

Abstract
Haptoglobin is a liver-secreted glycoprotein with four N-glycosylation sites. Its glycosylation was reported to change in several cancer diseases, which prompted us to examine site-specific glycoforms of haptoglobin in liver cirrhosis and hepatocellular carcinoma. To this end, we have used two-dimensional separation composed of hydrophilic interaction and nano-reverse phase chromatography coupled to QTOF mass spectrometry of the enriched glycopeptides. Our results show increased fucosylation of haptoglobin in liver disease with up to six fucoses associated with specific glycoforms of one glycopeptide. Structural analysis using exoglycosidase treatment and MALDI-MS/MS of detached permethylated glycans led to the identification of Lewis Y-type structures observed particularly in the pooled hepatocellular carcinoma sample. To confirm the increase of the Lewis Y structures observed by LC-MS, we have used immunoaffinity detection with Lewis Y-specific antibodies. The presence of multiply fucosylated Lewis Y glycoforms of haptoglobin in the disease context could have important functional implications.
Glycosylation of proteins during their passage through the endoplasmic reticulum and Golgi compartments is controlled by the activity of multiple glycosyltransferases, glycosidases, and their interacting partners (1–3). Changes in the expression of these enzymes were associated with numerous diseases, including cancer (4). Multiple studies published evidence that the modified glycosylation of proteins reflects disease progression and could have important functional impact (5, 6). Specifically, progression of liver disease to hepatocellular carcinoma (HCC)1 has been associated with changes in the expression of glycosyltransferases, including α-1,6-mannosylglycoprotein 6-β-N-acetylglucosaminyltransferase, and with changes in architecture of the organ (7).
Because liver is a major source of circulating blood proteins, the disease-associated changes are expected to alter circulating N-glycoproteins (8–10). Some studies suggest that changes in glycosylation of proteins are sufficiently sensitive and specific to serve as a diagnostic test for noninvasive monitoring of liver disease progression (11–13). The majority of the published reports quantify N-glycans following enzymatic de-glycosylation with PNGaseF (10, 11, 14, 15). The assumption of these studies is that the changes in the expression of glycosyltransferases result in a uniform change in specific types of glycans across all glycoproteins. This means that an “average” quantification across many proteins and glycosites would be sufficient to monitor the disease processes. MALDI-TOF screening of permethylated detached N-glycans or analysis of fluorescently labeled glycans coupled with capillary, graphitized carbon or HILIC separations was successfully implemented (10, 14–16). The alterations of glycans observed in liver disease can be divided into four major groups as follows: 1) bisecting glycans with one molecule of N-acetylglucosamine attached via β1–4 bond to the branched core mannose associated with increased activity of N-acetylglucosamine transferase III (17); 2) asialo- and agalactoglycans, truncated structures with terminal galactoses or N-acetylglucosamines (18); 3) (poly)-N-acetyllactosamines with β1–6 branching associated with increased activity of α-1,6-mannosylglycoprotein 6-β-N-acetylglucosaminyltransferase (19); and 4) fucosylated glycans, where fucose is attached at the C-3 position of outer arm N-acetylglucosamine or, less frequently, at C-6 core N-acetylglucosamine (20).
A major limitation of the analyses of detached N-glycans is the undefined influence of changes in protein concentration on the N-glycan distribution. For example, the bisecting and asialo/agalactosylated glycans observed in liver disease are associated primarily with immunoglobulins (10, 21). Specific proteins carry different types of glycans, and the concentration of many circulating glycoproteins change significantly in the disease context (22). In addition, glycans vary by site of attachment on a protein unless the distribution of glycans at each site of the protein is uniform (7). This suggests that an analysis of site-specific glycoforms of liver-secreted N-glycoproteins is expected to increase the specificity of detection and improve our understanding of the functional impact of the observed changes. In fact, α-fetoprotein-L3, the fucosylated version of α-fetoprotein, is a glycoproteomic marker used clinically for the detection of hepatocellular carcinoma. Even though the sensitivity of the test is not sufficient for an efficient early detection of HCC, specificity is improved compared with the measurement of α-fetoprotein without glycoform analysis (23–25).
Haptoglobin (Hp) is an abundant glycoprotein secreted into plasma primarily by the liver. Its major known biological role is the capture of released hemoglobin during intravascular hemolysis and the prevention of kidney damage by released iron (26). Hp-hemoglobin complex binds to macrophage scavenger receptor CD163 (27), and the structure of the porcine Hp-hemoglobin complex was recently determined (28). Increased binding of Hp to galectin-1 in breast cancer was attributed to changes in its glycosylation and altered the intracellular fate of the Hp-hemoglobin complex (6). Here, we describe changes in the site-specific glycoforms of Hp associated with liver cirrhosis and HCC. We expand our recently reported LC-MS/MS analysis of Hp (29) by exoglycosidase treatment of glycopeptides, immunoblotting, and MS/MS analysis of detached permethylated glycans in the context of liver disease.
EXPERIMENTAL PROCEDURES
Study Population
All participants were enrolled under the protocols approved by the Georgetown University Institutional Review Board. The HCC patients (n = 5), cirrhotic patients (n = 5), and healthy individuals (n = 5) were enrolled into the study in collaboration with the Department of Hepatology and Liver Transplantation, Georgetown University Hospital, Washington D. C. The diagnosis of HCC was made by the attending physician based on liver imaging and/or liver biopsy. All the HCC patients had early stage disease (stage 1 and 2) according to the 7th Edition of the American Joint Committee on Cancer Staging manual. The cirrhotic patients were slightly younger than the HCC patients and healthy controls. All the patients (HCC or cirrhosis) had chronic hepatitis C virus infection as the primary diagnosis and were selected to have detectable haptoglobin by Western blotting and comparable low level liver damage as measured by MELD scores. The basic characteristics of the study participants are summarized in supplemental Table 1.
Isolation of Haptoglobin from Plasma
Hp was purified from all samples by a combination of hemoglobin-Sepharose affinity and reverse phase fractionation. Pooled samples of plasma were created for the three groups (healthy controls, cirrhosis, and HCC) to isolate sufficient amounts of Hp for the analysis of site-specific glycoforms. The hemoglobin-Sepharose resin was prepared by coupling of 33 mg of hemoglobin (Sigma-Aldrich) to 1 g of lyophilized CNBr-activated Sepharose (Sigma-Aldrich) under conditions recommended by the manufacturer. Briefly, resin was loaded into 600- μl spin columns (Thermo Scientific, Rockford, IL) and washed several times with PBS. Human plasma (125 μl) was diluted with PBS, pH 7.4, to 500 μl and left to bind to 150 μl of the hemoglobin-Sepharose resin at room temperature for 2 h on a rotary mixer. After washing (five times with 500 μl of PBS), bound proteins were eluted three times with 300 μl of 0.1 m glycine, pH 2.5, and immediately neutralized with 1:10 (v/v) 1 m Tris-HCl, pH 9. After protein determination, the elution fractions were combined, and guanidine HCl (Sigma-Aldrich) was added to a 6 m final concentration. The samples were loaded on an HPLC ProSwift RP-1S column (Dionex, Sunnyvale, CA) in mobile phase A (2% ACN, 0.08% TFA) and separated at 30°C under a gradient of 1–75% B (98% ACN, 0.05% TFA) at 18 min at a flow rate of 1 ml/min. Under these conditions, Hp eluted with retention times of 12.2–12.6 min; the collected Hp fraction was dried on a SpeedVac and diluted in distilled H2O for further analysis.
Protein Digestion and Exoglycosidase Treatment
Hp was digested as described previously (29). Briefly, 2.5 μg of isolated Hp was resuspended in 20 μl of 50 mm NH4HCO3 at pH 7.8 (Sigma-Aldrich) with 0.05% RapiGest (Waters, Milford, MA), reduced with 5 mm DTT and alkylated with 15 mm iodoacetamide (Sigma-Aldrich). The tryptic digest (2.5 ng/μl) (Promega, Madison, WI) was carried out at 37°C in Barocycler NEP2320 (Pressure BioSciences, South Easton, MA) or with endoproteinase Glu-C (60 ng/μl) (Roche Applied Science) at 25°C overnight. The digests were desalted on a MicroTrap peptide cartridge (Michrom Bioresources, Auburn, CA) and washed three times with 250 μl of 0.1% aqueous TFA (Sigma-Aldrich). The peptides were eluted with 100 μl of 60% ACN with 0.1% TFA, and the eluate was dried using a SpeedVac concentrator.
