

Abstract
Label-free quantification using precursor-based intensities is a versatile workflow for large-scale proteomics studies. The method however requires extensive computational analysis and is therefore in need of robust quality control during the data mining stage. We present a new label-free data analysis workflow integrated into a multiuser software platform. A novel adaptive alignment algorithm has been developed to minimize the possible systematic bias introduced into the analysis. Parameters are estimated on the fly from the data at hand, producing a user-friendly analysis suite. Quality metrics are output in every step of the analysis as well as actively incorporated into the parameter estimation. We furthermore show the improvement of this system by comprehensive comparison to classical label-free analysis methodology as well as current state-of-the-art software.
Shotgun liquid chromatography (LC)1-MS/MS is widely used for quantitative proteomics research. There are several LC-MS/MS approaches that rely on chemical labels as internal standards, and additionally numerous label-free workflows exist. Of these options, label-free quantification has lately emerged as a viable approach because of its lack of limitations concerning the number and types of samples under investigation (1–3). Despite the advantages, there are a number of data analysis challenges (4) associated with the label-free methodology that may unnecessarily increase technical variation. This has led to an uncertainty toward the usage of the label-free workflows, to much extent because of difficulties in assessing how well the data analysis works for the experiment at hand. There is a large amount of software available for label-free data processing and recent studies have shown that the results of data analysis vary considerably with the choice of software (5–7).
Two major types of label-free quantification exist; spectral count and precursor-based. The former is based on the number of times a peptide has been subjected to fragmentation using MS/MS. The counts for the peptide content of a protein are subsequently recalculated into a protein count, which is related to the protein abundance (3). Although straightforward, this quantification strategy has proven less accurate than the precursor-based method as the number of spectra necessary to quantify more subtle expression changes increases exponentially (8). As quantification is coupled to identification, the dependence on the semirandom MS/MS sampling process leads to variability in protein abundance (9). The quantification coverage is further limited as the commonly employed dynamic exclusion for increasing the number of identifications introduces saturation effects into the analysis (8). In contrast, label-free quantification using precursor intensities shows great potential for large-scale proteomic analyses (1–3, 10–13).
Data from a label-free sample consists of LC-MS files that can be visualized individually as three-dimensional maps where the dimensions correspond to mass-to-charge ratio, retention time and intensity. Two fundamental steps that need to be performed in any precursor-based label-free pipeline are the extraction of peptide information from the maps (feature detection) and matching of corresponding peptides between maps for subsequent differential expression analysis (alignment).
A feature is a three-dimensional cluster of spectral peaks, detected in consecutive mass scans (the time dimension), and represents an eluting potential peptide at the MS level. Features are extracted based on defined criteria depending on the algorithm used, e.g., a high signal-to-noise ratio or a certain fit to a computational model of the isotopic envelope (14, 15). The output from feature detection algorithms consists of feature lists containing basic information for every feature such as mass-to-charge ratio, the start and end as well as apex time points for the elution profile, charge and some form of abundance measure (integrated and/or apex intensity). Using parallel MS/MS and identification using peptide fragment fingerprinting, a feature can be assigned a peptide identity. Feature detection is therefore the basis for peptide, and subsequently protein, quantification. The number and quality of features detected by different software suites have been investigated in (5) and possible approaches for dealing with missing features such as a combination of feature detection modules have been suggested in (6, 7).
Alignment is used to propagate peptide identities between features in different LC-MS maps, as the peptides identified by MS/MS can vary between files to a large extent. The alignment procedure will thus reduce the number of missing values in differential expression analysis. However, LC retention time drifts are common, and the alignment algorithm will need to handle such drifts, as well as ambiguities in matching for dense maps. The process can be divided into feature-based or profile-based approaches, where feature detection is performed before, or after, alignment, respectively (16). A further subdivision of algorithms can be performed based on whether the algorithm uses a reference map or not for the alignment. Use of a reference file to which the other maps are aligned facilitates implementation, but the choice of reference is crucial for the alignment outcome, and the wrong selection can considerably decrease the number of correctly aligned features (5). Errors in alignment in general lead to an important amount of missing values when features representing the same peptide are not matched between maps, as well as skewed quantification because of noncorresponding features being matched up.
