

Abstract
In dividing cells, the two aims a gene therapeutic approach should accomplish are efficient nuclear delivery and retention of therapeutic DNA. For stable transgene expression, therapeutic DNA can either be maintained by somatic integration or episomal persistence of which the latter approach would diminish the risk of insertional mutagenesis. As most monosystems fail to fulfill both tasks with equal efficiency, hybrid-vector systems represent promising alternatives. Our hybrid-vector system synergizes high-capacity adenoviral vectors (HCAdV) for efficient delivery and the scaffold/matrix attachment region (S/MAR)–based pEPito plasmid replicon for episomal persistence. After proving that this plasmid replicon can be excised from adenovirus in vitro, colony forming assays were performed. We found an increased number of colonies of up to sevenfold in cells that received the functional plasmid replicon proving that the hybrid-vector system is functional. Transgene expression could be maintained for 6 weeks and the extrachromosomal plasmid replicon was rescued. To show efficacy in vivo, the adenoviral hybrid-vector system was injected into C57Bl/6 mice. We found that the plasmid replicon can be released from adenoviral DNA in murine liver resulting in long-term transgene expression. In conclusion, we demonstrate the efficacy of our novel HCAdV-pEPito hybrid-vector system in vitro and in vivo.
Keywords: adenovirus, pEPito, persistent transgene expression, plasmid replicon
Introduction
There are two major strategies for long-term maintenance of the transgene within a transduced cell of which one is based on somatic integration and the other is based on extrachromosomal maintenance of the therapeutic DNA. Modern adenoviral vectors efficiently deliver the genetic information into the nucleus but due to the absence of efficient nuclear retention and replication, recombinant adenoviral DNA molecules remain episomal and transgene expression levels decline due to the loss of vector molecules in dividing tissue.1,2 So-called hybrid-vector systems are capable of maintaining the administered extrinsic DNA by merging efficient viral delivery with a second component for nuclear persistence of the therapeutic gene. In currently available versions of hybrid vectors, the viral vector encodes an integration machinery of diverse viral or non-viral offspring and the therapeutic DNA to be integrated into the host genome.2,3 Once expressed, the integration apparatus inserts the therapeutic gene into the chromosomes, allowing its duplication during cell cycle and long-term expression. The adenoviral capsid has already been packaged with integration machineries of the AAV rep protein,4,5 the Sleeping Beauty transposase system,6,7 and the bacteriophage-derived integrase PhiC31.8 However, adverse effects such as genotoxicity and insertional mutagenesis may be expected from hybrid vectors using recombinases for somatic integration analogous to integrating retroviral vectors that were demonstrated to result in serious adverse events in clinical trials.9 Furthermore, position effect variegation, for instance integration into transcriptionally inactive regions of the host genome, may result in vanishing expression of the therapeutic transgene. In contrast, hybrid-vector systems that preserve the therapeutic gene in an episomally replicating plasmid within the cell nucleus should not display any risk of insertional mutagenesis.
Toward this end, episomal adenoviral hybrid vectors based on the Epstein–Barr virus (EBV) replication and retention mechanisms have already been described.10,11,12,13 Two of these studies10,11 used high-capacity adenoviral vectors (HCAdV) for delivery of the EBV episome. HCAdV lacks all viral coding sequences and compared with other adenovirus-based vectors HCAdV features the highest packaging capacity for foreign DNA with up to 36 kb.14 Gene therapeutic applications of HCAdV were effectually described for various monogenic diseases.15,16,17,18,19,20 This type of vectors represent one of the most advanced versions for adenoviral gene delivery resulting in reduced toxicity attenuated immune responses in vivo.21,22
Our novel hybrid-vector synergizes the HCAdV technology for efficient delivery and the episomal plasmid replicon pEPito for nuclear retention and replication of the transgene. The pEPito vector23 is a promoter optimized and CpG-depleted derivative of the plasmid pEPI-1, which was constructed in 1999.24,25 Essential feature of both plasmids is a 2 kb S/MAR (scaffold/matrix attachment region) sequence23 that is derived from the 5′-region of the human β-interferon gene26 and comprises 69% of nucleotides A and T, extensive base unpairing regions and >94% binding-affinity to the nuclear matrix. Importantly, upstream transcription directed without any stop-signal into S/MAR is obligatory for the episomal persistence and replication of both pEPI-1 and pEPito, most likely because of conformational changes necessary for the S/MAR to operate.27,28 S/MAR-binding to nuclear matrix proteins and association with early replicating foci that is preserved over mitosis presumably mediates the equal distribution of pEPI-1 to daughter cells during mitosis by non-covalent attachment of the plasmids to metaphase chromosomes.24,29,30 pEPI-1 persists at a copy number of 2–10 per cell over hundreds of generations, and its replication proceeds once per cell cycle in early S phase with the prereplication complex being able to assemble at multiple sites of the plasmid.31 Compared with its ancestor pEPI-1, pEPito shows a stronger and more prolonged transgene expression in vitro and in vivo, although still sharing the characteristics of episomal replication and persistence.23 Attenuated epigenetic silencing and inflammatory response in vivo can be expected due to CpG-depletion of the pEPito promoter and the backbone.
Results
Functionality of HCAdV used to establish the hybrid-vector system
In this study, we aimed at establishing an adenoviral hybrid-vector system that combines high transduction efficiencies of adenoviral vectors with the plasmid replicon pEPito23 for episomally maintained stable transgene expression. Therefore, in contrast to other systems for somatic integration and long-term transgene expression, our system should preclude the risk of insertional mutagenesis. The principle of the system is based on delivery of the plasmid replicon pEPito into the target cell using HCAdV. After the adenoviral genome enters the nucleus, the plasmid replicon can be released from the adenoviral vector genome by FLPe-recombinase–mediated excision. The principle of the system is schematically shown in Figure 1a. Mitotic stability is then provided by its replication during S phase and linking the plasmid replicon to metaphase chromosomes during mitosis (Figure 1a). To establish the system, we cloned and produced several HCAdV constructs, and the DNA sequences contained in these adenoviral vectors are displayed in Figure 1b–e. HCAdV-pEPito-FRT2 corresponds to linearized pEPito flanked by two directed FRT sites and inserted into the HCAdV genome (Figure 1b). Coinfection with HCAdV-FLPe (Figure 1d) expressing the FLPe gene is required for functioning of HCAdV-pEPito-FRT2. FLPe-recombinase excises linearized pEPito out of HCAdV-pEPito-FRT2 and circularizes the episomal plasmid that begins to replicate and remains mitotically stable. The structure of negative control HCAdV-pEPito-ΔS/MAR-FRT2 lacks the S/MAR region and therefore there is no stability of the episomal plasmid replicon after FLPe-mediated excision (Figure 1c). As another control vector, the previously described vector HCAdV-hFIX (Figure 1e) was amplified.21 This irrelevant vector was used to ensure that equal total amounts of recombinant virus were used in all infection experiments.
Figure 1.
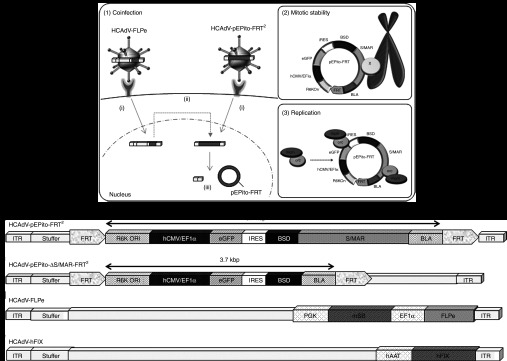
Principle of the adenovirus/pEPito hybrid vector and DNA sequences contained in the high-capacity adenoviral vectors to establish the hybrid-vector system. (a) Principle of the adenovirus/pEPito hybrid vector. (1) High-capacity adenoviral vectors HCAdV-pEPito-FRT2 and HCAdV-FLPe simultaneously (i) transduce the target cell. (ii) FLPe is expressed from HCAdV-FLPe and recombines HCAdV-pEPito-FRT2. (iii) Products of FLPe recombination are the recircularized pEPito insert containing one FRT site and the linked and linear HCAdV-backbone fragments which were flanking the pEPito insert. (2) Mitotic stability of recombined pEPito-FRT. pEPito-FRT linked to metaphase chromosomes by putative nuclear matrix proteins (X) mediates retention of the plasmid replicon. (3) Replication of pEPito-FRT. It is speculated that the prereplicative complex consisting of the origin recognition complex (orc) and the minichromosome maintenance complex (mcm) binds at multiple sites of the excised plasmid replicon pEPito-FRT. (b) DNA sequences within the adenoviral vector HCAdV-pEPito-FRT2 containing the plasmid replicon pEPito flanked by FRT sites for FLPe-mediated recombination and excision.21 (c) The control vector HCAdV-pEPito-ΔS/MAR-FRT2 contains identical DNA sequences as HCAdV-pEPito-FRT2 but lacks the S/MAR sequence. Therefore, the excised plasmid is not maintained during cell division. (d) The vector HCAdV-FLPe was used in our previous study6 and expresses FLPe under the control of the elongation factor-1α promoter (EF1α). In addition, this vector contains an expression cassette for an inactive version of Sleeping Beauty transposase (mSB) expressed under the control of the phosphor-glycerate-kinase promoter (PGK). (e) The irrelevant vector HCAdV-hFIX was used as an infection control. As described in a previous study,21 this vector encodes the human coagulation factor IX (hFIX) under the control of the human α1-antitrypsin promoter (hAAT). BLA, β-lactamase gene; BSD, blasticidin resistance gene; eGFP, enhanced green fluorescence protein; FRT, recognition site of FLPe-recombinase; hCMV/EF1, elongation factor α1 promoter with human cytomegalovirus enhancer; IRES, internal ribosomal entry site; ITR, left and right inverted terminal repeats of adenovirus serotype 5; S/MAR: scaffold/matrix attachment region; stuffer DNA, stuffer DNA contained in the HCAdV DNA molecule based on the previously described HCAdV production plasmid pAdFTC.
