

Abstract
We have used powerful HPLC-mass spectrometric approaches to characterize the secreted form of epidermal growth factor receptor (sEGFR). We demonstrated that the amino acid sequence lacked the cytoplasmic domain and was consistent with the primary sequence reported for EGFR purified from a human plasma pool. One of the sEGFR forms, attributed to the alternative RNA splicing, was also confirmed by transcriptional analysis (RNA sequencing). Two unusual types of glycan structures were observed in sEGFR as compared with membrane-bound EGFR from the A431 cell line. The unusual glycan structures were di-sialylated glycans (sialic acid attached to sialic acid) at Asn-151 and N-acetylhexosamine attached to a branched fucosylated galactose with N-acetylglucosamine moieties (HexNAc-(Fuc)Gal-GlcNAc) at Asn-420. These unusual glycans at specific sites were either present at a much lower level or were not observable in membrane-bound EGFR present in the A431 cell lysate. The observation of these di-sialylated glycan structures was consistent with the observed expression of the corresponding α-N-acetylneuraminide α-2,8-sialyltransferase 2 (ST8SiA2) and α-N-acetylneuraminide α-2,8-sialyltransferase 4 (ST8SiA4), by quantitative real time RT-PCR. The connectivity present at the branched fucosylated galactose was also confirmed by methylation of the glycans followed by analysis with sequential fragmentation in mass spectrometry. We hypothesize that the presence of such glycan structures could promote secretion via anionic or steric repulsion mechanisms and thus facilitate the observation of these glycan forms in the secreted fractions. We plan to use this model system to facilitate the search for novel glycan structures present at specific sites in sEGFR as well as other secreted oncoproteins such as Erbb2 as markers of disease progression in blood samples from cancer patients.
Cancers are disease-associated with considerable morbidity, such as disease recurrence, anxiety, and side effects of treatment and mortality (1). Early diagnosis often significantly improves survival rates compared with late stage cancer detection, such as for breast, lung, and colon cancers (2–4). Proteins in the blood hold enormous promise for early stage cancer diagnostic tests, but the complexity and dynamic range of blood have confounded the search for cancer biomarkers. Nevertheless, the pressing need for a clinical assay has prompted us to investigate a different approach toward discovering new breast cancer biomarkers circulating in blood (5–7). In addition, the use of a panel of cancer cell lines, representing cancers with different subtypes, could alleviate the difficulty of analyzing the plasma samples directly. Although there is no substitute for the direct study of clinical samples, genetic and molecular aberrations found in cell lines can be translated to similar dysregulations in tumors (8). Cell lines, through the proteins they secrete or shed, should be a complementary model system for the discovery of circulating blood markers. For cancer biomarkers, the change of gene or protein sequence, such as mutation, is often a preclusion for cancers. A similar argument could also be true for the change of protein glycan structures, which relates to alteration in expression of specific glycosyltransferases in cancers (9–11).
To investigate this hypothesis, we have studied an important oncoprotein, epidermal growth factor receptor (EGFR),1 from the A431 cell line, which is known to have high expression of EGFR and is thus suitable to be characterized extensively. EGFR has been utilized as a biomarker associated with lung, ovarian, and breast cancers (12–14). In this study, we have isolated secreted EGFR (sEGFR) from the cell line using a polyclonal antibody specific for the secreted form. The isolated form was shown to have a protein sequence comparable with that of circulating EGFR from plasma pool samples (no cytoplasmic domain as compared with the membrane-bound EGFR). The glycan structures for each site of sEGFR were then characterized using state of the art LC-MS techniques. In general, the glycan profile of sEGFR exhibited more branches with sialylation than the membrane-bound EGFR. These results are consistent with reports on alterations of glycosylation in membrane-bound proteins in cancer metastasis (15). In the future, these interesting glycan structure-associated sites (glycopeptides) of sEGFR, which can be compared and correlated to circulating glycoforms of EGFR in plasma, will be selected for development of a quantitative multistage reaction monitoring assay for patient samples.
EXPERIMENTAL PROCEDURES
A431 Media Collection
A431 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, 11965) supplemented with 10% fetal bovine serum (FBS) at 37°C in a humidified atmosphere in 5% (v/v) CO2. After the cells reached confluence, the media were exchanged, and the FBS concentration was reduced to 1%. Media were then collected after 24 h of culture, centrifuged at 800 rpm for 10 min, and filtered with a 0.22-μm membrane (Millipore).
Immunoaffinity Chromatography
5 ml of resin solution (UltraLink Immobilized NeutrAvidin, Pierce 53150) was packed in a column at room temperature, washed, and then equilibrated with 50 ml of PBS. 100 μg of biotinylated EGFR polyclonal antibody (R&D Systems, BAF231) diluted in 5 ml of total of PBS was loaded onto the column. The column was closed and kept overnight at 4°C. After washing and equilibrating the column with 50 ml of PBS, a total of 300 ml of A431 media containing inhibitor mixture (complete Mini EDTA-free, Roche Applied Science, 11836170001) was loaded onto the column two times. The column was washed with 100 ml of PBS, and elution was performed with 50 ml of elution buffer (Pierce, 21004). Eluted EGFR-enriched fraction was immediately neutralized with 1.5 m Tris-HCl, pH 8.8, and inhibitor mixture, and 0.5% octyl glucoside was added.
Plasma Experiments
Reference pools of plasma were used in the experiments described here and processed as described previously (32). Briefly, each pool was immunodepleted of the top six most abundant proteins using HU-6 columns (Agilent). The immunodepleted samples were then reduced and alkylated with acrylamide. Intact proteins were separated in the first dimension by anion-exchange chromatography. Collected fractions were further separated in a second dimension by reversed-phase chromatography. Resulting fractions were then lyophilized prior to LC-MS analysis.