Glycopeptides were resuspended in 20 μl of reaction buffer containing 50 mm sodium acetate (Sigma-Aldrich), 5 mm CaCl2, pH 5.5, for a double glycosidase digest using both α-2/3,6,8-neuraminidase from Clostridium perfringens overexpressed in Escherichia coli (New England Biolabs, Ipswich, MA) and β1,4-galactosidase from Bacteroides fragilis expressed in E. coli (New England Biolabs). Two microliters of each exoglycosidase (100 units of neuraminidase and 16 units of galactosidase) were added to the sample based on the manufacturer's recommendation and incubated at 37°C for 20 h. We have verified that each glycosidase reaction reaches completeness by inspection of the spectra (disappearance of the peaks) of known glycoforms.
For structural characterization of glycopeptides, Hp (2.5 μg) isolated from pooled plasma samples of HCC patients and healthy controls was digested with trypsin as described above, desalted, and treated with exoglycosidases in the following order: α2/3,6,8-neuraminidase (100 units) from C. perfringens overexpressed in E. coli (New England Biolabs); α1/2-fucosidase (20 units) from Xanthomonas manihotis (New England Biolabs); α1/3,4-fucosidase (16 microunits) from almond meal (Prozyme, Hayward, CA); β1,4-galactosidase (16 units) from B. fragilis expressed in E. coli (New England Biolabs); and β1,3-galactosidase (20 units) from X. manihotis expressed in E. coli (New England Biolabs). Between each exoglycosidase treatment, glycopeptides were desalted by a microtrap device, eluted with 50% ACN + 0.1% TFA, dried, and an aliquot was resuspended in solvent A (0.1% formic acid in 2% acetonitrile) for MS analysis as described below. Remaining sample was resuspended in 20 μl of digestion buffer and incubated 18–24 h at 37°C as recommended by the manufacturers.
For determination of sialic acid linkage, isolated Hp (2.5 μg) from pooled samples of HCC, cirrhosis, and healthy plasma was treated for 24 h at 37°C with α2/3-sialidase (20 microunits), from recombinant Streptococcus pneumoniae expressed in E. coli (Prozyme, Hayward, CA), in 20 μl of buffer as recommended by the manufacturer. After incubation, samples were desalted and analyzed as described below.
Separation of Glycopeptides by HILIC and Nano-reverse Phase C18 Chromatography
Ten micrograms of a tryptic digest of Hp isolated from all samples were resuspended in 20 μl of 50% ACN in water (Fisher Scientific, Fair Lawn, NJ) and injected on a HILIC XBridge column (5 μm, 2.1 × 100 mm) (Waters) as described previously (29). Briefly, the column was equilibrated in 90% of solvent B (0.01% TFA in acetonitrile (Fisher Scientific)), and separation was achieved using a 35-min linear gradient of 90 to 40% solvent B at 40°C (solvent A, 0.01% TFA in water; solvent B, 0.01% TFA in acetonitrile). Eluted glycopeptides were monitored at 214 nm, and collected fractions were dried in a SpeedVac concentrator. The enriched glycopeptides were resuspended in 20 μl of 2% acetonitrile, 0.1% formic acid and separated by nano-reverse phase chromatography (NanoAcquity, Waters) using a C18 column (1.7-μm particles, 75 μm inner diameter × 150 mm) (Waters Associates) for the LC-ES-MS/MS analysis on a QStar Elite mass spectrometer (Applied Biosystems, Foster City, CA). Two microliters of each fraction were injected, and glycopeptides were separated at a flow rate of 0.4 μl/min, using the following gradients: 0 min, 1% B; 35 min, 35% B; 37 min, 90% B; 42 min, 90% B; 44 min, 1% B; and 55 min, 1% B (solvent A, 0.1% formic acid in 2% acetonitrile; solvent B, 0.1% formic acid in 98% acetonitrile). The mass spectrometer was operated in a data-dependent mode; after a full scan (m/z 400 to m/z 1800) survey, the three most intense precursor ions were selected for collision-induced dissociation. Collision energy and MS/MS accumulation time were set automatically, and the MS/MS spectra were recorded from m/z 150 to m/z 2000. Dynamic exclusion was set at 15 s and five counts for two repeated precursors. The fragment intensity multiplier was set at 16 with maximum accumulation of 2 s, which resulted in a total cycle time of 6.5 s. In the case of direct analysis without prior HILIC enrichment, 2 pmol of Hp tryptic digest was injected onto a C18 column (1.7-μm particles, 75 μm inner diameter × 250 mm) (Waters Associates) without trapping using the following gradients: 5 min, 1% B; 105 min, 37% B; 107 min, 90% B; 112 min, 90% B; 114 min, 1% B; 150 min, 1% B (solvent A, 0.1% formic acid in 2% acetonitrile; solvent B,: 0.1% formic acid in 98% acetonitrile). The mass spectrometer was set as described above.
Determination of Glyco-site Occupancy
Occupancies of all four Hp glycosites were quantified by comparison of XIC precursor ion intensities of unoccupied N-(asparagine) peptides and deglycosylated D-(aspartic acid) peptides following PNGaseF de-glycosylation under [18O]water as described previously (30, 31). Briefly, Hp (2 pmol) was digested after reduction and alkylation by trypsin or endoproteinase Glu-C as described above. The Glu-C digest was split in half, and one portion was further digested with trypsin, and the other was processed as described below; this was done to generate singly glycosylated proteolytic peptides of all four Hp glycosites. All enzymatic digests (tryptic, Glu-C, and combination) were heated for 15 min at 90°C to deactivate proteolytic enzymes and evaporated to dryness using a SpeedVac. Separately, 2 μl of G7 reaction buffer and 0.5 μl of PNGaseF (New England Biolabs) were evaporated and diluted in 20 μl of [18O]water (Cambridge Isotope Laboratories, Andover, MA). This solution was used to dissolve the dried peptides for de-glycosylation (1.5 h at 37°C) using Barocycler NEP2320 (Pressure BioSciences, South Easton, MA). De-glycosylated 18O-labeled peptides (2 pmol) were analyzed directly after the PNGaseF enzymatic treatment on a QTOF mass analyzer using an IDA workflow. Peptides were separated by reverse phase chromatography (Tempo Eksigent-AB Sciex, Framingham, MA) on a ChromXP C18-CL (3 μm, 120 Å, 180 μm, 20 mm) trap column and ChromXP C18-CL (3 μm, 120 Å, 75 μm, 150 mm) HPLC capillary column (Eksigent-AB Sciex) interfaced with a 5600 TripleTOF mass analyzer (AB Sciex, Framingham, MA). Analysis was carried out by a 5-min trapping/washing step using 2% ACN, 0.1% formic acid at a 5 μl/min flow rate followed by a 30-min gradient elution of 0.1% formic acid in ACN. Starting conditions were set to 3% ACN, 0.1% formic acid; 3–50% ACN, 0.1% formic acid 5–25 min; 50–98% ACN, 0.1% formic acid 27–27.1 min; 98% ACN, 0.1% formic acid 27.1–30 min. Mass spectrometric conditions were set to curtain gas 25, ion spray voltage 2400 V, ion source gas 16, and interface heater temperature 180°C; and the instrument was operated in data-dependent mode; after a full scan (m/z 400 to m/z 1800) survey, the 25 most intense precursor ions were selected for collision-induced dissociation. Collision energy and MS/MS accumulation time were set automatically, and the MS/MS spectra were recorded from m/z 150 to m/z 1800. Dynamic exclusion was set at 5 s and 150 counts for two repeated precursors.
Analysis of Detached Permethylated Glycans
Purified Hp (20 μg) was reduced with DTT and alkylated with iodoacetamide. N-Glycans were detached with PNGaseF (New England Biolabs) overnight at 37°C, reduced with ammonium-borane complex (Sigma-Aldrich), and permethylated as described previously (9, 32). MALDI-TOF MS/MS spectra were acquired on a 4800 mass analyzer (AB Sciex) equipped with an Nd:YAG 355-nm laser. In addition, dried permethylated glycans were resuspended in 10 μl of 20% methanol, and 2 μl were injected on nano-reverse phase C18 column (1.7 μm particles, 75 μm inner diameter × 250 mm) (Waters Associates) coupled with a QStar Elite mass spectrometer under the following chromatographic conditions: 5 min, 30% B; 35 min, 65% B; 36 min, 95% B; 38 min, 95% B; 39 min, 30% B; 70 min, 30% B (solvent A, 0.1% formic acid in 2% acetonitrile; solvent B, 0.1% formic acid in 98% acetonitrile). The mass spectrometer was operated as described above except the collision energy was fixed at 30.