To alleviate the introduction of such technical bias, as well as to estimate the accuracy of the data analysis, quality control is necessary. There are a number of issues that need to be addressed during the development of a quality control pipeline for label-free data analysis. Quality control metrics should be easy to overview and independent of the data at hand so as to assess an absolute quality of the analysis that is comparable between different experiments as well as between different software solutions and parameter settings. Some recently introduced metrics require either manual validation (17) or spiked in peptides to assess quality (6, 18) and may even require user-defined parameters (6).
Precision and recall are metrics commonly used for evaluation of feature detection and alignment and can also be used to evaluate the whole analysis (5, 18–23). Precision controls the false positive rate, while recall controls the false negative rate, in which the exact definition of a false positive and false negative depends on the process evaluated. Although precision and recall are intuitive metrics with a well-defined range, the conventional application of them is simply as an output given at the end of the analysis, forcing the user to repeat the entire analysis to improve the quality of the performance. Such repeated analysis is not only time-consuming but can be difficult if many user-defined parameters need optimization.
Here, we present a new data analysis workflow for label-free LC-MS, which includes a novel alignment algorithm, and has been implemented within the Proteios Software Environment (24). The alignment algorithm estimates parameter settings from the data at hand, avoiding the issue of default settings that may bias results if used indiscriminately between data sets. The need for a reference run is circumvented because of a computed order of pairwise map matching based on shared peptide identifications. The alignment is furthermore not dependent on any particular feature detection algorithm, as it is based on standard feature list information and can be coupled with any feature detection module of choice. In addition, quality control has been incorporated into the algorithm, not merely as an output but also as a regulator of the parameters, guaranteeing the user an optimized performance based on the precision and recall metrics. Alignment quality is continuously evaluated for every pairwise matching as well as for the end result. The quantification is assessed as described in (5) where both feature detection and alignment is taken into account and the performance of the algorithm is compared with alignment using msInspect (25) and OpenMS (26). We show that a combination of an adaptive algorithm and rigorous quality control leads to more reliable and reproducible data analysis by extensive comparison to common label-free analysis approaches.
MATERIALS AND METHODS
Workflow Implementation
The label-free workflow was implemented in Java 1.6 within the Proteios Software Environment (24), and is illustrated schematically in Fig. 1A. Plug-ins were implemented for executing OpenMS (26) and msInpect (25) feature detection in batch, and for import of the features into a dedicated database feature table. Common parameters for feature detection can be set in the web browser user interface. Another plug-in was implemented for matching of peptide identifications to features. Identifications that pass a user-defined FDR cutoff after a combination of searches (27) from any number of the supported search algorithms (currently Mascot (http://www.matrixscience.com), X!Tandem (http://thegpm.org/tandem/) with native scoring, X!Tandem with k-score (28), and OMSSA (29)), are matched with features of the same charge state and within a certain m/z and retention time tolerance. The latter is required because MS/MS identifications are sometimes acquired outside the feature boundary reported by the feature detection algorithm. Another plug-in was implemented for the novel alignment algorithm described below. The algorithm assigns features to clusters, in which a unique cluster ID defines features that have been matched over multiple files. Features are also assigned a peptide sequence if any of the features in the cluster has been identified. The user can select whether samples from different fractions should be matched or not in the case of fractionated samples. In the case of between-fraction matching being disabled, fractions are aligned and matched separately, and global quality metrics are reported for each fraction. The plug-in reports similarity scores for every pair of files to detect possible outlier files. Furthermore, precision and recall for the pairwise matching and for the global alignment (see alignment section) as well as the total increase in sequence coverage for every file after alignment is reported. Finally, a report plug-in generates a spreadsheet type report for all samples and feature clusters in the project, which can be imported to dedicated statistics software. The source code and binaries are available at http://www.proteios.org.
Fig. 1.
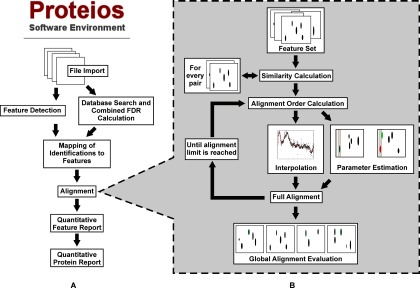
Schematic of the Proteios label-free analysis workflow (A) and alignment (B).