Reversed-phase Chromatography
The 50 ml (∼1 μg) of EGFR immunoaffinity-enriched fraction was concentrated to 5 ml with an Amicon Ultra system filter and subjected to a reversed-phase separation to further purify the EGFR protein. A POROS R1-perfusion chromatography (Applied Biosystems) column was used. Buffer A consisted of 0.1% TFA and buffer B was 90% acetonitrile, 0.095% TFA. Chromatography was carried out at a flow rate of 2 ml/min. The gradient consisted of 20% solvent B for 10 min and 25–100% solvent B for 60 min. One fraction per min was collected.
Total RNA Isolation and cDNA Synthesis
A431 cells were harvested and stored in TRIzol at −80°C until use. Total RNA was isolated by partitioning the RNA into the aqueous phase with the addition of chloroform to the TRIzol. The aqueous phase was transferred to another tube, and an equal volume of 70% ethanol was added. This solution was used as the starting material for RNA isolation using the RNeasy Plus kit (Qiagen). Total RNA was quantitated using a Nanodrop spectrophotometer. Total RNA (500 ng) was used with the SuperScript III first-strand synthesis system (Invitrogen) to generate cDNA in a 20-μl reaction. Reactions were diluted 1:10 with diethyl pyrocarbonate-treated water prior to qRT-PCR assays.
qRT-PCR
Primer sequences for genes analyzed in this study are presented in supplemental Table S3. Triplicate reactions (5 μl each) containing 1.25 μl of diluted cDNA, 1.25 μl of primer pair mix (125 μm final concentration), and 2.5 μl of iQTM SYBR Green Supermix (Bio-Rad) were assembled in 96-well microliter plates. A Realplex2 real time PCR system (Eppendorf) was used for amplification with the following cycling conditions: 95°C for 3 min, followed by 40 cycles of 95°C for 10 s (denaturing), 65°C for 45 s (annealing), 78°C for 20 s (data collection). Following the thermal cycling and data collection steps, amplimer products were analyzed using a melt curve program (95°C for 1 min, 55°C for 1 min, and then increasing 0.5°C per cycle for 80 cycles of 10 s each). Ribosomal Protein L4 (RPL4, NM_024212) was included on each plate to control for run variation and to normalize individual gene expression. Average relative gene expression levels were determined as described previously by normalizing transcript abundances to RPL4 and scaling the data so that a value of 1 × 10−6 was the lower limit of detection (33). Error bars represent 1 S.D. from the mean value.
RNA Sequencing
The cell line A431was used as the sample type. Strand-specific RNA-Sequencing libraries were prepared and sequenced using the Illumina HiSeq 2000 instrument to obtain transcript data (34). For analysis of isoforms, Tophat/Cufflinks (version 1.4.0 and 1.3.0, respectively) was run with Ensembl (GRCh37) as a reference.
SDS-PAGE and Western Blot
Reverse phase fractions were resuspended in electrophoresis buffer (0.125 m Tris, pH 6.8, 4% SDS, 20% glycerol, and 2% DTT) after lyophilization and were loaded in 12% acrylamide gels (8.5 × 13.5 cm, Bio-Rad) and run at 30 mA/gel. Gels were stained with Coomassie (Pierce) or transferred for 2 h (100 V/gel) to PVDF membranes (Bio-Rad) to localize EGFR protein by Western blotting. Recombinant EGFR (R&D Systems, 1095-ER) produced from a DNA sequence encoding the extracellular domain of human EGFR (Met-1 to Ser-645) and expressed in a mouse myeloma cell line was also loaded to gels. PVDF membranes were blocked overnight with 5% nonfat dry milk (Bio-Rad) in PBS and then incubated for 2 h with anti-EGFR polyclonal antibody (R&D Systems, AF231) at a dilution of 1:500 in PBS containing 0.1% Tween 20, at room temperature (RT). After 1 h of washing with PBS, 0.1% Tween 20 (six times for 10 min), membranes were incubated with a 1:10,000 dilution of anti-goat IgG (Jackson ImmunoResearch, 205-035-108) in PBS, 0.1% Tween 20 for 1 h at RT. Membranes were then washed for 1 h, and the chemiluminescence immunodetection was performed with ECL reagents (Amersham Biosciences). Hyperfilms (Amersham Biosciences) were exposed for 30 s to optimal image visualization.
In-gel Analysis
One-dimensional bands were excised directly from gels or PVDF membranes and extensively washed with 50 mm ammonium bicarbonate containing 50% acetonitrile, vacuum-dried, and then incubated with trypsin digestion solution (12.5 ng/μl trypsin in 50 mm ammonium bicarbonate, pH 8.0) for 30–35 min at 4°C, followed by a further incubation overnight at 37°C. For a Lys-C digestion, the endoproteinase Lys-C (10 ng/μl Lys-C in 50 mm ammonium bicarbonate, pH 8.0) was used and incubated the same way as trypsin digestion. For a Lys-C plus peptide:N-glycosidase F digestion, the Lys-C solution was added with peptide:N-glycosidase F (10 units/mg) and incubated the same way as trypsin digestion. The digested peptides were extracted from the gel with 25 mm ammonium bicarbonate and then acetonitrile (37°C for 15 min) and further extracted with 5% formic acid at 37°C for 5 min. All supernatants were collected and concentrated for the subsequent LC-MS analysis. An aliquot of 2 μg of the enzyme digest was analyzed per LC-MS run.
