

Impaired respiration is a potent stimulus for biofilm formation, yet how cells sense environmental signals that trigger bacterial multicellularity is not well understood. Kolodkin-Gal et al. now identify two histidine kinases that mediate the response to impaired respiration in B. subtilis. Membrane protein KinB forms a complex with the oxidative stress machinery. KinA is activated by a decrease in the NAD+/NADH ratio and directly binds NAD+. This study thus uncovers two independent pathways that regulate biofilm production.
Keywords: Bacillus subtilis, biofilms, histidine kinase, respiration, cytochromes
Abstract
Bacillus subtilis forms organized multicellular communities known as biofilms wherein the individual cells are held together by a self-produced extracellular matrix. The environmental signals that promote matrix synthesis remain largely unknown. We discovered that one such signal is impaired respiration. Specifically, high oxygen levels suppressed synthesis of the extracellular matrix. In contrast, low oxygen levels, in the absence of an alternative electron acceptor, led to increased matrix production. The response to impaired respiration was blocked in a mutant lacking cytochromes caa3 and bc and markedly reduced in a mutant lacking kinase KinB. Mass spectrometry of proteins associated with KinB showed that the kinase was in a complex with multiple components of the aerobic respiratory chain. We propose that KinB is activated via a redox switch involving interaction of its second transmembrane segment with one or more cytochromes under conditions of reduced electron transport. In addition, a second kinase (KinA) contributes to the response to impaired respiration. Evidence suggests that KinA is activated by a decrease in the nicotinamide adenine dinucleotide (NAD+)/NADH ratio via binding of NAD+ to the kinase in a PAS domain A-dependent manner. Thus, B. subtilis switches from a unicellular to a multicellular state by two pathways that independently respond to conditions of impaired respiration.
Bacterial cells are constantly faced with the challenge of having to adapt to changes in their environment. One such adaptation is the formation of multicellular communities known as biofilms in which cells are held together by a self-produced extracellular matrix (Branda et al. 2005; Kolter and Greenberg 2006). Cells in biofilms are typically more resistant to various environmental stresses than are free-living bacteria (Costerton et al. 1999; Mah and O'Toole 2001). Many bacteria are capable of producing biofilms. However, the mechanisms by which cells sense and respond to environmental cues by switching to a multicellular state are poorly understood. A highly accessible experimental system for studying biofilm formation is Bacillus subtilis. This spore-forming, Gram-positive bacterium forms architecturally complex biofilms on solid surfaces and at air–liquid interfaces (such floating biofilms are known as pellicles) (Branda et al. 2001; Aguilar et al. 2007). B. subtilis biofilms are characterized by a high degree of wrinkling, which is a consequence of, and represents a direct readout for, matrix production. The matrix consists of an exopolysaccharide, which is produced by enzymes encoded by the 15-gene epsA-O operon (henceforth simply eps) and amyloid-like fibers specified by the tapA–sipW–tasA operon (henceforth tapA) (Branda et al. 2004; Chu et al. 2006; Romero et al. 2010, 2011). These operons are under the negative control of a dedicated regulator of matrix production, the repressor SinR, and a global repressor of genes expressed in stationary phase AbrB (Chu et al. 2006; Chai et al. 2008). Relief from repression is mediated by the response regulator Spo0A, which, when phosphorylated, turns on the synthesis of a SinR anti-repressor, SinI, and represses the gene for AbrB (as well as turning on the synthesis of an AbrB anti-repressor, AbbA) (Banse et al. 2011). Anti-repression of SinR also derepresses the synthesis of a second SinR anti-repressor, SlrR, which in turn sets up a self-reinforcing cycle of SlrR production and matrix operon expression (Chu et al. 2008; Chai et al. 2010a,b). Thus, the key determinant for matrix production is Spo0A∼P. This phosphoprotein is also the master regulator for entry into the process of spore formation; indeed, the original focus of studies of Spo0A∼P was in the context of sporulation (Burbulys et al. 1991). Low to intermediate levels of Spo0A∼P turn on the gene for SinI and turn off the gene for AbrB and hence derepress matrix operons. However, when Spo0A∼P reaches high levels, production of SinI is repressed, and genes that initiate sporulation, whose promoters have a relatively low affinity for the phosphoprotein, are switched on (Chai et al. 2008, 2011). Here we are concerned with the environmental signals that lead to the phosphorylation of Spo0A and the mechanisms by which these signals are sensed and transduced into the activation of the response regulator.
Phosphorylation of Spo0A is governed by a phosphorelay that transfers phosphoryl groups from histidine kinases via two intermediary proteins (Spo0F and Spo0B) to the response regulator. Five histidine kinases (KinA–E) feed phosphoryl groups into the relay, but only four (KinA, KinB, KinC, and KinD) contribute significantly to biofilm formation, and only two (KinA and KinB) play an important role in spore formation (Burbulys et al. 1991; LeDeaux et al. 1995; Tzeng et al. 1998; McLoon et al. 2011). Evidence indicates that KinD directly or indirectly responds to an unknown small molecule signal released from the roots of tomato plants (which B. subtilis colonizes to form a protective barrier against pathogens) (Chen et al. 2012a,b). KinD independently can detect the presence of the extracellular matrix presumably because it is an osmosensor that responds to the osmolarity caused by exopolysaccharide production (Aguilar et al. 2010; Rubinstein et al. 2012). KinC responds to both membrane damage and potassium ion leakage caused by a self-produced surfactant, surfactin (Lopez et al. 2009; Shemesh et al. 2010). However, the nature of the signals to which KinA and KinB respond has been mysterious. It had been argued that KinA binds to and responds to ATP (Stephenson and Hoch 2001), but this could not be confirmed in a subsequent investigation (Lee et al. 2008). Although KinA has three possible ligand-binding PAS domains, it has also been argued that KinA does not respond to a signal at all but rather is activated by an increase in KinA levels brought about by conditions of nutrient depletion (Eswaramoorthy et al. 2010).
Recent work has identified impaired respiration as a cue for biofilm formation by the Gram-negative bacterium Pseudomonas aeruginosa. Dietrich et al. (2008, 2013) have shown that P. aeruginosa produces redox-active small molecules (phenazines), which shuttle electrons out of the biofilm, where the limited availability of oxygen prevents it from serving as an electron acceptor. Interestingly, in the absence of phenazines, P. aeruginosa biofilms, like those of B. subtilis (which does not produce phenazines), become highly wrinkled, thereby increasing the surface-to-volume ratio of the colony and allowing cells in the biofilm greater access to oxygen. However, the mechanisms by which P. aeruginosa senses and responds to impaired respiration are unknown. Here we provide evidence that matrix production and the consequent colony wrinkling by B. subtilis are a response to impaired respiration and that impaired respiration is sensed by the KinA and KinB kinases. We present evidence indicating that KinB, which is an integral membrane protein, is in a complex with the aerobic respiratory apparatus and is activated by impaired electron transport through the cytochrome chain. We also show that KinA contributes to the response to impaired respiration and present evidence suggesting that it does so by a mechanism involving a decrease in the cellular ratio of nicotinamide adenine dinucleotide (NAD+) to NADH and, importantly, a direct interaction with NAD+.
Results
Impaired respiration stimulates wrinkling and matrix gene expression
To investigate the hypothesis that impaired cellular respiration is a cue for triggering matrix production, we determined the effect of changes in the atmospheric oxygen concentration on colony wrinkling when cells were grown in biofilm-inducing medium. Oxygen concentration was controlled using an environmental chamber, with 22% oxygen representing the normal level of atmospheric oxygen (Materials and Methods). Decreasing the oxygen concentration from 22% to 5% had the reproducible effect of increasing the number of wrinkles on the colony (Fig. 1A). In contrast, increasing oxygen concentration to 30% and 35% markedly suppressed wrinkling, resulting in relatively flat, featureless colonies (Fig. 1A).