Before elaborate production of adenoviruses HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2, shuttle plasmids pHM5-pEPito-FRT2 and pHM5-pEPito-ΔS/MAR-FRT2 (Supplementary Figure S1) containing plasmid replicons flanked by FRT sites for final HCAdV cloning were characterized in detail to ensure that essential DNA sequences are functional. Toward this end, we performed provisional experiments in a plasmid context successfully matching the main characteristics of our adenoviral hybrid-vector system. Replicon pEPito has never been substrate of recombinational events, thus evaluation of FLPe-recombinase activity was necessary by excluding any negative influence on pEPito and its functional cassettes. After cotransfection of pHM5-pEPito-FRT2 and pHM5-pEPito-ΔS/MAR-FRT2 with a plasmid encoding FLPe, we show functional enhanced green fluorescent protein (eGFP) expression 48 hours after transfection (Supplementary Figure S1), and we verified that FLPe recombination occurs for both substrates using a PCR-based approach (Supplementary Figure S1). Moreover, we performed colony forming assays using blasticidin selection showing that the plasmid pHM5-pEPito-FRT2 resulted in an increased number of blasticidin resistant expressing colonies that express eGFP with and without FLPe recombination compared the control plasmid pHM5-pEPito-ΔS/MAR-FRT2 (Supplementary Figure S1).
After confirming the functionality of DNA sequences contained in the shuttle vectors, adenoviral vectors HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2 were produced according to our recently published protocol for HCAdV production.32 Accurate genome sequences of HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2 in final vector preparations were proven by PCR and diagnostic restriction enzyme digests (Supplementary Figure S2). Physical titers and transducing units were measured by quantitative PCR, and the infectious titer was also determined by transducing human embryonic kidney (HEK) 293 cells at various multiplicity of infections (MOIs) (Supplementary Figure S3).
To analyze the functionality of the different components contained in the adenoviral vectors, we generated the stably FLPe-expressing cell line PurFLPe-R. To prove the functionality of FLPe-recombinase in this cell line, cells were transfected with a HCAdV plasmid containing two FRT sites for FLPe-mediated excision8 (Supplementary Figure S4). On the basis of a previously published PCR approach,8 we could demonstrate the FLPe-mediated recombination of the plasmid pFTC-attB-TCM-FRT (Supplementary Figure S4). After infection of PurFLPe-R cells with adenoviruses HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2 (Figure 1), the plasmid replicon should be excised from the adenoviral genome by FLPe-mediated recombination, which in concordance to our plasmid-based approach (Supplementary Figure S1) can be visualized by a specific PCR reaction. After infection of PurFLPe-R cells at an MOI of 50 resulting into transduction efficiencies of up to 30% (Supplementary Figure S5), we isolated genomic DNA 48 hours after infection and performed a PCR reaction specifically detecting excised DNA circles (Supplementary Figure S5). The PCR product of 530 bp in size spans the FRT site of the excised plasmid replicon pEPito.
As commonly used cell lines lack FLPe-recombinase expression, we designed a coinfection model using U87 cells. U87 cells are derived from a human glioblastoma-astrocytoma and were used in previous studies exploring stability of HCAdV genomes in vitro.33 Therefore, we first optimized infection conditions for this particular cell line when using the human adenovirus serotype 5, the serotype used for encapsidation of the recombinant viral vectors containing the plasmid replicon in this study. We tested different MOIs and found that an MOI of 100 is sufficient to transduce up to 90% of all cells after a single infection either with HCAdV-pEPito-FRT2 or HCAdV-pEPito-ΔS/MAR-FRT2 (data not shown). To optimize coinfection conditions, we coinfected cells at a ratio of 1:1 using the adenoviral vector HCAdV-FLPe and either the vector HCAdV-pEPito-FRT2 containing the active plasmid replicon or the negative control vector HCAdV-pEPito-ΔS/MAR-FRT2 without S/MAR sequence. A total dose of MOI of 600 (MOI of 300 for each virus respectively) resulted into transduction efficiencies of up to 87.6 and 76.5% as determined by flow cytometry (Supplementary Figure S3). To prove the excision of the DNA replicon mediated by FLPe-mediated recombination, we coinfected U87 cells with viruses HCAdV-pEPito-FRT2 or HCAdV-pEPito-ΔS/MAR-FRT2 along with the FLPe encoding virus HCAdV-FLPe or the control vector HCAdV-hFIX. Simultaneous infection resulted in 62.2 and 46.93% eGFP positive U87 cells after transduction with HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2, respectively (Figure 2a).
Figure 2.

Optimization of infection conditions and basic features of the hybrid-vector system. (a) U87 cells were infected with HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2 at MOI of 200 and either coinfected with the stuffer virus HCAdV-hFIX (see Figure 1e) at MOI of 200 or the FLPe-encoding virus HCAdV-FLPe at MOI of 200, respectively (total MOI of 400 for each coinfection experiment). Seventy-two hours after infection percentages of eGFP-expressing U87 cells were analyzed by flow cytometry (top panel). Mean values of three independent experiments (±SD) are shown; *P < 0.05. To prove excision, a PCR was performed with primers Recomb-fw and Recomb-rev on genomic DNA from U87 cells 72 hours after infection (bottom panel). The expected PCR product is 530 bp in length. For details of the PCR setup, please also refer to Supplementary Figure S5. (b) Plasmid rescue of excised plasmid replicons pEPito-FRT and pEPito-ΔS/MAR-FRT. Total genomic DNA was isolated and recircularized pEPito-FRT and pEPito-ΔS/MAR-FRT were rescued from U87 cells 72 hours after infection. Rescued plasmids were analyzed by restriction enzyme digest using the restriction enzyme XhoI. Exemplary XhoI-digests of two rescued pEPito-FRT and two pEPito-ΔS/MAR-FRT plasmids are shown. M, DNA ladder as marker; MOI, multiplicity of infections.
FLPe-recombination excises pEPito-FRT out of HCAdV-pEPito-FRT2 and pEPito-ΔS/MAR-FRT out of HCAdV-pEPito-ΔS/MAR-FRT2 genome, respectively (Supplementary Figure S5). Subsequently, the two remaining linear HCAdV-backbone fragments are ligated tail to head to form linear HCAdV-FRT that carries the second FRT site (Supplementary Figure S5). Recombined plasmids as products of FLPe recombination could be gained from genomic DNA isolated 72 hours after infection and determined using our established recombination PCR specifically detecting excised plasmids from the HCAdV genome (Figure 2a and Supplementary Figure S5). In addition, we were able to rescue the excised plasmid replicon pEPito-FRT and the respective negative control without S/MAR (pEPito-ΔS/MAR-FRT) 72 hours after infection. Restriction enzyme digests revealed a restriction pattern as expected, demonstrating that no rearrangement processes occurred during the processing of the vector in the cell (Figure 2b) and showing that the eGFP-expressing plasmid replicon can be released from the adenoviral DNA molecule within a cell. Therefore, in a further step, we wanted to investigate in long-term experiments that whether the plasmid replicon and transgene expression can be maintained during further cell divisions in vitro.
Persistence of the plasmid replicon after release from the adenoviral vector genome
To analyze the persistence of the plasmid replicon after it was released from the adenoviral DNA molecule, we cotransduced adenoviruses HCAdV-pEPito-FRT2 or HCAdV-pEPito-ΔS/MAR-FRT2 with the FLPe-encoding vector HCAdV-FLPe at increasing MOIs (MOIs of 10, 30, and 100). Colony forming assays of which the outline is schematically shown in Figure 3a were performed in U87 cells. After 6 weeks of blasticidin selection, we found that cells that received the vector HCAdV-pEPito-FRT2 along with the FLPe-encoding virus showed significantly increased numbers of colonies for all analyzed MOIs which was in contrast to the group that received the virus HCAdV-pEPito-ΔS/MAR-FRT2 releasing the plasmid without functional S/MAR sequence (Figure 3b).