LC-MS
An Ultimate 3000 nano-LC pump (Dionex, Mountain View, CA) and a self-packed C18 column (Magic C18, 200-Å pore and 5-μm particle size, 75 μm inner diameter × 15 cm) (Michrom Bioresources, Auburn, CA) was coupled on line to an LTQ-FTICR mass spectrometer (Thermo Fisher Scientific, San Jose, CA) through a nanospray ion source (New Objective, Woburn, MA). Mobile phase A was 0.1% formic acid in water, and mobile phase B was 0.1% formic acid in acetonitrile. The peptides were eluted at 200 nl/min using a linear gradient from 2 to 65% B in 65 min, followed by 65 to 80% B for 10 min. The LTQ-FTICR mass spectrometer was operated as follows: survey full-scan MS spectra (m/z 400–2000) were acquired in the Orbitrap cell with a mass resolution of 100,000 at m/z 400, followed by eight sequential CID-MS2 scans using the LTQ portion in a data-dependent mode. For an inadequate assignment, the analysis was repeated by targeting the desired ions to gain additional information. If necessary, the ions of interest obtained with CID-MS2 were further targeted for CID-MS3. For proteomic analysis, acquired MS/MS scans were converted into DTA files by Extract-MSn (version 4.0, Thermo Fisher Scientific) and searched against the SwissProt human database (release 2010_06 downloaded in July, 2010, 20,342 entries including common contaminants) combined with a database containing reversed sequences using the Sequest algorithm (cluster version 27, revision 12, Thermo Fisher Scientific). The search results were stored in a Computational Proteomics Analysis System (CPAS) (version 9.10 LabKey, Seattle, WA). The peptide mass search tolerance was set to 1.4 Da, and the fragment ion mass tolerance was 1.0 Da. Full Lys-C or trypsin enzyme specificity was selected with up to two missed cleavage sites allowed. Cysteine carbamidomethylation was considered as a fixed modification. The search results (identified peptides) were filtered by Xcorr ≥1.9 for charge state +1, ≥2.2 for charge state +2, and ≥3.8 for charge state +3 and by PeptideProphet (Institute for System Biology, Seattle, WA) using a peptide probability ≥0.95. ProteinProphet (Institute for System Biology) was used to assign peptides to protein groups with acceptance criteria specified using a probability of ≥0.9 resulting in the false discovery rate of <1% at the protein level.
Glycan Structure Identification
Theoretical masses of glycan structures were added to the enzymatic peptide backbone. The anticipated glycopeptide masses with different charges were thus obtained to match the observed masses in the LC-MS chromatogram. The matched masses (with ≤5 ppm mass accuracy) were further confirmed by the corresponding CID-MS2 fragmentation. For EGFR glycopeptides, the likely glycan structure in a glycopeptide was initially assigned by applying the mass obtained from the difference of a glycopeptide and its deglycosylated counterpart to match against the masses of the glycans in the Glycosuite database (Proteome Systems, Sydney, Australia). Among these likely glycostructure candidates, the best assignment was then selected from the preferred fragmentation patterns obtained in the related MSn spectra.
RESULTS AND DISCUSSION
Isolation of sEGFR and Circulating EGFR
Media from A431 cultured cells were collected and flowed through an immunoaffinity column containing a polyclonal anti-EGFR antibody. A total protein staining of the A431 total media, recombinant EGFR, and product of the EGFR purification is shown in supplemental Fig. S1A, panel A. Bands at the expected molecular weight for the extracellular domain of EGFR are circled in supplemental Fig. S1A, panel A. The Western blot using anti-EGFR polyclonal antibody for the A431 total media, EGFR recombinant protein standard, and product of EGFR purification are also shown in supplemental Fig. S1A, panel B, along with glycoprotein staining for recombinant EGFR and the product of the EGFR purification (supplemental Fig. S1A, panel C). In the figure for glycoprotein staining, both bands corresponding to EGFR appear to be glycosylated, and possibly one band contains sEGFR, and the other was a proteolytic cleavage form of EGFR. Others have described that the sEGFR, caused by alternative RNA splicing, often contains additional unique amino acids at its C terminus that are unrelated to the full-length EGFR, whereas the protease-cleaved form has identical amino acids to the extracellular domain of the full-length EGFR (15, 16). In addition, transcriptional analysis (RNA sequencing) also indicated that the splice variants of EGFR exist in an A431 cell line (see supplemental Fig. S2). Purified EGFR was further subjected to analysis by LC-MS. EGFR was identified with 20.4% sequence coverage, along with the identified peptide sequence, precursor charge, and m/z (supplemental Table S1). The immunopurified EGFR was further subjected to separation and purification by reversed-phase chromatography. The resulting chromatogram is shown in supplemental Fig. S1B, panel A. Aliquots of the reversed-phase fractions were subject to Western blot analysis to identify EGFR-containing fractions (supplemental Fig. S1B, panel B). For circulating EGFR from plasma pool samples, the isolation was done first by immunodepleting the top six most abundant proteins from plasma, followed by anion-exchange chromatography as the first dimension, and then by the reversed-phase chromatography as the second dimension, which is the same as for sEGFR as shown in supplemental Fig. S1C.
After purification, the tryptic digest of various EGFR-containing fractions (in supplemental Fig. S1B) were subjected to analysis by LC-MS. The protein coverage for the various fractions can be seen in Fig. 1. Peptides corresponding to most of the extracellular but not cytoplasmic domain of EGFR were identified from the A431 media and also fractions isolated from plasma pools. The expression of a truncated version of the receptor has been reported in other members of the EGF receptor family, ErbB2, ErbB3, and ErbB4 as well. These secreted forms are attributed to the alternative RNA splicing or metalloprotease cleavage of the plasma membrane form (17–19). For the plasma samples, intact proteins (without enzymatic digestion) were separated in two dimensions, first by anion-exchange chromatography, followed by reversed-phase fractionation. The supplemental Fig. S1C clearly shows the trailing of EGFR elution in the reversed-phase dimension, which is consistent with the protein existing in a complex mixture of glycosylated forms.
Fig. 1.