Figure 1.

Respiration suppresses wrinkling and matrix gene expression. (A) Colony wrinkling increases with decreasing oxygen concentration, except when nitrate is used as a terminal electron acceptor. The wild strain 3610 was grown on solid biofilm-inducing medium containing 250 μM iron chloride at the indicated oxygen concentrations for 4 d in an environmental chamber. The medium contained 20 mM KNO3 for the colony grown on nitrate. (B,C) Matrix gene expression increases with decreasing oxygen concentration, except in the presence of nitrate. Gene expression was monitored using a PtapA-lacZ-containing strain (RL4582) (B) and a PtapA-yfp-containing strain (RL5477) (C). Colonies were grown in the environmental chamber for 48 h. For B, colonies were harvested in triplicate, and PtapA-lacZ activity was measured. (MU) Miller units. For C, colonies were examined using a dissecting fluorescence microscope (Zeiss).
If the cells sense oxygen concentration via its effects on respiration, then restoring respiration using an alternative terminal electron acceptor ought to reverse the effects of reduced oxygen concentration. For this purpose, we used nitrate, since B. subtilis is able to use nitrate as a terminal electron acceptor (Nakano and Zuber 1998). The results shown in Figure 1A demonstrate that when the medium was supplemented with nitrate (20 mM), colony wrinkling was almost completely suppressed even when the oxygen concentration was 5%. That this effect was due to nitrate reduction was confirmed using a mutant (ΔnarG-I) of nitrate reductase, which mediates the use of nitrate as a terminal electron acceptor (Hoffmann et al. 1995). In contrast to the wild type, the mutant remained wrinkled at 5% oxygen concentration even when nitrate was added to the medium (Supplemental Fig. S1).
We next asked whether the effects of respiration were exerted at the level of the expression of genes involved in matrix production. If so, then changes in oxygen concentration should have little effect on colony wrinkling in cells mutant for the matrix operon repressor SinR (Kearns et al. 2005; Chai et al. 2008). SinR mutant colonies exhibit a hyperwrinkly phenotype due to constitutive expression of the matrix operons. Indeed, increases or decreases in oxygen concentration had no effect on wrinkling in colonies of SinR mutant cells (data not shown). To directly monitor the effect of oxygen concentration on matrix gene expression, we used lacZ and gfp reporter genes fused to the promoter (PtapA) for the operon (tapA–sipW–tasA) specifying the protein component (TasA) of the matrix. The results show that transcription from PtapA-lacZ was markedly increased by reduced oxygen availability, as determined by measuring β-galactosidase levels in cells harvested from solid biofilm-inducing medium (Fig. 1B). Likewise, fluorescence from a PtapA-yfp fusion was strikingly more intense in a colony grown on biofilm-inducing medium when the oxygen concentration was 5% rather than 22% (Fig. 1C). In both cases, the effect of low oxygen concentration was reversed by the presence of nitrate as an alternative electron acceptor (Fig. 1B,C). In addition, because the nitrate reductase genes are not expressed at 22% oxygen, there is no effect of adding nitrate on tapA transcription at that oxygen concentration (Fig. 1B; Cruz Ramos et al. 1995).
Wrinkling and matrix gene expression are optimal at high iron concentrations
The results presented above are consistent with the hypothesis that a reduction in the efficiency of respiration is a signal for matrix production. Because of the importance of iron to the function of heme-containing cytochromes in the respiratory apparatus (Gaballa et al. 2008), we wondered whether matrix production would be sensitive to levels of iron in the medium. While iron clearly can have many effects on cellular physiology, we reasoned that one of the effects of having low amounts of iron accessible to cells might lead to defects in the assembly of the cytochromes in the respiratory apparatus. We imagined that iron availability might be a particular issue for cells within and near the top of colonies, a fact that is supported by a greater expression of low-iron-inducible genes in the tops of colonies (data not shown). We therefore sought to examine the effect on matrix production of adjusting the concentration of iron in the medium over a wide range. Indeed, when the Fe+3 concentration in the medium was reduced from 50 μM (the concentration we traditionally used in past investigations) to 5 μM, wrinkling was markedly suppressed (Fig. 2A). In addition, increasing the Fe+3 concentration to a high level (250 μM) caused a small but reproducible increase in wrinkling. Furthermore, decreasing the Fe+3 concentration from 250 to 50 to 5 μM progressively reduced matrix gene expression in colonies (Fig. 2B) without impairing cell growth in shaking liquid cultures (Fig. 2C). Reinforcing the observations that iron exerts its effect at the level of matrix gene expression, the decrease in matrix gene expression observed when lowering the Fe+3 concentration was reversed in a mutant lacking SinR (Fig. 2B).
Figure 2.

Iron promotes wrinkling and matrix gene expression. (A) Colonies of the PtapA-lacZ-containing strain RL4582 grown on biofilm-inducing medium containing the indicated concentration of iron chloride. (B) PtapA-lacZ activity in pellicles of wild-type (RL4582) and sinR mutant (RL4585) cells grown in the presence of the indicated concentrations of iron chloride. (C) The growth of RL4582 in shaking liquid biofilm-inducing medium containing 5 μM (triangles), 50 μM (boxes), or 250 μM (diamonds) iron chloride, each grown in triplicate.
To determine whether lowering the iron concentration was indeed leading to defects in the respiratory machinery, we measured the ratio of oxidized to reduced forms of NAD+/NADH, which indicates the cellular redox state (Kohanski et al. 2007; Dietrich et al. 2013) and reflects the efficiency of respiration as a function of concentration of oxygen (see Supplemental Fig. S2). Under conditions of high iron concentration (250 μM), expected to maximize iron availability for cells within the colony, cells were able to use oxygen for cellular respiration, as reflected in a NAD+/NADH ratio of ∼1.2, indicating that their aerobic respiratory apparatus was fully functional (Fig. 3A). However, at low iron levels (5 μM), the NAD+/NADH ratio decreased to ∼0.5 (Fig. 3A). These foregoing results suggest that in low iron, some of the cells do not have functional cytochromes, perhaps phenocopying the effects of cytochrome deletion mutants as discussed below.
Figure 3.
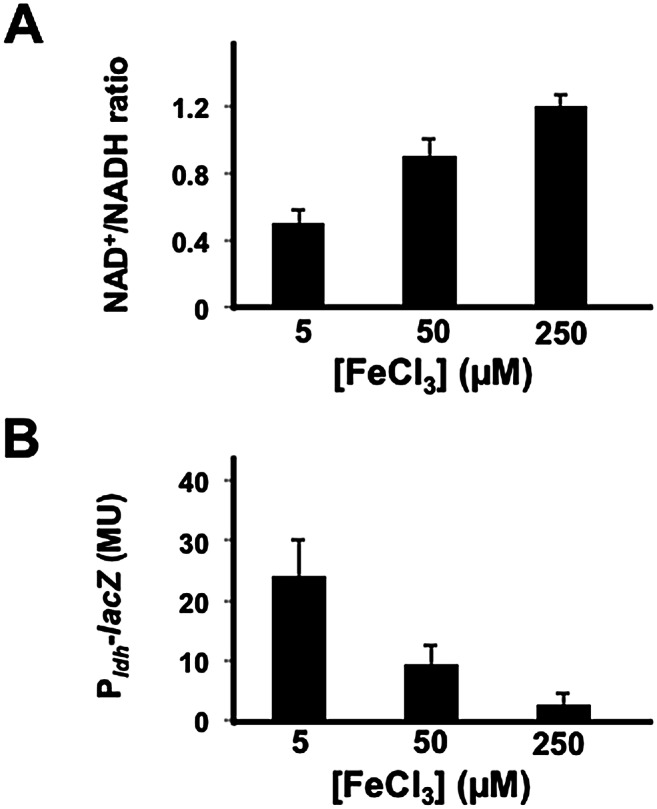
The NAD+/NADH ratio increases with increasing iron concentration. (A) The NAD+/NADH ratio for the wild strain 3610 grown for 48 h on solid biofilm-inducing medium containing the indicated levels of iron. Pairs of colonies were harvested and extracted under either acidic or basic conditions for measurements of NAD+ and NADH, respectively. The NAD+/NADH ratios are the average for six colonies (representing three acidic and three basic extractions) for each iron concentration. (B) Expression of Pldh-lacZ in a wild strain carrying the fusion (AE276). Cells were grown on solid biofilm-inducing medium containing the indicated concentration of iron for 48 h. After 48 h, colonies were harvested, and Pldh activity was measured. The results are the average for three colonies for each iron concentration.