Figure 3.

Establishment of the plasmid replicon after infection with the hybrid-vector system. (a) Experimental setup for long-term evaluation of the adenovirus/pEPito hybrid-vector system. U87 cells were coinfected with the FLPe-encoding virus HCAdV-FLPe and either HCAdV-pEPito-FRT2 or HCAdV-pEPito-ΔS/MAR-FRT2 at MOIs of 10, 30, and 100 for each virus. Cells were selected for 6 weeks using blasticidin, surviving cell colonies were stained with methylene blue and molecular analysis was performed. (b) Colony forming assay 6 weeks after blasticidin selection. Surviving colonies were stained with methylene blue. (c) Recombination PCR was performed with primers Recomb-fw and Recomb-rev on genomic DNA from U87 cells coinfected with HCAdV-pEPito-FRT2 or HCAdV-pEPito-ΔS/MAR-FRT2 and the FLPe-encoding virus HCAdV-FLPe. The expected PCR product was 530 bp in length. M, DNA ladder as marker. (d) Plasmid rescue 6 weeks after infection under 6 weeks of selection pressure. Genomic DNA was isolated and transformed into competent bacteria to grow single colonies of episomal plasmid replicons. The left panel shows a control digest of plasmid pEPito-FRT using the single-cutter BamHI serving as a positive control. The right panel shows exemplarily 5 rescued plasmids (R1-R5) of the group that received virus HCAdV-pEPito-FRT2 and HCAdV-FLPe. M, DNA ladder as marker. (e) Overlapping PCR after 6 weeks of selection procedure. Principle of PCR (left panel): Two overlapping fragments I and II were amplified covering the complete pEPito-FRT plasmid. PCR was performed on genomic DNA from U87 cells coinfected with HCAdV-pEPito-FRT2 and HCAdV-FLPe (right panel). Expected length of PCR product I amplified with primers Recomb-fw and BSD-IRES-rev-BamHI was 2.9 kb and the expected length of PCR product II amplified with primers Recomb-rev and BSD-IRES-fw-BglII is 3.9 kb.
Next, we investigated the episomal status of excised plasmids by means of PCR and plasmid rescue. The recombination PCR was carried out using the primers Recomb-fw and Recomb-rev (Supplementary Figure S5) on genomic DNA of U87 cells infected with an MOI of 100 for each virus. The specific excision PCR product of 530 bp in length was amplified for the group that received vectors HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2 (Figure 3c). Moreover, we performed a plasmid rescue to recover the episomally maintained plasmid replicon pEPito-FRT. A diagnostic BamHI digest of rescued plasmids was performed, and as shown in Figure 3d, the plasmid replicon was recovered. Furthermore, we confirmed the presence of the excised plasmid replicon pEPito-FRT by an overlapping PCR approach which covers the complete pEPito-FRT plasmid (Figure 3e). Notably, we also rescued a limited amount of episomal DNA from the group that received the vector without S/MAR sequence. After detailed characterization of these plasmids by diagnostic restriction enzyme digest, PCR and sequencing, we found that these rescued plasmids in the ΔS/MAR group equaled the original version of the excised pEPito-ΔMAR-FRT plasmid (Supplementary Figure S6).
At the beginning of the experiment (72 hours after infection), 18 and 13% of cells were eGFP positive as determined by flow cytometry, and at the completion of experiment, 89.8% of U87 cells infected with HCAdV-pEPito-FRT2 (MOI of 30) and 92.6% infected with HCAdV-pEPito-ΔS/MAR-FRT2 (MOI of 30) were positive for eGFP expression, respectively (Figure 4a, left panel). Intensity of eGFP expression was comparable in both groups for both time points (Figure 4a, right panel). Representative histograms of the early time point (72 hours after infection) and the later time points (6 weeks after Blasticidin selection) are displayed in Figure 4b. Representative eGFP positive colonies after selection are shown in Figure 4c.
Figure 4.
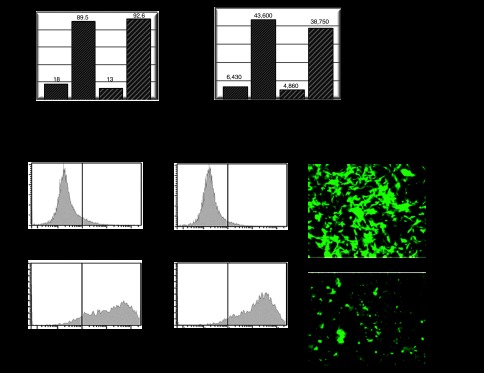
Analysis of persistence of transgene expression after delivery of the hybrid-vector system. eGFP expression was measured 72 hours after infection and 6 weeks after blasticidin selection. U87 cells were coinfected with the FLPe-encoding virus HCAdV-FLPe and either HCAdV-pEPito-FRT2 or HCAdV-pEPito-ΔS/MAR-FRT2 at MOI of 30 for each virus. Cells were selected for 6 weeks using blasticidin and surviving cell colonies were analyzed. (a) Flow cytometer analysis measuring percentages of eGFP-expressing U87 cells before and after treatment with blasticidin for 6 weeks (left panel). The right panel shows the mean intensity of eGFP expression of the same experiment. Exemplary results of one experiment are shown. (b) Original histograms of exemplary samples after flow cytometry showing eGFP-expressing U87 cells 72 hours (top panel) and 6 weeks (bottom panel) after infection and selection pressure using blasticidin. (c) Fluorescence microscopy 6 weeks after infection and selection pressure using blasticidin. Groups which received the adenoviral vectors HCAdV-pEPito-FRT2 or HCAdV-pEPito-δS/MAR-FRT2 along with the FLPe-encoding virus were analyzed. MOI, multiplicity of infections.
Performance of the adenoviral hybrid-vector system without FLPe recombination
As a further step, we aimed at investigating the system without codelivery of the FLPe-recombinase–encoding virus. To address this question, we infected U87 cells either with HCAdV-pEPito-FRT2 or with HCAdV-pEPito-ΔS/MAR-FRT2 at MOIs of 10 and 100 and replaced the FLPe-encoding vector HCAdV-FLPe by codelivering the previously described irrelevant control vector HCAdV-hFIX (Figure 1e) expressing the human coagulation factor IX.21 We speculated that without any recombination reaction for release of the plasmid replicon, linear HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2 should not result in episomal persistence similar to that of conventional non-hybrid HCAdV vectors. We performed colony forming assays to analyze stability of blasticidin and eGFP expression, and unexpectedly, we found that even without FLPe recombination the virus HCAdV-pEPito-FRT2 seems to persist when compared with the experimental group that received the virus HCAdV-pEPito-ΔS/MAR-FRT2 (Figure 5a). Seventy-two hours after coinfection, we measured eGFP expression in 40.2% of cells that received the viruses HCAdV-pEPito-FRT2 and HCAdV-hFIX and in 29.4% of cells that were coinfected with viruses HCAdV-pEPito-ΔS/MAR-FRT2 and HCAdV-hFIX (Figure 5b). The recombination PCR retrieving the recombination products pEPito-FRT and pEPito-ΔS/MAR-FRT resided negative 72 hours after infection and also after 6 weeks of treatment with blasticidin that excluded any spontaneous recombination via the FRT sites that could explain the observed persistency (Figure 5c). No plasmids or other low molecular weight DNA molecules could be rescued from both groups at the initiation and completion of the experiment, excluding that spontaneous excision of the plasmid replicon occurred. To investigate the molecular forms of vectors genomes in experiments performed without FLPe recombination, we pursued a PCR strategy detecting covalent linkage of inverted terminal repeats (ITRs) and therefore circularized or covalently linked HCAdV genomes. A similar approach was also used in our previous study analyzing the molecular status of HCAdV DNA molecules after cellular transduction.43 In our PCR approach, a PCR product of 144 bp in length was expected when using a primer binding to the ITR of the adenoviral genome. As a positive control resulting in a PCR product of 3,048 bp in length, we used a previously described HCAdV genome34 that was cloned into a plasmid backbone separating the ITRs. As displayed in Figure 5d, we observed several bands at higher molecular weight on the agarose gel and no PCR products that refer to expected circular forms of adenoviral vector genomes.
Figure 5.