Western Blot
To confirm the presence of Hp, 1 μl of plasma was diluted in 10 μl of 50 mm Tris-HCl buffer, pH 7.4, and reduced by the addition of 50 mm DTT. Samples were separated on BisTris 4–12% polyacrylamide gel (Invitrogen) and transferred to a PVDF membrane (Invitrogen). Anti-human Hp antibody (Sigma-Aldrich) was used as a primary antibody, and anti-rabbit IgG-HRP (Sigma-Aldrich) was used as a secondary antibody; immobilized complexes were visualized by a chemiluminescent assay (GE Healthcare). To detect the Le(y) structure, 1 μg of Hp, as estimated by a standard BCA assay according to the manufacturer's instructions (Pierce), underwent desialylation as described above. Desialylated Hp was reduced by 50 mm DTT, separated on BisTris 4–12% polyacrylamide gel, and transferred to a PVDF membrane. Anti-blood group Lewis Y monoclonal antibody (F3) (Thermo Scientific, Rockford, IL) was used as primary antibody, and biotinylated IgM (Vector Laboratories, Burlingame, CA) was used as secondary antibody. Biotin was detected by streptavidin-HRP and visualized by a chemiluminescent assay (GE Healthcare). To confirm the presence of Hp, the membrane was stripped and incubated with primary antibody against the Hp protein as described above.
RESULTS
Purification of Haptoglobin
Detailed structural characterization of glycosylated Hp requires purification of the protein because confident assignment of the minor glycoforms in a complex sample was not feasible. We have optimized affinity chromatography on hemoglobin-Sepharose beads followed by monolithic reverse phase chromatography of the partially purified Hp. We have selected these methods because neither the hemoglobin-Hp interaction nor the interaction of the protein with the reverse phase was affected by the glycosylation of the protein (6). The reverse phase, however, separates Hp based on different complexes of its phenotypes (Hp1-1, Hp2-2, and Hp2-1), which elute at RT in 12.2–12.6 min (Fig. 1). We have confirmed that Hp is the major component of all the peaks in this cluster, and we collect the forms as one fraction for further analysis. Deglycosylation of the isolated Hp with PNGaseF shifts the molecular mass of Hp by ∼10 kDa which, together with the analysis of glycopeptides and detached glycans, shows that the glycoforms co-elute under our chromatographic conditions.
Fig. 1.

Isolation of haptoglobin. A, scheme of the Hp isolation procedure; B, SDS electrophoresis of Hp isolated from the sera of two of the healthy controls before and after deglycosylation with PNGaseF; C, UV traces of the Hp complexes containing β and α1 subunits (upper trace) and a mixture of β, α1, and α2 subunits (bottom trace).
Characterization of Hp Glycoforms
We have used enrichment of glycopeptides by HILIC chromatography, combined digest with trypsin and Glu-C, processing of glycopeptides with neuraminidases, fucosidases, and galactosidases, and MS/MS analysis of glycopeptides and detached permethylated glycans to characterize the disease-related site-specific glycoforms of Hp. The two-dimensional HILIC and nano-reverse phase C18 chromatography was coupled to a QTOF mass spectrometer as described previously (29). This configuration achieves an efficient enrichment of glycopeptides and sensitive detection of the glycoforms of the individual peptides. The following example highlights the usefulness of the workflow in the assignment of the site-specific glycoforms. The extracted ion chromatogram at m/z value 1220.0 (4+) corresponds to the first monoisotopic peak of the T3 peptide with glycan composition A4G4S1F3 and also to the second isotopic peak of the T3 peptide with glycan composition A4G4S2F1 with observed monoisotopic mass at m/z 1219.7 (4+) (Fig. 2). We adopt the N-glycan nomenclature from the NIBRT GlycoBase to enumerate the identified glycoforms (Table I) (34). The mass difference of these glycoforms is relatively small, and their chromatographic resolution on the C18 resin helps with their identification. The most abundant ions in the collision-induced dissociation (CID) spectra of the A4G4S1F3 and A4G4S2F1 glycopeptides correspond to glycan fragments (oxonium ions) at m/z 204.1 (HexNAc), 366.1 (Hex-HexNAc), 512.2 (Hex-HexNAc-Fuc), 657.2 (NeuAc-Hex-HexNAc), and 803.3 (NeuAc-Hex-HexNAc-Fuc); but the intensity ratio of the oxonium ions at m/z 512.2 and m/z 366.1 is substantially higher in case of the triply fucosylated glycopeptide (Fig. 3). The other important observed ions are glycopeptide fragments from the N-glycan core such as peptide-HexNAc, peptide-2HexNAc, or peptide-2HexNAc-Hex and fragments derived from neutral loss of the precursor ion. For some glycopeptides, especially when the precursor ion is abundant, y and b fragments from the peptide backbone were observed as well, but this is not frequent, and the ions tend to be of low intensity. As shown in Fig. 2, difference of the quadruply charged precursor ions of the two glycopeptides is only 0.25 m/z units and 0.4 min in retention time. Even smaller mass differences would be present in glycoforms containing more than three fucoses or their sialylated counterparts. We find that the exoglycosidase treatments and careful examination of the spectra improve our ability to resolve these glycoforms.
Fig. 2.

Extracted ion chromatogram of multiply sialylated and fucosylated glycoforms of the T3 peptide isolated from pooled plasma of HCC patients and separated by nano-reverse phase C18 chromatography after HILIC enrichment. The mass difference between the isoforms is 1 Da (0.25 m/z units for a quadruply charged precursor).
Table I. Observed glycoforms of Hp glycopeptides in patients with HCC, cirrhosis, and in healthy controls.
The presence of glycoforms is shown as a percentage of the sum of glycopeptide intensities for each peptide and glycosidase treatment. N represents the number of glycoforms observed for each peptide and glycosidase treatment.

Fig. 3.

CID spectra of Hp T3 glycopeptide isolated from pooled plasma of HCC patients with two sialic acids and one fucose (A) and with one sialic acid and three fucoses (B). The intensity of oxonium ion at m/z 512.2 (Le(x)-type structure) is significantly elevated in the glycopeptide with three fucoses. The triply fucosylated structure is observed also in the pooled cirrhosis sample but not in the pooled healthy control sample.
Processing of Glycopeptides with Proteases and Exoglycosidases
Digestion of Hp with trypsin does not allow separation of each glycosylation site as the tryptic peptide NLFLNHSENATAK (T2) contains two N-glycosylation sites. We have observed tryptic glycoforms composed of A2G2+A2G2, A2G2+A3G3, A2G2+A4G4, or A3G3+A3G3 and A3G3+A4G4, A4G4+A4G4, and their fucosylated forms. Fucosylated forms with up to seven fucoses were identified at the T2 glycopeptide from the HCC sample bearing two tetra-antennary glycans. All combinations of glycoforms identified at the T2 peptide after neuraminidase treatment are shown in supplemental Table 2.
Identification of the glycoforms of the doubly glycosylated tryptic peptide NLFLNHSENATAK (T2) was achieved using a combined protein digest with trypsin and/or endoproteinase Glu-C. The Glu-C protease separates the two glycosites and makes assignment of the glycoforms feasible (Table I). We were not able to detect the small glycopeptide NATAK resulting from the trypsin Glu-C double digest likely due to its hydrophilicity. However, we have detected efficiently the glycoforms of the Glu-C peptide NATAKDIAPTLTLYVGKKQLVE. The singly glycosylated T2-1 and T2-2 peptides separated by endoproteinase Glu-C and other tryptic peptides of Hp (T1 and T3) were characterized by a combination of exoglycosidases and CID MS/MS fragmentation.
