and 454 reads (fivefold genomic coverage; 200-bp- to 1.5-kb-long single-end reads). Raw reads were first quality-filtered and assembled into contig sequences without any gaps. We used ALLPATHS-LG (Gnerre et al. 2011) for Illumina reads and Newbler (Roche) for 454 reads. Resulting contigs from both sources were combined by Minimus (Sommer et al. 2007) and then linked into longer scaffolds by SSPACE (Boetzer et al. 2011). Scaffolds were finally gap-patched by a prerelease of PyGap software from Wes Warren's group at Washington University. We assembled a draft genome with much better continuity (scaffold N50: 1029 kb) and integrity (contig N50: 60 kb, 2.9% gap sequences) compared with a previous version (Zhou and Bachtrog 2012). Scaffolds were joined into chromosomes using D. pseudoobscura chromosomal sequences (version 2.27; http://www.flybase.org) as a reference. We also reannotated the genome and built consensus gene models for the D. miranda genome using both D. pseudoobcura proteins (version 2.29) as homology evidence and D. miranda RNA sequencing (RNA-seq) data from nine different tissues/stages as experimental evidence with MAKER (Cantarel et al. 2008).
Generation of a transgenic DmirMSL3-TAP D. miranda strain
We constructed a D. miranda MSL3-TAP transgenic construct in a P-element transformation vector, pGREENi, marked with eyeless-driven GFP. D. miranda embryos were injected by Genetic Services, Inc., and the location of inserts was mapped by inverse PCR. Two transgenic lines were recovered (MSH22_22-1: neo-Y; MSH22_22-11: chr 2). The stock with a fortuitous insertion on the neo-Y was chosen for ChIP analyses. We also created D. melanogaster transgenic lines to test functionality of the DmirMSL3-TAP transgene in an msl3 mutant background. Polytene chromosomes from such DmirMSL3TAP-expressing males displayed consistent labeling of the X, and the mutant males were fully rescued.
Transgenic assays
We PCR-amplified 1 to 1.5-kb DNA fragments encompassing putative CESs from the D. miranda MSH22 strain (sequences available on request) cloned each into SalI-digested piB-GFP and used recombination-mediated cassette exchange (RMCE) to place each construct into a landing site at 37B7 in D. melanogaster, as described (Alekseyenko et al. 2008). Binding of the MSL complex to transgenic CESs was assayed cytologically in females expressing MSL2 and mutant for msl3 (y w; 37B7[CES-X]; H83M2-6I, msl3) (Alekseyenko et al. 2008) because msl3 mutant male chromosomes exhibit poor morphology. CESs from all three X chromosome arms were tested: from position 17673571 to 17675136 on XL, from position 10843911 to 10845207 on XR, and from position 2520435 to 2521917 on the neo-X and its homologous region from the neo-Y.
Chromatin preparation and sequencing
Approximately 10 g of mixed-sex third instar larvae with the insertion of pGREENi-DmirMSL3-TAP on the neo-Y was used for ChIP. Chromatin was cross-linked with formaldehyde and sheared by sonication. Chromatin pull-down with IgG agarose beads (Sigma, A2909) was performed as described previously (Alekseyenko et al. 2006, 2008). The following antibodies against histone modifications were used for ChIP-seq experiments: (1) anti-H3K36me3 (3 μL per immunoprecipitation; Abcam, ab9050) and (2) anti-H4K16ac (5 μL per immunoprecipitation; Millipore, 07-329). Immunoprecipitated and input DNAs from sexed larvae were purified as described (Larschan et al. 2007) and processed according to the standard paired-end Solexa library preparation protocol. Paired-end 100-bp DNA sequencing was performed on the Illumina Genome Analyzer located at University of California at Berkeley Vincent J. Coates Genomic Sequencing Facility.
ChIP-seq data analysis
The sequenced fragments, both ChIP-DNA and input DNA sequences, were aligned against the D. miranda genome assembly using bowtie2 (Langmead et al. 2009). To identify binding positions, a binding score was calculated for all positions in the genome as the ratio of mapped MSL3-IP versus Input control DNA, and peaks were called using MACS (Zhang et al. 2008). A subset of peaks with the highest MSL occupancy were classified as CESs. Using a threshold of 9.19-fold enrichment of MSL3-IP versus Input, we identified CESs on XL, XR, and the neo-X. A sequence motif was determined using MEME (Bailey et al. 2009) on the 500-bp region surrounding these top peaks.