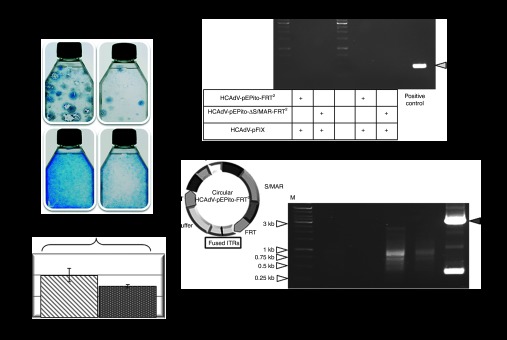
Evaluation of HCAdVs without FLPe recombination. (a) Colony forming assay performed in U87 cells to analyze maintenance of the transgene during selection pressure without FLPe recombination. Cells were infected with viruses HCAdV-pEPito-FRT2 or HCAdV-pEPito-ΔS/MAR-FRT2 at MOIs of 10 and 100. After 6 weeks under blasticidin selection pressure cell colonies were stained with methylene blue. (b) Flow cytometry 72 hours after infection with the adenoviruses HCAdV-pEPito-FRT2 or HCAdV-pEPito-ΔS/MAR-FRT2 at MOI of 50 with stuffer virus HCAdV-hFIX at MOI of 50 (total MOI of 100). The percentage of U87-expressing eGFP cells is shown. Mean values (±SD) of two independent experiments are presented. (c) Recombination PCR. U87 cells were infected at MOI of 100 for each virus. PCR was performed with primer pair Recomb-fw and Recomb-rev on genomic DNA isolated 72 hours after infection and 6 weeks after continuous blasticidin selection. As shown for the positive control a 530 bp PCR-band was expected. (d) PCR excluding ITR-fused HCAdV circles. U87 cells were either infected with HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2 or coinfected with the FLPe-encoding virus most likely excluding ITR-fused circles. The possible structure of HCAdV-pEPito-FRT2-genome circularized via ITR-linkage is shown in the left panel. To address the question whether giant circles of the complete adenoviral genome are formed, the primer ITR-rev binding to the ITR sequence was used. The expected PCR product spanning the potentially covalently linked adenoviral genome at the ITR sequences was 144 bp in length. The positive control shows PCR results of a PCR reaction in which the ITRs were linked by a plasmid backbone. The expected PCR product of the positive control was 3,048 bp in length (gray arrow). ITR, inverted terminal repeat; M, DNA ladder as marker; MOI, multiplicity of infections.
Toward in vivo application of the adenoviral hybrid-vector system
To test efficacy of the adenoviral hybrid-vector system in vivo, C57Bl/6 mice were treated with the hybrid-vector system by coinjecting the adenoviral vectors HCAdV-pEPito-FRT2 and HCAdV-FLPe. The goal was to prove that release of the plasmid replicon from the adenoviral vector genome can also occur in vivo in murine liver. To directly compare the adenovirus-based system with a non-viral system, a second group of mice received the plasmid replicon pEPito as naked DNA. Adenoviral vectors were delivered by tail vein injection into C57BL/6 mice (1.5 × 109 transducing units of HCAdV-pEPito-FRT2 and 1.5 × 109 transducing units of HCAdV-FLPe per mouse), and for plasmid transduction of the liver, we used the previously described hydrodynamic delivery of plasmid DNA.35,36 The outline of the experimental approach is shown in Figure 6a. We measured liver alanine aminotransferase (ALT) levels in mouse serum 1 and 7 days after injection. One day after injection, induced liver toxicity was significantly increased after hydrodynamic delivery of the plasmid in comparison with adenoviral delivery for which ALT levels remained in a normal range. One week after injection, ALT levels returned to a normal level (Figure 6b).
Figure 6.

Evaluation of the adenovirus/pEPito hybrid-vector system in vivo. The adenoviral vector HCAdV-pEPito-FRT2 (1.5 × 109 transducing units) was coinjected with the FLPe-encoding virus HCAdV-FLPe (1.5 × 109 transducing units) into C57BL/6 mice (n = 5 per group). As a direct comparison, 30 µg of the plasmid replicon pEPito51 were administered using hydrodynamic injection via the tail vein into C57BL/6 mice (n = 4 per group). ALT, alanine aminotransferase levels; CCl4, carbon-tetrachloride; eGFP, enhanced version of green fluorescent protein. (a) Outline of the experimental approach. (b) To evaluate liver toxicity, alanine aminotransferase (ALT) levels were measured 1 and 7 days after injection. Normal levels for mice range from 20 to 80 international units per liter (IU/l). Plasmid injected mice (triangle) and adenovirus infected mice are shown (square). Mean values of the viral approach (n = 5) and the plasmid approach (n = 4) are shown. (c) Analysis of eGFP expression 5 weeks after injection (one exemplary mouse is shown for each setup) and 6 months after adenovirus administration and three sequential carbon-tetrachloride (CCl4) injections for induction of rapid cell cycling in murine liver. For the adenovirus injected group 4 mice (mouse 1–4) are shown and for the plasmid injected group 3 mice (mouse 1–3) are shown. Whole organs were analyzed for eGFP expression. (d) The plasmid replicon pEPito-FRT can be released from the adenoviral vector genome in vivo. The excision PCR described in Supplementary Figure S5 was performed to detect the excised plasmid replicon in the group that received the adenoviral vector and for detection of the plasmid replicon pEPito. Amplification product for pEPito-FRT presents with bigger size compared with pEPito because of added FRT site during recombination process with FLPe.
Five weeks after injection, mouse liver was analyzed for eGFP expression, and we found a stronger expression for livers which received the plasmid replicon in a non-viral approach (Figure 6c, top panel). To investigate stability of the plasmid replicon during cell division, we injected the liver toxin carbon tetrachlorid (CCl4) for induction of cell cycling in liver. This liver toxin that causes massive liver damage resulting into cell cycling and regeneration of murine liver and only integrated or episomally maintained DNA such as the plasmid replicon pEPito should be remained in dividing cells. After three applications of CCl4, livers were analyzed for eGFP expression, and we found that in both groups, eGFP expression was detectable (Figure 6c, lower panel). To show that the plasmid replicon pEPito-FRT was released from the adenoviral vector, we performed the excision PCR spanning the FRT site in adenovirus-treated mice and a regular PCR to detect the plasmid replicon pEPito (Figure 6d). For both groups, specific PCR bands were amplified.
Discussion
The novel adenoviral hybrid-vector system established in this study combines the efficient transport of genetic information by an adenoviral vector with its stable maintenance by the chromosomal retention element S/MAR. This was the first evidence that the plasmid replicon can be released and stably maintained in eukaryotic cells after adenoviral delivery. Our system was designed as a two-vector system requiring coinfection with HCAdV-FLPe for FLPe-mediated excision of pEPito-FRT which then persists episomally inside the target cells. Herein, we successfully investigated all functional elements of the hybrid-vector system and stable maintenance of transgene expression in vitro, and we show adenovirus-based delivery of the plasmid replicon in vivo.
In contrast to other previously described adenoviral hybrid-vector systems,3,37 our vector releases a genetic element that can be maintained episomally after excision based on FLPe recombination. In this study, we demonstrated that the plasmid replicon pEPito can function as a substrate for FLPe recombination and, at the same time, maintain its function as a plasmid replicon for stable transgene expression. Therefore, no additional recombinases for somatic integration such as transposases,6,7 integrases,8 or other recombinases are required. Due to these reasons, we believe that our system may be superior to other systems for somatic integration because these are associated with the risk of genotoxicity due to insertional mutagenesis.
In previous studies, other hybrid-vector systems for episomal maintenance of the therapeutic DNA were developed using the EBV-based replication and retention mechanisms.10,11,12,13 In concordance to our work, these systems were also designed as two vector systems and used either FLPe or Cre-recombinase for excision of circular episomes carrying the EBV retention and replication signals. However, in contrast to our study, the latter HCAdV hybrid vectors require viral sequences such as the EBV protein EBNA1 for proper function. Disadvantages may arise from the viral nature of the maintenance device, because EBNA1 was found to be associated with the ability to transform cells and therefore, the formation of cancer.38,39
We also rescued a limited amount of episomal DNA from the group that received the vector without S/MAR sequence (Supplementary Figure S6). Detailed characterization of these plasmids revealed that this DNA equaled the original version of the excised pEPito-ΔMAR-FRT plasmid. One potential explanation for this observation could be that 6 weeks of selection pressure and cell division was not sufficient to lose all episomal DNA molecules.
We investigated the behavior of HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2 without cotransducing HCAdV-FLPe and therefore, without excision of the plasmid replicon from the adenoviral DNA molecule. Interestingly, these groups without FLPe recombination showed higher colony forming numbers compared with experimental groups that also received FLPe. As spontaneous recombination via FRT sites could not be verified, we reasoned that spontaneous formation of linked circular HCAdV DNA molecules may have occurred. It is of note that a previous study10 also observed an outstanding performance of a control group without FLPe recombination which was due to self-circularization of the HCAdV/EBNA–hybrid-vector genome via ITR sequences. However, our PCR specifically detecting linkage at the ITRs of the HCAdV genome did not reveal clear results (Figure 5d), because no expected PCR product of 144 bp in length was detected. Although we did observe a mixture of larger PCR products for the group that received the vector HCAdV-pEPito-S/MAR-FRT2 without FLPe (Figure 5d) potentially suggesting that recombination events between the ITRs occurred, we observed the same bands for the control group that received the ΔS/MAR version. Somatic integration of HCAdV-pEPito-FRT2 or HCAdV-pEPito-ΔS/MAR-FRT2 in experimental groups without cotransduction of HCAdV-FLPe as another mechanism for stability of the adenoviral genome was not analyzed. However, we assume that spontaneous integration of the HCAdV genome was not responsible for the results without FLPe recombination, because the natural integration efficiency of recombinant adenoviral vectors is low.33,40,41,42 In our previous study, we could show that only if replication of the HCAdV genome occurs, concatemers or circular DNA forms can be generated.43 Therefore, further studies need to explore whether replication of the complete hybrid-vector genome occurs potentially after circle formation. This question could be experimentally addressed by using methylated vector genomes, a strategy that was used in our previous study.43 Another possibility for the improved features of the hybrid vectors without FLPe recombination could be a synergistic effect between the S/MAR sequence and the mechanism responsible for persistence of adenoviral vector genomes, which may be lost when FLPe recombination excises the transgene expression cassette. Independent of the molecular mechanism responsible for stability of the hybrid-vector HCAdV-pEPito-FRT2 without excision of the plasmid replicon, this phenomenon is an important feature of the hybrid-vector system, because without FLPe recombination, the hybrid-vector system would not rely on an additional recombinase and cotransduction of one cell with two adenoviral vectors.