To reduce the complexity of glycoforms, glycopeptides were treated with nonspecific neuraminidase that cleaves sialic acids with α(2–3,6,8) linkage. We remove the sialic acids because our data and results of other groups point to the importance of glycopeptide fucosylation in liver disease, which we wanted to examine in detail; this does not imply that sialylation is not important. De-sialylation improves signal intensity of the fucosylated glycopeptides due to the fusion of multiple sialoforms and improved ionization efficiency of the de-sialylated peptides. The de-sialylation revealed the presence of glycoforms with up to six fucoses in the HCC (Fig. 4) and up to four fucoses in the cirrhotic samples on the VVLHPNYSQVDIGLIK tryptic glycopeptide (T3) (Table I). These glycoforms would be below the detection limit without the de-sialylation. Glycoforms with only three fucoses were identified on the glycopeptide MVSHHNLTTGATLINEQWLLTTAK (T1), and four fucoses were observed on its truncated form MVSHHNLTTGATLINE after the trypsin Glu-C double digestion. The NLFLNHSE (T2-1) trypsin Glu-C fragment and NATAKDIAPTLTLYVGKKQLVE (T2-2) Glu-C peptide of the T2 glycopeptide carry glycoforms with up to five fucoses in HCC and three fucoses in cirrhosis. It is interesting to note that we have observed a maximum of one fucose on any glycopeptide in healthy controls.
Fig. 4.

MS of the T3 glycopeptide after neuraminidase treatment with tetra-antennary glycans bearing up to six fucoses. The spectrum represents a sum of the MS scans in the pooled HCC sample in a 1-min retention window.
The reduction of glycan complexity after neuraminidase treatment allows detection of minor glycoforms, but there was still a relatively high number of possible isobaric glycopeptides consistent with each observed molecular composition. To further investigate the structure of multiply fucosylated glycopeptides and to test the association of specific glycoforms with a particular disease state, we have performed further digests with β1,4-galactosidase (Table I). The treatment with β1,4-galactosidase cleaves specifically terminal galactoses except when fucose is attached to the subterminal N-acetylglucosamine (35). This reduces the number of possible isoforms and allows identification of specific linkages, but we have performed further digests to confirm the linkage assignments. To this end, glycopeptides were first treated with α2/3,6,8-neuraminidase to comprehensively de-sialylate the glycopeptides followed by α1/2- and α1/3,4-fucosidases to comprehensively remove all fucoses. A2G2 at m/z 1139.9, A3G3 at m/z 946.4, and A4G4 at m/z 1037.7 represent all the resulting glycoforms except a small fraction of a tetra-antennary product A4G4F1 at m/z 1074.2 (its intensity is less than 1% of the A4G4 intensity) (supplemental Fig. 3B). Subsequent β1/4-galactosidase treatment produced the A2 at m/z 774.2, A3 at m/z 824.9, A4 at m/z 875.7, and in HCC sample A4G1 at m/z 916.2 with ion intensity about 5% of the A4 intensity (supplemental Fig. 3C). Not even traces of the galactose were detectable in the bi- and tri-antennary glycoforms. An additional 1/3 galactosidase treatment did not cleave the remaining galactose from the A4G1 glycoform; we repeated this step twice to ensure complete digestion. Intensity of the A4 glycoform is substantially lower in control samples than in the HCC samples, and the intensity of the A4G1 glycoform, in control samples, is below the detection limit of the analysis.
An example of Hp-T3 glycoforms with three outer arm N-acetylglucosamines and two fucoses (after the combined α2/3,6,8-neuraminidase and β1,4-galactosidase treatment) consistent with all the generated exoglycosidase-MS/MS information is shown in Fig. 5. Le(y)-type structures were observed on the T3 glycopeptide in the pooled HCC sample, but a different linkage dominates the cirrhosis sample. The presence of Le(y)-type structures was confirmed by immunostaining using an anti-blood group Lewis Y monoclonal antibody and by fragmentation of detached permethylated glycans (Fig. 5). In the case of the A3G1F2 glycoform, the sodiated m/z 834.3 fragment (Le(y)-type structure) and the m/z 1647.8 fragment (loss of Le(y)) were observed (Fig. 5C).
Fig. 5.

A, select site-specific glycoforms of the T3 peptide at m/z 938.4 and at m/z 978.9 after neuraminidase and β1,4-galactosidase treatment consistent with the observed results in pooled HCC (left) and pooled cirrhosis (right) samples. The presented glycoforms with A3G1F2 and A3G2F2 composition are the only structures showing the correct fucose distribution based on the data obtained after consecutive exoglycosidase treatment with neuraminidase and β1,4-galactosidase. B, Western blot confirms the presence of Le(y)-type structures with higher intensity in the HCC sample. C, MALDI-MS/MS spectrum of detached permethylated glycan A3G1F2 showing the fragment ion at m/z 834.3 corresponding to the Le(y) structure. Inset shows the precursor ion at m/z 2459.2. Additional structure confirming digests with α1,2- and α1–3,4-fucosidase and β1,3-galactosidase are presented in supplemental Fig. 3.
Finally, we have used treatment of Hp glycopeptides with α2/3-sialidase to further resolve sialic acid linkage to galactose. The α2/3-sialidase left the glycans virtually intact in all three samples (HCC, cirrhosis, and controls), which shows that the majority of sialic acids in the Hp-glycopeptides are linked to galactose via the α2/6 bond (data not shown).
Core Versus Outer Arm Fucose
The CID fragmentation of all the Hp glycopeptides in our dataset is consistent with outer arm fucosylation. We did not observe core fucosylation, and we have therefore performed an experiment comparing CID MS/MS of the tryptic digest of Hp, after neuraminidase treatment, with the tryptic digest of IgG1 (P01857), which was previously determined to be core-fucosylated (36). The results show that the glycopeptide derived from IgG1 contains the fragment peptide-HexNAc-Fuc at m/z 1538.5 (2+), which is absent in the Hp glycopeptide (+2 fragment expected at m/z 1072.5) (supplemental Fig. 2). In fact, we did not observe the peptide-HexNAc-Fuc fragment in any of the glycopeptide spectra of Hp. In addition, we have observed in the Hp spectra the 512.2 oxonium ion, indicative of outer arm fucosylation, of substantially higher intensity compared with the 528.2 oxonium ion (HexNAc-Hex-Hex), although the IgG1 glycopeptide contains the 512.2 fragment at low intensity. We did not expect to see the 512.2 fragment in the CID MS/MS of IgG1 because previous analyses report core fucosylation (36), and the rearrangement of fucoses between core and outer arm is not likely (37). We do not know the exact reason for the presence of this fragment in the IgG1 glycopeptide, but the experiment shows that we would be able to detect core fucosylation in the CID MS/MS of Hp if such glycoforms contributed significantly to the pool of Hp glycopeptides. The inhibition of β1,4-galactosidase with the outer arm fucoses provides further evidence for the lack of core fucosylation. As mentioned above, this enzyme is inhibited by outer arm fucosylation on neighboring N-acetylglucosamine, which is consistent with the outer arm fucosylation data in our experiments.
It is important to point out that an oxonium ion at m/z 658.3 (Hex-HexNAc-Fuc-Fuc) was clearly observed in the CID spectra of multiple glycopeptides with glycans containing two or more fucoses. One could assign this ion to an Le(y)-type structure, but a more likely explanation than Le(y) is fucose rearrangement between the outer arms. The rearrangement of fucose between outer arms during CID fragmentation was described previously, although migration between the core and outer arms was not observed (37). The outer arm rearrangement in our experiments was confirmed by CID fragmentation of detached permethylated glycans where the fragment ions corresponding to Le(y) were not observed (Fig. 6). Interestingly, CID spectra of glycopeptides bearing glycans with four and more fucoses contain an oxonium ion at m/z 804.3 (Hex-HexNAc-Fuc-Fuc-Fuc) (supplemental Fig. 1). This further supports the likelihood of fucose rearrangement between the outer arms (37). We have therefore used the galactosidase treatment, fragmentation of permethylated detached glycans, and immunostaining as a positive confirmation of Le(y) in some of the glycopeptides (Fig. 5). Because these intriguing liver disease-associated N-glycan alterations with up to six fucoses per glycopeptide and Le(y)-type structures were best observed on the tryptic glycopeptide VVLHPNYSQVDIGLIK (T3), we have focused on this glycopeptide in the quantitative analyses comparing the healthy control, cirrhosis, and HCC groups as described in the accompanying paper (57).
Fig. 6.