Accession numbers
The ChIP-seq data are available from the Short Read Archive (SRA) with accession numbers SRS402820 and SRS402821. The Whole Genome Shotgun data are deposited at DNA Data Bank of Japan/EMBL/GenBank under the accession AJMI00000000. The version described in this study is the current update, AJMI02000000.
Acknowledgments
We thank E. Brown for the heat map in Supplemental Figure S1A, and C. Lim for critical reading of the manuscript. This work was supported by grants from the NIH (GM45744 to M.I.K., and GM076007 and GM093182 to D.B.) and a Packard Fellowship (to D.B.). A.A.A. and A.A.G. generated transgenic D. miranda and D. melanogaster and performed immunostaining and ChIP experiments. C.E.E. and Q.Z. generated the genome assembly, and Q.Z. and V.B.K. generated the genome annotation. C.E.E., N.T., and S.P. performed the ChIP-seq data analysis. Q.Z. and Z.W. prepared all genomic and RNA-seq libraries. D.B., M.I.K., and P.J.P. supervised the project. A.A.A., C.E.E., D.B., and M.I.K. wrote the manuscript in consultation with all coauthors.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.215426.113.
References
- Alekseyenko AA, Larschan E, Lai WR, Park PJ, Kuroda MI 2006. High-resolution ChIP–chip analysis reveals that the Drosophila MSL complex selectively identifies active genes on the male X chromosome. Genes Dev 20: 848–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Peng S, Larschan E, Gorchakov AA, Lee OK, Kharchenko P, McGrath SD, Wang CI, Mardis ER, Park PJ, et al. 2008. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell 134: 599–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Ho JW, Peng S, Gelbart M, Tolstorukov MY, Plachetka A, Kharchenko PV, Jung YL, Gorchakov AA, Larschan E, et al. 2012. Sequence-specific targeting of dosage compensation in Drosophila favors an active chromatin context. PLoS Genet 8: e1002646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D 2008. Positive selection at the binding sites of the male-specific lethal complex involved in dosage compensation in Drosophila. Genetics 180: 1123–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D, Charlesworth B 2002. Reduced adaptation of a non-recombining neo-Y chromosome. Nature 416: 323–326 [DOI] [PubMed] [Google Scholar]
- Bachtrog D, Hom E, Wong KM, Maside X, de Jong P 2008. Genomic degradation of a young Y chromosome in Drosophila miranda. Genome Biol 9: R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS 2009. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell O, Conrad T, Kind J, Wirbelauer C, Akhtar A, Schubeler D 2008. Transcription-coupled methylation of histone H3 at lysine 36 regulates dosage compensation by enhancing recruitment of the MSL complex in Drosophila melanogaster. Mol Cell Biol 28: 3401–3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W 2011. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27: 578–579 [DOI] [PubMed] [Google Scholar]
- Bone JR, Kuroda MI 1996. Dosage compensation regulatory proteins and the evolution of sex chromosomes in Drosophila. Genetics 144: 705–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bone JR, Lavender J, Richman R, Palmer MJ, Turner BM, Kuroda MI 1994. Acetylated histone H4 on the male X chromosome is associated with dosage compensation in Drosophila. Genes Dev 8: 96–104 [DOI] [PubMed] [Google Scholar]
- Cantarel BL, Korf I, Robb SM, Parra G, Ross E, Moore B, Holt C, Sanchez Alvarado A, Yandell M 2008. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18: 188–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B 1978. Model for evolution of Y chromosomes and dosage compensation. Proc Natl Acad Sci 75: 5618–5622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csankovszki G, McDonel P, Meyer BJ 2004. Recruitment and spreading of the C. elegans dosage compensation complex along X chromosomes. Science 303: 1182–1185 [DOI] [PubMed] [Google Scholar]
