

Abstract
Deciphering the network of signaling pathways in cancer via protein-protein interactions (PPIs) at the cellular level is a promising approach but remains incomplete. We used an in situ proximity ligation assay to identify and quantify 67 endogenous PPIs among 21 interlinked pathways in two hepatocellular carcinoma (HCC) cells, Huh7 (minimally migratory cells) and Mahlavu (highly migratory cells). We then applied a differential network biology analysis and determined that the novel interaction, CRKL-FLT1, has a high centrality ranking, and the expression of this interaction is strongly correlated with the migratory ability of HCC and other cancer cell lines. Knockdown of CRKL and FLT1 in HCC cells leads to a decrease in cell migration via ERK signaling and the epithelial-mesenchymal transition process. Our immunohistochemical analysis shows high expression levels of the CRKL and CRKL-FLT1 pair that strongly correlate with reduced disease-free and overall survival in HCC patient samples, and a multivariate analysis further established CRKL and the CRKL-FLT1 as novel prognosis markers. This study demonstrated that functional exploration of a disease network with interlinked pathways via PPIs can be used to discover novel biomarkers.
Hepatocellular carcinoma (HCC)1 is the third leading cause of cancer-related deaths worldwide and is a global health concern (1). The malignant phenotype of HCC may result in part from the disruption and dysregulation of several biological pathways (2). Moreover, metastasis is one of the main causes of mortality from solid tumors, and metastasis is a poor prognostic factor for HCC. Understanding protein-protein interactions (PPIs) may uncover the generic organization of functional networks in cancer cells when both the spatial and temporal aspects of the interactions are considered (3). Recently, several studies applied a protein network-based approach and a differential network-based approach to identify markers to predict patient prognosis (4, 5). These computational approaches demonstrated great potential and could be further enhanced if more thorough PPI and pathway information is available, especially at the cellular level, and analyzed with a more sophisticated method.
Cancer can be considered as perturbations of highly interlinked cellular networks. Our hypothesis is that uncovering new PPI links within or between, referred to as interlinked PPIs (cross-talk PPIs), different signaling pathways could recapitulate the relationship between the genotype and phenotype in HCC. Multiple signaling cascades are interlinked in cancer cells via a variety of cross-talk connections with other pathways leading to several of the hallmarks of cancer (e.g. proliferative signaling, angiogenesis, invasion, and survival) (6, 7). Therefore, targeting these interlinked pathways could provide an opportunity for therapeutic application (6–10). Here, we present a systems approach that computationally infers the interlinked pathways from numerous PPIs in HCC up-regulated genes and empirically detects endogenous PPIs using an in situ proximity ligation assay (PLA), which allows quantitative and localized detection of endogenous PPIs in cells (11). Empirically, we validated 67 endogenous PPIs within or between signaling pathways in HCC. To the best of our knowledge, applying in situ PLA to this scale in cancer cells is unprecedented.
We demonstrate its effectiveness with the identification of a prioritized interaction, CRKL-FLT1, which links the c-Met, IGF1, PDGFR-α, and VEGFR1/VEGFR2 pathways together. CRKL-FLT1 was identified as a hub in the PPI network in HCC and is crucial for migration in HCC cells. CRKL, an adaptor protein important in the regulation of several GTPases, is involved in many intracellular signaling cascades (12, 13) and mediates cell morphology and movement (14). Recently, a genomic and functional analysis identified CRKL as an oncogene that is amplified in lung cancer (15). FLT1, a VEGFR family member, mediates the migration of endothelial cells and monocytes/macrophages (16, 17). Our analysis result shows the expression of CRKL-FLT1 is strongly correlated with the migratory ability of cancer cell lines. Moreover, we demonstrate that CRKL and FLT1 was involved in the ERK pathway and regulated the epithelial-mesenchymal transition (EMT) process in migration of HCC. Furthermore, we demonstrate that, via an immunohistochemistry analysis, high expression levels of either CRKL alone or CRKL-FLT1 combined strongly correlate with reduced overall and disease-free survival in 192 HCC tissue samples. In summary, this study provides broad insight into potential therapeutic and prognosis biomarkers by building an interlinked pathway map via PPIs in HCC.
EXPERIMENTAL PROCEDURES
Identification of HCC-related Pathways
In this over-representation analysis, N represents the total number of genes in the background population; n represents the number of HCC-related genes; and m denotes the number of genes within the given pathways. The number of genes that overlapped with both HCC-related genes and this pathway is denoted as k. A p value was calculated by the cumulative hyper-geometric distribution to evaluate statistical significance of pathways as shown in Equation 1,
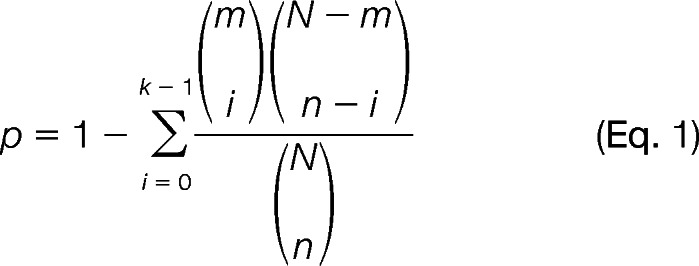 |
This is a one-sided test for over-representation. In this study, a pathway is considered significantly enriched for selection as a node if its q is less than 0.05 with an adjustment of q using a false discovery rate. Our HCC gene signatures n is derived from the Encyclopedia of Hepatocellular Carcinoma Genes Online 2 (18, 19), an integrative platform to systematically collect and identify 4,020 HCC-related signatures. The core of the Encyclopedia of Hepatocellular Carcinoma Genes Online 2 is a collection of 14 HCC-related gene sets from a wide range of sources, including text-mining results from PubMed, reports of high throughput studies, computational predictions, and validations. All of the pathways with their m gene members are derived from an integrated pathway database, ConsensusPathDB (20), which integrated 12 pathway databases with 2,100 pathways. We prioritized 60 pathways that were statistically significant by this test for rewiring pathways via PPIs; 12 out of 60 pathways were reported as important therapeutic pathways (10).
Interlinking of HCC-related Pathways via PPIs
Here, we present the formal definition of the interlinked PPIs among HCC-related pathways. The interlinked PPIs that occur in HCC were identified by pathway information and PPI databases. We represent the interlinked PPIs among M HCC-related pathways as an undirected simple graph G = (VG, EG), where VG = (v1, v2, · · ·, vM) a finite set of M vertices representing the M pathways, and EG ⊂ VG × VG is the set of edges representing the pairs of interlinked pathways. Each such graph can equivalently be represented by a symmetric M × M adjacency matrix AG, if pathway Vi has at least interlinked PPIs with pathway Vj and 0 and otherwise by the following Equations 2 and 3. To detect interlinked PPIs among pathway profiles, we first calculated the score IVi,Vj(X,Y) of each PPI from PPI databases.
 |
Where (X,Y) denotes the pair of PPI, and Vi,Vj denotes the pair of pathway. IE(K) is the function to extract the expression for each protein from the profile of HCC-related genes. Finally, we generated a binary outcome y by calculating Interlink(X,Y,Vi,Vj) in which interlinked PPIs are matched to overexpression/overexpression pattern.
We applied the overexpression patterns stored in Encyclopedia of Hepatocellular Carcinoma Genes Online 2 to estimate IE(K) in HCC and identified 375 interlinked PPIs from POINeT (59,639 binary PPIs (21)) and PIPS (37,606 binary PPIs (22)), respectively.
Detection of Protein-Protein Interaction by in Situ Proximity Ligation Assay
Recently, the in situ PLA was developed to detect and visualize endogenous PPIs and post-translational modifications of proteins with a high sensitivity and specificity (11, 23). To detect PPIs, the dual targets of primary antibody pairs were added. If an antibody pair is in close proximity, secondary antibodies with oligonucleotides will be close enough to serve as templates for the ligation of two additional linear oligonucleotides into a DNA circle. The DNA circle can be amplified with the oligonucleotide in one of the secondary antibodies using rolling circle amplification. Rolling circle amplification can then be hybridized with fluorescent-labeled oligonucleotides to reveal dot-signal representing both their subcellular locations and the frequency of the PPI occurrences (11, 23). This technique opens new opportunities to accurately quantify PPIs in cells.