Alignment
The new alignment workflow is outlined in Fig. 1B. Initially, a set of features with shared peptide identities between every possible file pair is determined. This set is used to estimate a similarity measure for the separate file pairs in the batch, which determines the order in which the pairs are aligned. Pairwise alignment is subsequently conducted in two steps: First, the set of features with shared peptide identities is partitioned into two equal sized sets. The first set is used as a basis for a retention time correction function, which eliminates the need for user-defined tolerances for the correction. The second step is to find the entire set of features that match within given m/z and retention time tolerances between the aligned maps. The Proteios alignment algorithm estimates these tolerances by maximizing two quality metrics, precision and recall, using the second feature set. The tolerances set in this stage are critical and can introduce an important amount of incorrect feature correspondences that can bias statistical analysis downstream. On matching of the entire feature complement of the files, clusters of features that are aligned between multiple files are built, and peptide identities are transferred to aligned features that were previously missing peptide identity. Finally, the quality of the alignment is evaluated using the entire set of files.
Quality Measures
To evaluate and optimize alignment performance, quality control metrics were used. They are based on the precision and recall metrics described in (5, 23) and quality estimation is performed pairwise during the alignment and globally for all files after alignment. The number of True Positives (TPs), False Positives (FPs), and False Negatives (FNs) are used to calculate the metrics. FPs are features that have been erroneously linked to a cluster. If a peptide identity is associated to the feature or the cluster, it will be propagated throughout the cluster and will impact both qualitative and quantitative results. FNs are features that have been erroneously left out of a cluster. A high FN rate indicates that there is an important amount of clusters with missing features after alignment.
The recall metric is defined as TP/(TP + FN), which in an alignment context controls the ratio of features that are aligned to the features that are known to correspond. Precision is defined as TP/(TP + FP) and controls the assumed one-to-one correspondence of features between files. A decrease in precision can be caused by errors in alignment but also to peptide peaks being split into multiple features during feature detection (peak splitting). The latter can be overcome by rerunning the feature detection or by performing an additional data analysis step to merge split peaks (30). Computation of the metrics requires knowledge of the underlying feature correspondences and here a set of shared identified features between files is used.
There are two types of precision and recall computed during the alignment; identity-based and occurrence-based, taking the true peptide identity into account or not. These measures are computed both for a file pair during alignment and for all files in the batch after alignment. The occurrence-based metrics represent the evaluation of alignment of features without peptide identities, where matching of any feature within the set m/z and retention time tolerances signify a correct alignment. The identity-based metrics take peptide identification information into account when distinguishing between correctly and incorrectly aligned features, as illustrated in Fig. 2. Based on these underlying assumptions, the quality metrics can be interpreted as follows:
Fig. 2.
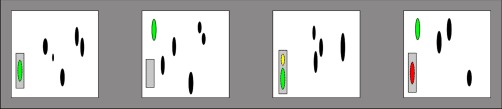
Global precision and recall computed for one feature aligned between four LC-MS maps. The ovals represent detected features. The colors represent a priori knowledge of the underlying correspondences. The green features are known to represent the same peptide based on peptide identity information. The gray boxes surrounding the features represent a feature cluster after matching; the features contained in the boxes are aligned. The size of the box represents the m/z and retention time tolerance used after retention time correction. An empty box indicates a missing value, i.e., there is no feature matched in the cluster for that file. The yellow feature is a false positive for both identity- and occurrence-based metrics, violating the assumption of a one-to-one correspondence between features. The red feature on the other hand, would be considered a false negative for the identity-based recall, as false positive for the identity-based precision, but a true positive for the occurrence-based metrics. This leads to an identity-based recall of 2/(2 + 2) = 0.5 and precision of 2/(2 + 2) = 0.5 whereas corresponding occurrence-based metrics equal 3/(3 + 1) = 0.75 and 3/(3 + 1) = 0.75, respectively. Every feature with an associated identity that is shared between all files before alignment is evaluated after the process as described above, leading to overall estimates of the alignment performance.