- Gilfillan GD, Straub T, de Wit E, Greil F, Lamm R, van Steensel B, Becker PB 2006. Chromosome-wide gene-specific targeting of the Drosophila dosage compensation complex. Genes Dev 20: 858–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al. 2011. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci 108: 1513–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada FN, Park PJ, Gordadze PR, Kuroda MI 2005. Global regulation of X chromosomal genes by the MSL complex in Drosophila melanogaster. Genes Dev 19: 2289–2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker A, Hilfiker-Kleiner D, Pannuti A, Lucchesi JC 1997. mof, a putative acetyl transferase gene related to the Tip60 and MOZ human genes and to the SAS genes of yeast, is required for dosage compensation in Drosophila. EMBO J 16: 2054–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley RL, Meller VH, Gordadze PR, Roman G, Davis RL, Kuroda MI 1999. Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell 98: 513–522 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larschan E, Alekseyenko AA, Gortchakov AA, Peng S, Li B, Yang P, Workman JL, Park PJ, Kuroda MI 2007. MSL complex is attracted to genes marked by H3K36 trimethylation using a sequence-independent mechanism. Mol Cell 28: 121–133 [DOI] [PubMed] [Google Scholar]
- Larschan E, Bishop EP, Kharchenko PV, Core LJ, Lis JT, Park PJ, Kuroda MI 2011. X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature 471: 115–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MT, Holloway AK, Arshad U, Begun DJ 2007. Pervasive and largely lineage-specific adaptive protein evolution in the dosage compensation complex of Drosophila melanogaster. Genetics 177: 1959–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi JC 1978. Gene dosage compensation and the evolution of sex chromosomes. Science 202: 711–716 [DOI] [PubMed] [Google Scholar]
- Marin I, Franke A, Bashaw GJ, Baker BS 1996. The dosage compensation system of Drosophila is co-opted by newly evolved X chromosomes. Nature 383: 160–163 [DOI] [PubMed] [Google Scholar]
- Meller VH, Rattner BP 2002. The roX genes encode redundant male-specific lethal transcripts required for targeting of the MSL complex. EMBO J 21: 1084–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller VH, Wu KH, Roman G, Kuroda MI, Davis RL 1997. roX1 RNA paints the X chromosome of male Drosophila and is regulated by the dosage compensation system. Cell 88: 445–457 [DOI] [PubMed] [Google Scholar]
- Pinter SF, Sadreyev RI, Yildirim E, Jeon Y, Ohsumi TK, Borowsky M, Lee JT 2012. Spreading of X chromosome inactivation via a hierarchy of defined Polycomb stations. Genome Res 22: 1864–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez MA, Vermaak D, Bayes JJ, Malik HS 2007. Species-specific positive selection of the male-specific lethal complex that participates in dosage compensation in Drosophila. Proc Natl Acad Sci 104: 15412–15417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ER, Allis CD, Lucchesi JC 2001. Linking global histone acetylation to the transcription enhancement of X-chromosomal genes in Drosophila males. J Biol Chem 276: 31483–31486 [DOI] [PubMed] [Google Scholar]
- Sommer DD, Delcher AL, Salzberg SL, Pop M 2007. Minimus: A fast, lightweight genome assembler. BMC Bioinformatics 8: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinemann M, Steinemann S 1992. Degenerating Y chromosome of Drosophila miranda: A trap for retrotransposons. Proc Natl Acad Sci 89: 7591–7595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinemann M, Steinemann S, Turner BM 1996. Evolution of dosage compensation. Chromosome Res 4: 185–190 [DOI] [PubMed] [Google Scholar]
- Straub T, Gilfillan GD, Maier VK, Becker PB 2005. The Drosophila MSL complex activates the transcription of target genes. Genes Dev 19: 2284–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub T, Grimaud C, Gilfillan GD, Mitterweger A, Becker PB 2008. The chromosomal high-affinity binding sites for the Drosophila dosage compensation complex. PLoS Genet 4: e1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobel E, Pelling C, Arnheim N 1978. Incomplete dosage compensation in an evolving Drosophila sex chromosome. Proc Natl Acad Sci 75: 931–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sural TH, Peng S, Li B, Workman JL, Park PJ, Kuroda MI 2008. The MSL3 chromodomain directs a key targeting step for dosage compensation of the Drosophila melanogaster X chromosome. Nat Struct Mol Biol 15: 1318–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BM, Birley AJ, Lavender J 1992. Histone H4 isoforms acetylated at specific lysine residues define individual chromosomes and chromatin domains in Drosophila polytene nuclei. Cell 69: 375–384 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Bachtrog D 2012. Sex-specific adaptation drives early sex chromosome evolution in Drosophila. Science 337: 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]