The cells were washed with PBS and fixed in 3.7% paraformaldehyde for 30 min on ice. After washing with PBS, the cells were permeabilized with 0.2% Triton X-100 in PBS for 3 min at room temperature. To reduce the nonspecific signal, the cells were incubated with a blocking solution (OLINK Bioscience) for 30 min at 37°C. Then, primary 1× antibody Diluent (OLINK Bioscience) with two primary antibodies (1:50 dilution for mouse monoclonal antibody and 1:1200 for rabbit polyclonal antibody) were added to the cells and incubated overnight at 4°C. The negative control was performed by only one primary antibody (rabbit polyclonal antibody) into cells for incubation at 4°C overnight. All of the procedures were performed according to the manufacturer's instructions. The images of the cells were acquired using an Olympus BX61 microscope (Olympus, Uppsala University, Sweden). Images for each slide with an in situ PLA sample were acquired at five different fields with two z axis images. Then the images were analyzed with Blob Finder Version 3.2, which automatically counts the number of dots per cell. The detection of a dot signal with signal ratio was defined as SR = (Spos/Cpos) (Sneg/Cpos)/(Sneg/Cneg). The PPI was determined to be a positive PPI only if SR >20 (Spos/Cpos) >10; Spos was the signal, and Cpos was the cell number for dual recognition with one rabbit polyclonal antibody and one mouse monoclonal antibody; Sneg was the signal and Cneg was the cell number for the negative control, in which only one rabbit polyclonal antibody was added. All the experiments were repeated at least three times, except fro Fig. 1C (the screening of PPIs). The antibody pairs for the in situ PLA for detection of the PPIs are listed in supplemental Table 1.
Fig. 1.

Schematic illustration of the major steps in our combined computational and experimental approaches to identify interlinked PPIs in signaling pathways. A, Step 1, identification of HCC-related pathways. We identified 60 HCC-related pathways (443 proteins) by applying a hypergeometric test with the HCC gene signatures set and pathway databases. Step 2, interlinking of PPIs in signaling pathways. The proteins belonging to the HCC-related pathways (gray nodes on left side) indicated whether any PPIs (red dotted line on left side) with overexpressed patterns could link each pathway and enable potential interlinking. Eventually, we identified 194 PPIs with the potential for interlinking. This topological property suggests that interlinked PPIs can be categorized into two classes as follows: r1) between/within pathway PPIs, and r2) between pathway PPIs. Step 3, PPIs that had available antibody pairs were identified and further tested in HCC cell lines with in situ PLA technology. We validated 67 PPIs in this step. B, MAPK14-AKT1 (top) interaction was highly expressed in Huh7 cells and in Mahlavu cells; the CRKL-SOS1 (bottom) interaction was higher in Mahlavu cells than Huh7 cells. C, histogram of PLA signals for 67 validated PPIs showed a differential expression pattern between Huh7 and Mahlavu cells. D, huge PPI network from up-regulated genes (1062 nodes and 2777 edges). E, evaluation of the expression of PPIs from the in situ PLA assay in HCC. The color of each link in the sub-network (49 nodes and 67 edges) indicates whether its interaction can be observed through a manual curation of protein interactions from the literature (blue) or not (red). The interactions with red color are novel interactions. The node sizes correlate with the number of interactions. The combined computational and experimental approaches can construct effectively a concise empirical HCC PPI network (E) from a huge PPI network of up-regulated genes (D).
Clustering and Visualization
The information structure embedded in our database for positive PPIs, pathways, and cell lines is highly dimensional and very complex in nature. We adopted a clustering and matrix visualization environment in generalized association plots (24, 25) to explore and summarize this information structured at various levels as follows. First, we elucidated the tripartite interactions of positive PPIs, pathways, and cell lines. In all, 67 of the 194 PPIs tested via the in situ PLA method were considered positive PPIs in one or both of the cell lines (Mahlavu or Huh7). Thus, the positive PPIs can be grouped into three types as follows: 1) Mahlavu only; 2) Huh7 only, and 3) both positive. Matrix M (Fig. 2A) was prepared to store information regarding the 67 PPIs (rows), 21 pathways (columns), and cell lines as follows: for Mij = 0, the ith PPI does not engage the jth pathway (displayed in white in Fig. 2A); 1) the ith PPI engages the jth pathway only in Huh7 (displayed in magenta in Fig. 2A); 2) the ith PPI engages the jth pathway only in Mahlavu (displayed in cyan in Fig. 2A); 3) the ith PPI engages the jth pathway in both cell lines (displayed in purple in Fig. 2A), where i = 1,···,67 and j = 1,···,21.
Fig. 2.

Tripartite association of PPIs, pathways and cell lines, and analysis of the differential interaction hubs in the PPI networks. Sixty seven positive PPIs within 21 pathways are displayed as a matrix map using generalized association plots (24, 25) with a measurement of modified SMC and hierarchical clustering. A, PPIs that are specific in Huh7, Mahlavu, both cell lines, or none are color coded as cyan, magenta, purple, or white, respectively; the 67 positive PPIs can also be partitioned into eight clusters (M1, M2, B1, B2, H1, H2, A1, and A2). The PPIs in the M1 and M2 clusters were observed only in Mahlavu cells, whereas the PPIs in the H1 and H2 clusters were observed only in Huh7 cells. The PPIs in the B1 and B2 clusters were observed in both cell lines. The PPIs in the A1 and A2 clusters were observed in either one cell line or both cell lines, but they did not belong to larger clusters because of the similarity of the corresponding pathways. B, similarity of the pathways is to measure the similarity of between-PPI using a modified SMC, which ranges from 0 (dissimilarity/no association, dark blue) to 1 (high similarity/perfect association, dark red). Abbreviations for the pathway names were used and different sets of colors were generated to represent branching structure of different hierarchical clustering trees. The shortened pathway names make the links between the matrix and clustering tree stand out much better, and the grouping (clustering) patterns are now visually more readily apparent. C, similarity of the PPIs is to measure the similarity of between-pathway using modified SMC and reveals the hierarchical linkage of the PPIs. D, inferred interlinked pathway map via interlinked PPIs. The nodes in this network represent pathways, and the edges in the network indicate whether at least one interlinked PPI can be observed through in situ PLA. The line colors reflect the number of the interlinked PPIs that appear within the pathway pairs. E, scatterplot shows the number of interactions in highly migratory cells (Mahlavu cell) and the number of interactors in minimally migratory cells (Huh7 cell) for each protein in this study. The proteins that only have one interactor in either Huh7 cells or Mahlavu cells are not labeled in this figure. F, Venn diagram showing the overlap of protein interactions and differential protein interactions in CRKL (top) and FLT1 (bottom). Three of the 11 CRKL interaction partners occurred in both minimally migratory cells and highly migratory cells, but the remaining eight partners only occurred in highly migratory cells. FLT1 has seven interaction partners, and the only partner that occurs in highly migratory cells is CRKL.
Simple matching coefficient (SMC) is a value between 0 and 1 representing how completely similar two objects are. The value is calculated by counting the number of exact matches of the same status (presence or attributes) between two objects. SMC = 1 means two objects are identical in every attribute, and SMC = 0 means two objects have nothing in common. In this study, we adopted a modified version of SMC, SMC0, which excludes SMC counts for attributes with both objects belonging to the nonexistent state (Mi,j = 0, below) in the calculation for both denominator and numerator. SMC0 is more robust than SMC for sparse data (data with many nonexistent states), as in matrix M (Fig. 2A). SMC0 can also be termed the nominal version of the Jaccard coefficient.