As a further test of the idea that the low iron concentration resulted in a dysfunctional aerobic respiratory machinery, we measured gene expression under the control of Rex. Rex is an NADH-sensitive repressor that inhibits the expression of genes under its control when conditions are aerobic (Wang et al. 2008). When the NADH level rises upon oxygen limitation, Rex becomes inactive, leading to derepression of target genes. Using the reporter lacZ fused to the promoter for the gene (ldh) for lactate dehydrogenase, which is under the negative control of Rex (Pldh-lacZ), we found that expression increased with decreasing iron concentration at atmospheric oxygen levels (Fig. 3B) and confirmed that this iron-dependent effect on ldh expression is Rex-dependent (Supplemental Fig. S3). In toto, the above results are consistent with the idea (but not to the exclusion of other possible interpretations) that iron limitation within the colony results in a dysfunctional respiratory machinery. Indeed, it is likely that iron is having many other effects on cellular physiology. However, as we show below through the use of mutants, the most likely explanation of the foregoing results is that under low iron conditions, the respiratory apparatus is not functional, and thus the cells cannot respond by making more matrix. This lack of functionality might be the result of impaired assembly or simply lack of heme incorporation. Indeed, we can partially regain functionality in low iron by extracellular addition of hemin (data not shown). The important point, however, is that in the absence of a functional respiratory apparatus, the cells cannot mount a response. In the experiments presented below, we go on to prove this idea by the use of cytochrome mutants. Thus, the varying iron concentration experiments shown thus far as well as those presented subsequently serve simply as a proxy for lack of cytochromes because low iron phenocopies mutants that lack certain cytochromes.
The response to high iron concentration depends on kinases KinA and KinB
Critical to matrix production is the phosphorylation of the response regulator Spo0A, which turns on the synthesis of SinI, the anti-repressor of SinR (Branda et al. 2001; Kearns et al. 2005; Chai et al. 2010b). Phosphorylation of Spo0A is governed by five histidine kinases (KinA–KinE) that feed phosphoryl groups into a multicomponent phosphorelay that mediates phosphorylation of the response regulator. To investigate the role of these kinases in the response to impaired respiration, we measured the expression of sinI under various iron concentrations in single and double mutants of the kinases using a lacZ fusion (Fig. 4A). The results showed that sinI expression was modestly impaired in single mutants of KinA, KinC, and KinD as well as in a double mutant of KinC and KinD. In contrast, expression was markedly impaired in a KinB single mutant and more severely so in a KinA KinB double mutant. The effects of the mutations on sinI expression were observed under all three iron concentrations tested but were most dramatic in high iron concentration (250 μM) because of the high levels of expression observed in the wild type. We also measured sinI expression using a PsinI-lux luminescent reporter fusion. The results show that under high iron concentrations, expression was severely impaired in a KinA KinB double mutant but not so severely in a KinC KinD double mutant (Fig. 4B). The function of KinB is also known to depend on a lipoprotein KapB (Dartois et al. 1997). We confirmed that sinI expression at high iron concentrations depended, as expected, on KapB as well as KinB but did not investigate the role of the lipoprotein further in what follows (data not shown).
Figure 4.

The response to iron depends on kinases KinA and KinB. (A) The levels of PsinI-lacZ expression in a wild strain (RL4577) and the following lacZ fusion bearing single and double mutants—kinA (RL5426), kinB (RL5429), kinC (KG19), kinD (KG20), kinE (RL5423), kinA kinB (KG22), and kinC kinD (KG23)—that had been grown on solid medium containing the indicated iron concentrations for 48 h. (B) The time course of light emission from colonies of the wild type (RL5268) and the indicated double mutants harboring PsinI-lux at iron concentrations of 5 μM (open symbols) and 250 μM (closed symbols) (see the Materials and Methods). (C) Colony morphology for the wild type and the indicated double mutants grown for 48 h on medium containing the indicated iron levels.
Finally, we examined the pattern of colony wrinkling at 5 μM and 250 μM Fe+3 concentrations in the wild type and in the two pairs of kinase double mutants. It was known previously that at 50 μM Fe+3, the contribution of KinC and KinD is principally restricted to the peripheral zone of the colony (McLoon et al. 2011). Indeed, at high iron concentration, the KinC KinD double mutant exhibited a highly wrinkled central zone and a flat peripheral zone (Fig. 4C). In contrast, the KinA KinB double mutant formed relatively flat and featureless colonies, comparable with that seen for the wild type at low iron concentration (Fig. 4C). Thus, at the high iron concentration used here, KinA and KinB contribute to wrinkling throughout the colony.
KinB depends on and is in a complex with cytochromes
The observation that high iron concentrations are critical for robust biofilm formation and the fact that the cellular respiratory apparatus depends on iron via its heme-containing cytochromes led us to hypothesize that KinA or KinB senses impaired respiration (as a consequence of low oxygen concentration in the absence of another terminal electron acceptor) via the cytochrome chain. To investigate this hypothesis, we tested the effects of mutations ΔctaCDEF, ΔqcrABC, and ΔqoxABCD in the genes for cytochrome caa3, cytochrome bc, and cytochrome aa3, respectively (van der Oost et al. 1991; Azarkina et al. 1999). Single deletions of ctaCDEF and qcrABC showed somewhat decreased expression of sinI-lacZ in an iron-dependent manner (Fig. 5A), whereas a deletion mutant of qoxABCD had no significant effect (data not shown). However, in a double deletion mutant of ctaCDEF and qcrABC, iron-dependent sinI expression was reduced to the levels seen for a kinB mutant (Fig. 5A). This mimic of the kinB effect by the double cytochrome mutant was apparent in their inability to respond to lowering oxygen concentration (Supplemental Fig. S4). Moreover, when a kinA mutation was combined with mutations of ctaCDEF and qcrABC, the resulting mutant was as severely impaired in sinI-lacZ expression as was a kinB kinA double mutant. In addition, colonies lacking both caa3 and bc cytochromes were significantly defective in wrinkling, even under conditions of high iron concentration (Fig. 5B). As expected, these effects were through the control of expression of the matrix genes because their flat colony phenotype was lost when the sinR mutation was introduced (Supplemental Fig. S5).
Figure 5.

KinB interacts with and depends on cytochromes Cta and Qcr. (A) Levels of PsinI-lacZ expression in a fusion-bearing wild strain (RL4577) and fusion-bearing single and multiple mutants. In every case, “qcr” is used to designate the three-gene deletion ΔqcrABC, and “cta” indicates the four-gene deletion ΔctaCDEF. Colonies were harvested after growth for 48 h on biofilm-inducing medium. (B) Colony morphology for the wild type (3610) and a cta qcr double mutant (KG11) grown for 72 h on medium containing the indicated iron levels, focusing on the colony centers. (C) Stained bands from a gel of proteins from an immunoprecipitation with anti-GFP antibodies of proteins solubilized from the membrane from cells carrying GFP fused to KinB (RL5474) or to KinC (KG13) or strain 3610 bearing no GFP. The cells were grown in liquid MSgg medium for 48 h. (D) Stained bands of a KinB-GFP immunoprecipitation from fusion-bearing cells of the wild type or of qcr and cta mutants. The labeled bands in C and D correspond in size to that expected for QcrC or CtaC subunits. These proteins were predominant in the immunoprecipitation of KinB-GFP from wild-type cells as indicated by high-accuracy mass spectrometry.