The hybrid-vector system was also evaluated in vivo in immunocompetent mice. Regarding safety issues, systemic administration of HCAdV-pEPito-FRT2 with HCAdV-FLPe was devoid of hepatotoxicity. Integrity of eGFP transgene could be demonstrated 6 months after injection. Proof of FLPe recombination via PCR was performed and excised pEPito-FRT was detected at the termination of the experiment. The plasmid replicon persisted even after repeated induction of rapid liver cell cycling induced by CCl4-application. Therefore, these experiments show that a plasmid replicon can be released from an adenoviral vector and that the episomal DNA can be maintained even during rapid cell cycling and repopulation in murine liver. In another study, Gil and colleagues11 evaluated an HCAdV/EBV-hybrid vector system using Cre-recombinase in immunocompetent mice. However, in contrast to our study, retention of vector vehicles was not challenged via rapid liver cell cycling. At the time, we can only state that the plasmid replicon can be released and maintained in murine liver even after the induction of rapid cell cycling. However, future studies have to address the efficacy of the system in comparison with the respective control groups and a therapeutic transgene such as the coagulation factor IX could be analyzed. Also, replication of the excised plasmid replicon needs to be studied for instance using an approach based on methylated vector genomes.43
Comparing colony forming numbers (Figure 3 and Supplementary Figure S1) and transgene expression levels in mice (Figure 6) obtained after delivery of the plasmid replicon pEPito and the adenovirus-based system, it seems that the latter system is less efficient. However, it needs to be emphasized that commonly used transfection protocols for naked DNA usually result in hundreds of plasmid molecules per cell with up to 30–80% transduction efficiencies dependent on the cell line and the transfection eGFP expression levels in mice after injection of the pEPito plasmid if directly reagent. In contrast, although 100% of cells can be transduced, adenovirus delivery in general results into significantly less adenovirus genome copy numbers per cell. Although this feature of the plasmid replicon remains to be analyzed, it has been shown that the establishment of S/MAR vectors is a stochastic event, similar to that observed for other eukaryotic replicons,44 and it seems reasonable to assume that delivery of a larger number of vector molecules to the nucleus will result in a higher establishment rate.29 We observed higher eGFP expression levels compared with mice that received the adenoviral vectors (Figure 6). However, it is of note that mice received 30 µg of the plasmid pEPito by hydrodynamic tail vein injection which equals a total of 5 × 1012 pEPito molecules per mouse. In contrast, we injected of a total dose of 3 × 109 infectious adenoviral particles per mouse. Given the fact that a mouse liver approximately contains 1 × 108 hepatocytes, in theory, 5 × 104 plasmid molecules and 30 infectious adenoviral particles could have reached one hepatocyte which could account for the increased eGFP expression levels in mice after plasmid delivery. It was also shown in previous studies that plasmid injection results into up to 80 plasmid molecules per cell45 and adenovirus delivery results in up to 0.1–1 copies per cell.46 It is of note that toxicity after high pressure tail vein injection is a well-described phenomenon which was shown in many previous studies. This delivery method results in massive liver toxicity due to the high pressure and volume and as a consequence liver regeneration and cell division occurs.
Especially, for in vivo applications, the viral hybrid-vector system may be advantageous compared with approaches based on non-viral DNA. Generally, viruses are still more efficient in transducing a broad variety of target cells compared with naked plasmid DNA. This is the first study showing that a non-viral plasmid replicon without any viral DNA sequences can be delivered by a viral vector, and in the future, this strategy may also be used for other viral vectors such as adeno-associated virus vectors or non-integrating lentiviral vectors. In summary, we have successfully established an episomal hybrid-vector system depending only on human chromosomal element S/MAR. Its basic features are functional in vitro and in vivo.
Materials and Methods
Construction and production of HCAdV. Generation of plasmid replicon pEPito and the control plasmid pEPito-ΔS/MAR was described elsewhere.23 For the production of constructs used for HCAdV production, the previously described plasmid pHM5-FRT28 was used which is derived from the cloning vector pHM5.47,48 pEPito and pEPito-ΔS/MAR that were linearized by the restriction enzyme MscI were inserted into the restriction enzyme site SnaBI of pHM5-FRT2 which is located between the two FRT sites. The resulting constructs were designated as pHM5-pEPito-FRT2 and pHM5-pEPito-ΔS/MAR-FRT2, respectively. DNA sequences contained in the final constructs are shown in Supplementary Figure S1. Recombinant DNA contained in the recombinant adenoviral vectors HCAdV-pEPito-FRT2 and the negative control virus HCAdV-pEPito-ΔS/MAR-FRT2 was cloned using a cloning technology based on bacterial artificial chromosomes. In brief, a PCR performed on plasmids pHM5-pEPito-FRT2 and pHM5-pEPito-ΔS/MAR-FRT2 amplified the pEPito and pEPito-ΔS/MAR insert including surrounding FRT sites. Subsequently, the PCR products pEPito-FRT and pEPito-ΔS/MAR-FRT were inserted via a sequence specific homologous recombination mechanism near the 5′-ITR of the bacterial artificial chromosomes pBHCA49 (Supplementary Figure S2). The resulting constructs were designated as pBHCA-pEPito-FRT2 and pBHCA-pEPito-ΔS/MAR-FRT2, respectively. Details on the bacterial artificial chromosomes cloning technique can be obtained upon request.
Adenoviral vectors HCAdV-FLPe and HCAdV-hFIX were described in our previous studies.7,21 HCAdV adenoviral particles were generated in 116 cells as preciously described.32,50 Briefly, for the production of HCAdV-pEPito- FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2, HCAdV DNA was excised from pBHCA-pEPito-FRT2 and pBHCA-pEPito-ΔS/MAR-FRT2 using the restriction enzyme PmeI (Supplementary Figure S2). Linearized HCAdV genomes were transfected into 116 cells and preamplified in cell culture dishes in successive amplification steps using helper virus AdNG163R-2.50 Preamplified viral particles containing HCAdV were purified with Vivapure Adenopack20 column (Sartorius Stedim, Goettingen, Germany). These viral stocks were applied for proliferation in high volume spinner flasks. Subsequently, viral particles were purified via ultracentrifugation in cesium chloride gradients and dialyzed.32,50
To verify DNA sequences contained in the viral particles, 25 µl of viral particles were decapsidated by adding proteinase K in 475 µl lysis buffer (1 mol/l TE, 0.1% SDS). DNA was extracted using an equal volume of phenol/chloroform/isoamylalcohol (Carl Roth, Germany), precipitated with 1 ml ethanol (100%) and 50 µl sodium acetate (3 mol/l, pH 5) (Carl Roth), washed with 500 µl of ethanol (70%), air dried and resolved in 50 µl of water. Subsequently, DNA sequences of pEPito and pEPito-ΔS/MAR contained in the respective HCAdVs were confirmed by amplifying overlapping PCR fragments of the corresponding vectors (Supplementary Figure S2). Toward this end, 100 ng of vector DNA was applied for each reaction using 0.2 mmol/l of each primer in combination with KOD Hot Start Polymerase (Novagen, Darmstadt, Germany) according to manufacturer's instruction in a total volume of 25 µl supplemented with MgSO4 (2 mmol/l). All PCR reactions were performed in a Biometra T professional thermal cycler (Biometra, Göttingen, Germany) applying 5 minutes of initial polymerase activation at 95 °C and 30 cycles of 95 °C for 20 seconds, 60 °C for 20 seconds, and 72 °C for 60 seconds. For HCAdV-pEPito-FRT2, three overlapping fragments (1–3) were amplified using the primer sets pICeu and TQ-eGFP-RH-rv, TQ-eGFP-RH-fw, and BSD-IRES-rev-BamHI, as well as BSD-IRES-fw-BglII and pPISce, respectively (Supplementary Figure S2). For HCAdV-pEPito-ΔS/MAR-FRT2, three overlapping fragments (4–6) were amplified using the primer sets BCHA-18365seq5′ and TQ-eGFP-RH-rev, TQ-eGFP-RH-fw, and BSD-IRES-rev-BamHI, as well as BSD-IRES-fw-BglII and BHCA-PiSce3′, respectively (Supplementary Figure S2). Sequences of all primers used in this study are summarized at the end of the materials and methods section.