To have a more structural and visual representation, we used SMC0 for representing the between rows (PPIs) and between columns (pathways) association structure and to identify PPI clusters with pathway groups. First, we sorted the rows (PPIs) and columns (pathways) as follows. Two proximity matrices C (Fig. 2B) and R (Fig. 2C) were calculated using the modified simple matching coefficient representing the between rows (PPIs) and between columns (pathways) association structure as shown in Equation 4,
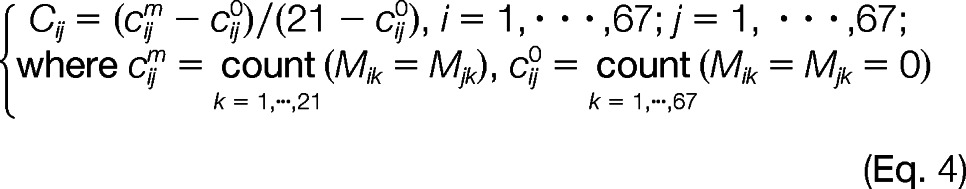 |
and Equation 5,
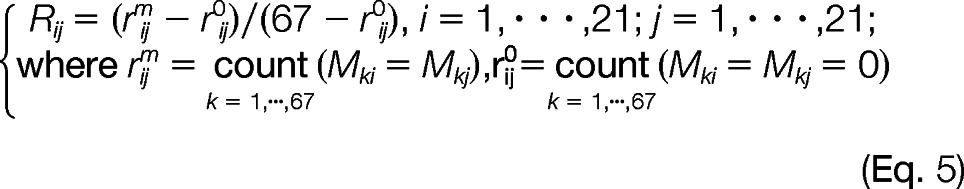 |
Both Cij (1 ≤ i, j ltequ] 67) and Rij (1 ≤ i, j ltequ] 21) range between zero and one with Cij = 1 (Rij = 1) (color-coded in dark red in Fig. 2B) indicating two PPIs i and j share identical pathway profiles (two pathways i and j share identical PPI patterns), whereas Cij = 0 (Rij = 0) (color-coded in blue in Fig. 2B) representing two PPIs i and j share no common pathways (two pathways i and j share no common PPIs). Two generalized association plot-divisive hierarchical clustering trees (24, 25) TR and TC were built on R and C and to sort similar PPIs (PPIs with higher Cij in Fig. 2B) and related pathways (pathways with higher Rij in Fig. 2C) into clusters of PPIs and groups of pathways using relative positions for terminal nodes of TR and TC. Rows (PPIs) and columns (pathways) of M (Fig. 1A) were also rearranged using corresponding orders in R (Fig. 2C) and C (Fig. 2B).
Estimation of the Degree Centrality for Differential Hub
The degree of a vertex in a network is the number of edges attached to it. A graph can be represented by an adjacency matrix A, where Aij = 1 if there is an edge between nodes i and j and 0 otherwise. The degrees centrality (DC) of a node i are defined as Equation 6,
 |
Migration Assay
For Mahlavu stable clones (vehicle, shCRKL, and shFLT1), 1 × 104 Mahlavu cells were suspended in 200 μl of DMEM without serum and were seeded into the upper chamber, whereas 750 μl of DMEM containing 10% FBS was added to the outer side of the chamber. For measuring migratory ability in different HCC cell lines (HepG2, PLC5, Huh7, SK-Hep1, and Mahlavu), 1 × 105 cells were seeded into the upper chamber with 200 μl of DMEM without serum. After being cultured in a 37°C, 5% CO2, 95% air environment and allowed to adhere for 12–16 h and then incubated, cells on the upper surface of the membrane were removed by a cotton tip applicator, and migratory cells on the lower membrane surface were fixed by methanol and stained with Giemsa (Sigma-Aldrich). Cell migration values were determined by counting from three independent membranes and then normalized using vehicle cells to give a relative ratio.
Viral Infection
Short hairpin RNAs (shRNA) targeting CRKL and FLT1 were cloned into the pGIPZ lentiviral vector (Open Biosystems). Targeting sequences were as follows: shCRKL-1, ATGTAACTAAAGGAATCTGAAA; shCRKL-2, AGGTGAGATCCTAGTGATAATA; shFLT1–1, CGGCTACTCGTTAATTATCAAA; and shFLT1–2: ACACAGTTAACAAGTTCTTATA. Mahlavu cells were infected with lentiviruses expressing either a nonsilencing hairpin control (vehicle) or the CRKL- or FLT1-targeting hairpins. The infected populations were selected in puromycin.
MTT Assay for Cell Growth
Mahlavu cells were seeded in 96-well plates in DMEM containing 10% FBS for 24, 48 and 72 h followed by MTT assay to quantify the cell growth. Data were normalized against OD570 value on day 1 of cells.
Immunoprecipitation and Immunoblotting
Transient transfection of HEK293T was performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After 48 h of transfection, the HEK293T cell pellets collected were lysed in the Nonidet P-40 lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, 1 mm EGTA, 1 mm phenylmethylsulfonyl fluoride, and 10 μg/ml each of leupeptin, aprotinin, and chymostatin). Lysates were centrifuged and incubated with protein A/G beads (Santa Cruz Biotechnology) at 4°C for 1 h. The precleared lysates were then incubated with anti-FLAG M-agarose affinity gel (Sigma-Aldrich) at 4°C overnight. Beads were recovered by centrifugation, washed three times with the TBST buffer, resuspended in SDS sample buffer, heated at 95°C, analyzed by SDS-PAGE, and immunoblotted with appropriate antibodies. The antibodies used in this study are listed in supplemental Table 2.
Patient Clinicopathological Data
For this study, which was approved by our institutional review board (201010021IC), we obtained archived formalin-fixed, paraffin-embedded material from surgically resected HCC specimens containing tumor and adjacent liver tissues from the Taipei Veterans General Hospital. Tissue specimens collected between 1990 and 2006 from 192 liver cancer tumors were used to construct tissue microarrays. In addition, clinical data and pathological data were obtained through a detailed retrospective review of the medical records of the 192 HCC patients. The median age of these patients was 63 years (range, 21–83 years; mean, 60.9 years). Follow-up was possible for all cases, and the follow-up period ranged from 0.7 to 172 months (median, 51 months; mean, 58.2 months). During the follow-up period, 151 patients exhibited evidence of disease recurrence, and 120 patients died. The latest survival data were collected on August 31, 2011. The total survival rate was 71.8% at 5 years and 45.4% at 10 years.
Tissue Microarray Construction and Immunohistochemical Staining
The hematoxylin and eosin staining of all 192 samples were reviewed by the pathologist, and the HCC tissue microarrays were constructed by obtaining three 1-mm-diameter cores from each tumor and the paired adjacent liver tissue. The hematoxylin and eosin stain of constructed tissue microarrays were reviewed and confirmed again by the pathologist. The specimens had been fixed in formalin and embedded in paraffin before they were archived. We used the archived specimens for immunohistochemical staining. The immunohistochemical staining was performed using Bond-Max autostainer (Leica Microsystems). The hematoxylin and eosin stains of all 192 samples were reviewed by the pathologist, and the slides were stained with antibodies directed against CRKL (1:50 dilution, Abcam) or FLT1 (1:250 dilution, Abcam). Briefly, specimens from the paraffin-embedded blocks were cut into 5-μm sections. The sections were dewaxed in a 60°C oven and then deparaffinized in xylene, rehydrated through serial dilutions of alcohol, and washed in phosphate-buffered saline (pH 7.2). Immunohistochemical staining was performed using the fully automated Bond-Max autostainer using the onboard, heat-induced antigen retrieval in citrate buffer with the ER1 protocol for 20 min and a VBS refine polymer detection system (Leica Microsystems). Diaminobenzidine was used as the chromogen (Leica Microsystems).