These observations suggested that the ability of KinB to sense a change in respiratory activity requires the presence of the cytochromes, thus linking the activity of the respiratory apparatus to matrix gene expression. We wondered whether the effect of the cytochromes on KinB, which (unlike KinA) is an integral membrane protein, is the result of direct interaction with the respiratory apparatus. To investigate this possibility, we created a fusion of KinB to GFP and used an anti-GFP antibody to immunoprecipitate the fusion protein from a membrane preparation (Fig. 5C, left lane) and subjected KinB-GFP and associated proteins to high-accuracy mass spectrometry. Mass spectrometry revealed that the KinB-GFP immunoprecipitate was specifically enriched (as compared with a control lacking the fusion) for components of the respiratory apparatus, including, most prominently, cytochromes caa3, bc, and aa3 and the F1F0-ATPase, together with NADH dehydrogenase components. As controls, we carried out immunoprecipitations using a strain harboring a fusion of GFP to KinC, which is also a membrane protein (Fig. 5C, middle lane), and a strain that lacked the GFP fusion (Fig. 5C, right lane) and subjected the antibody-precipitated proteins to gel electrophoresis. Proteins corresponding in size to the CtaC subunit of caa3 and the QcrC subunit of bc could be seen in the immunoprecipitates obtained with KinB-GFP but not with the controls. To further investigate the identity of the proteins corresponding in size to CtaC and QcrC, we carried out additional immunoprecipitation experiments from membrane preparations using KinB-GFP with wild-type cells and with cells mutant for ctaCDEF and qcrABC (Fig. 5D). Proteins corresponding in size to CtaC (Fig. 5D, top panel) and QcrC (Fig. 5D, bottom panel) were specifically absent in the immunoprecipitates from the corresponding mutant strains.
These results are consistent with the following: (1) The activity of KinB is controlled by its interaction with the respiratory apparatus. (2) Impaired electron transport through the chain stimulates KinB activity, but this stimulation requires the presence of functional (but not fully functioning) cytochromes in that KinB was not active in a mutant lacking caa3 and bc (or when oxygen levels were high). In other words, KinB must be in a complex with a functional apparatus to be able to be activated. However, KinB is only activated when the respiratory activity of that functional apparatus is low.
Transmembrane segment 2 of KinB plays a critical role in sensing aerobic respiration
To identify KinB residues critical for sensing the activity of the respiratory machinery, we selected for hyperactive mutants that would form biofilms under a condition of low iron using this low iron concentration as a surrogate for low activity of the respiratory apparatus. To do so, we took advantage of the observation that the formation of floating biofilms (pellicles) in liquid medium containing 5 μM Fe+3 was markedly delayed. We therefore selected for mutants that would form pellicles without delay under these conditions. In a complementary approach, we screened for mutants that formed wrinkly colonies on solid medium under low iron conditions. Among the mutants so identified, a small proportion contained point mutations in kinB (none contained mutations in kinA). One of these mutations in kinB was in codon 42, resulting in a threonine-to-methionine change, and another mutation was in codon 44, resulting in a phenylalanine-to-cysteine change. KinB contains six predicted transmembrane segments, and both of these amino acid substitutions lie in transmembrane segment 2 (Fig. 6A). These mutants showed slightly increased wrinkling relative to the wild type when grown in 5 μM FeCl3 (Fig. 6B).
Figure 6.

KinB function depends on transmembrane segments 1 and 2. (A) The structure of the KinB sensor kinase. (B) Colony morphologies for the wild-type strain and the indicated mutants (obtained as described in the Supplemental Material) after 72 h of growth on biofilm-inducing medium containing the indicated iron levels. (C) Colony morphologies for a kinA kinB double mutant containing a wild-type copy of kinB at amyE or the indicated mutant alleles (obtained as described in the Supplemental Material). Colonies were grown for 72 h on medium containing 250 μM iron. (D) Levels of PsinI-lacZ expression in a kinA kinB double mutant complemented with either wild-type kinB or the indicated kinB mutant alleles.
We also mutagenized kinB via error-prone PCR and screened for mutations that resulted in flat, featureless colonies under high iron concentrations (250 μM). To increase the robustness of the screen, we started with a kinA kinB double mutant background in which we reintroduced the wild-type and mutated kinB genes at the amyE locus. In addition to mutations that introduced stop codons and a mutation in the codon for the active site histidine, we recovered a mutation in codon 52 that caused a cysteine-to-serine change in transmembrane segment 2 (Fig. 6A). This allele resulted in flat colonies and reduced sinI expression even at 250 μM iron (Fig. 6C,D). Immunostaining using anti-GFP antibodies showed that the levels of the mutant protein in the membrane fraction of cells harboring a KinB-GFP fusion were not measurably affected by the C52S substitution (data not shown). Thus, it is unlikely that the substitution was causing degradation or substantial misfolding of KinB, although we cannot rule out this possibility entirely.
Finally, we created a series of in-frame deletion mutations that removed pairs of transmembrane segments (see Fig. 6A). Again, the mutant genes were introduced into the chromosome at amyE in a kinA kinB double mutant. Removal of transmembrane segments 3 and 4 and segments 5 and 6 had only modest effects on biofilm formation, whereas removal of segments 1 and 2 had a severe effect (Fig. 6C,D). As above, we confirmed that the removal of segments 1 and 2 did not significantly reduce the level of the protein in the membrane fraction using a GFP fusion.
In toto, these data suggest that KinB senses the activity of the respiratory apparatus via its transmembrane segment 2. We note that cysteine residues are redox-sensitive. Because one of the gain-of-function mutations introduced a cysteine in segment 2 and a loss-of-function mutation removed a cysteine, it is tempting to speculate that the respiratory chain controls the activity of KinB by a molecular redox switch involving one or more cysteines.
KinA binds NAD+ in a PAS domain-dependent manner
As we observed (Fig. 4), the response to impaired respiration depends on KinA as well as KinB. What, then, is the signal that KinA senses? In contrast to KinB, KinA is a cytoplasmic protein and hence is unlikely to interact directly with the respiratory machinery. Instead, KinA possesses three PAS domains, which are potential binding sites for small molecule ligands (Moglich et al. 2009). Indeed, Dago et al. (2012) suggested that KinA is activated by a mechanism involving ligand binding. We therefore reasoned that KinA is activated by a small molecule ligand that accumulates under conditions of impaired respiration or, alternatively, inhibited by a ligand that decreases in abundance when respiration is impaired. Given that impaired respiration was associated with a decrease in the NAD+/NADH ratio (Fig. 3), an appealing hypothesis is that either NAD+ or NADH is the ligand for one or more of the PAS domains.
To investigate this possibility, we created internal, in-frame deletion mutants that separately removed each of the PAS domain-coding regions from kinA and introduced the mutated genes into the chromosome in place of wild-type kinA at its native locus. In agreement with the results of Wang et al. (2008), we found that removal of PASA and PASC, but not of PASB, impaired sporulation (data not shown). Moreover and importantly, removal of PASA and PASC (but, not so severely, removal of PASB) reduced the amount of colony wrinkling and sinI gene expression (Fig. 7A,B). Immunoblot analysis confirmed that the deletion mutations had not lowered the levels of mutant proteins (data not shown).
Figure 7.