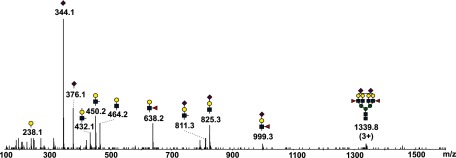
CID spectrum of triply protonated permethylated glycan with two outer arm fucoses obtained from Hp isolated from pooled plasma of HCC patients. Fragment ion at m/z 638.2 corresponds to Le(x)-type structure, ion at 999.3 to SLe(x)-type structure. Fragment ions related to core fucosylation were not observed in any of the pooled samples.
DISCUSSION
We describe in this study unusual multiply fucosylated site-specific glycoforms of Hp isolated from the plasma of patients with HCC and liver cirrhosis. We have used a combination of proteases and exoglycosidases, HILIC enrichment, LC-MS/MS of the enriched glycopeptides, CID fragmentation of detached permethylated glycans, and immunoaffinity-based detection to characterize the microheterogeneity of Hp glycoforms. The evidence for the presence of the hyper-fucosylated glycoforms and a comparison with other diseases is summarized in the following discussion.
Glycosylation of Hp in Cancer and Other Diseases
Hp is a liver-secreted glycoprotein composed of two subunits (α and β) linked by disulfide bonds (38). There are three human phenotypes of Hp (Hp1-1, Hp2-2, and Hp2-1) with an identical larger β subunit and a truncated α-chain in the Hp1 variant (39). The β subunit contains all four N-glycosylation sites whose glycoforms create additional phenotypic variants (40). Changes in Hp glycosylation have been recently studied in patients with cancer and other diseases (7). Increased fucosylation is one of the frequently observed alterations of Hp glycoforms in cancer diseases, including the progression of liver disease to HCC. Shu et al. (41) reported increased Le(x)-type singly fucosylated glycoforms of Hp in liver cirrhosis compared with healthy individuals; these changes were not sufficient to distinguish cirrhosis from hepatocellular carcinoma. Nakano et al. (7) reported that patients with pancreatic cancer have elevated expression of singly fucosylated Le(x) structures that distinguish pancreatic cancer from controls. The overall concentrations of fucosylated glycans were increased in patient samples, and the site-specific analysis revealed higher concentration of tri-antennary singly fucosylated Le(x)-type glycans at the Asn-211 (T2-1) N-glycosylation site in patient samples. The authors also detected traces of doubly fucosylated Le(y)-type structures in tetra-antennary glycans. Lin et al. (42) reported increased core fucosylation in patients with pancreatic cancer; both core and outer arm fucoses were detected among the doubly fucosylated glycans. Dimeric Lewis-A type (Le(a) on Le(a)) structures of Hp were found increased in colon cancer (43). Bi-antennary N-acetyllactosamine glycans with increased binding to galectin-1 have been detected on Hp in metastatic breast cancer (6).
Our optimized workflow combining exoglycosidase treatments with LC-MS/MS analysis improved the resolution of Hp glycoforms compared with our previously published study (29). Using the optimized workflow, we have identified disease-related glycoforms of Hp with up to six fucoses per glycosite. This is to our knowledge the first report of such glycoforms of Hp or any liver-secreted glycoprotein. Multiply fucosylated N-glycans were detected in seminal plasma (44). One of the proteins extensively fucosylated in seminal plasma is protein-C inhibitor, but this protein in plasma carries N-glycans with at most two fucoses (45, 46). Zona pellucida and sperm N-glycans with extensive fucosylation were described to modify protein interactions in the reproductive context, although a recent report described hyper-fucosylated lactosamines with up to six fucoses on N-linked glycans in pancreatic cyst fluids primarily associated with α-amylase and triacylglycerol lipase (47, 48). The pancreatic cyst N-glycans have again a different tissue source and seem structurally different from the liver-secreted Hp glycoforms described in our study. Collectively, these studies suggest that tissue- and disease-specific alterations in protein N-glycoforms could be used efficiently for the monitoring of (patho)physiological processes and that differential binding of such glycoforms could have functional impact on disease progression.
Microheterogeneity of Hp Glycoforms
We have characterized the microheterogeneity of Hp primarily by site-specific LC-MS/MS analysis of its glycopeptides. To provide a good overview of the microheterogeneity, we have first optimized Hp isolation from human plasma and the enrichment of glycopeptides by HILIC chromatography (29). This procedure is not biased regarding the selection of Hp glycoforms, and this enabled us to enrich the low abundance glycopeptides for the LC-MS/MS identification. In the first dimension (HILIC), glycopeptides were enriched primarily based on the hydrophilic interaction of the stationary phase with the glycan portion of the glycopeptides. The HILIC-enriched glycopeptides were further separated by reversed phase chromatography based on the hydrophobic character of their peptide backbone (29, 49).
We have analyzed two singly glycosylated tryptic peptides (T1 and T3), and the analysis of the doubly glycosylated tryptic peptide (T2) was further simplified by a digest with proteinase Glu-C. Our results show that glycosite occupancies of samples in all three groups of healthy controls, cirrhosis, and HCC patients for all four glycosites are in the 95–98% range (supplemental Table 4). This shows that the liver disease-related changes in glycoform distributions are not caused by changes in site occupancy, and we did not adjust for site occupancy in our analyses. We have detected 34 glycoforms of Hp in healthy controls, which is comparable with recent papers using different approaches, including LC-ESI-MS of glycopeptides, separation of pyridyl amine-labeled N-glycans on normal phase-HPLC, or site-specific characterization of Hp glycopeptides by porous layer open tubular liquid chromatography-CID/electron transfer dissociation LC-MS/MS (7, 30, 50). The increased number of glycoforms in cirrhosis (n = 56) and HCC (n = 62) is primarily due to increased fucosylation; sialylation adds to the variability of glycoforms, but we did not pursue the changes in sialylation (Table I). We have, in fact, decided to remove the sialic acid comprehensively with neuraminidase to define better the fucosylated peptide glycoforms.
In healthy controls, all four glycosites carry complex sialylated glycans with a maximum of one fucose residue per glycopeptide. The structure of glycosylated human Hp was not yet determined, but structure-based models suggest that all four glycosylation sites are surface-exposed (39). The accessibility to the endoplasmic reticulum and Golgi glycosidases is probably the reason for the lack of high mannose and hybrid glycans. In addition, we have evidence that the two glycosylation sites at the tryptic T2 peptide are occupied simultaneously with tetra-antennary multiply fucosylated glycoforms (supplemental Table 2), but the composition of the N-glycans on the doubly glycosylated tryptic T2 peptide is difficult to interpret. The double protease digest with trypsin and/or Glu-C helped us determine that both sites are glycosylated with glycoforms bearing up to four and five fucoses, respectively, in HCC. It seems that the close proximity of glycosylation sites does not interfere with the synthesis of branched fucosylated glycans.
The distribution of the glycoforms seems to be site-specific (Table I and supplemental Table 3). Semi-quantitative comparison of the ion intensities allows only estimates of glycoform contributions but shows clearly re-distribution of intensities with disease. In healthy controls, we have observed an ∼60% contribution of bi-antennary N-glycans to T1, T2-1, and T3 glycopeptides but only 20% to the T2-2 glycopeptide. Tetra-antennary N-glycans are hardly detectable on T1 and T2-1 but represent up to 10% on T2-2 and T3. In liver disease, tetra-antennary glycans increase on the T1 and T2-1 glycosites to 10%, and the T2-2 and T3 glycosites carry up to 40% of these N-glycans. This is in agreement with published data suggesting that tetra-antennary N-glycans increase in the serum of patients with HCC (51). Fucosylated T1 and T2-1 glycopeptides represent up to 50% intensity in liver disease, whereas fucosylated glycans on the T3 glycopeptide reach nearly 70%. The bi-antennary glycans never carry more than one fucose under any disease state; it appears that the branching is required for addition of multiple fucoses. The T3 site has the highest variety of glycoforms (27 in HCC) with a higher percentage of the tri- and tetra-antennary glycans and up to six fucoses per glycan.
Most of the hyperfucosylated glycoforms have low intensities (Table I). Relative quantification based on the intensity of precursor ions in the QTOF spectra provides only estimates of the distributions and especially differences between the comparison groups because of lack of normalization standards. However, the differences observed between the healthy controls and the disease groups are convincing, and the differences between cirrhosis and HCC groups in case the T3 glycopeptide further encouraged us to develop an LC-MS-multiple reaction monitoring method for improved comparison of quantitative changes between the disease groups, as described in the accompanying paper (57).