RESULTS
Identification of Interlinked PPIs in Cross-talk Pathways in Human Hepatocellular Carcinoma
We systematically collected ∼3,000 differentially expressed HCC-related signatures (18, 19) and examined ∼2,100 pathways from an integrated pathway database (ConsensusPathDB) (20). Then we prioritized 60 HCC-related pathways according to the HCC-related signatures and pathway datasets using a hypergeometric test (Fig. 1A, Step 1). The proteins belonging to the HCC-related pathways (gray nodes on the left) were detected to determine whether collected PPIs (POINeT) (21) or predicted PPIs (PIPS) (22) (red dotted line in Step 2 of Fig. 1A) with overexpressed patterns can link each pathway and enable potential interlinking. Specifically, each pathway member (single gene) among the different HCC-related pathways was used to map each single protein for each PPI pair in the PPI dataset, and thus it was possible to identify the cross-talk relationship (protein 1 and protein 2 in supplemental Table 3). Because many genes/proteins are involved in multiple pathways, most interlinked PPIs map to several different pathways (Pathway 1 and Pathway 2 in supplemental Table 3). These PPIs might link the cross-talk pathways together. In this step, we identified 97 proteins that participate in 375 PPIs occurring in overexpression patterns from HCC gene expression profiles, which revealed the pattern of interlinking among the 60 HCC-related pathways (Fig. 1A, Step 2). The interlinked PPIs among the signaling pathways can be categorized by topological characteristics (Fig. 1A, Step 2) as follows: r1, between/within pathway PPIs, which connect different pathways and are located in the same pathway(s); r2, between pathway PPIs, which serve as connections among different pathways.
An in situ proximity ligation assay (PLA) with the available paired antibodies was used to detect, validate, and quantify the endogenous presence of 194 PPIs in two HCC cell lines, Huh7 (minimally migratory cells) and Mahlavu (highly migratory cells). We observed 67 PPIs (49 proteins) among 21 pathways (supplemental Table 4) in either Huh7 or Mahlavu cells (Fig. 1A, Step 3, and B and C, and supplemental Fig. 1 and Table 5). There are four key features of these datasets. First, according to a literature survey, 17 PPIs (including 11 PPIs from PIPS) of the 67 PPIs are novel PPIs identified in this study (Fig. 1E and supplemental Table 6). Second, there are contrastingly different distributions of the in situ PLA signal for the 67 validated PPIs between Huh7 and Mahlavu. For example, the interaction signals for CRKL-SOS1 were much higher in Mahlavu cells than in Huh7 cells (Fig. 1, B and C), providing the opportunity to apply differential network biology to characterize tumor invasion. Third, ∼69% of between/within the pathway PPIs (r1) not only reflect the current knowledge on pathways but also suggest that these PPIs can interlink the components of unknown pathways. Moreover, the rest of the ∼31% of the PPIs belong to the between pathway PPI group (r2), implying that these corresponding pathways can be merged into more complete pathways by these PPIs (Fig. 1A, Step 2). Finally, by integrating bioinformatics identification and empirical validation, we can effectively construct a concise empirical HCC PPI network from a huge PPI network of up-regulated genes (Fig. 1D) to a concise empirical network (Fig. 1E).
In addition to revealing individual PPIs, our purpose was to provide a global view of the 67 analyzed PPIs and 21 pathways in two HCC cell lines (with different migratory abilities). Briefly, the pairwise modified version of simple matching coefficient (SMC0) was employed to construct both the between-pathway proximity matrix C (with C221 = 210 pairs in Fig. 2B) and the between-PPI proximity R (with C267 = 2211 pairs in Fig. 2C) in the first step. In the second step, we applied two dendrograms (hierarchical clustering trees) to sort the 21 pathways in C into five clusters of pathways with 7, 5, 3, 3, or 3 pathways each (P1 to P5 in Figs. 2B and 2A) and 67 PPIs in R as eight groups of PPIs (M1, M2, B1, B2, H1, H2, A1, and A2 in Figs. 2C and 2A). Then we created three heatmaps to elucidate the tripartite interactions (Fig. 2, A–C). PPIs that are specific in Huh7, Mahlavu, and either cell lines or none are color-coded as cyan, magenta, purple, or white.
For example, the CRKL-FLT1 interaction was observed in the M1 cluster, which might involve the c-Met pathway, IGF1 pathway, PDGFR-α signaling pathway, and VEGFR pathway. Specifically, the M1, H1, and B1 clusters contain PPIs involved in both P1 and P2, which include signaling pathways such as mammalian target of rapamycin, PI3K/AKT, and c-Met among others. Although the PPIs in the M1, H2, and B1 clusters are involved in pathways from the P1 group only, PPIs in the M2, H1, and B2 clusters are involved in pathways from clusters P1 and P2 simultaneously. One interesting note is that in this study the P2 pathways are found to never work alone and are observable only in the presence of the P1 pathways but not vice versa. Unlike P1 and P2 that complicate pathway clusters, P3 pathways are connected by only one or two PPIs from the M1, B1, or B2 clusters. Finally, the P4 and P5 pathways are related to DNA replication and the cell cycle. P4 contains three DNA replication-related pathways that, with CDC6 related PPIs, are involved in Huh7, Mahlavu, or both cell lines. P5 contains three pathways that are connected by CCNB1-related PPIs, mostly in Mahlavu cells or in both cell lines. A1 and A2 only occur in the clusters of P4 and P5. Overall, the clustering analysis shows that most of the PPIs are involved in the P1 pathway group, suggesting pathways in P1 group might play an important role in hepatocarcinogenesis. PPIs in M1 can only be observed in Mahlavu cells (highly migratory cells) and belong to the P1 pathway group. Therefore, this allows for intuitive selection of candidate PPIs (e.g. CRKL-FLT1) from M1 for further functional characterization. In addition to the similarity among the validated PPIs, pathways, and cell lines, we show these interlinked pathways as an undirected graph with no self-loops and with weights on the edges (number of involved PPIs) among the vertices (pathways) (Fig. 2D). The color of the edges in this graph also provides the quantitative information for the PPIs among the pathways. The edges with different colors indicate a different density of PPIs cross-talking between the two pathways, e.g. the CRKL-EGFR interactions (supplemental Table 3) connect the cMet and EGFR pathways. Moreover, the graph revealed that the c-Met, IGF1, PDGFR-β, and VEGFR pathways are highly connected with other pathways in HCC, suggesting the possibility of an HCC therapeutic strategy for disrupting the interlinked PPIs among these four pathways.
Analysis of the Differential Interaction Hubs in PPI Networks
A graphic theoretical analysis of PPI networks can reveal the essential genes/proteins that are over-represented among proteins with high centralities. Such hubs (the highly connected nodes) are of general interest because of their central roles, and pathway errors related to hubs usually cause lethality. In addition, previous studies suggest that hubs are more likely to be cancer-related genes than proteins with few interaction partners (27, 28). Moreover, a recent study suggests that a differential network biology approach, such as differential interaction hubs, is a promising approach to dynamic network discovery under multiple conditions, such as environment, tissue type, disease state, development, or speciation (29, 30).
These observations led us to measure the number of interactions to identify the differential interaction hubs for ranking essential genes/proteins/pairs, which might be involved in migration in HCC from this PPI network (Fig. 2E). For each protein in the differentially expressed network, CRKL is the hub with the highest degree of centrality in Mahlavu cells (Fig. 2E). CRKL interacted with the other 11 proteins. Eight of the 11 CRKL-interacting proteins (FLT1, HCK, PDGFRB, PTPN11, RAF1, PIK3R1, MAPK1, and GAB1) were detected empirically by in situ PLA assay in Mahlavu cells, but not in Huh7 cells, whereas CRKL interacted with EGFR, SOS1 and PTK2 in both Mahlavu and Huh7 cells (Fig. 2F). This topological analysis suggests that CRKL may play a crucial role in the malignant network, especially in the Mahlavu cells. FLT1, another hub with seven partners in Huh7 cells and six partners in Mahlavu cells, ranks second in degree centrality (Fig. 2E). These two hubs, CRKL and FLT1, interact with each other in the network of Mahlavu cells. Interaction between CRKL and FLT1 thus has a higher connectivity than other interactions, suggesting that the CRKL-FLT1 interaction might be important. More importantly, FLT1 has seven interaction partners but only CRKL occurred in highly migratory cells (Fig. 2F), implying an unusual relationship between these two proteins. According to the pathway analysis, the CRKL-FLT1 interaction was involved in the c-Met, IGF1, PDGFR-α, VEGFR1, and VEGFR2 pathways, which are well known invasion and metastasis pathways in HCC (2). Consequently, CRKL-FLT1 might be a crucial interaction among the 67 PPIs in the cross-talk pathway map of highly migratory HCC cells.