The PASA domain of KinA is required for sensing impaired respiration. (A) Colony morphologies for the wild type (3610) and mutants of KinA lacking PASA (AE253), PASB (AE254), or PASC (AE255). Colonies were grown for 72 h on MSgg medium containing 250 μM iron chloride. (B) Expression of PsinI-lacZ in colonies that had been grown in liquid MSgg medium for 48 h.
To identify the ligands bound by KinA, we affinity-purified an N-terminal His6-tagged KinA. Next, we treated the purified protein with chloroform and subjected the aqueous phase, in which NAD+ and NADH would be located, to reverse-phase high-pressure liquid chromatography (RP-HPLC) analysis using a protocol optimized for NAD+ and NADH detection (Sporty et al. 2008). Indeed, a small molecule that eluted at the same time (∼12 min) as bona fide NAD+ (and not NADH) was found in the sample coming from the chloroform extraction of the His6-tagged KinA (Fig. 8A). This compound was then subjected to high-resolution mass spectrometry analysis. The molecule showed the same mass-to-charge ratio (m/z) as NAD+ (664.1150 as compared with 664.1164 for bona fide NAD+) (Fig. 8B). Moreover, comparing the tandem mass spectrometry (MS/MS) spectrum of the recovered molecule with that in the METLIN database showed that it matched perfectly with the known fragmentation pattern for NAD+ (Fig. 8C; Tautenhahn et al. 2012). The results also demonstrated that NAD+ was specifically associated with KinA in vivo, as it was only found in samples from His6-KinA immunoprecipitates but not in control experiments with immunoprecipitates from cells lacking the His6-tagged protein or from cells harboring His6-tagged Spo0A (Fig. 8A).
Figure 8.

KinA associates with NAD+. (A) The HPLC chromatogram for KinA-associated molecules and standards for NAD+ (10 μg) and NADH (9.36 μg) as well as a negative control (Spo0A-associated molecules). (B) The m/z (mass-to-charge) ratio of the peak collected at 12 min from the KinA purification using high-resolution mass spectrometry. (C) The MS/MS spectrum of the same peak analyzed in B, which matches the known fragmentation pattern for bona fide NAD+ in the METLIN database.
Next, we created His6-tagged derivatives of the truncated KinA proteins and tested for the presence of NAD+ after affinity purification. We found that binding of NAD+ was specifically dependent on PASA, as we were not able to detect NAD+ associated with a truncated KinA mutant lacking PASA but could detect NAD+ with KinA lacking PASC (Supplemental Fig. S6).
Finally, we considered the possibility that KinA actually binds NADH rather than NAD+ and that the reduced form of the dinucleotide had been converted to NAD+ by oxidation during the isolation of the protein. To investigate this possibility, we subjected pure NADH to the same protocol that we used in purifying KinA and tested for the presence of NAD+. HPLC analysis showed that only a small proportion of the NADH had been converted to NAD+ and hence that little oxidation had occurred during our extraction procedure (data not shown). We conclude that it is unlikely that NADH was being oxidized during the extraction and hence that the ligand is NAD+.
Discussion
We discovered that matrix production is robustly induced in B. subtilis by impaired respiration. B. subtilis biofilms are characteristically highly wrinkled. This wrinkling could be expected to increase the surface-to-volume ratio and hence the accessibility of cells in the community to oxygen. Indeed, experiments with P. aeruginosa have shown that wrinkles do indeed provide increased access to oxygen to cells in the biofilm (Dietrich et al. 2008, 2013). We therefore surmise that B. subtilis responds to conditions of impaired respiration as a compensatory mechanism that in effect helps cells in the biofilm to “breathe” under conditions of reduced oxygen availability. In keeping with the idea that cells in the biofilm face the challenge of coping with reduced oxygen availability, we note that the lut operon, which is responsible for oxidation of lactate, is under the direct control of the regulatory pathway governing matrix production in B. subtilis (Chai et al. 2009). Derepression of the operon during biofilm formation in the presence of lactate would provide an alternative electron acceptor under conditions of impaired access to oxygen.
The principal contribution of the current investigation is the discovery that B. subtilis responds to impaired respiration via interaction of a particular histidine kinase, KinB, with the aerobic respiratory apparatus. We showed that KinB activation depends on cytochromes caa3 and bc and that KinB is in a complex with multiple components of the respiratory apparatus, including caa3 and bc. Interestingly, a recent study indicates that these two cytochromes are in a complex with each other (Garcia Montes de Oca et al. 2012).
Activation of KinB requires both caa3 and bc and conditions of impaired respiration. We therefore conclude that the respiratory chain is not inhibiting KinB. Rather, it activates the kinase but only when respiration is impaired. We imagine that KinB in a complex with the respiratory apparatus is subject to a switch that turns on kinase activity under conditions of low electron transport and silences it under conditions of high electron transport. We hypothesize that the switch is mediated by a redox mechanism involving transmembrane segment 2 of KinB based on the following observations: (1) An amino acid substitution introducing a cysteine into segment 2 at residue 44 caused KinB to be active under conditions in which the function of the respiratory apparatus was inhibited (by virtue of low iron concentration). (2) An amino acid substitution removing a cysteine from segment 2 at residue 52 inactivated the kinase (under conditions of high iron concentration). Conceivably, electron transfer from the respiratory machinery to KinB in the complex reduces Cys 52, preventing activation of the kinase. Discovering whether and how such a redox mechanism operates will require a detailed understanding of the interaction of KinB with components of the respiratory apparatus. As a precedent for such a redox switch, the ArcB sensor kinase of Escherichia coli is silenced by quinone-mediated oxidation of two cysteines that participate in disulphide bond formation (Malpica et al. 2004).
The response to impaired respiration was not solely mediated by KinB. Under conditions of high iron concentration, at which the response to impaired respiration was most robust, a KinB mutant was largely but not entirely blocked in expression of the biofilm-inducing gene sinI, whereas a KinA KinB double mutant was almost completely blocked. A KinA single mutant and a KinC KinD double mutant were only modestly impaired under these conditions. We therefore conclude that KinA also contributes to sensing impaired respiration.
Although KinB was our main focus, we obtained a clue as to how KinA responds to impaired respiration. KinA is not a membrane protein and is unlikely to interact with the respiratory apparatus. Instead, we discovered that KinA associates with NAD+ and does so in a manner that depends on one of its three PAS domains: PASA. We therefore hypothesize that KinA is inhibited by NAD+ when respiration levels are high and is released from inhibition when the NAD+/NADH ratio drops due to conditions of impaired respiration. Efforts to test this hypothesis biochemically by investigating the effect of NAD+ on KinA activity are in progress. If our hypothesis is correct, then NAD+ can be added to the list of small molecule ligands known to be recognized by PAS domains. These include FAD, heme, metals, malonate, succinate, citrate, and glycerol (Henry and Crosson 2011).
In sum, we obtained evidence for impaired respiration as a signal for triggering matrix production and hence multicellularity in B. subtilis. Our findings support the hypothesis that biofilm wrinkling is in part an adaptation to reduced atmospheric oxygen, increasing the surface-to-volume ratio and thereby increasing accessibility to oxygen (Xavier and Foster 2007; Dietrich et al. 2008, 2013). (Other work has shown that wrinkles also create channels that facilitate nutrient flow within the colony [Wilking et al. 2012]). Importantly, we showed that impaired respiration is sensed by KinB, which does so by interacting with the respiratory apparatus (Fig. 9). KinA also contributes to the response to impaired respiration, and evidence suggests that it does so by sensing a drop in the NAD+/NADH ratio. Thus, KinA and KinB respond to impaired respiration by complementary mechanisms. KinA and KinB are also responsible for driving Spo0A∼P to the high levels needed for entry into sporulation during nutrient exhaustion. We therefore suggest that a drop in NAD+ levels and impaired oxidative phosphorylation are the direct physiological signals for sporulation as well as for biofilm formation (Fig. 9).