Identification of EGFR in A431 total media and reference plasma pools. The extracellular domain sequence of EGFR (629 amino acids) is shown at the top (bars indicate tryptic digestion peptides). Blue boxes indicate peptides with known N-linked glycosylation sites. Green boxes indicate peptides identified in a series of LC-MS/MS experiments on reference plasma pools. Yellow boxes were the sequence identified from secreted EGFR in A431.
Glycopeptide Analysis of sEGFR
As reported in our previous study (20), membrane-bound EGFR has 12 potential N-linked glycosylation sites, and 10 of these sites are glycosylated with either high mannose or complex-type structures. As expected, all of these glycosylation sites are located at the outer membrane surface and are present in sEGFR. The corresponding enzymatic peptide fragments for these sites are listed in supplemental Table S2. Among the 10 glycosylation sites in membrane-bound EGFR, three of these sites, located at Asn-328, Asn-337, and Asn-599, contained high mannose structures, although the other seven glycosylation sites (Asn-32, -151, -389, -420, -504, -544, and -579) contained complex-type glycans. One additional site at Asn-615 can be potentially glycosylated in sEGFR (with additional NGS consensus sequence due to alternative splicing) (16). Although most glycan structures were similar and have been characterized for both the membrane-bound and secreted EGFR (16, 20), there were some differences in terms of glycan structure and relative ratio in glycan distributions. The different types of glycan, which are unusual and primarily in sEGFR, are identified by our LC-MS approach in the following section.
Di-sialylated Glycans
In the analysis of the complex-type glycans at Asn-151, a different type of glycan structure consisting of di-antennary branches with three terminal sialic acids was identified from the Lys-C digestion of sEGFR (Fig. 2). As shown, the glycopeptide was identified in an extracted ion chromatogram at 35 min (Fig. 2A); the precursor ion (m/z 1291.64, 7+) was measured by FTMS (middle panel, Fig. 2B), and the precursor ion was fragmented by CID-MS2 (Fig. 2C). The ions fragmented by CID illustrated that the fragile sialic acids were dissociated preferentially from the precursor ion, yielding the precursor minus 1, 2, and 3 sialic acids (see the high abundance ions in Fig. 2C). Other glycan variants at this position were also detected at similar retention times, such as a bi-antennary glycan with four terminal sialic acids (Fig. 3). The disialylated species have been consistently found in the measurements from two repeat preparations in cell culture media and were also consistently observed with different charge states. CID fragmentation of these hyper-sialylated glycan structures at different charge states are also consistent for the assignment as shown in supplemental Figs. S3–S6 for 6+ to 9+ charges of the peptide with three terminal sialic acids and supplemental Figs. S7–S9 for 6+ to 8+ charges of the peptide with four terminal sialic acids. The peptide backbone was identified after peptide:N-glycosidase F treatment (supplemental Fig. S10). This observation was also supported by the measurement of the relative transcript abundance as measured by qRT-PCR of members of glycosyltransferase family 29, ST8SIA2 (STX), and ST8SIA4 (PST), which are involved in the synthesis of polysialic acid structures (Fig. 4).
Fig. 2.

Identification of a tri-sialylated glycan at Asn-151 from sEGFR. A, extracted ion chromatogram (XIC) of the glycopeptide containing Asn-151; B, mass and charge of the Lys-C-digested peptide with the anticipated glycan structure; and C, CID-MS2 spectrum of the precursor ion from B. In the glycan structures, the green circle represents mannose; the yellow circle represents galactose; the blue square represents N-acetylglucosamine; the red triangle represents fucose, and the purple diamond represents sialic acid.
Fig. 3.

Identification of a tetra-sialylated glycan at Asn-151 from sEGFR. Similar to Fig. 2, only the CID-MS2 spectrum of the anticipated glycopeptides precursor ion is shown.
Fig. 4.
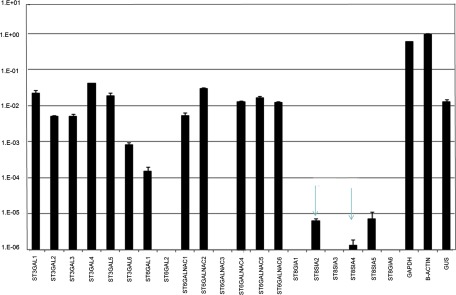
Relative transcript abundance of GT29 family genes in A431 cells. The ST8SiA2/ST8SiA4 genes encoded for synthesizing polysialic acids are indicated with arrows.
Polysialylated glycans have been reported in neuron and tumor cell membranes (21, 22) as well as embryonal polylactosaminyl structures (23). The STX and PST genes have been shown to be capable of forming di-sialic acid structures (24). The di-sialo structures, in mammalian brain, have been suggested to relate to aging or cerebellar diseases (25). The function of such glycan structures in sEGFR is unknown, but we can hypothesize that more negatively charged glycans could promote secretion of membrane-bound EGFR and thus be potential targets for blood-based measurements.
Branched Fucosylated Galactosyl Glycans
In the analysis of Asn-420, using both the MS2 and MS3 data, a branched fucosylated galactose structure was assigned (Fig. 5), which consists of the HexNAc-(Fuc)Gal-GlcNAc epitope. The branched linkage at the epitope is also detected by permethylated N-glycan analysis (Fig. 6). The exact connectivity of the epitope was clearly determined by MSn analysis (MS3, MS4, and MS5), as shown in supplemental Figs. S11–S14. The distally fucosylated structures capped with HexNAc (perhaps blood group A structures) were detected with and without core fucose (supplemental Fig. S11). Unlike the di-sialic acid (10% at the site), the HexNAc-(Fuc)Gal-GlcNAc epitope was detected with a significant amount (more than 50% at the site). This unusual branched glycan structure was detected in a minor amount in membrane-bound EGFR but highly up-regulated in sEGFR. It has been reported that the removal of the glycans at the Asn-420 site can abolish EGF binding, and thus the up-regulated glycan moieties at this site can be of significance (26). The formation of the branched (bulky) epitopes at either one or both arms (supplemental Fig. S11) could also contribute to the secretion of sEGFR.