Functional and Histopathological Analysis of CRKL and FLT1 in HCC
Because of the high degree of similarity of CRKL and CRK, we have confirmed the specificity of CRKL antibody by Western blotting with overexpression of CRKL or CRK in HEK293T cells (supplemental Fig. 2). The role of CRKL, FLT1, and the prioritized CRKL-FLT1 interaction, in HCC remains unclear. Thus, we measured CRKL-FLT1 interaction in hepatocyte and five different HCC cell lines (HepG2, Huh7, PLC5, SK-Hep1, and Mahlavu) (Fig. 3A). Compared with the migratory ability of five HCC cell lines, the CRKL-FLT1 interaction correlates with the migratory ability of the cells analyzed (Fig. 3B), which is consistent with our hypothesis. The expression of EMT markers in the tested cell lines also consists of the migratory ability (supplemental Fig. 3). Moreover, we extend the measurement of CRKL-FLT1 interaction and migratory ability in other cancer cells, including cervical cancer (HeLa), lung adenocarcinoma (A549), colon cancer (HT29), and prostate cancer (PC3) (Fig. 3, C and D, and supplemental Fig. 4). The intensity of in situ PLA for CRKL-FLT1 interaction and migrated cells/high power field showed a positive correlation with a correlation coefficient of 0.886 (p < 0.001), as estimated by the p value by t test for Pearson product-moment correlation when we observed all nine cancer cells (Fig. 3E). Taken together, the expression of CRKL-FLT1 interaction is correlated with cell migration not only in HCC but also in other cancer cells.
Fig. 3.

Characterization of the CRKL-FLT1 interaction in HCC cell lines. A, images (left) and quantification (right) of in situ PLA signal for CRKL-FLT1 interaction in hepatocytes and five HCC cell lines (HepG2, Huh7, PLC5, SK-Hep1, and Mahlavu) are shown. B, migratory ability and CRKL-FLT1 interaction were evaluated in five HCC cell lines. The Mahlavu cell is highly migratory cells among five HCC cell lines. The CRKL-FLT1 interaction via in situ PLA (C) and migratory ability (D) was measured in other cancers, including prostate cancer cells (PC3), colon cancer cells (HT29), lung cancer cells (A549), and cervical cancer (HeLa). E, CRKL-FLT1 expression was correlated with cell migration. The intensity of in situ PLA for CRKL-FLT1 interaction and migrated cells/high power field showed a positive correlation with a correlation coefficient = 0.886 (p < 0.001) as analyzed for Pearson product-moment correlation when we observed all nine cancer cells. F, five different stable clones of CRKL and FLT1 Mahlavu cells (vehicle (scramble control), shCRKL-1, shCRKL-2, shFLT1–1, and shFLT1–2) with knockdown of CRKL or FLT1 were established. The expression of CRKL-FLT1 interaction was decreased in Mahlavu stable clones with knockdown of CRKL or FLT1 compared with vehicle control cells. G, expression of CRK was not affected by knockdown of CRKL in Mahlavu cells. The shCRKL-1 and shCRKL-2 specifically target CRKL, but not CRK. H, knockdown of CRKL or FLT1 reduced migration ability compared with vehicle control cells. I, lysates from Mahlavu cells with CRKL or FLT1 knockdown were subject to Western blotting for mesenchymal markers and several markers of signaling pathways. Western blot analysis showed that both CRKL and FLT1 participated in the ERK but not in JNK or p38 signaling pathway and that they were also involved in EMT process. Vehicle, scramble control.
Next, we established five different Mahlavu stable clones with knockdown of CRKL or FLT1 to investigate the biochemical function of CRKL and FLT1. The specificity of knockdown of CRKL was confirmed by in situ PLA for CRKL-FLT1 interaction (Fig. 3F) and immunoblotting (Fig. 3G). We then tested the effect of reducing CRKL and FLT1 on migration and proliferation in highly migratory Mahlavu cells. Depletion of CRKL and FLT1 decreased the migration of Mahlavu cells ∼60 and 40%, respectively (Fig. 3H), but did not affect the proliferation rate of Mahlavu cells (supplemental Fig. 5). Our Western blotting analysis of p-ERK at Thr-202/Tyr-204 and mesenchymal markers, e.g. N-cadherin, Vimentin, Snail, and Twist, suggested that CRKL and FLT1 are involved in the migration process via ERK phosphorylation to regulate the mechanism of EMT (Fig. 3I). Moreover, the ablation of CRKL reduced FLT1 expression, implying that CRKL and FLT1 might have a feedback regulation relationship via phosphorylation and protein stability. Finally, we also showed the SH2, SH3(N), and SH3(C) domains of CRKL interacted with FLT1 by immunoprecipitation and in situ PLA (supplemental Fig. 6). Together, CRKL and FLT1 may play a crucial role in the regulation of metastasis in HCC.
CRKL Is a Novel Marker and Is Associated with FLT1 Expression and Poor Prognosis in HCC
To provide an independent validation of the prognostic value of the CRKL and CRKL-FLT1 pair, their expression levels were measured using a 192 pairwise HCC tissue microarrays. Both CRKL and FLT1 were expressed significantly higher in tumors than in the adjacent normal tissue (data not shown). The relationships between the expression levels of CRKL and FLT1 with the clinicopathological characteristics of HCC are summarized in Table I. High CRKL expression levels (scores of 2 and 3) in tumors were strongly correlated with reduced disease-free (p = 0.002) and overall survival (p = 0.001) relative to low CRKL expression levels (scores of 0 and 1) (Fig. 4, A and B). Our univariate and multivariate analyses showed that advanced stage major vessel invasion and CRKL overexpression were significant, independent prognostic indicators for disease-free and overall survival rates (Table II and supplemental Table 7). High FLT1 expression was significantly correlated with disease-free survival (p = 0.047) and showed trends with overall survival (Fig. 4C). The FLT1 score, however, was not significantly correlated with disease-free (p = 0.156) and overall survival (p = 0.185), as assessed by a multivariate analysis (Table II). In summary, a high level of CRKL can be used as a novel prognostic marker in HCC.
Table I. Relationship between CRKL and FLT1 expression and clinicopathological factors in 192 HCC patients.
| Characteristics | CRKL expression |
FLT1 expression |
||||
|---|---|---|---|---|---|---|
| Low |
High |
p valuea | Low |
High |
p valuea | |
| (n = 81) | (n = 111) | (n = 81) | (n = 111) | |||
| Age | 0.694 | 0.759 | ||||
| Years (mean ± S.D.) | 61.19 ± 12.53 | 61.77 ± 11.61 | 61.95 ± 11.15 | 61.21 ± 11.9 | ||
| Gender | 0.269 | 0.502 | ||||
| Male | 73 | 94 | 72 | 95 | ||
| Female | 8 | 17 | 9 | 16 | ||
| AFPc | 0.163 | 0.079 | ||||
| <400 ng/ml | 68 | 84 | 69 | 83 | ||
| ≥400 ng/ml | 13 | 27 | 12 | 28 | ||
| Stage | 0.432 | 0.432 | ||||
| I + II | 59 | 75 | 59 | 75 | ||
| III | 22 | 36 | 22 | 36 | ||
| Hepatitis B virus | 0.507 | 0.058 | ||||
| Negative | 30 | 36 | 34 | 32 | ||
| Positive | 51 | 75 | 47 | 79 | ||
| Hepatitis C virus | 0.975 | 0.049b | ||||
| Negative | 60 | 82 | 54 | 88 | ||
| Positive | 21 | 29 | 27 | 23 | ||
| Recurrence status | 0.089 | 0.316 | ||||
| No | 23 | 20 | 21 | 22 | ||
| Yes | 58 | 91 | 60 | 89 | ||
| Major vessel invasion | 0.096 | 0.224 | ||||
| No | 75 | 94 | 74 | 95 | ||
| Yes | 6 | 17 | 7 | 16 | ||
a p value for age was derived from Student's t test; other p values were derived with a two-tailed Pearson χ2 test. S.D. represents standard deviation.
b p < 0.05.
c AFP represents α-fetoprotein.