Figure 9.

Model for how impaired respiration controls matrix/sporulation gene expression (description in the text).
Materials and methods
Construction of strains and plasmids
Strains and plasmids were constructed using standard methods (Sambrook and Russell 2001). Oligonucleotides used for PCR in this study are listed in Supplemental Table S2. Transcriptional fusion with lacZ was obtained by cloning the ldh upstream region, including the start codon, into pAH124 using the corresponding restriction sites (Camp and Losick 2009). All deletion mutations were generated by long-flanking PCR mutagenesis (Wach 1996). Complementation of kinA was performed by PCR amplification of fragments carrying the promoter and entire gene coding sequence. These PCR fragments were cloned using the appropriate restriction enzymes into pDG1662 (Guerout-Fleury et al. 1996). The B. subtilis strains used in this work are listed in Supplemental Table S1. All strains were derived from NCIB 3610, a wild strain that forms robust biofilms (Branda et al. 2001). Transformation of B. subtilis PY79 (Youngman et al. 1984) with double-stranded PCR fragments, plasmid, or genomic DNA was done as described (Wilson and Bott 1968). SPP1 phage-mediated transduction was used to introduce antibiotic resistance-linked mutations from PY79 derivatives into NCIB 3610 (Yasbin and Young 1974).
Environmental chamber
An acrylic portable glove box (2100 series, Cleatech) was modified to produce an atmosphere of varying oxygen/nitrogen ratios. Specifically, a cylinder of compressed oxygen (medical grade) and nitrogen (CP grade) were connected via two-stage regulators to electronic mass flow controllers (MFCs) (SmartTrak 100, Sierra Instruments, Inc) that were capable of delivering gas at a rate of 200 SSC (standard cubic centimeter)/min. Polyvinylchloride (PVC) tubing (6.25-mm outer diameter, 3.12-mm inner diameter) was used to couple the two MFCs via a compression “tee” fitting (Jaco, Inc). The resulting gas mixture was directed into the chamber through PVC tubing. To enable continuous flushing of the environmental chamber, a gastight check valve with a 0.5 psi cracking pressure was positioned on the opposite wall of the chamber and continuously exhausted the gas mix. Prior to operation, the chamber was flushed for 24 h at 200 SSC/min of a predetermined oxygen:nitrogen ratio to achieve the desired percentage of atmospheric oxygen composition. The selected gas ratio and flow rate were maintained for the duration of the incubation to minimize changes in atmospheric composition. An oxygen sensor (Cleatech, Inc) was used to monitor the concentration of oxygen within the chamber throughout the experiment. In this configuration, oxygen concentration varied by no more than 2% for the duration of each experiment.
Real-time measurements of gene expression using luciferase
Starter LB cultures of desired strains were diluted 1:50 in double-distilled water (ddH2O), and then 112.5 μL was plated onto 1.5 mL of solid MSgg medium in each well (white with a clear bottom) of a 24-well polystyrene Visiplate (Wallac). The cultures were then grown for 24 h at 30°C in a BioTek Synergy 2 plate reader, and the OD at 600 nm (OD600) and luminescence (sensitivity setting, 200) were measured every 30 min. The data shown in the figures are averages for two replicate wells from a single representative experiment.
β-Galactosidase assays
Biofilms were formed in solid MSgg medium (Branda et al. 2001). The colonies were collected, resuspended with 1 mL of PBS, and sonicated to remove the extracellular matrix. OD600 had been measured. Cells (108–109) were taken for the assay. Cells were spun down, and pellets were resuspended in 1 mL of Z buffer (40 mM NaH2PO4, 60 mM Na2HPO4, 1 mM MgSO4, 10 mM KCl, 38 mM β-mercaptoethanol) supplemented with 200 μg mL−1 freshly made lysozyme. Resuspensions were incubated for 15 min at 30°C. Reactions were started by adding 200 μL of 4 mg mL−1 ONPG (2-nitrophenyl β-D-galactopyranoside) and stopped by adding 500 μL of 1 M Na2CO3. The soluble fractions were transferred to cuvettes (VWR), and OD420 values of the samples were recorded using a Pharmacia Ultraspectrometer 2000. The β-galactosidase-specific activity was calculated according to the equation [(OD420/(time × OD600)] × dilution factor × 1000. Assays were conducted in duplicate.
Measuring the NAD+/NADH ratio
The concentration of the steady-state pools of NAD+ and NADH were determined by the cycling assay of Bernofsky and Swan 1973) using an adaptation kindly provided to us by N. Sullivan and D. Newman. (1) Six colonies of each sample were harvested into 1 mL of PBS and subjected to mild sonication to separate cells' extracellular matrix. The cells were pelleted by centrifugation at 15,000 rpm for 1 min and resuspended in 500 μL of PBS, and the OD600 was measured. (2) In an anaerobic chamber, three samples (one from each colony) were resuspended for each condition in 300 μL of either 0.1 M HCL (for NADH extraction) or 0.1 M NaOH (for NAD+ extraction). NADH and NAD+ stocks were prepared in an anaerobic chamber using anaerobic water. (3) Outside the chamber, cells were incubated for 10 min at 50°C and then cooled down on ice for 10 min. Samples were neutralized by adding drop-wise while vortexing at medium speed of 300 μL of either HCL (for NADH extraction) or NaOH (for NAD+ extraction). (4) Samples were centrifuged at 15,000 rpm for 5 min, and the supernatant fluids were collected in fresh tubes and frozen at −80°C. (5) A reagent mixture was prepared in an anaerobic chamber and consisted of 200 μM Bicine buffer (1.0 M Bicine at pH 8), 200 μM 40 mM EDTA (pH 8), 200 μL of 100% ethanol, 200 μM 4.2 mM MTT (thiazolyle blue, Sigma), and 400 μM 16.6 mM phenazineethosulfate (Sigma). (6) In anaerobic chamber plates, we mixed 30 μL of anaerobic double-distilled water, 5 μL of serial dilutions of colony extract diluted up to 10−4, and 60 μL of the reagent mixture above, 5 μL of alcohol dehydrogenase, and 100 μg/mL Bicine buffer (Sigma). Each reaction was conducted on a separate well in 96-well polystyrene plates (VWR). (7) Outside the chamber, we took kinetic readings at 570 nm for 6 min at 30-sec intervals at 30°C. (8) For data analysis, we determined the linear range and used this time frame to calculate slopes for standard curves (0–0.1 mM, steps of 10 μM) of NAD+ and, separately, NADH and the serial dilution of the samples. (9) Finally, we normalized by OD600 of the initial samples.
Mutagenic PCR
PCR was performed with AcuStart PCR Supermix (Quanta Biosciences) on PCR product amplifying kinB with the restriction sites of BamHI and EcoRI. Reaction mixtures were cycled for 2 min at 94°C, followed by 10 cycles of 1 min at 94°C, 1 min at 65°C–1°C per cycle, and 5 min at 72°C. Finally, 25 cycles of 1 min at 94°C, 1 min at 55°C , and 5 min at 72°C were run. Pooled PCR products were ligated into cut plasmid PDG1728 (a gift from Win Chai) and transformed directly into strain RL4573 as described previously.