We have observed glycoforms bearing up to four fucoses in the pooled sample of patients with HCC before exoglycosidase treatments. The complexity of the sialylated multiply fucosylated glycoforms at the T3 peptide and the suboptimal behavior of sialylated glycans and glycopeptides in chromatographic and mass spectrometric analyses led to our decision to remove the sialic acid with neuraminidase. The use of neuraminidase allowed us to detect additional minor glycoforms with up to six fucoses on the T3 peptide. It also allowed us to further resolve linkage isoforms by subsequent galactosidase treatment of the glycopeptides. The branched multiply fucosylated glycoforms is one of the structural groups we wanted to elucidate because experimental evidence points to their up-regulation in cancer diseases (20, 52–54). The fucosidase and galactosidase treatments uncovered the presence of Le(y) structures and possibly polylactosamine chains (in the tetra-antennary glycoforms) primarily in the HCC sample. The exoglycosidases allowed us to limit the potential linkage isoforms consistent with the observed data. Comprehensive de-sialylation and de-fucosylation revealed the remainder of a glycoform bearing one fucose (A4G4F1) but only in the tetra-antennary glycan group. Its intensity was less than 1% of the A4G4 intensity, which suggested either incomplete digestion or the presence of a trace of core fucose. Subsequent treatment with specific galactosidases revealed virtually exclusive β1/4-galactose linkage. All galactoses were removed from the de-fucosylated glycopeptide with β1/4-galactosidase treatment, except an ∼5% fraction of the A4G1 in the HCC sample. This fraction is resistant to 1/3 galactosidase, which suggests a nonterminal galactose in poly-N-acetyllactosamine of this small fraction of the tetra-antennary glycoforms. Residual galactose was not observed on bi- or tri-antennary glycoforms which, together with MS/MS data of detached glycans, further limits the number of possible structures of the fucosylated tri-antennary glycans observed in HCC sample after sialidase and galactosidase treatment (Fig. 5). The β1/4-galactosidase treatment of fucosylated de-sialylated glycopeptides confirmed the presence of Le(y) structures and excluded the presence of Le(b) and Le(a) structures. For example, the A3G3F2 glycopeptide is resolved into glycans with composition A3G1F2 (Lewis Y-type structure), dominant in the HCC sample, and A3G2F2, dominant in the cirrhosis sample (Fig. 5). Finally, the minimal removal of sialic acid with α2/3-sialidase, observed uniformly in healthy, cirrhosis, and HCC groups, indicates dominant α2/6 linkage of sialic acids in all three samples.
Fucose Rearrangement and Core Versus Outer Arm Fucose
We have observed an oxonium ion at m/z values of 658.3 in CID spectra of glycopeptides with more than two outer arm fucoses. This ion could derive from Le(y)-type structures; however, the glycopeptide data must be interpreted with caution because of the previously described fucose rearrangement between outer arm fucoses during CID analysis of glycans (37). We have in fact evidence that some samples showing ions at m/z values of 658.3 cannot be confirmed to contain permethylated detached N-glycans of the Le(y)-type structures (Fig. 6). The permethylation stabilizes the glycans and prevents the fucose rearrangement as described by Wuhrer et al. (37). In addition, in quadruply and more fucosylated glycopeptides, we have also observed an oxonium ion at an m/z value of 804.3 corresponding to a N-acetyllactosamine with three fucoses (supplemental Fig. 1). This type of structure is not known to exist in the mammalian glycome and further suggests the likelihood of rearrangement during the CID fragmentation. This means that the CID fragmentation of fucosylated glycopeptides needs to be interpreted with caution. To further confirm the presence of the Le(y)-type structures, detected in the permethylated detached N-glycans derived from the liver disease samples, we have carried out immunoaffinity detection using Le(y) antibodies. This independent methodology further confirms the presence of this structural glycoform in the samples of patients with liver disease.
Because of the recently described elevation of core-fucosylated glycoproteins in some liver diseases, we have looked specifically for core-fucosylated Hp glycoforms in our samples (55, 56). We were not been able to confirm the presence of core fucosylation (such as the glycopeptide fragment peptide-HexNAc-Fuc or peptide-2HexNAc-Fuc), and all the data are consistent with multiply fucosylated glycoforms of Hp bearing outer arm fucose(s). However, we cannot definitively exclude the presence of minor core-fucosylated glycoforms in a mixture with the outer arm-fucosylated glycoforms. To show that the lack of core fucoses is not due to limitations of our experimental procedure, we have performed analysis of a tryptic glycopeptide of IgG1 known to be core-fucosylated (36). We have detected clearly core fucosylation on the IgG1 (peptide-glycan fragment corresponding to peptide-GlcNAc-Fuc) but not on the Hp (supplemental Fig. 2). In the case of Hp, we have rather observed the peptide-glycan fragment corresponding to peptide-GlcNAc-GlcNAc. Interestingly, we have observed the oxonium ion 512, Le(x)-type structure, in the fragmentation spectrum of the IgG1 glycopeptide. The intensity of this ion was lower in the IgG1 than in Hp but was clearly detected. This could be explained either by the presence of outer arm fucosylation on the IgG1 or by fucose rearrangement between outer arm and core. Such rearrangement is less likely but was observed by Mazurier et al. (33, 37).
In summary, we have detected multiply fucosylated glycoforms of Hp with up to six fucoses associated with the presence of Le(y)-type glycoforms of the T3 glycopeptide in the HCC disease group. Analysis of detached permethylated N-glycans and immunostaining with the anti-Le(y) antibody confirmed the presence of this glycoform. The function of these unusual minor glycoforms is at present unknown; further studies would be required to establish the association of the glycoforms with viral infection or liver disease. The hyper-fucosylation likely affects interactions with lectins and other binding partners that could impact the disease progression of viral infection to HCC.
Supplementary Material
Acknowledgments
We thank Dr. Martin Gilar for insightful discussions on HILIC and Drs. Tomas Rejtar and Marina Hincapie for introduction to the concepts of site-specific glycopeptide quantification.