Fig. 4.

Overexpression of CRKL is a marker of poor prognosis in HCC. A, CRKL and FLT1 protein expression in a representative HCC specimen. The expression levels of CRKL and FLT1 were quantified according to the staining intensity. The results were stratified into two groups according to the intensity of staining; in the low expression group, either no staining was present (staining intensity score 0) or positive staining was detected in <10% of the cells (staining intensity score 1); in the high expression group, positive immunostaining was present in 10–30% (staining intensity score 2) or >30% of the cells (staining intensity score 3). A Kaplan-Meier curve of disease-free (up) and overall (down) survival in 192 HCC patients was stratified by CRKL (B) and FLT1 (C) expression. D, representative IHC staining of endogenous CRKL and FLT1 in serial paraffin sections of HCC tissue is shown. Note the significant positive correlation between the level of CRKL and that of FLT1 (correlation coefficient = 0.506, p < 0.001 using Spearman's nonparametric correlation test). E, disease-free (left) and overall (right) survival of 192 HCC patients according to the combination analysis of CRKL and FLT1 expression. F, disease-free survival analysis of HCC patients stratified by CRKL and FLT1 status.
Table II. Cox multivariate regression analysis of prognostic factors of CRKL, FLT1, or CRKL/FLT1 expression for disease-free survival (DFS) and overall survival (OS) in 192 HCC patients.
| Factors | CRKL |
FLT1 |
CRKL/FLT1 |
|||
|---|---|---|---|---|---|---|
| HRb (95% CIc) | p value | HR (95% CI) | p value | HR (95% CI) | p value | |
| Disease-free survival | ||||||
| CRKL (low vs. high) | 1.579 (1.125–2.216) | 0.008a | ||||
| FLT1 (low vs. high) | 1.275 (0.912–1.783) | 0.156 | ||||
| CRKL/FLT1 (others vs. high/high) | 1.271 (1.043–1.549) | 0.018a | ||||
| Stage (I and II vs. III) | 1.687 (1.153–2.469) | 0.007a | 1.706 (1.161–2.507) | 0.007a | 1.679 (1.146–2.458) | 0.008a |
| AFPd (<400 vs. ≥400 ng/ml) | 1.147 (0.737–1.786) | 0.543 | 1.076 (0.687–1.686) | 0.750 | 1.090 (0.699–1.699) | 0.703 |
| Major vessel invasion (yes vs. no) | 1.904 (1.117–3.246) | 0.018a | 2.040 (1.204–3.457) | 0.008a | 1.954 (1.148–3.325) | 0.014a |
| Overall survival | ||||||
| CRKL (low vs. high) | 1.747 (1.186–2.573) | 0.005a | ||||
| LT1 (low vs. high) | 1.289 (0.886–1.876) | 0.185 | ||||
| CRKL/FLT1 (others vs. high/high) | 1.313 (1.052–1.638) | 0.016a | ||||
| Stage (I and II vs. III) | 1.694 (1.139–2.517) | 0.009a | 1.686 (1.130–2.514) | 0.01a | 1.687 (1.134–2.509) | 0.010a |
| AFP (<400 vs. ≥400 ng/ml) | 1.326 (0.857–2.051) | 0.205 | 1.297 (0.833–2.019) | 0.249 | 1.280 (0.825–1.984) | 0.271 |
| Major vessel invasion (yes vs. no) | 1.694 (1.054–2.571) | 0.031a | 1.881 (1.111–3.185) | 0.019a | 1.799 (1.060–3.053) | 0.029a |
a p < 0.05.
b HR represents hazard ratio.
c CI represents confidence interval.
d AFP represents α-fetoprotein.
To investigate the interplay between CRKL and FLT1 in HCC patients, we subsequently performed an IHC analysis of CRKL and FLT1 in serial sections of HCC tissues. The representative IHC staining for CRKL and FLT1 (Fig. 4D) showed a positive correlation with a correlation coefficient = 0.506 (p < 0.001), as analyzed by the Spearman's nonparametric correlation test. This result is consistent with the biological findings in HCC cells. Moreover, a subgroup analysis using both CRKL and FLT1 expression levels further showed that patients with high CRKL and FLT1 levels had significantly poorer disease-free (p = 0.001) and overall survival (p = 0.002) compared with the group of patients with low CRKL and FLT1 levels (Fig. 4E). An IHC score of CRKL and FLT1 ≥3 remains an independent predictor of shortened time of disease-free (HR = 1.271, 95% CI, 1.0–1.549, p = 0.018) and overall (HR = 1.313, 95% CI, 1.052–1.638, p = 0.016) survival after analyses by multivariate Cox regression (Table II). To clarify this issue, we stratified patients into four groups as follows: CRKL high/FLT1 high, CRKL high/FLT1 low, CRKL low/FLT1 high, and CRKL low/FLT1 low, and performed Kaplan-Meier analysis of disease-free survival. The result showed that patients with CRKL high/FLT1 high indeed had the most dismal outcome (Fig. 4F, red line). However, if we analyze this issue from the perspective of FLT1, combining both CRKL and FLT1 status can further enhance the prognostic value, which implies the interaction of both proteins was indeed associated with HCC progression (Fig. 4, C and E). These results indicate that CRKL expression is clinically associated with FLT1 expression and that such expression can be used as an independent prognostic factor to predict the treatment outcomes of patients with HCC.
DISCUSSION
Our study was inspired by the notion that cross-talk connections (interlinked PPIs) with new pathways may lead to hallmark capabilities (6) in cancer and that the interlinking of signaling networks drives phenotypic alterations while maintaining the robustness of the network during tumor progression (32). Therefore, we applied a cost-effective computational method using existing PPI databases to infer the candidates are likely to be involved in HCC migration. We subsequently validated these candidates using an empirical method to boost the accuracy, followed by computational analysis to prioritize the candidates for a functional assay. This is the first systematic and quantitative measurement of PPIs at the cellular level via an in situ PLA assay. The resulting PPI data from highly migratory and minimally migratory HCC cells were used in differential network biology to study the fundamental biological responses and to reveal the architecture of an interactome, which can be massively rewired during a cellular or adaptive response (29, 30). Finally, the identification and characterization of the CRKL-FLT1 interaction in HCC cell lines and patient specimens establish the CRKL and CRKL-FLT1 pair as new prognosis markers in HCC. We anticipate that this scalable and general approach will be applicable to other cancer types and complex diseases.
FLT1 is a therapeutic target for several clinically utilized tyrosine kinase inhibitors (e.g. Vatalanib, Cediranib, and Sorafenib) and is detected on bone marrow-derived hematopoietic progenitor cells, which home to tumor-specific pre-metastatic sites and form clusters (33). CRKL is a molecular bridge connecting tyrosine kinases and their substrates to many cellular signaling processes (34). A recent study revealed that CRKL may be a potential druggable target for tumors resistant to kinase inhibitors (35) and that its CRKL amplification may be a mechanism of primary or acquired resistance to EGFR kinase inhibitors in non-small cell lung cancer (36). We also found that the knockdown of FLT1 in Mahlavu cells decreased the phosphorylation level of CRKL at Tyr-207 (data not shown), which is a well known site for regulating the signaling cascade triggered by CRKL. This finding suggests that FLT1 could regulate CRKL phosphorylation. Moreover, when CRKL was depleted, FLT1 expression was also down-regulated. These data indicate that CRKL and FLT1 might provide feedback regulation at a functional level in HCC; this finding warrants further investigation. Thus, a combination therapy of a kinase inhibitor and targeting CRKL may result in a better anti-cancer clinical response.