Immunoprecipitation of KinB
Biofilms were grown in a liquid biofilm-inducing medium for 48 h without shaking in six-well plates. For each sample, six plates were harvested in a total volume of 48 mL. After 48 h, cells were harvested by centrifugation (8000 rpm for 10 min at 4°C). Membrane protein purification was performed according to Eymann et al. (2004). GFP antibodies (Abcam) were cross-linked to protein A magnetic beads (Dynabeads, Life Technologies) according to the manufacturer's instructions. Then, membrane protein extracts were incubated with cross-linked antibodies under continuous rotation on a Stuart rotator SB3 at 30 rpm for 1 h at 4°C. After incubation, the beads were washed seven times with 1 mL of washing buffer (150 mM NaCl, 0.01% Triton X-100, 50 mM Tris-HCl at pH 8) for 3 min on the rotator at 30 rpm. The beads were collected with the magnetic rack, and the supernatants were discarded. Finally, the pure bead pellet was mixed with 100 μL of SDS sample buffer (Laemmli 1970) and denatured for 10 min at 90°C. Twenty-microliter aliquots were subjected to SDS-PAGE, and proteins in fractions from the gel were subjected to identification by high-accuracy mass spectrometry. The mass spectrometry analysis was performed at the Harvard Mass Spectrometry and Proteomics Resource Laboratory, FAS Center for Systems Biology, by microcapillary RP-HPLC nanoelectrospray MS/MS (μLC/MS/MS) on a Thermo LTQ-Orbitrap mass spectrometer.
Purification of KinA and HPLC/MS detection of NAD+
Cells harboring an N-terminal His6 tag for KinA(His6-KinA) were grown in DS medium and were collected 1 h after entry into stationary phase by centrifugation at 8000 rpm for 10 min at 4°C. Cells were washed twice in 50 mM Tris/HCl (pH 8), 200 mM NaCl, 20 mM imidazole and lysed using a cell disrupter. Cell debris was removed by centrifugation at 15,000 rpm at 4°C for 30 min. KinA was purified using Ni-NTA agarose (Quiagen) according to the manufacturer's protocol. The soluble supernatant was incubated with the agarose under continuous rotation on a Stuart rotator SB3 at 30 rpm for 1 h at 4°C. The supernatant, including the beads, was then passed through a flow-through polypropylene column (Bio-Rad). The column was washed with 10 bed volumes of buffer. The purified protein was eluted from the column with 0.5 mL of elution buffer (50 mM Tris-HCl at pH 8.0, 150 mM NaCl, 250 mM imidazole). Purified KinA was then subjected to a protocol established for NAD+ and NADH extraction (Sporty et al. 2008). HPLC measurements were performed using a Hydrosphere C18 column (5 μm, 150 mm × 4.6 mm inner diameter) (Waters) on an Agilent 1100 HPLC (Hewlett-Packard) according to a published protocol optimized for NAD+/NADH detection (Sporty et al. 2008).
Acknowledgments
We thank Sunia Trauger (Harvard FAS Center for Systems Biology) for help with MS analysis of NAD+, and A.L. Sonenshein for advice on the manuscript. I.K.-G. was a fellow of the Human Frontier Science Program, and A.E. was an EMBO post-doctoral fellow. This work was supported by grants from the NIH to R.L. (GM18568) and R.K. (GM58213) and from Advanced Research Projects Agency-Energy (ARPA-E) to P.R.G. (DE-AR 0000079).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.215244.113.
References
- Aguilar C, Vlamakis H, Losick R, Kolter R 2007. Thinking about Bacillus subtilis as a multicellular organism. Curr Opin Microbiol 10: 638–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar C, Vlamakis H, Guzman A, Losick R, Kolter R 2010. KinD is a checkpoint protein linking spore formation to extracellular-matrix production in Bacillus subtilis biofilms. mBio 1: e00035-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azarkina N, Siletsky S, Borisov V, von Wachenfeldt C, Hederstedt L, Konstantinov AA 1999. A cytochrome bb′-type quinol oxidase in Bacillus subtilis strain 168. J Biol Chem 274: 32810–32817 [DOI] [PubMed] [Google Scholar]
- Banse AV, Hobbs EC, Losick R 2011. Phosphorylation of Spo0A by histidine kinase KinD requires the lipoprotein Med in Bacillus subtilis. J Bacteriol 15: 3949–3955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernofsky C, Swan M 1973. An improved cycling assay for nicotinamide adenine dinucleotide. Anal Biochem 53: 452–458 [DOI] [PubMed] [Google Scholar]
- Branda SS, Gonzalez-Pastor JE, Ben-Yehuda S, Losick R, Kolter R 2001. Fruiting body formation by Bacillus subtilis. Proc Natl Acad Sci 98: 11621–11626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branda SS, Gonzalez-Pastor JE, Dervyn E, Ehrlich SD, Losick R, Kolter R 2004. Genes involved in formation of structured multicellular communities by Bacillus subtilis. J Bacteriol 186: 3970–3979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branda SS, Vik S, Friedman L, Kolter R 2005. Biofilms: The matrix revisited. Trends Microbiol 13: 20–26 [DOI] [PubMed] [Google Scholar]
- Burbulys D, Trach KA, Hoch JA 1991. Initiation of sporulation in B. subtilis is controlled by a multicomponent phosphorelay. Cell 64: 545–552 [DOI] [PubMed] [Google Scholar]
- Camp AH, Losick R 2009. A feeding tube model for activation of a cell-specific transcription factor during sporulation in Bacillus subtilis. Genes Dev 23: 1014–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Chu F, Kolter R, Losick R 2008. Bistability and biofilm formation in Bacillus subtilis. Mol Microbiol 67: 254–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Kolter R, Losick R 2009. A widely conserved gene cluster required for lactate utilization in Bacillus subtilis and its involvement in biofilm formation. J Bacteriol 191: 2423–2430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Kolter R, Losick R 2010a. Reversal of an epigenetic switch governing cell chaining in Bacillus subtilis by protein instability. Mol Microbiol 78: 218–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Norman T, Kolter R, Losick R 2010b. An epigenetic switch governing daughter cell separation in Bacillus subtilis. Genes Dev 24: 754–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Norman T, Kolter R, Losick R 2011. Evidence that metabolism and chromosome copy number control mutually exclusive cell fates in Bacillus subtilis. EMBO J 30: 1402–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Cao S, Chai Y, Clardy J, Kolter R, Guo JH, Losick R 2012a. A Bacillus subtilis sensor kinase involved in triggering biofilm formation on the roots of tomato plants. Mol Microbiol 85: 418–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yan F, Chai Y, Liu H, Kolter R, Losick R, Guo JH 2012b. Biocontrol of tomato wilt disease by Bacillus subtilis isolates from natural environments depends on conserved genes mediating biofilm formation. Environ Microbiol. 15: 848–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu F, Kearns DB, Branda SS, Kolter R, Losick R 2006. Targets of the master regulator of biofilm formation in Bacillus subtilis. Mol Microbiol 59: 1216–1228 [DOI] [PubMed] [Google Scholar]
- Chu F, Kearns DB, McLoon A, Chai Y, Kolter R, Losick R 2008. A novel regulatory protein governing biofilm formation in Bacillus subtilis. Mol Microbiol 68: 1117–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costerton JW, Stewart PS, Greenberg EP 1999. Bacterial biofilms: A common cause of persistent infections. Science 284: 1318–1322 [DOI] [PubMed] [Google Scholar]
- Cruz Ramos H, Boursier L, Moszer I, Kunst F, Danchin A, Glaser P 1995. Anaerobic transcription activation in Bacillus subtilis: Identification of distinct FNR-dependent and -independent regulatory mechanisms. EMBO J 14: 5984–5994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dago AE, Schug A, Procaccini A, Hoch JA, Weigt M, Szurmant H 2012. Structural basis of histidine kinase autophosphorylation deduced by integrating genomics, molecular dynamics, and mutagenesis. Proc Natl Acad Sci 109: E1733–E1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dartois V, Djavakhishvili T, Hoch JA 1997. KapB is a lipoprotein required for KinB signal transduction and activation of the phosphorelay to sporulation in Bacillus subtilis. Mol Microbiol 26: 1097–1108 [DOI] [PubMed] [Google Scholar]