Footnotes
* This work was supported, in whole or in part, by National Institutes of Health Grants U01 CA168926, RO1 CA115625, and RO1 CA135069 (to R.G.) and CCSG Grant P30 CA51008 (to Lombardi Comprehensive Cancer Center supporting the Proteomics and Metabolomics Shared Resource).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- HCC
- hepatocellular carcinoma
- HexNAc
- N-acetylhexosamine
- HILIC
- hydrophilic interaction chromatography
- Hp
- haptoglobin
- Le(a)
- Lewis A
- Le(b)
- Lewis B
- Le(x)
- Lewis X
- Le(y)
- Lewis Y
- MSMS
- tandem mass spectrometry
- PNGaseF
- peptide:N-glycosidase F
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Kornfeld R., Kornfeld S. (1985) Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 54, 631–664 [DOI] [PubMed] [Google Scholar]
- 2. Roth J., Taatjes D. J., Lucocq J. M., Weinstein J., Paulson J. C. (1985) Demonstration of an extensive trans-tubular network continuous with the Golgi apparatus stack that may function in glycosylation. Cell 43, 287–295 [DOI] [PubMed] [Google Scholar]
- 3. Lairson L. L., Henrissat B., Davies G. J., Withers S. G. (2008) Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem. 77, 521–555 [DOI] [PubMed] [Google Scholar]
- 4. Meany D. L., Chan D. W. (2011) Aberrant glycosylation associated with enzymes as cancer biomarkers. Clin. Proteomics 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nakagawa T., Uozumi N., Nakano M., Mizuno-Horikawa Y., Okuyama N., Taguchi T., Gu J., Kondo A., Taniguchi N., Miyoshi E. (2006) Fucosylation of N-glycans regulates the secretion of hepatic glycoproteins into bile ducts. J. Biol. Chem. 281, 29797–29806 [DOI] [PubMed] [Google Scholar]
- 6. Carlsson M. C., Cederfur C., Schaar V., Balog C. I., Lepur A., Touret F., Salomonsson E., Deelder A. M., Fernö M., Olsson H., Wuhrer M., Leffler H. (2011) Galectin-1-binding glycoforms of haptoglobin with altered intracellular trafficking and increase in metastatic breast cancer patients. PLoS One 6, e26560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakano M., Nakagawa T., Ito T., Kitada T., Hijioka T., Kasahara A., Tajiri M., Wada Y., Taniguchi N., Miyoshi E. (2008) Site-specific analysis of N-glycans on haptoglobin in sera of patients with pancreatic cancer: a novel approach for the development of tumor markers. Int. J Cancer 122, 2301–2309 [DOI] [PubMed] [Google Scholar]
- 8. Wang M., Long R. E., Comunale M. A., Junaidi O., Marrero J., Di Bisceglie A. M., Block T. M., Mehta A. S. (2009) Novel fucosylated biomarkers for the early detection of hepatocellular carcinoma. Cancer Epidemiol. Biomarkers Prev. 18, 1914–1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goldman R., Ressom H. W., Varghese R. S., Goldman L., Bascug G., Loffredo C. A., Abdel-Hamid M., Gouda I., Ezzat S., Kyselova Z., Mechref Y., Novotny M. V. (2009) Detection of hepatocellular carcinoma using glycomic analysis. Clin. Cancer Res. 15, 1808–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bekesova S., Kosti O., Chandler K. B., Wu J., Madej H. L., Brown K. C., Simonyan V., Goldman R. (2012) N-Glycans in liver-secreted and immunoglobulin-derived protein fractions. J. Proteomics 75, 2216–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vanderschaeghe D., Laroy W., Sablon E., Halfon P., Van Hecke A., Delanghe J., Callewaert N. (2009) GlycoFibroTest is a highly performant liver fibrosis biomarker derived from DNA sequencer-based serum protein glycomics. Mol. Cell. Proteomics 8, 986–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mehta A. S., Long R. E., Comunale M. A., Wang M., Rodemich L., Krakover J., Philip R., Marrero J. A., Dwek R. A., Block T. M. (2008) Increased levels of galactose-deficient anti-Gal immunoglobulin G in the sera of hepatitis C virus-infected individuals with fibrosis and cirrhosis. J. Virol. 82, 1259–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blomme B., Van Steenkiste C., Callewaert N., Van Vlierberghe H. (2009) Alteration of protein glycosylation in liver diseases. J. Hepatol. 50, 592–603 [DOI] [PubMed] [Google Scholar]
- 14. Klein A., Michalski J. C., Morelle W. (2010) Modifications of human total serum N-glycome during liver fibrosis-cirrhosis, is it all about immunoglobulins? Proteomics Clin. Appl. 4, 372–378 [DOI] [PubMed] [Google Scholar]
- 15. Bones J., Mittermayr S., O'Donoghue N., Guttman A., Rudd P. M. (2010) Ultra performance liquid chromatographic profiling of serum N-glycans for fast and efficient identification of cancer associated alterations in glycosylation. Anal. Chem. 82, 10208–10215 [DOI] [PubMed] [Google Scholar]
- 16. Vanderschaeghe D., Debruyne E., Van Vlierberghe H., Callewaert N., Delanghe J. (2009) Analysis of γ-globulin mobility on routine clinical CE equipment: exploring its molecular basis and potential clinical utility. Electrophoresis 30, 2617–2623 [DOI] [PubMed] [Google Scholar]
- 17. Mori S., Aoyagi Y., Yanagi M., Suzuki Y., Asakura H. (1998) Serum N-acetylglucosaminyltransferase III activities in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 13, 610–619 [DOI] [PubMed] [Google Scholar]
- 18. Tsutsumi M., Wang J. S., Takada A. (1994) Microheterogeneity of serum glycoproteins in alcoholics: is desialo-transferrin the marker of chronic alcohol drinking or alcoholic liver injury? Alcohol. Clin. Exp. Res. 18, 392–397 [DOI] [PubMed] [Google Scholar]
- 19. Sasaki K., Kurata-Miura K., Ujita M., Angata K., Nakagawa S., Sekine S., Nishi T., Fukuda M. (1997) Expression cloning of cDNA encoding a human β-1,3-N-acetylglucosaminyltransferase that is essential for poly-N-acetyllactosamine synthesis. Proc. Natl. Acad. Sci. U.S.A. 94, 14294–14299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Campion B., Léger D., Wieruszeski J. M., Montreuil J., Spik G. (1989) Presence of fucosylated triantennary, tetraantennary, and pentaantennary glycans in transferrin synthesized by the human hepatocarcinoma cell line Hep G2. Eur. J. Biochem. 184, 405–413 [DOI] [PubMed] [Google Scholar]
- 21. Blomme B., Van Steenkiste C., Grassi P., Haslam S. M., Dell A., Callewaert N., Van Vlierberghe H. (2011) Alterations of serum protein N-glycosylation in two mouse models of chronic liver disease are hepatocyte and not B cell driven. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G833–G842 [DOI] [PubMed] [Google Scholar]
- 22. Saldova R., Royle L., Radcliffe C. M., Abd Hamid U. M., Evans R., Arnold J. N., Banks R. E., Hutson R., Harvey D. J., Antrobus R., Petrescu S. M., Dwek R. A., Rudd P. M. (2007) Ovarian cancer is associated with changes in glycosylation in both acute-phase proteins and IgG. Glycobiology 17, 1344–1356 [DOI] [PubMed] [Google Scholar]
- 23. Du M. Q., Hutchinson W. L., Johnson P. J., Williams R. (1991) Differential α-fetoprotein lectin binding in hepatocellular carcinoma. Diagnostic utility at low serum levels. Cancer 67, 476–480 [DOI] [PubMed] [Google Scholar]
- 24. Taketa K., Sekiya C., Namiki M., Akamatsu K., Ohta Y., Endo Y., Kosaka K. (1990) Lectin-reactive profiles of α-fetoprotein characterizing hepatocellular carcinoma and related conditions. Gastroenterology 99, 508–518 [DOI] [PubMed] [Google Scholar]
- 25. Sterling R. K., Jeffers L., Gordon F., Sherman M., Venook A. P., Reddy K. R., Satomura S., Schwartz M. E. (2007) Clinical utility of AFP-L3% measurement in North American patients with HCV-related cirrhosis. Am. J. Gastroenterol. 102, 2196–2205 [DOI] [PubMed] [Google Scholar]
- 26. Dobryszycka W. (1997) Biological functions of haptoglobin–new pieces to an old puzzle. Eur. J. Clin. Chem. Clin. Biochem. 35, 647–654 [PubMed] [Google Scholar]
- 27. Kristiansen M., Graversen J. H., Jacobsen C., Sonne O., Hoffman H. J., Law S. K., Moestrup S. K. (2001) Identification of the haemoglobin scavenger receptor. Nature 409, 198–201 [DOI] [PubMed] [Google Scholar]
- 28. Andersen C. B., Torvund-Jensen M., Nielsen M. J., de Oliveira C. L., Hersleth H. P., Andersen N. H., Pedersen J. S., Andersen G. R., Moestrup S. K. (2012) Structure of the haptoglobin-haemoglobin complex. Nature 489, 456–459 [DOI] [PubMed] [Google Scholar]