Clinically, CRKL amplification at the 22q11.21 is frequently detected in lung cancer patients with elevated CRKL expression in tumor cells (15). In previous studies, CRKL was demonstrated to facilitate migration/invasion in non-small cell lung cancer (15), colon cancer (37), and breast cancer (38). However, to our knowledge there are no data indicating that CRKL participates in the migration and progression of HCC. Despite the high degree of similarity of CRKL and CRK, recent structural investigations show that the intramolecular assembly of CRKL is entirely distinct from that of CRK (39). The review article also indicates that CRKL and CRK are expected to have different signaling characteristics (34). We characterized CRKL participating in migration via the ERK pathway in HCC, which is consistent with a previous study in hematopoietic cells. CRKL is also known to be a key effector in Bcr-Abl activity and a prognostic indicator for chronic myelogenous leukemia (41). However, the clinical relevance of CRKL in HCC remains unknown. We demonstrate that CRKL acts as a novel prognostic marker and that CRKL may be a therapeutic target in HCC. In some cancers, FLT1-mediated signaling could promote tumor invasion (42, 43). The prognostic impact of FLT1 expression is significantly related to disease-free survival and trends toward overall survival. A recent study shows that FLT1 expression is significant for recurrence-free and overall survival in HCC patients who underwent surgical resection (44), but their study included a smaller patient cohort than our study as reported here. Overexpression of CRKL and FLT1 was also identified as a significant prognostic factor for HCC by our multivariate analysis and was associated with significantly worse disease-free and overall conditions. Collectively, these results indicate that CRKL expression is clinically associated with FLT1 expression and that such expression could be used as an independent prognostic factor to predict the treatment outcomes of patients with HCC.
Notably, HCC is frequently resistant to systemic therapies and recurs even after aggressive local therapies. The need for molecular markers to identify which groups of patients have a high recurrence probability and which groups have a high Sorafenib-resistant rate is an important unmet medical need (45). The feasibility of using CRKL or CRKL-FLT1 for risk stratification in clinical practice needs to be investigated.
Other interactions in the network might also contribute to the malignant phenotype of HCC. For example, a prioritized CRKL-EGFR interaction was found and may explain why combining Erlotinib (EGFR inhibitor) and Bevacizumab (VEGF inhibitor) in an HCC phase II study was more effective than single drug treatment (31). Another example is the HCK-CRKL interaction, which is a prioritized PPI and ranks as a third prioritization of pairwise degree centrality in the HCC PPI network (supplemental Fig. 7). In addition, we found that HCK had several novel interaction partners, including CRKL, FLT1, EGFR, SOS1, PLCG1, and PIK3CB, in our PPI network. A recent study indicates that HCK mediates tumor-associated macrophage migration and matrix remodeling in the migratory capacity of tumor cells within the tumor environment in breast cancer (40). Although there is no evidence of a migratory role for HCK in HCC, functional analyses of these interactions may allow us to better characterize its role in cancer cell behaviors and responses.
In summary, we anticipate that our integrated approach and analysis will improve the interpretation of interlinked PPIs and pathways and facilitate the development of novel prognosis markers and drug targets in HCC research.
Supplementary Material
Acknowledgments
We thank Wilber Huang (Abnova Corp.) and OLINK Bioscience (Uppsala, Sweden) for technical support.
Footnotes
* This work was supported by National Science Council Grants NSC99-3112-B-010-005-CC1 and NSC100-2627-B-010-005-; Taipei Veterans General Hospital Grant V100E2-010; National Health Research Institutes Grant NHRI-EX101-10029BI; Ministry of Economic Affairs Grant 100-EC-17-A-17-S1-152; the Ministry of Education, Aim for the Top University Plan of National Yang Ming University (to C.Y.H.); Taipei Veterans General Hospital Grants V100E2-005 and 101DHA0100653 (to M.H.C.); National Core Facility Program for Biotechnology of Taiwan Grant NSC100-2319-B-010-002 from the Bioinformatics Consortium of Taiwan (to C.H.C.); Hsing-Tian-Kong Long Term Scholarship for Talented Students; National Science Council/Interchange Association of Japan (2009 Summer Program); National Science Council/German Academic Exchange Service (2010 Summer Institute Program); and National Science Council 2011 Graduate Students Study Abroad Program (to C.H.L.).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- HCC
- hepatocellular carcinoma
- PPI
- protein-protein interaction
- EMT
- epithelial-mesenchymal transition
- PLA
- proximity ligation assay
- PDGFR
- platelet-derived growth factor receptor
- SMC
- simple matching coefficient
- IHC
- immunohistochemistry
- SH
- Src homology
- VEGFR
- VEGF receptor
- EGFR
- EGF receptor.
REFERENCES
- 1. Jemal A., Siegel R., Ward E., Hao Y., Xu J., Murray T., Thun M. J. (2008) Cancer Statistics, 2008. CA-Cancer J. Clin. 58, 71–96 [DOI] [PubMed] [Google Scholar]
- 2. Whittaker S., Marais R., Zhu A. X. (2010) The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 29, 4989–5005 [DOI] [PubMed] [Google Scholar]
- 3. Barabási A.-L., Gulbahce N., Loscalzo J. (2011) Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 12, 56–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taylor I. W., Linding R., Warde-Farley D., Liu Y., Pesquita C., Faria D., Bull S., Pawson T., Morris Q., Wrana J. L. (2009) Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nat. Biotechnol. 27, 199–204 [DOI] [PubMed] [Google Scholar]
- 5. Chuang H.-Y., Lee E., Liu Y.-T., Lee D., Ideker T. (2007) Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 3, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 7. Vogelstein B., Kinzler K. W. (2004) Cancer genes and the pathways they control. Nat. Med. 10, 789–799 [DOI] [PubMed] [Google Scholar]
- 8. Sawyers C. (2004) Targeted cancer therapy. Nature 432, 294–297 [DOI] [PubMed] [Google Scholar]
- 9. Hopkins A. L. (2008) Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 4, 682–690 [DOI] [PubMed] [Google Scholar]
- 10. Llovet J. M., Bruix J. (2008) Molecular targeted therapies in hepatocellular carcinoma. Hepatology 48, 1312–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 12. Ahmad S., Alsayed Y. M., Druker B. J., Platanias L. C. (1997) The type I interferon receptor mediates tyrosine phosphorylation of the CrkL adaptor protein. J. Biol. Chem. 272, 29991–29994 [DOI] [PubMed] [Google Scholar]
- 13. Smit L., van der Horst G., Borst J. (1996) Sos, Vav, and C3G participate in B cell receptor-induced signaling pathways and differentially associate with Shc-Grb2, Crk, and Crk-L adaptors. J. Biol. Chem. 271, 8564–8569 [DOI] [PubMed] [Google Scholar]
- 14. Feller S. M. (2001) Crk family adaptors-signalling complex formation and biological roles. Oncogene 20, 6348–6371 [DOI] [PubMed] [Google Scholar]
- 15. Kim Y. H., Kwei K. A., Girard L., Salari K., Kao J., Pacyna-Gengelbach M., Wang P., Hernandez-Boussard T., Gazdar A. F., Petersen I., Minna J. D., Pollack J. R. (2010) Genomic and functional analysis identifies CRKL as an oncogene amplified in lung cancer. Oncogene 29, 1421–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barleon B., Sozzani S., Zhou D., Weich H. A., Mantovani A., Marmé D. (1996) Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 87, 3336–3343 [PubMed] [Google Scholar]