Fig. 5.

Identification of the glycopeptides with HexNAc-(Fuc)Gal-GlcNAc epitope at Asn-420. A, base peak chromatogram of Lys-C-digested sEGFR peptide map; B, mass and charge of the Lys-C-digested peptide with the anticipated glycan structure; C, CID-MS2 spectrum of the precursor ion from B, and D, CID-MS3 spectrum of the precursor ion from Fig. 8C. The glycan symbols are the same as Fig. 2, except the gray square represents N-acetylhexosamine or HexNAc.
Fig. 6.
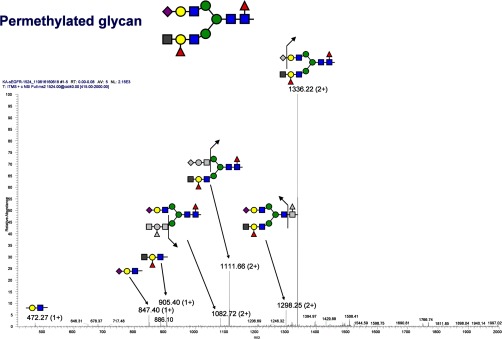
Analysis of the permethylated glycan with HexNAc-(Fuc)Gal-GlcNAc epitope. The CID-MS2 spectrum of the precursor ion with m/z 1524.00 (2+) is shown. In the spectrum, the charge state of 2+ is labeled for m/z containing the glycans denoted with split arrows, and 1+ is labeled for m/z containing glycan only.
Three Sites of High Mannose Glycans
There are three sites (Asn-328, Asn-337, and Asn-599) that contain high mannose structures in membrane-bound EGFR. For sEGFR, in the analysis of Asn-328, a high mannose (Man8) structure was identified as shown in Fig. 7, which illustrates the location of the glycopeptide in the LC-MS map (Fig. 7A), the accurate precursor ion (m/z 1439.7003, 2+) (Fig. 7B), and the fragmentation of the precursor ion by CID-MS2 (Fig. 7C). The accurate mass measured in the precursor ion spectrum is consistent with the observed peptide backbone with mannose fragmentation in the MS/MS spectrum for the assignment. There are also Man7 and Man6 structures associated with this site but less abundant than Man8 (data not shown). Interestingly, a complex-type structure at this site was found in the human plasma pool samples (circulating EGFR) so that this site may contain glycan structural variability that is sample-specific (7). In this study, only one glycopeptide at Asn-328 was characterized because it contains the same cleavage using either trypsin or Lys-C digestion. The plasma samples were from a different laboratory using trypsin digestion for identification of peptides (not glycopeptides) to confirm the existence of circulating EGFR. Because only a minute amount of EGFR from a patient sample was available, we will recollect and redo the experiment with Lys-C digestion in the future.
Fig. 7.
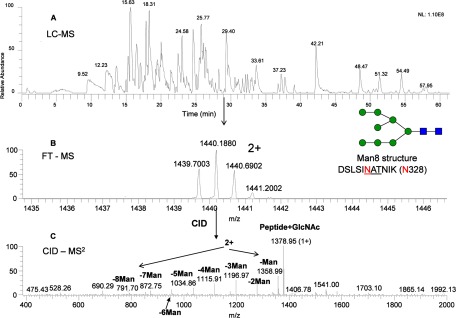
Identification of the glycopeptides with high mannose structure at Asn-328. A, base peak chromatogram of Lys-C-digested sEGFR (from HPLC fraction 37) peptide map; B, mass and charge of the Lys-C-digested peptide with the anticipated high mannose structure; C, CID-MS2 spectrum of the precursor ion from B.
Another high mannose site, Asn-337, was detected as mainly the Man8, along with Man7, Man6, and Man5 glycoforms in sEGFR (see supplemental Fig. S15). In membrane-bound EGFR, a significant amount of a Man9 glycoform was observed but not in sEGFR. In addition, additional complex-type glycan, bi-antennary with one terminal sialic acid (with or without core fucose), was observed only in sEGFR (supplemental Fig. S16, A and B).
For the third high mannose site, Asn-599, we could not observe any high mannose-containing peptide in sEGFR. Because this site is close to the C-terminal end of sEGFR, this portion could be cleaved by metalloprotease and thus could not be detected. For the alternative splicing variant, this C-terminal end contains additional 13 amino acids after Lys-C digestion (see the last sequence in supplemental Table S2), with an additional consensus site (Asn-615) that could be glycosylated in the same Lys-C-digested peptide. This long peptide with two possible glycosylation sites could be masked from our detection procedure (e.g. mass beyond the m/z range of MS system). Nevertheless, a short form (miscleavage) of this Lys-C-digested peptide was observed as the glycoforms of tri-antennary with two and three terminal sialic acids (see supplemental Fig. S17, A and B). A previous study also reported a complex type, not high mannose glycans, in this region for secreted EGFR (16). The glycans terminated with sialic acid usually are more stable (longer half-life) than high mannose-type glycans. This characteristic may also help EGFR stabilize in a culture media or bloodstream.