- 29. Pompach P., Chandler K. B., Lan R., Edwards N., Goldman R. (2012) Semi-automated identification of N-glycopeptides by hydrophilic interaction chromatography, nano-reverse-phase LC-MS/MS, and Glycan Database Search. J. Proteome Res. 11, 1728–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang D., Hincapie M., Rejtar T., Karger B. L. (2011) Ultrasensitive characterization of site-specific glycosylation of affinity-purified haptoglobin from lung cancer patient plasma using 10 μm i.d. porous layer open tubular liquid chromatography-linear ion trap collision-induced dissociation/electron transfer dissociation mass spectrometry. Anal. Chem. 83, 2029–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Z., Cao J., He Y., Qiao L., Xu C., Lu H., Yang P. (2010) Tandem O-18 stable isotope labeling for quantification of N-glycoproteome. J. Proteome Res. 9, 227–236 [DOI] [PubMed] [Google Scholar]
- 32. Alley W. R., Jr., Madera M., Mechref Y., Novotny M. V. (2010) Chip-based reversed-phase liquid chromatography-mass spectrometry of permethylated N-linked glycans: a potential methodology for cancer-biomarker discovery. Anal. Chem. 82, 5095–5106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mazurier J., Dauchez M., Vergoten G., Montreuil J., Spik G. (1991) Molecular modeling of a disialylated monofucosylated biantennary glycan of the N-acetyllactosamine type. Glycoconj. J. 8, 390–399 [DOI] [PubMed] [Google Scholar]
- 34. Royle L., Campbell M. P., Radcliffe C. M., White D. M., Harvey D. J., Abrahams J. L., Kim Y. G., Henry G. W., Shadick N. A., Weinblatt M. E., Lee D. M., Rudd P. M., Dwek R. A. (2008) HPLC-based analysis of serum N-glycans on a 96-well plate platform with dedicated database software. Anal. Biochem. 376, 1–12 [DOI] [PubMed] [Google Scholar]
- 35. Lucka L., Fernando M., Grunow D., Kannicht C., Horst A. K., Nollau P., Wagener C. (2005) Identification of Lewis x structures of the cell adhesion molecule CEACAM1 from human granulocytes. Glycobiology 15, 87–100 [DOI] [PubMed] [Google Scholar]
- 36. Selman M. H., McDonnell L. A., Palmblad M., Ruhaak L. R., Deelder A. M., Wuhrer M. (2010) Immunoglobulin G glycopeptide profiling by matrix-assisted laser desorption ionization Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 82, 1073–1081 [DOI] [PubMed] [Google Scholar]
- 37. Wuhrer M., Koeleman C. A., Hokke C. H., Deelder A. M. (2006) Mass spectrometry of proton adducts of fucosylated N-glycans: fucose transfer between antennae gives rise to misleading fragments. Rapid Commun. Mass Spectrom. 20, 1747–1754 [DOI] [PubMed] [Google Scholar]
- 38. Gordon S., Cleve H., Bearn A. G. (1968) An improved method of preparing haptoglobin polypeptide chains using guanidine hydrochloride. Proc. Soc. Exp. Biol. Med. 127, 52–59 [DOI] [PubMed] [Google Scholar]
- 39. Boonyapranai K., Tsai H. Y., Chen M. C., Sriyam S., Sinchaikul S., Phutrakul S., Chen S. T. (2011) Glycoproteomic analysis and molecular modeling of haptoglobin multimers. Electrophoresis 32, 1422–1432 [DOI] [PubMed] [Google Scholar]
- 40. Polticelli F., Bocedi A., Minervini G., Ascenzi P. (2008) Human haptoglobin structure and function–a molecular modelling study. FEBS J. 275, 5648–5656 [DOI] [PubMed] [Google Scholar]
- 41. Shu H., Zhang S., Kang X., Li S., Qin X., Sun C., Lu H., Liu Y. (2011) Protein expression and fucosylated glycans of the serum haptoglobin-β subunit in hepatitis B virus-based liver diseases. Acta Biochim. Biophys. Sin. 43, 528–534 [DOI] [PubMed] [Google Scholar]
- 42. Lin Z., Simeone D. M., Anderson M. A., Brand R. E., Xie X., Shedden K. A., Ruffin M. T., Lubman D. M. (2011) Mass spectrometric assay for analysis of haptoglobin fucosylation in pancreatic cancer. J. Proteome Res. 10, 2602–2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Park S. Y., Yoon S. J., Hakomori S. I., Kim J. M., Kim J. Y., Bernert B., Ullman T., Itzkowitz S. H., Kim J. H. (2010) Dimeric Le(a) (Le(a)-on-Le(a)) status of β-haptoglobin in sera of colon cancer, chronic inflammatory disease, and normal subjects. Int. J. Oncol. 36, 1291–1297 [DOI] [PubMed] [Google Scholar]
- 44. Clark G. F., Grassi P., Pang P. C., Panico M., Lafrenz D., Drobnis E. Z., Baldwin M. R., Morris H. R., Haslam S. M., Schedin-Weiss S., Sun W., Dell A. (2012) Tumor biomarker glycoproteins in the seminal plasma of healthy human males are endogenous ligands for DC-SIGN. Mol. Cell. Proteomics 11, Available at: 10.1074/mcp.M111.008730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sun W., Grassi P., Engström A., Sooriyaarachchi S., Ubhayasekera W., Hreinsson J., Wånggren K., Clark G. F., Dell A., Schedin-Weiss S. (2011) N-Glycans of human protein C inhibitor: tissue-specific expression and function. PLoS One 6, e29011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sun W., Parry S., Panico M., Morris H. R., Kjellberg M., Engström A., Dell A., Schedin-Weiss S. (2008) N-Glycans and the N terminus of protein C inhibitor affect the cofactor-enhanced rates of thrombin inhibition. J. Biol. Chem. 283, 18601–18611 [DOI] [PubMed] [Google Scholar]
- 47. Pang P. C., Chiu P. C., Lee C. L., Chang L. Y., Panico M., Morris H. R., Haslam S. M., Khoo K. H., Clark G. F., Yeung W. S., Dell A. (2011) Human sperm binding is mediated by the sialyl-Lewis(x) oligosaccharide on the zona pellucida. Science 333, 1761–1764 [DOI] [PubMed] [Google Scholar]
- 48. Mann B. F., Goetz J. A., House M. G., Schmidt C. M., Novotny M. V. (2012) Glycomic and proteomic profiling of pancreatic cyst fluids identifies hyperfucosylated lactosamines on the N-linked glycans of overexpressed glycoproteins. Mol. Cell. Proteomics Available at: 10.1074/mcp.M111.015792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gilar M., Jaworski A. (2011) Retention behavior of peptides in hydrophilic-interaction chromatography. J. Chromatogr. A 1218, 8890–8896 [DOI] [PubMed] [Google Scholar]
- 50. Nilsson J., Rüetschi U., Halim A., Hesse C., Carlsohn E., Brinkmalm G., Larson G. (2009) Enrichment of glycopeptides for glycan structure and attachment site identification. Nat. Methods 6, 809–811 [DOI] [PubMed] [Google Scholar]
- 51. Mehta A. S., Norton P., Liang H., Comunale M. A., Wang M., Rodemich-Betesh L., Koszycki A., Noda K., Miyoshi E., Block T. (2012) Increased levels of tetra-antennary N-linked glycan but not core fucosylation are associated with hepatocellular carcinoma tissue. Cancer Epidemiol. Biomarkers Prev. 21, 925–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Miyoshi E., Nakano M. (2008) Fucosylated haptoglobin is a novel marker for pancreatic cancer: detailed analyses of oligosaccharide structures. Proteomics 8, 3257–3262 [DOI] [PubMed] [Google Scholar]
- 53. Fujimura T., Shinohara Y., Tissot B., Pang P. C., Kurogochi M., Saito S., Arai Y., Sadilek M., Murayama K., Dell A., Nishimura S., Hakomori S. I. (2008) Glycosylation status of haptoglobin in sera of patients with prostate cancer versus benign prostate disease or normal subjects. Int. J. Cancer 122, 39–49 [DOI] [PubMed] [Google Scholar]
- 54. Okuyama N., Ide Y., Nakano M., Nakagawa T., Yamanaka K., Moriwaki K., Murata K., Ohigashi H., Yokoyama S., Eguchi H., Ishikawa O., Ito T., Kato M., Kasahara A., Kawano S., Gu J., Taniguchi N., Miyoshi E. (2006) Fucosylated haptoglobin is a novel marker for pancreatic cancer: a detailed analysis of the oligosaccharide structure and a possible mechanism for fucosylation. Int. J Cancer 118, 2803–2808 [DOI] [PubMed] [Google Scholar]
- 55. Taketa K., Endo Y., Sekiya C., Tanikawa K., Koji T., Taga H., Satomura S., Matsuura S., Kawai T., Hirai H. (1993) A collaborative study for the evaluation of lectin-reactive α-fetoproteins in early detection of hepatocellular carcinoma. Cancer Res. 53, 5419–5423 [PubMed] [Google Scholar]
- 56. Comunale M. A., Rodemich-Betesh L., Hafner J., Wang M., Norton P., Di Bisceglie A. M., Block T., Mehta A. (2010) Linkage-specific fucosylation of α-1-antitrypsin in liver cirrhosis and cancer patients: implications for a biomarker of hepatocellular carcinoma. PLoS One 5, e12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sanda M., Pompach P., Brnakova Z., Wu J., Makambi K., Goldman R., (2013) Quantitative LC-MS-MRM analysis of site-specific glycoforms of haptoglobin in liver disease. Mol Cell Proteomics. 12, 1294–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.