- 17. Kanno S., Oda N., Abe M., Terai Y., Ito M., Shitara K., Tabayashi K., Shibuya M., Sato Y. (2000) Roles of two VEGF receptors, Flt-1 and KDR, in the signal transduction of VEGF effects in human vascular endothelial cells. Oncogene 19, 2138–2146 [DOI] [PubMed] [Google Scholar]
- 18. Hsu C. N., Lai J. M., Liu C. H., Tseng H. H., Lin C. Y., Lin K. T., Yeh H. H., Sung T. Y., Hsu W. L., Su L. J., Lee S. A., Chen C. H., Lee G. C., Lee D. T., Shiue Y. L., Yeh C. W., Chang C. H., Kao C. Y., Huang C. Y. (2007) Detection of the inferred interaction network in hepatocellular carcinoma from EHCO (Encyclopedia of Hepatocellular Carcinoma Genes Online). BMC Bioinformatics 8, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen M. H., Yang W. L., Lin K. T., Liu C. H., Liu Y. W., Huang K. W., Chang P. M., Lai J. M., Hsu C. N., Chao K. M., Kao C. Y., Huang C. Y. (2011) Gene expression-based chemical genomics identifies potential therapeutic drugs in hepatocellular carcinoma. PLoS ONE 6, e27186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kamburov A., Stelzl U., Lehrach H., Herwig R. (2012) The ConsensusPathDB Interaction Database: 2013 update. Nucleic Acids Res., 41, D793–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee S. A., Chan C. H., Chen T. C., Yang C. Y., Huang K. C., Tsai C. H., Lai J. M., Wang F. S., Kao C. Y., Huang C. Y. (2009) POINeT: protein interactome with sub-network analysis and hub prioritization. BMC Bioinformatics 10, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McDowall M. D., Scott M. S., Barton G. J. (2009) PIPs: human protein-protein interaction prediction database. Nucleic Acids Res. 37, D651–D656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jarvius M., Paulsson J., Weibrecht I., Leuchowius K.-J., Andersson A.-C., Wählby C., Gullberg M., Botling J., Sjöblom T., Markova B., Ostman A., Landegren U., Söderberg O. (2007) In situ detection of phosphorylated platelet-derived growth factor receptor β using a generalized proximity ligation method. Mol. Cell. Proteomics 6, 1500–1509 [DOI] [PubMed] [Google Scholar]
- 24. Chen C. H. (2002) Generalized association plots: Information visualization via iteratively generated correlation matrices. Statistica Sinica 12, 7–30 [Google Scholar]
- 25. Wu H. M., Tien Y. J., Chen C. (2010) GAP: A graphical environment for matrix visualization and cluster analysis. Computational Statistics & Data Analysis 54, 767–778 [Google Scholar]
- 26.Deleted in proof
- 27. Jonsson P. F., Bates P. A. (2006) Global topological features of cancer proteins in the human interactome. Bioinformatics 22, 2291–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goh K. I., Cusick M. E., Valle D., Childs B., Vidal M., Barabási A. L. (2007) The human disease network. Proc. Natl. Acad. Sci. U.S.A. 104, 8685–8690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ideker T., Krogan N. J. (2012) Differential network biology. Mol. Syst. Biol. 8, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bandyopadhyay S., Mehta M., Kuo D., Sung M. K., Chuang R., Jaehnig E. J., Bodenmiller B., Licon K., Copeland W., Shales M., Fiedler D., Dutkowski J., Guénolé A, van Attikum H., Shokat K. M., Kolodner R. D., Huh W. K., Aebersold R., Keogh M. C., Krogan N. J., Ideker T. (2010) Rewiring of genetic networks in response to DNA damage. Science 330, 1385–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thomas M. B., Morris J. S., Chadha R., Iwasaki M., Kaur H., Lin E., Lin E., Kaseb A., Glover K., Davila M., Abbruzzese J. (2009) Phase II trial of the combination of bevacizumab and erlotinib in patients who have advanced hepatocellular carcinoma. J. Clin. Oncol. 27, 843–850 [DOI] [PubMed] [Google Scholar]
- 32. Barabási A. L., Oltvai Z. N. (2004) Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 5, 101–113 [DOI] [PubMed] [Google Scholar]
- 33. Kaplan R. N., Riba R. D., Zacharoulis S., Bramley A. H., Vincent L., Costa C., MacDonald D. D., Jin D. K., Shido K., Kerns S. A., Zhu Z., Hicklin D., Wu Y., Port J. L., Altorki N., Port E. R., Ruggero D., Shmelkov S. V., Jensen K. K., Rafii S., Lyden D. (2005) VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438, 820–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sriram G., Birge R. B. (2010) Emerging roles for Crk in human cancer. Genes Cancer 1, 1132–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johannessen C. M., Boehm J. S., Kim S. Y., Thomas S. R., Wardwell L., Johnson L. A., Emery C. M., Stransky N., Cogdill A. P., Barretina J., Caponigro G., Hieronymus H., Murray R. R., Salehi-Ashtiani K., Hill D. E., Vidal M., Zhao J. J., Yang X., Alkan O., Kim S., Harris J. L., Wilson C. J., Myer V. E., Finan P. M., Root D. E., Roberts T. M., Golub T., Flaherty K. T., Dummer R., Weber B. L., Sellers W. R., Schlegel R., Wargo J. A., Hahn W. C., Garraway L. A. (2010) COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468, 968–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cheung H. W., Du J., Boehm J. S., He F., Weir B. A., Wang X., Butaney M., Sequist L. V., Luo B., Engelman J. A., Root D. E., Meyerson M., Golub T. R., Jänne P. A., Hahn W. C. (2011) Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancers. Cancer Discovery, 1, 608–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chiu S. T., Chang K. J., Ting C. H., Shen H. C., Li H., Hsieh F. J. (2009) Over-expression of EphB3 enhances cell-cell contacts and suppresses tumor growth in HT-29 human colon cancer cells. Carcinogenesis 30, 1475–1486 [DOI] [PubMed] [Google Scholar]
- 38. Shen Q., Rahn J. J., Zhang J., Gunasekera N., Sun X., Shaw A. R., Hendzel M. J., Hoffman P., Bernier A., Hugh J. C. (2008) MUC1 initiates Src-CrkL-Rac1/Cdc42-mediated actin cytoskeletal protrusive motility after ligating intercellular adhesion molecule-1. Mol. Cancer Res. 6, 555–567 [DOI] [PubMed] [Google Scholar]
- 39. Kobashigawa Y., Inagaki F. (2012) Structural biology: CrkL is not Crk-like. Nat. Chem. Biol. 8, 504–505 [DOI] [PubMed] [Google Scholar]
- 40. Guiet R., Van Goethem E., Cougoule C., Balor S., Valette A., Al Saati T., Lowell C. A., Le Cabec V., Maridonneau-Parini I. (2011) The process of macrophage migration promotes matrix metalloproteinase-independent invasion by tumor cells. J. Immunol. 187, 3806–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hemmeryckx B., van Wijk A., Reichert A., Kaartinen V., de Jong R., Pattengale P. K., Gonzalez-Gomez I., Groffen J., Heisterkamp N. (2001) Crkl enhances leukemogenesis in BCR/ABL P190 transgenic mice. Cancer Res. 61, 1398–1405 [PubMed] [Google Scholar]
- 42. Fan F., Wey J. S., McCarty M. F., Belcheva A., Liu W., Bauer T. W., Somcio R. J., Wu Y., Hooper A., Hicklin D. J., Ellis L. M. (2005) Expression and function of vascular endothelial growth factor receptor-1 on human colorectal cancer cells. Oncogene 24, 2647–2653 [DOI] [PubMed] [Google Scholar]
- 43. Wey J. S., Fan F., Gray M. J., Bauer T. W., McCarty M. F., Somcio R., Liu W., Evans D. B., Wu Y., Hicklin D. J., Ellis L. M. (2005) Vascular endothelial growth factor receptor-1 promotes migration and invasion in pancreatic carcinoma cell lines. Cancer 104, 427–438 [DOI] [PubMed] [Google Scholar]
- 44. Li T., Zhu Y., Ren W., Xu S., Yang Z., Fang A., Qin C. (2012) High co-expression of vascular endothelial growth factor receptor-1 and Snail is associated with poor prognosis after curative resection of hepatocellular carcinoma. Med. Oncol. 29(4), 2750–2761 [DOI] [PubMed] [Google Scholar]
- 45. Villanueva A., Hoshida Y., Toffanin S., Lachenmayer A., Alsinet C., Savic R., Cornella H., Llovet J. M. (2010) New strategies in hepatocellular carcinoma: genomic prognostic markers. Clin. Cancer Res. 16, 4688–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.