In summary, we have used the power of our LC-MS approach to identify the glycan heterogeneity present in sEGFR. This approach allows the characterization of the population of glycan structures at individual sites (major glycoforms at each site are listed in Table I). Although there is no clear mechanism for the roles of these glycans in ligand binding and signal transduction, nevertheless, these glycosylation sites are distributed exclusively in all of the four ligand-binding subdomains (see Fig. 8). In these subdomains, domains II and IV contain cysteine-rich region (cysteine knots). The amino acid sequences in domains I, II, and IV involve the heterodimerization with Erbb2 receptor (27), and domain III provides the binding to the growth factors such as EGF and TGF-α (28–31). The glycosylation sites in domain III have been studied by point mutation (Asn to Gln) to eliminate the oligosaccharides, and we found that only the elimination at Asn-420 affected the receptor dimerization (26). So far, there is no study that describes the effect of the point mutations on glycosylation in domains I, II, and IV. Moreover, these point mutation studies demolish the glycans totally and may not truly reflect the subtle effect of glycosylation in the disease state, which often presents as a glycosylation pattern or ratio change. We therefore believe that it is important to measure detailed glycan heterogeneity for each site, which may shed light on the secretion mechanism (i.e. through alternative splicing) or ligand binding mechanism to provide a basis for changes in the disease state. The unusual glycans identified specifically at sEGFR could provide us with valuable information on unique markers related to the secretion process or altered glycosyltransferase expression in diseases.
Table I. Major glycans at each site of sEGFR and full-length EGFR.
| Site | Major glycans for sEGFR | Major glycans for full-length EGFRa | Occupancy |
|---|---|---|---|
| Asn-32 | Tetra-antennary with 1 core fucose and 1 terminal sialic acid (20%) | Tri-antennary with 1 core fucose and 1 terminal sialic acid (10%) | Partial |
| Asn-151b | Tri-antennary with 1 core fucose and 1 terminal sialic acid (25%). Found significant amounts of di-sialylated glycoforms (10%) | Tri-antennary with 1 core fucose and 1 terminal sialic acid complex (30%) | Full |
| Asn-328c | Man6 (55%) | Man8 (40%) | Full |
| Asn-337d | Man8 (55%), no Man9, contained complex type glycans | Man8 (65%), contains 13% Man9 | Full |
| Asn-389 | Tri-antennary with 1 core fucose and 1 terminal sialic acid (15%) | Bi-antennary with 1 core fucose and 1 terminal sialic acid (13%) | Partial |
| Asn-420b | Bi-antennary, tri-antennary, and tetra-antennary with 1 core fucose and 1–4 terminal sialic acids with additional epitope extension as HexNAc-(Fuc)Gal-GlcNAc (60%) | Bi-antennary with 1 core fucose and 1 terminal sialic acid (55%) | Full |
| Asn-504 | Tetra-antennary with 1 core fucose and 3 terminal sialic acids (lacking 1 terminal galactose) (35%) | Tetra-antennary with 1 core fucose and 2 terminal sialic acid (lacking terminal galactose) (25%) | Full |
| Asn-544 | Tetra-antennary with 1 core fucose and 3 terminal sialic acids and 1 NGln-Gal repeat (25%) | Tetra-antennary with 1 core fucose and 2 terminal sialic acid and 1 NGln-Gal repeat complex partial (15%) | Partial |
| Asn-579 | Tri-antennary with 1 fucose (terminal end) and 2 sialic acid and with 4 NGln-Gal repeat (20%) | Bi-antennary with 1 fucose (terminal end) and 1 sialic acid and with 4 NGln-Gal repeat (15%) | Partial |
| Asn-599 | Tri-antennary without core fucose and with 3 terminal sialic acids | Man7 (40%) | Full |
| Asn-615e | Unknown | Not a glycosylation site | Unknown |
a Data were obtained from the analysis of full-length EGFR in a previous study (24).
b We found unusual (or up-regulated) glycan structures in sEGFR (as compared with the full-length EGFR).
c We found complex-type glycan structure in circulating EGFR.
d We found complex-type glycan structure in sEGFR.
e Glycosylation only occurred in sEGFR, in which both Asn-599 and Asn-615 are located in the same Lys-C-digested peptide, and the Asn-615 glycan structure has yet to be determined.
Fig. 8.
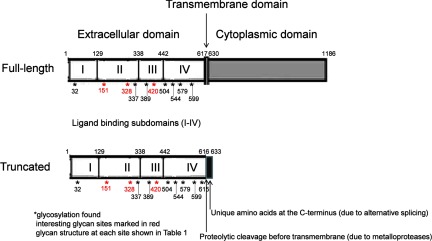
Structural diagram of full-length and truncated EGFR.
In future studies, we plan to use the glycopeptide structures identified from sEGFR in the cell line as a starting point for the development of a mass spectrometric assay (based on extraction ion chromatograms and multiple stage reaction monitoring) of plasma or serum clinical samples. We have shown in this study that the peptide backbones of glycopeptides, which contributes much more than the polar glycan moiety for retention on a typical reversed-phase chromatograph, are the same for EGFR samples isolated from cell lines or plasma. Thus, we can use the same mass in a similar retention time window to quickly examine if the same glycostructures exist for individual clinical samples. Any new masses present in that retention window can be taken into account by calculating glycan masses that fit with the observed mass shift (e.g. sialic acid with 292 Da and GlcNAc with 203 Da, etc.) and thus develop new glycostructures. The potential structures (matched masses) can be further confirmed by analysis of MS/MS or MSn spectra. In this manner, we can assign and monitor a new glycostructure such as the ones observed for Asn-328 in human plasma pool samples.