Occurrence-based Recall
Missing values are the only form of FNs possible as seen in the second file in Fig. 2. The occurrence-based recall therefore measures the introduction of missing values into the analysis. Because the feature set used for estimation is known to have correspondences albeit not which ones, missing values is an error introduced by the alignment.
Occurrence-based Precision
The occurrence-based precision only decreases if the one-to-one correspondence between features is violated, which is exemplified in the third file in Fig. 2. This could be because of a co-eluting compound with similar m/z, but also because of peak splitting. Because the evaluation is performed on the basis of identified peptides, known to be relatively high abundant and therefore more prone to peak splitting, the decrease in occurrence-based precision will to a large extent reflect this feature detection artifact.
Identity-based Recall and Precision
FNs are features that have not been aligned to other features with the same identity. This could be caused by either a missing value or incorrect matching with a feature of another or no identity. The latter will be counted as a FP and decrease the identity-based precision. As the identifications represent known correspondences in the data, the identity-based metrics give an indication of the true performance of the analysis.
In Fig. 2 the computation of global occurrence- and identity-based metrics for one feature cluster in four aligned LC-MS maps is illustrated.
Similarity Estimation
To determine which files are biologically most similar, a pairwise ratio of the number of shared unique (peptide sequence and charge) feature identifications to the total number of unique feature identifications is computed, i.e., 2* (# shared unique identifications between file 1 and 2)/(# unique identifications file 1 + # unique identifications file 2). This is performed for every possible file pair. The combination of precursor charge and peptide sequence is used so the same peptide identification will be handled separately for each precursor charge state. A ratio of one would indicate that a file pair share all identifications. The feature set sharing unique identifications constitutes the basis for the alignment. To avoid errors when estimating the retention time correction function, outlier removal based on the interquartile range of retention time differences is performed. When a file pair has less than 20 features in common, no alignment is performed for that file pair. This lower limit restricts matching of distantly related samples, but did not come into use in the present study.
Alignment Limit and Order
The alignment is terminated when every file has been aligned a certain number of times. This limit is empirically derived and illustrated in Supplemental Figs. S1 and S2. In addition, an upper limit for the number of alignments per file is set to obtain a uniform distribution and so ensure that no file will be aligned more times than the rest and subsequently introduce bias into the analysis. Inspection of the number of features aligned per run showed that the pairwise ratio of features already sharing a cluster to new features added into clusters saturated rapidly (supplemental Fig. S1B) and continued alignment of a file would only marginally increase recall at a relatively high cost of precision (supplemental Fig. S2).
The computed order of pairwise alignments is based on the calculated similarities; the pair with the highest similarity consisting of at least one file not yet aligned will be next in line. If no alignment limit were set the order would be irrelevant, as all possible file pairs would be processed. However, because the number of alignments is restricted, the biologically most similar files are selected to minimize possible errors in alignment. Specifically, the probability of two unidentified features within tolerance limits actually corresponding to the same peptide and not different ones sharing similar m/z and retention time is higher if the files are biologically more similar.
When a file pair has been selected for alignment, the set of features with common identifications are uniformly partitioned into two sets that are used for retention time correction and for parameter estimation, respectively.
Retention Time Correction
The apex retention time points of the shared features are used for spline interpolation to temporarily adjust retention times of features in the map being aligned to those of the target map in a file pair. First, a local regression smoothing is performed (LOWESS (31)) to reduce the effect of variation and outliers. A cubic spline function is subsequently interpolated between the time points. The Apache Commons Mathematics Library v. 2.2 (http://commons.apache.org/math/) was used for this section of the analysis.
Parameter Estimation
For two features to belong to the same cluster, they have to share charge as well as fall within certain m/z and retention time limits after the spline function is applied. The m/z tolerance is set as the largest deviation seen in the set of features sharing identity. The other half of the partitioned set is used to determine the retention time tolerance. The shared identity features of one file are matched to all features detected in the second file, and matches based solely on charge and m/z tolerance are computed. The retention time differences of these matches are stored in two lists; one containing the smallest retention time difference found for every feature and one list containing all other matches. These lists are subsequently sorted and the precision and recall metrics are computed as shown in Fig. 3. The retention time difference at which both metrics are simultaneously as large as possible is set as retention time tolerance for this file pair. It should be noted that the precision and recall used for the estimated tolerance optimizes the total number of TPs with respect to FNs and FPs for the pairwise alignment. Using the set tolerances, the precision and recall used for comparison to the global values described below are calculated as in Fig. 2. A high agreement between pairwise and global metrics implies that the file batch has been aligned as expected and subsequently, as the quality metrics are maximized, as optimally as possible.