To determine the transducing units of HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2, serial dilutions of final virus preparations were added to 293 cells grown to 90% confluency in 60 mm2 dishes. After 2 hours, cells were trypsinized for 5 minutes to denature viral particles in the medium and on the cell surface. Cells were then flushed off, washed with Dulbecco's phosphate buffered saline (DPBS) (PAA, Pasching, Austria), and resuspended in 200 µl PBS. Cells were treated with 20 µl proteinase K (20 mg/ml) in 200 µl lysis buffer (10 mmol/l Tris pH = 7.5, 10 mmol/l EDTA, 1.7% SDS), and shaked overnight at 55 °C. Then, 2 µl RNaseI (10 mg/ml) (Carl Roth) was added and incubated for 30 minutes at 37 °C. Finally, genomic DNA was extracted and 100 ng was applied for real-time PCR using 0.2 mmol/l of each primer TQ-eGFP-RH-fw and TQ-eGFP-RH-rev in combination with 25 µl Fast Start Universal SYBR Green Master reagent (Roche, Mannheim, Germany) in a final volume of 45 µl. Real-time PCR was done in a 7500 Fast Real-Time PCR System light cycler (Applied Biosystems, Darmstadt, Germany) following a protocol that was published before.32
Cell culture. Glioblastoma derived U87 cells, HEK293 cells, and 116 cells were cultured as previously described.32,33 PurFLPe-R cells were established using the plasmid pIRESpuro (Clontech, Mountain View, CA) into which the FLPecDNA was cloned upstream of the IRES sequence resulting into the plasmid pIRESpuro-FLPe. To stably transduce HEK293 cells plasmid pIRESpuro-FLPe was linearized using the restriction enzyme MluI which was then transfected (2 µg plasmid) into HEK293 cells plated in six-well tissue culture plates using FuGENE6 transfection reagent (Promega, Mannheim, Germany). Three days after transfection puromycin selection was started at a concentration of 600 ng/ml. Two weeks later single cell clones were amplified using cloning rings and amplified under selection pressure. Single cell clones were analyzed for FLPe expression by PCR and a positive clone (PurFLPe-R) was selected for further experiments.
Detection of FLPe recombination by specific PCR. To investigate FLPe recombination of the plasmid constructs, equal molar amounts of the following constructs were cotransfected into HEK293 cells (Supplementary Figure S1): pHM5-pEPito-FRT2 and pOG-FLPe6, pHM5-pEPito-FRT2 and irrelevant plasmid stuffer DNA, pHM5-pEPito-ΔS/MAR-FRT2 and pOG-FLPe6, pHM5-pEPito-ΔS/MAR-FRT2 along with irrelevant plasmid stuffer DNA. FLPe recombination of pHM5-pEPito-FRT2 excises pEPito-FRT and re-ligates the vector backbone resulting into pHM5-FRT. FLPe recombination of pHM5-pEPito-ΔS/MAR-FRT2 results in the formation of pEPito-ΔS/MAR-FRT and pHM5-FRT. Two days after infection and after 4 weeks of blasticidin selection, excised plasmids were detected via PCR spanning the FRT sites of excised pEPito-FRT and pEPito-ΔS/MAR-FRT, respectively resulting in a specific PCR product of 50 bp in length. In case of non-recombined constructs, no PCR products can be detected. For the PCR setup, 100 ng of total DNA isolated from transfected cells was used for the PCR reaction using 0.2 mmol/l of each primer in combination with KOD Hot Start Polymerase (Novagen) in a total volume of 25 µl supplemented with MgSO4 (2 mmol/l) following manufacturer's instructions. Recombination within plasmids was detected using the primers Pep-r-fw-FRT and Pep-l-rev-FRT in a touch-down PCR with 5 minutes initial denaturation at 95 °C followed by 35 cycles of 95 °C, an initial annealing temperature of 60 °C dropping by 0.3 °C in each following cycle, and 72 °C for 5 seconds.
FLPe recombination within the adenoviral genomes HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2 excising the plasmid replicon pEPito-FRT and the ΔS/MAR version pEPito-ΔS/MAR-FRT was detected using the primers Recomb-fw and Recomb-rev. PCR reaction included 5 minutes of initial denaturation at 95 °C followed by 25 cycles of 95 °C for 20 seconds, 60 °C for 20 seconds, and 72 °C for 10 seconds. A PCR product of 530 bp in length was expected if FLPe recombination occurred.
Detection of eGFP expression by flow cytometry. To determine eGFP expression after transfection (FuGENE6; Roche) of excised plasmid replicons pEPito-FRT and pEPito-ΔS/MAR-FRT, and pHM5-based plasmids pHM5-pEPito-FRT2 and pHM5-pEPito-ΔS/MAR-FRT2 into HEK293 cells grown in six-well tissue culture plates, eGFP fluorescence was measured in using the flow cytometer BD FACS Canto II (BD Bioscience, Franklin Lakes, NJ) 48 hours after transfection. To confirm long-term eGFP expression, cells were kept in six-well plates and treated with 7–9 µg/ml blasticidin (PAA) for 4 weeks before fluorescence activated cell sorting analyses. Medium and blasticidin were changed every 2–3 days.
To determine eGFP expression from HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2, U87 cells and HEK293 were grown in six-well tissue culture plates and infected at the depicted MOIs. Seventy-two hours after infection, cells were trypsinized, analyzed by flow cytometry. To confirm long-term eGFP expression, 1 × 106 U87 cells were seeded into T-25 flasks (BD Biosciences, Heidelberg, Germany) coated with Collagen A (Biochrom AG, Berlin, Germany). Infection with respective adenoviral vectors at an MOI of 30 was performed in 1–1.5 ml standard medium for 3–4 hours. After infection, the volume of medium was increased to 3–5 ml. Two days after infection, blasticidin selection started at a concentration of 4 µg/ml. In concordance with the plasmid-based experiments, medium and blasticidin were changed every 2–3 days. After 6 weeks of selection, eGFP expression was evaluated a second time by flow cytometry.
Colony forming assays. For colony forming assay using plasmid constructs, HEK293 cells were grown to 60–80% confluency in six-well plates. Cotransfection of 1 µg of the respective vector DNA, from the various plasmid groups, was carried out with FuGENE6 according to the producer's instructions. Two days later, cells were split 1:2 into 10 cm tissue culture dishes. One day after passaging, blasticidin was added to a final concentration of 7–9 µg/ml to select for plasmid containing cell clones. Cells were kept under blasticidin selection pressure for 28 days. Tissue medium including blasticidin was replaced every 2–3 days.
For colony forming assay using the adenoviral vectors HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2, 1 × 106 U87 cells were plated in collagen coated T-25 flasks. Infection with the respective HCAdV was performed at MOIs of 10, 30, and 100 in 1–1.5 ml of medium for 3–4 hours. Blasticidin selection was started 72 hours after infection at a concentration of 4 µg/ml and transduced cells were kept under blasticidin selection pressure for 6 weeks while changing the medium including the blasticidin every 2–3 days. Surviving cells and colonies were washed with DPBS, fixed with 4% paraformaldehyde (Sigma Aldrich, Munich, Germany) in DPBS and stained with methylene blue (Sigma Aldrich). Colonies formed under selection pressure were counted and pictured.
Plasmid rescue of pEPito-FRT and pEPito-ΔS/MAR-FRT. To confirm FLPe-mediated excision of pEPito-FRT and pEPito-ΔS/MAR-FRT from adenoviral vectors, genomic DNA of the respective cells was isolated 72 hours and 6 weeks after transduction. Isolated DNA was transformed into chemical competent GT115-E.coli. Transformed bacteria were grown in Luria-Bertani (LB) medium for 3–4 hours and plated on LB-Agar-dishes containing ampicillin (Carl Roth) at a concentration of 100 µg/ml. Next day single colonies were picked and cultured overnight. Until plasmid DNA was isolated using a plasmid Miniprep Kit (Qiagen, Hilden, Germany) and analyzed by XhoI and BamHI restriction enzyme digests.
Detection of potential circular molecular forms of HCAdV vector genomes by PCR. To investigate if circular hybrid AdV vector concatemers have formed during infection, 100 ng genomic DNA isolated from infected was applied for PCR using the Primer ITR-rev in combination with KOD Hot Start Polymerase (Novagen) according to manufacturer's instruction. As ITRs at the 5′- and 3′-end of HCAdV DNA molecules are identical, but inversely orientated, one single primer was sufficient for this reaction. PCR was executed applying 5 minutes initial denaturation at 95 °C followed by 35 cycles of 95 °C for 20 seconds, 60 °C for 20 seconds, and 72 °C for 40 seconds.