CONCLUSIONS
In this study, we analyzed glycans linked to specific sites (as glycopeptides) present in the secreted form of EGFR. Although EGFR contains many different glycans at multiple sites and the majority of them are quite similar between membrane-bound and -secreted forms, the approach we developed is sufficiently sensitive to differentiate the subtle structural changes. The key difference we observed, the up-regulation of fucosylated galactose and di-sialic acids at two specific sites in sEGFR, has not been previously reported. These unusual glycans with either more negative charges or bulky branched structures could be important glycan markers for secretion or cancer metastasis. We also showed that the protein sequence of secreted EGFR from the A431 cell line corresponded to the extracellular domain reported for EGFR and was consistent with previous observations of circulating EGFR present in a human plasma pool. We thus hypothesized that the micro-heterogeneity of glycans observed in proteins secreted from a cancer-related cell line could be analogous to corresponding glycoproteins present in the circulation in patients and observable in plasma or serum clinical samples. An important advantage of the characterization of glycosylation forms secreted from cell lines can be conducted at a depth that enables the characterization of unusual glycan structures. EGFR is secreted in the A431 cell line with high abundance, which enabled us to characterize the glycans extensively, and thus provided us a good foundation to explore secreted EGFR from other sources such as different breast cancer cell lines or patients. In future studies, we will use the glycan structures characterized in glycoproteins secreted from a cell line as a guide to develop mass spectrometric assays of novel glycan forms present in circulating glycoproteins. Thus, in this study we have implemented a unique workflow for the characterization of glycans present at a given site in secreted forms, which can be developed as potential markers for monitoring serum samples from cancer patients.
Supplementary Material
Acknowledgments
We are grateful to Drs. Kelly W. Moremen, Alison V. Nairn, Kazuhiro Aoki, Michael Tiemeyer, and Mike Pierce of the P41 Resource Center for performing analyses of permethylated glycans and transferases. We also thank Dr. Milos Novotny and John Geotz for collaborations in glycan measurements.
Footnotes
* This work was supported, in whole or in part, by National Institutes of Health Grant 5UO1CA128427 from NCI. This work was also supported by Korean Research World Class University Grant R31-2008-000-10086. This paper is Contribution Number 1024 from the Barnett Institute.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- sEGFR
- secreted form of epidermal growth factor receptor
- EGFR
- EGF receptor
- qRT-PCR
- quantitative real time reverse transcription-PCR
- CID
- collision-induced dissociation.
REFERENCES
- 1. Cancer Facts and Figures (2011) Am. Cancer Soc., [Google Scholar]
- 2. Kramer B. S. (2004) The science of early detection. Urol. Oncol. 22, 344–347 [DOI] [PubMed] [Google Scholar]
- 3. Nishizawa S., Kojima S., Teramukai S., Inubushi M., Kodama H., Maeda Y., Okada H., Zhou B., Nagai Y., Fukushima M. (2009) Prospective evaluation of whole-body cancer screening with multiple modalities including [18F]fluorodeoxyglucose positron emission tomography in a healthy population: a preliminary report. J. Clin. Oncol. 27, 1767–1773 [DOI] [PubMed] [Google Scholar]
- 4. Croswell J. M., Kramer B. S., Kreimer A. R., Prorok P. C., Xu J. L., Baker S. G., Fagerstrom R., Riley T. L., Clapp J. D., Berg C. D., Gohagan J. K., Andriole G. L., Chia D., Church T. R., Crawford E. D., Fouad M. N., Gelmann E. P., Lamerato L., Reding D. J., Schoen R. E. (2009) Cumulative incidence of false-positive results in repeated, multimodal cancer screening. Ann. Fam. Med. 7, 212–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ross J. S., Symmans W. F., Pusztai L., Hortobagyi G. N. (2005) Pharmacogenomics and clinical biomarkers in drug discovery and development. Am. J. Clin. Pathol. 124, S29–S41 [DOI] [PubMed] [Google Scholar]
- 6. Floyd E., Mcshane T. M. (2004) Development and use of biomarkers in oncology drug development. Toxicol. Pathol. 32, 106–115 [DOI] [PubMed] [Google Scholar]
- 7. Hanash S. M., Pitteri S. J., Faca V. M. (2008) Mining the plasma proteome for cancer biomarkers. Nature 452, 571–579 [DOI] [PubMed] [Google Scholar]
- 8. Chin L., Gray J. W. (2008) Translating insights from the cancer genome into clinical practice. Nature 452, 553–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lewandoski M. (2001) Conditional control of gene expression in the mouse. Nat. Rev. Genet. 2, 743–755 [DOI] [PubMed] [Google Scholar]
- 10. Gingrich J. A., Hen R. (2000) The broken mouse: the role of development, plasticity, and environment in the interpretation of phenotypic changes in knockout mice. Curr. Opin. Neurobiol. 10, 146–152 [DOI] [PubMed] [Google Scholar]
- 11. Fuster M. M., Esko J. D. (2005) The sweet and sour of cancer: Glycans as novel therapeutic targets. Nat. Rev. Cancer 5, 526–542 [DOI] [PubMed] [Google Scholar]
- 12. Ivanovic V. (2005) Aberrations of growth factors as biomarkers of cancer progression. Arch. Oncol. 13, 121 [Google Scholar]
- 13. Hanash S. M., Baik C. S., Kallioniemi O. (2011) Emerging molecular biomarkers—blood-based strategies to detect and monitor cancer. Nat. Rev. Clin. Oncol. 8, 142–150 [DOI] [PubMed] [Google Scholar]
- 14. Baron A. T., Lafky J. M., Boardman C. H., Cora E. M., Buenafe M. C., Liu D., Rademaker A., Fishman D. A., Podratz K. C., Reiter J. L., Maihle N. J. (2009) Soluble epidermal growth factor receptor: a biomarker of epithelial ovarian cancer. Cancer Treat. Res. 149, 189–202 [DOI] [PubMed] [Google Scholar]