Fig. 3.

Example retention time parameter estimation in a set containing four features sharing identifications (“A,” “B,” “C,” “D”) between a file pair. The correct match known beforehand is marked with a corresponding letter in the top table. Features without identity are denoted N/A. The time difference for all possible matches, based on the set m/z tolerance and charge between the four features from the first file and all features detected in the second file, are shown in the panel to the right (eight in total). For feature “B,” the match with the smallest time difference is not the feature with corresponding identity. Precision and recall is computed in the bottom panel for four different retention time tolerances. They are equivalent to the list of the smallest retention time differences for each feature, as these represent the minimum cost in terms of FPs for including a TP. Both identity- and occurrence-based metrics are computed as seen in the table. Abbreviations used in the table: Tol, Tolerance; Prec, Precision; Occ, Occurrence-based; Id, Identity-based. When two settings result in the same sum of precision and recall, the setting that results in the highest recall is selected. Here, the retention time tolerance used for subsequent alignment would be set to 5 min.
Global Evaluation
During the similarity calculations, features with unique identifications common to all files in a sample cohort before alignment are saved. At the end of the process, these features are extracted and precision and recall are computed as seen in Fig. 2. This is performed for every feature cluster with a unique identification and the average is used as global evaluation of the alignment.
Experimental Data
Two sets of samples were used for the results in this study. Potato secretome samples were prepared by extraction from three potato clones; Desiree, Sarpo Mira, and SW93–1015. Two data sets were collected before and after infection with Phytophthora infestans. The first consisted of an uninfected sample and one sample collected 3 days after infection from the same potato clone (Desiree), referred to as the TimePoint data set (Table I). The other data set consisted of samples from two different potato clones, Sarpo Mira and SW93–1015, collected 6 h post-infection, referred to as the Clone data set (Table II). Secretome isolation and Phytophthora infections were performed according to our previously described experimental setup (32). Thirty microliters of the secretome sample was dissolved in 6 × SDS-PAGE buffer containing dithiothreitol and separated for 1 cm with SDS-PAGE. After staining with Coomassie, the gel lane from each sample was cut into about 1 mm2 sized pieces and subjected to in-gel digestion with trypsin (modified sequencing grade; Promega, Madison, WI) overnight at 37 °C. The peptides obtained were extracted in 50–80% acetonitrile. Acetonitrile was vaporized using vacuum with centrifugation and desalting was performed using UltraMicro spin columns (Nest group). Samples were analyzed pure and mixed 1:2 or 2:1, giving four mix-points for each data set. To evaluate the reproducibility of the analysis when scaling the experiments, two biological replicates from each time point (0 h, 6 h, 24 h, and 3 days) for each pure potato secretome sample (Ali et al. in preparation), totaling 24 files, were added.
Table I. The first data set used to evaluate the alignment algorithm, consisting of linear mixes of the Desiree potato clone pre- and three days postinfection, respectively, with Phytophthora infestans. The technical replicates are ordered in each ratio cohort on date of acquisition. Two of the technical replicates in the first ratio cohort and one in the second are run almost 5 months after the rest.
| TimePoint | ||
|---|---|---|
| 0h Desiree + 3d Desiree | ||
| Technical Replicate | ||
| Ratio 1 | 0h | 1 |
| 2 | ||
| 3 | ||
| Ratio 2 | 2/3 0h + 1/3 3d | 1 |
| 2 | ||
| 3 | ||
| Ratio 3 | 1/3 0h + 2/3 3d | 1 |
| 2 | ||
| 3 | ||
| Ratio 4 | 3d | 1 |
| 2 | ||
| 3 |
Table II. The second data set used to evaluate the alignment algorithm, consisting of linear mixes of the Sarpo Mira and SW93–1015 potato clones 6 hours postinfection with Phytophthora infestans. The technical replicates are ordered in each ratio cohort on date of acquisition and run within a week's time.