In vivo experiments. To prove excision of the pEPito-FRT from the HCAdV genome and to analyze stability of transgene expression in murine liver, C57Bl/6 mice (n = 5) were coinjected by tail vein injection with 1.5 × 109 transducing units of HCAdV-pEPito-FRT2 and 1.5 × 109 transducing units of HCAdV-FLPe in a total volume of 200 µl dissolved in DPBS. To compare our adenovirus-based system with a non-viral system, liver cells of a second group of mice (n = 4) were transduced with 30 µg of pEPito as naked DNA by hydrodynamic delivery of plasmid DNA. This method was described elsewere.35,36
At day 1 and 7 after transduction, liver ALT levels in mouse serum were quantified using a Vitrospec 3000-Spectrometer (Pharmacia, Stockholm, Sweden) in combination with an ALT-kit (Randox, Crumlin, UK) according to manufacturer's instructions.
To investigate the stability of released plasmid pEPito-FRT from the adenoviral vector and the pEPito plasmid, we injected the liver toxin carbon tetrachlorid (CCl4; Sigma Aldrich) for induction of cell cycling in liver. Five weeks and 6 months after injection and three applications of CCl4, mice were analyzed for eGFP expression in the liver using a VivoVision IVIS Lumina Luminometer (Xenogen, Alameda, CA) applying default parameters recommended by the manufacturer.
At the termination of the experiment (6 months after injection), mice were euthanized and 100 mg of liver tissue was used for DNA isolation. Tissue was homogenized in 2.3 ml lysis buffer (40 mmol/l NaCl, 10 mmol/l Tris pH 7.5, 10 mmol/l EDTA, 1.3% SDS). Then, 10 µl proteinase K (20 mg/ml, Qiagen) was added and the mixture was shook at 56 °C overnight. Next day, 15 µl RNaseA (10 mg/ml; New England Biolabs, Ipswich, MA) was added and incubated for another 30 minutes at 37 °C. DNA was separated using consecutive rounds of phenol/chloroform/isoamylalcohol extraction. DNA was precipitated by transferring the supernatant to three volumes of ice cold ethanol (100%) supplemented with sodium acetate (3 mol/l, pH = 5). After centrifugation, the DNA pellet was washed with ethanol (70%), air dried, and resolved in 300 µl of water. Subsequently, 100 ng of liver DNA of adenovirus treated mice was subjected to PCRs spanning the FRT sites for detection of excision. For the pEPito plasmid replicon, a regular PCR detecting a 450 bp band was conducted.
Statistical analysis. Data are reported as mean ± SD. Statistical comparison was made using the two-tailed Student's t-test, and a value of P < 0.05 was considered to be relevant compared with the respective control group.
PRIMER LIST

SUPPLEMENTARY MATERIAL Figure S1. Evaluation of functionality of essential components contained in the hybrid-vector system. Figure S2. Construction of BAC clones pBHCA-pEPito-FRT2 and pBHCA-ΔS/MAR-FRT2 for generation of recombinant adenoviruses HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2. Figure S3. Characterization of adenoviral vectors HCAdV-pEPito-FRT2 and HCAdV-pEPito-ΔS/MAR-FRT2. Figure S4. FLPe mediated recombination in the stably Flpe expressing cell line Pur FLPe-R. Figure S5. Excision of the plasmid replicon in the stably FLPe expressing cell line Pur FLPe-R. Figure S6. Characterisation of the excised plasmids in groups which which were co-infected with HCAdV- pEPito-ΔS/MAR-FRT2 and the FLPe encoding virus HCAdV-FLPe.
Acknowledgments
The authors thank Philip Ng for providing the producer cell line 116 and the helper virus AdNG163R-2. This work was supported in part by DFG grants SPP 1230 (A.E. and A.B.) and EH 192/4-1 to A.E. (Heisenberg-Programme), the Wilhelm Sander-Foundation, EU Framework Programme 7 (Persistent Transgenesis) the FöFoLe programme of the Ludwig-Maximilians-UniversitätMünchen (A.E.), and a UWH research grant of the University Witten/Herdecke (A.E. and H.J.L.). W.Z. was supported by a fellowship from the Chinese Scholarship Council (CSC) in cooperation with Northwestern A&F University, Yangling, China. The authors declare no conflict of interest.
Supplementary material
Evaluation of functionality of essential components contained in the hybrid-vector system.
Construction of BAC clones pBHCA-pEPito-FRT2 and pBHCA-δS/MAR-FRT2 for generation of recombinant adenoviruses HCAdV-pEPito-FRT2 and HCAdV-pEPito-δS/MAR-FRT2.
Characterization of adenoviral vectors HCAdV-pEPito-FRT2 and HCAdV-pEPito-δS/MAR-FRT2.
FLPe mediated recombination in the stably Flpe expressing cell line Pur FLPe-R.
Excision of the plasmid replicon in the stably FLPe expressing cell line Pur FLPe-R.
Characterisation of the excised plasmids in groups which which were co-infected with HCAdV- pEPito-ΔS/MAR-FRT2 and the FLPe encoding virus HCAdV-FLPe.
References
- Thomas C.E., Ehrhardt A., &, Kay M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003;4:346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- Ehrhardt A., Xu H., &, Kay M.A. Episomal persistence of recombinant adenoviral vector genomes during the cell cycle in vivo. J. Virol. 2003;77:7689–7695. doi: 10.1128/JVI.77.13.7689-7695.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauschhuber C., Noske N., &, Ehrhardt A. New insights into stability of recombinant adenovirus vector genomes in mammalian cells. Eur. J. Cell Biol. 2012;91:2–9. doi: 10.1016/j.ejcb.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Recchia A., Perani L., Sartori D., Olgiati C., &, Mavilio F. Site-specific integration of functional transgenes into the human genome by adeno/AAV hybrid vectors. Mol. Ther. 2004;10:660–670. doi: 10.1016/j.ymthe.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Gonçalves M.A., Holkers M., van Nierop G.P., Wieringa R., Pau M.G., &, de Vries A.A. Targeted chromosomal insertion of large DNA into the human genome by a fiber-modified high-capacity adenovirus-based vector system. PLoS ONE. 2008;3:e3084. doi: 10.1371/journal.pone.0003084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausl M.A.et al. Hyperactive sleeping beauty transposase enables persistent phenotypic correction in mice and a canine model for hemophilia B Mol. Ther. 181896–1906.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant S.R., Ehrhardt A., Mikkelsen J.G., Meuse L., Pham T., &, Kay M.A. Transposition from a gutless adeno-transposon vector stabilizes transgene expression in vivo. Nat. Biotechnol. 2002;20:999–1005. doi: 10.1038/nbt738. [DOI] [PubMed] [Google Scholar]
- Ehrhardt A., Yant S.R., Giering J.C., Xu H., Engler J.A., &, Kay M.A. Somatic integration from an adenoviral hybrid vector into a hot spot in mouse liver results in persistent transgene expression levels in vivo. Mol. Ther. 2007;15:146–156. doi: 10.1038/sj.mt.6300011. [DOI] [PubMed] [Google Scholar]
- Biasco L., Baricordi C., &, Aiuti A. Retroviral integrations in gene therapy trials. Mol. Ther. 2012;20:709–716. doi: 10.1038/mt.2011.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreppel F., &, Kochanek S. Long-term transgene expression in proliferating cells mediated by episomally maintained high-capacity adenovirus vectors. J. Virol. 2004;78:9–22. doi: 10.1128/JVI.78.1.9-22.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil J.S., Gallaher S.D., &, Berk A.J. Delivery of an EBV episome by a self-circularizing helper-dependent adenovirus: long-term transgene expression in immunocompetent mice. Gene Ther. 2010;17:1288–1293. doi: 10.1038/gt.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorigo O.et al. Development of a novel helper-dependent adenovirus-Epstein-Barr virus hybrid system for the stable transformation of mammalian cells J. Virol. 786556–6566.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan B.T., Wu L., &, Berk A.J. An adenovirus-Epstein-Barr virus hybrid vector that stably transforms cultured cells with high efficiency. J. Virol. 1999;73:7582–7589. doi: 10.1128/jvi.73.9.7582-7589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer D.J., &, Ng P. Helper-dependent adenoviral vectors for gene therapy. Hum. Gene Ther. 2005;16:1–16. doi: 10.1089/hum.2005.16.1. [DOI] [PubMed] [Google Scholar]
- Brunetti-Pierri N.et al. Sustained phenotypic correction of canine hemophilia B after systemic administration of helper-dependent adenoviral vector Hum. Gene Ther. 16811–820.2005 [DOI] [PubMed] [Google Scholar]
- Brunetti-Pierri N.et al. Efficient, long-term hepatic gene transfer using clinically relevant HDAd doses by balloon occlusion catheter delivery in nonhuman primates Mol. Ther. 17327–333.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt A., Xu H., Dillow A.M., Bellinger D.A., Nichols T.C., &, Kay M.A. A gene-deleted adenoviral vector results in phenotypic correction of canine hemophilia B without liver toxicity or thrombocytopenia. Blood. 2003;102:2403–2411. doi: 10.1182/blood-2003-01-0314. [DOI] [PubMed] [Google Scholar]