- 15. Dall'olio F. (1996) Protein glycosylation in cancer biology: an overview. Clin. Mol. Pathol. 49, M126–M135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhen Y., Caprioli R. M., Staros J. V. (2003) Characterization of glycosylation sites of the epidermal growth factor receptor. Biochemistry 42, 5478–5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scott G. K., Robles R., Park J. W., Montgomery P. A., Daniel J., Holmes W. E., Lee J., Keller G. A., Li W. L., Fendly B. M. (1993) A truncated intracellular HER2/neu receptor produced by alternative RNA processing affects growth of human carcinoma cells. Mol. Cell. Biol. 13, 2247–2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee H., Maihle N. J. (1998) Isolation and characterization of four alternate c-erbB3 transcripts expressed in ovarian carcinoma-derived cell lines and normal human tissues. Oncogene 16, 3243–3252 [DOI] [PubMed] [Google Scholar]
- 19. Cheng Q. C., Tikhomirov O., Zhou W., Carpenter G. (2003) Ectodomain cleavage of ErbB-4: characterization of the cleavage site and m80 fragment. J. Biol. Chem. 278, 38421–38427 [DOI] [PubMed] [Google Scholar]
- 20. Wu S. L., Kim J., Bandle R. W., Liotta L., Petricoin E., Karger B. L. (2006) Dynamic profiling of the post-translational modifications and interaction partners of epidermal growth factor receptor signaling after stimulation by epidermal growth factor using extended range proteomic analysis (ERPA). Mol. Cell. Proteomics 5, 1610–1627 [DOI] [PubMed] [Google Scholar]
- 21. Foley D. A., Swartzentruber K. G., Lavie A., Colley K. J. (2010) Structure and mutagenesis of neural cell adhesion molecule domains. J. Biol. Chem. 285, 27360–27371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bogenrieder T., Herlyn M. (2003) Axis of evil: molecular mechanisms of cancer metastasis. Oncogene 22, 6524–6536 [DOI] [PubMed] [Google Scholar]
- 23. Fukuda M. N., Dell A., Oates J. E., Fukuda M. (1985) Embryonal lactosaminoglycan: the structure of branched lactosaminoglycans with novel disialosyl (sialyl α2—-9 sialyl) terminals isolated from PA1 human embryonal carcinoma cells. J. Biol. Chem. 260, 6623–6631 [PubMed] [Google Scholar]
- 24. Kitazume-Kawaguchi S., Kabata S., Arita M. (2001) Differential biosynthesis of polysialic or disialic acid structure by ST8Sia II and ST8Sia IV. J. Biol. Chem. 276, 15696–15703 [DOI] [PubMed] [Google Scholar]
- 25. Rinflerch A. R., Burgos V. L., Hidalgo A. M., Loresi M., Argibay P. F. (2012) Differential expression of disialic acids in the cerebellum of senile mice. Glycobiology 22, 411–416 [DOI] [PubMed] [Google Scholar]
- 26. Tsuda T., Ikeda Y., Taniguchi N. (2000) The Asn-420-linked sugar chain in human epidermal growth factor receptor suppresses ligand-independent spontaneous oligomerization. Possible role of a specific sugar chain in controllable receptor activation. J. Biol. Chem. 275, 21988–21994 [DOI] [PubMed] [Google Scholar]
- 27. Kumagai T., Davis J. G., Horie T., O'Rourke D. M., Greene M. I. (2001) The role of distinct p185neu extracellular subdomains for dimerization with the epidermal growth factor (EGF) receptor and EGF-mediated signaling. Proc. Natl. Acad. Sci. U.S.A. 98, 5526–5531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Greenfield C., Hiles I., Waterfield M. D., Federwisch M., Wollmer A., Blundell T. L., McDonald N. (1989) Epidermal growth factor binding induces a conformational change in the external domain of its receptor. EMBO J. 8, 4115–4123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lax I., Mitra A. K., Ravera C., Hurwitz D. R., Rubinstein M., Ullrich A., Stroud R. M., Schlessinger J. (1991) Epidermal growth factor (EGF) induces oligomerization of soluble, extracellular, ligand-binding domain of EGF receptor. A low resolution projection structure of the ligand-binding domain. J. Biol. Chem. 266, 13828–13833 [PubMed] [Google Scholar]
- 30. Zhou M., Felder S., Rubinstein M., Hurwitz D. R., Ullrich A., Lax I., Schlessinger J. (1993) Real-time measurements of kinetics of EGF binding to soluble EGF receptor monomers and dimers support the dimerization model for receptor activation. Biochemistry 32, 8193–8198 [DOI] [PubMed] [Google Scholar]
- 31. Lemmon M. A., Bu Z., Ladbury J. E., Zhou M., Pinchasi D., Lax I., Engelman D. M., Schlessinger J. (1997) Two EGF molecules contribute additively to stabilization of the EGFR dimer. EMBO J. 16, 281–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Faca V., Pitteri S. J., Newcomb L., Glukhova V., Phanstiel D., Krasnoselsky A., Zhang Q., Struthers J., Wang H., Eng J., Fitzgibbon M., McIntosh M., Hanash S. (2007) Contribution of protein fractionation to depth of analysis of the serum and plasma proteomes. J. Proteome Res. 6, 3558–3565 [DOI] [PubMed] [Google Scholar]
- 33. Nairn A. V., dela Rosa M., Moremen K. W. (2010) Transcript analysis of stem cells. Methods Enzymol. 479, 73–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen R., Mias G. I., Li-Pook-Than J., Jiang L., Lam H. Y., Chen R., Miriami E., Karczewski K. J., Hariharan M., Dewey F. E., Cheng Y., Clark M. J., Im H., Habegger L., Balasubramanian S., O'Huallachain M., Dudley J. T., Hillenmeyer S., Haraksingh R., Sharon D., Euskirchen G., Lacroute P., Bettinger K., Boyle A. P., Kasowski M., Grubert F., Seki S., Garcia M., Whirl-Carrillo M., Gallardo M., Blasco M. A., Greenberg P. L., Snyder P., Klein T. E., Altman R. B., Butte A. J., Ashley E. A., Gerstein M., Nadeau K. C., Tang H., Snyder M. (2012) Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 148, 1293–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.