- Dietrich LE, Teal TK, Price-Whelan A, Newman DK 2008. Redox-active antibiotics control gene expression and community behavior in divergent bacteria. Science 321: 1203–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich LE, Okegbe C, Price-Whelan A, Sakhtah H, Hunter RC, Newman DK 2013. Bacterial community morphogenesis is intimately linked to the intracellular redox state. J Bacteriol 195: 1371–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswaramoorthy P, Duan D, Dinh J, Dravis A, Devi SN, Fujita M 2010. The threshold level of the sensor histidine kinase KinA governs entry into sporulation in Bacillus subtilis. J Bacteriol 192: 3870–3882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eymann C, Dreisbach A, Albrecht D, Bernhardt J, Becher D, Gentner S, Tam LT, Büttner K, Buurman G, Scharf C, et al. 2004. A comprehensive proteome map of growing Bacillus subtilis cells. Proteomics 4: 2849–2876 [DOI] [PubMed] [Google Scholar]
- Gaballa A, Antelmann H, Aguilar C, Khakh SK, Song KB, Smaldone GT, Helmann JD 2008. The Bacillus subtilis iron-sparing response is mediated by a Fur-regulated small RNA and three small, basic proteins. Proc Natl Acad Sci 105: 11927–11932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia Montes de Oca LY, Chagolla-Lopez A, Gonzalez de la Vara L, Cabellos-Avelar T, Gomez-Lojero C, Gutierrez Cirlos EB 2012. The composition of the Bacillus subtilis aerobic respiratory chain supercomplexes. J Bioenerg Biomembr 44: 473–486 [DOI] [PubMed] [Google Scholar]
- Guerout-Fleury AM, Frandsen N, Stragier P 1996. Plasmids for ectopic integration in Bacillus subtilis. Gene 180: 57–61 [DOI] [PubMed] [Google Scholar]
- Henry JT, Crosson S 2011. Ligand-binding PAS domains in a genomic, cellular, and structural context. Annu Rev Microbiol 65: 261–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann T, Troup B, Szabo A, Hungerer C, Jahn D 1995. The anaerobic life of Bacillus subtilis: Cloning of the genes encoding the respiratory nitrate reductase system. FEMS Microbiol Lett 131: 219–225 [DOI] [PubMed] [Google Scholar]
- Kearns DB, Chu F, Branda SS, Kolter R, Losick R 2005. A master regulator for biofilm formation by Bacillus subtilis. Mol Microbiol 55: 739–749 [DOI] [PubMed] [Google Scholar]
- Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130: 797–810 [DOI] [PubMed] [Google Scholar]
- Kolter R, Greenberg EP 2006. Microbial sciences: The superficial life of microbes. Nature 441: 300–302 [DOI] [PubMed] [Google Scholar]
- Laemmli UK 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685 [DOI] [PubMed] [Google Scholar]
- LeDeaux JR, Yu N, Grossman AD 1995. Different roles for KinA, KinB, and KinC in the initiation of sporulation in Bacillus subtilis. J Bacteriol 177: 861–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Tomchick DR, Brautigam CA, Machius M, Kort R, Hellingwerf KJ, Gardner KH 2008. Changes at the KinA PAS-A dimerization interface influence histidine kinase function. Biochemistry 47: 4051–4064 [DOI] [PubMed] [Google Scholar]
- Lopez D, Fischbach MA, Chu F, Losick R, Kolter R 2009. Structurally diverse natural products that cause potassium leakage trigger multicellularity in Bacillus subtilis. Proc Natl Acad Sci 106: 280–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah TF, O'Toole GA 2001. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol 9: 34–39 [DOI] [PubMed] [Google Scholar]
- Malpica R, Franco B, Rodriguez C, Kwon O, Georgellis D 2004. Identification of a quinone-sensitive redox switch in the ArcB sensor kinase. Proc Natl Acad Sci 101: 13318–13323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLoon AL, Kolodkin-Gal I, Rubinstein SM, Kolter R, Losick R 2011. Spatial regulation of histidine kinases governing biofilm formation in Bacillus subtilis. J Bacteriol 193: 679–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moglich A, Ayers RA, Moffat K 2009. Structure and signaling mechanism of Per-ARNT-Sim domains. Structure 17: 1282–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano MM, Zuber P 1998. Anaerobic growth of a ‘strict aerobe’ (Bacillus subtilis). Annu Rev Microbiol 52: 165–190 [DOI] [PubMed] [Google Scholar]
- Romero D, Aguilar C, Losick R, Kolter R 2010. Amyloid fibers provide structural integrity to Bacillus subtilis biofilms. Proc Natl Acad Sci 107: 2230–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero D, Vlamakis H, Losick R, Kolter R 2011. An accessory protein required for anchoring and assembly of amyloid fibres in B. subtilis biofilms. Mol Microbiol 80: 1155–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein SM, Kolodkin-Gal I, McLoon A, Chai L, Kolter R, Losick R, Weitz DA 2012. Osmotic pressure can regulate matrix gene expression in Bacillus subtilis. Mol Microbiol 86: 426–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW 2001. Molecular cloning: A laboratory manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Shemesh M, Kolter R, Losick R 2010. The biocide chlorine dioxide stimulates biofilm formation in Bacillus subtilis by activation of the histidine kinase KinC. J Bacteriol 192: 6352–6356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporty JL, Kabir MM, Turteltaub KW, Ognibene T, Lin SJ, Bench G 2008. Single sample extraction protocol for the quantification of NAD and NADH redox states in Saccharomyces cerevisiae. J Sep Sci 31: 3202–3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson K, Hoch JA 2001. PAS-A domain of phosphorelay sensor kinase A: A catalytic ATP-binding domain involved in the initiation of development in Bacillus subtilis. Proc Natl Acad Sci 98: 15251–15256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautenhahn R, Cho K, Uritboonthai W, Zhu Z, Patti GJ, Siuzdak G 2012. An accelerated workflow for untargeted metabolomics using the METLIN database. Nat Biotechnol 30: 826–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng YL, Zhou XZ, Hoch JA 1998. Phosphorylation of the Spo0B response regulator phosphotransferase of the phosphorelay initiating development in Bacillus subtilis. J Biol Chem 273: 23849–23855 [DOI] [PubMed] [Google Scholar]
- van der Oost J, von Wachenfeld C, Hederstedt L, Saraste M 1991. Bacillus subtilis cytochrome oxidase mutants: Biochemical analysis and genetic evidence for two aa3-type oxidases. Mol Microbiol 5: 2063–2072 [DOI] [PubMed] [Google Scholar]
- Wach A 1996. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 12: 259–265 [DOI] [PubMed] [Google Scholar]
- Wang E, Bauer MC, Rogstam A, Linse S, Logan DT, von Wachenfeldt C 2008. Structure and functional properties of the Bacillus subtilis transcriptional repressor Rex. Mol Microbiol 69: 466–478 [DOI] [PubMed] [Google Scholar]
- Wilking JN, Zaburdaev V, De Volder M, Losick R, Brenner MP, Weitz DA 2012. Liquid transport facilitated by channels in Bacillus subtilis biofilms. Proc Natl Acad Sci 110: 848–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GA, Bott KF 1968. Nutritional factors influencing the development of competence in the Bacillus subtilis transformation system. J Bacteriol 95: 1439–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier JB, Foster KR 2007. Cooperation and conflict in microbial biofilms. Proc Natl Acad Sci 104: 876–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasbin RE, Young FE 1974. Transduction in Bacillus subtilis by bacteriophage SPP1. J Virol 14: 1343–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman P, Perkins JB, Losick R 1984. Construction of a cloning site near one end of Tn917 into which foreign DNA may be inserted without affecting transposition in Bacillus subtilis or expression of the transposon-borne erm gene. Plasmid 12: 1–9 [DOI] [PubMed] [Google Scholar]