- Chuah M.K.et al. Therapeutic factor VIII levels and negligible toxicity in mouse and dog models of hemophilia A following gene therapy with high-capacity adenoviral vectors Blood 1011734–1743.2003 [DOI] [PubMed] [Google Scholar]
- Kiang A.et al. Fully deleted adenovirus persistently expressing GAA accomplishes long-term skeletal muscle glycogen correction in tolerant and nontolerant GSD-II mice Mol. Ther. 13127–134.2006 [DOI] [PubMed] [Google Scholar]
- Brown B.D., Shi C.X., Powell S., Hurlbut D., Graham F.L., &, Lillicrap D. Helper-dependent adenoviral vectors mediate therapeutic factor VIII expression for several months with minimal accompanying toxicity in a canine model of severe hemophilia A. Blood. 2004;103:804–810. doi: 10.1182/blood-2003-05-1426. [DOI] [PubMed] [Google Scholar]
- Ehrhardt A., &, Kay M.A. A new adenoviral helper-dependent vector results in long-term therapeutic levels of human coagulation factor IX at low doses in vivo. Blood. 2002;99:3923–3930. doi: 10.1182/blood.v99.11.3923. [DOI] [PubMed] [Google Scholar]
- Schiedner G.et al. Genomic DNA transfer with a high-capacity adenovirus vector results in improved in vivo gene expression and decreased toxicity Nat. Genet. 18180–183.1998 [DOI] [PubMed] [Google Scholar]
- Haase R.et al. pEPito: a significantly improved non-viral episomal expression vector for mammalian cells BMC Biotechnol. 1020.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baiker A.et al. Mitotic stability of an episomal vector containing a human scaffold/matrix-attached region is provided by association with nuclear matrix Nat. Cell Biol. 2182–184.2000 [DOI] [PubMed] [Google Scholar]
- Piechaczek C., Fetzer C., Baiker A., Bode J., &, Lipps H.J. A vector based on the SV40 origin of replication and chromosomal S/MARs replicates episomally in CHO cells. Nucleic Acids Res. 1999;27:426–428. doi: 10.1093/nar/27.2.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode J.et al. Biological significance of unwinding capability of nuclear matrix-associating DNAs Science 255195–197.1992 [DOI] [PubMed] [Google Scholar]
- Jenke A.C.et al. Nuclear scaffold/matrix attached region modules linked to a transcription unit are sufficient for replication and maintenance of a mammalian episome Proc. Natl. Acad. Sci. U.S.A. 10111322–11327.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehle I.M., Scinteie M.F., Baiker A., Jenke A.C., &, Lipps H.J. Exploiting a minimal system to study the epigenetic control of DNA replication: the interplay between transcription and replication. Chromosome Res. 2003;11:413–421. doi: 10.1023/a:1024962308071. [DOI] [PubMed] [Google Scholar]
- Stehle I.M., Postberg J., Rupprecht S., Cremer T., Jackson D.A., &, Lipps H.J. Establishment and mitotic stability of an extra-chromosomal mammalian replicon. BMC Cell Biol. 2007;8:33. doi: 10.1186/1471-2121-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenke B.H.et al. An episomally replicating vector binds to the nuclear matrix protein SAF-A in vivo EMBO Rep. 3349–354.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaarschmidt D., Baltin J., Stehle I.M., Lipps H.J., &, Knippers R. An episomal mammalian replicon: sequence-independent binding of the origin recognition complex. EMBO J. 2004;23:191–201. doi: 10.1038/sj.emboj.7600029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager L., Hausl M.A., Rauschhuber C., Wolf N.M., Kay M.A., &, Ehrhardt A. A rapid protocol for construction and production of high-capacity adenoviral vectors. Nat. Protoc. 2009;4:547–564. doi: 10.1038/nprot.2009.4. [DOI] [PubMed] [Google Scholar]
- Hillgenberg M., Tönnies H., &, Strauss M. Chromosomal integration pattern of a helper-dependent minimal adenovirus vector with a selectable marker inserted into a 27.4-kilobase genomic stuffer. J. Virol. 2001;75:9896–9908. doi: 10.1128/JVI.75.20.9896-9908.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shayakhmetov D.M., Li Z.Y., Ternovoi V., Gaggar A., Gharwan H., &, Lieber A. The interaction between the fiber knob domain and the cellular attachment receptor determines the intracellular trafficking route of adenoviruses. J. Virol. 2003;77:3712–3723. doi: 10.1128/JVI.77.6.3712-3723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Song Y., &, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–1266. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- Zhang G., Budker V., &, Wolff J.A. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum. Gene Ther. 1999;10:1735–1737. doi: 10.1089/10430349950017734. [DOI] [PubMed] [Google Scholar]
- Müther N., Noske N., &, Ehrhardt A. Viral hybrid vectors for somatic integration - are they the better solution. Viruses. 2009;1:1295–1324. doi: 10.3390/v1031295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Q., Young L.S., Woodman C.B., &, Murray P.G. Epstein-Barr virus (EBV) and its associated human cancers–genetics, epigenetics, pathobiology and novel therapeutics. Front. Biosci. 2006;11:2672–2713. doi: 10.2741/2000. [DOI] [PubMed] [Google Scholar]
- Young L.S., &, Rickinson A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer. 2004;4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- Harui A., Suzuki S., Kochanek S., &, Mitani K. Frequency and stability of chromosomal integration of adenovirus vectors. J. Virol. 1999;73:6141–6146. doi: 10.1128/jvi.73.7.6141-6146.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen S.L., Sivanandam V.G., &, Kochanek S. Homologous and heterologous recombination between adenovirus vector DNA and chromosomal DNA. J. Gene Med. 2008;10:1176–1189. doi: 10.1002/jgm.1246. [DOI] [PubMed] [Google Scholar]
- Stephen S.L.et al. Chromosomal integration of adenoviral vector DNA in vivo J. Virol. 849987–9994.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager L., &, Ehrhardt A. Persistence of high-capacity adenoviral vectors as replication-defective monomeric genomes in vitro and in murine liver. Hum. Gene Ther. 2009;20:883–896. doi: 10.1089/hum.2009.020. [DOI] [PubMed] [Google Scholar]
- Leight E.R., &, Sugden B. Establishment of an oriP replicon is dependent upon an infrequent, epigenetic event. Mol. Cell. Biol. 2001;21:4149–4161. doi: 10.1128/MCB.21.13.4149-4161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.Y., Yant S.R., He C.Y., Meuse L., Shen S., &, Kay M.A. Linear DNAs concatemerize in vivo and result in sustained transgene expression in mouse liver. Mol. Ther. 2001;3:403–410. doi: 10.1006/mthe.2001.0278. [DOI] [PubMed] [Google Scholar]
- Sharma A., Bangari D.S., Tandon M., Pandey A., HogenEsch H., &, Mittal S.K. Comparative analysis of vector biodistribution, persistence and gene expression following intravenous delivery of bovine, porcine and human adenoviral vectors in a mouse model. Virology. 2009;386:44–54. doi: 10.1016/j.virol.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi H., &, Kay M.A. Efficient construction of a recombinant adenovirus vector by an improved in vitro ligation method. Hum. Gene Ther. 1998;9:2577–2583. doi: 10.1089/hum.1998.9.17-2577. [DOI] [PubMed] [Google Scholar]
- Mizuguchi H., &, Kay M.A. A simple method for constructing E1- and E1/E4-deleted recombinant adenoviral vectors. Hum. Gene Ther. 1999;10:2013–2017. doi: 10.1089/10430349950017374. [DOI] [PubMed] [Google Scholar]
- Shayakhmetov D.M.et al. Genome size and structure determine efficiency of postinternalization steps and gene transfer of capsid-modified adenovirus vectors in a cell-type-specific manner J. Virol. 7810009–10022.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer D., &, Ng P. Improved system for helper-dependent adenoviral vector production. Mol. Ther. 2003;8:846–852. doi: 10.1016/j.ymthe.2003.08.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Evaluation of functionality of essential components contained in the hybrid-vector system.
Construction of BAC clones pBHCA-pEPito-FRT2 and pBHCA-δS/MAR-FRT2 for generation of recombinant adenoviruses HCAdV-pEPito-FRT2 and HCAdV-pEPito-δS/MAR-FRT2.
Characterization of adenoviral vectors HCAdV-pEPito-FRT2 and HCAdV-pEPito-δS/MAR-FRT2.
FLPe mediated recombination in the stably Flpe expressing cell line Pur FLPe-R.
Excision of the plasmid replicon in the stably FLPe expressing cell line Pur FLPe-R.
Characterisation of the excised plasmids in groups which which were co-infected with HCAdV- pEPito-ΔS/MAR-FRT2 and the FLPe encoding virus HCAdV-FLPe.