| Clone | ||
|---|---|---|
| 6 h Sarpo Mira + 6h SW93–1015 | ||
| Technical Replicate | ||
| Ratio 1 | SW93–1015 | 1 |
| 2 | ||
| 3 | ||
| Ratio 2 | 2/3 SW-1015 + 1/3 Sarpo Mira | 1 |
| 2 | ||
| 3 | ||
| Ratio 3 | 1/3 SW-1015 + 2/3 Sarpo Mira | 1 |
| 2 | ||
| 3 | ||
| Ratio 4 | Sarpo Mira | 1 |
| 2 | ||
| 3 |
MS analysis was performed on a LTQ orbitrap XL mass spectrometer (Thermo Electron, Bremen, Germany) interfaced with an Eksigent nano-LC system (Eksigent technologies, Dublin, CA). A 5 μl sample was injected onto a fused silica emitter, 75 μm × 16 cm (PicoTipTM Emitter, New Objective, Inc.Woburn, MA), packed in-house with Reprosil-Pur C18-AQ resin (3 μm Dr. Maisch, GmbH, Germany) at a constant flow rate of 600 nl/min in three technical replicates for each ratio cohort. The peptides were subsequently eluted at a flow rate of 300 nl/min in a 90 min gradient of 3 to 35% (v/v) acetonitrile in water, containing 0.1% (v/v) formic acid. The LTQ-Orbitrap was operated in data-dependent mode to automatically switch between Orbitrap-MS (from m/z 400 to 2000) and LTQ-MS/MS acquisition. Four MS/MS spectra were acquired in the linear ion trap per each FT-MS scan, which was acquired at 60,000 FWHM (Full Width at Half Maximum) nominal resolution settings using the lock mass option (m/z 445.120025) for internal calibration. The dynamic exclusion list was restricted to 500 entries using a repeat count of two with a repeat duration of 20 s and with a maximum retention period of 120 s. Precursor ion charge state screening was enabled to select for ions with at least two charges and rejecting ions with undetermined charge state. The normalized collision energy was set to 35%, and one micro scan was acquired for each spectrum. Some of the injections were performed several months after the others, to simulate large projects that can typically not be run back-to-back. Information about acquisition time points is given with the raw data, available at the Swestore repository, as listed in Supplemental Table S1.
Files were converted to mzML (33) and Mascot Generic Format (MGF) using Proteowizard (34), and the mzML files have been deposited in the Swestore repository. MGF files were used for MS/MS identification, and mzML files for feature detection using msInspect and OpenMS. Identification searches were performed in Mascot 2.3.01 and X!Tandem Tornado 2008.12.01.1 with native scoring in a database consisting of all Solanum proteins in UniProt as of 2011–08-24 (http://www.uniprot.org) and all annotated proteins from the potato genome project (http://www.potatogenome.net (35)) plus reverse sequences and 11 common proteins, totaling 248627 sequences. One missed cleavage was allowed. Search tolerances were 5 ppm for precursors and 0.5 Da for MS/MS fragments. Fixed carbamidomethylation of cysteines and variable methionine oxidation were considered as modifications. After import of search results into Proteios, FDR was calculated for the combined searches using reverse sequences in Proteios (27). msInspect as well as OpenMS feature detection was performed directly from Proteios and the features were imported and matched to identifications within the same LC-MS/MS runs in Proteios using a retention time tolerance of 0.2 min, and a m/z tolerance of 0.005 Da as well as an FDR cutoff of 0.01. Alignment was performed directly in Proteios, and a report of the features was exported and further analyzed in MATLAB R2011a v. 7.12.0.635 (http://www.mathworks.org).
Comparison to msInspect and OpenMS
Feature lists were obtained using both the msInspect (build 633) and OpenMS (1.9.0) feature detection modules from within Proteios, and were used for the respective solution's alignment algorithms, as well as for the Proteios alignment.
msInspect feature detection was performed with default settings. For the alignment, the feature files were exported and aligned with the “–optimize” option, where the mass- and scan windows were set automatically to 0.025 Da and 400 scans, respectively, for both data sets. The resulting details.tsv file was used for further analysis.
OpenMS feature detection was run in two steps; the PeakPicker module was run with the “high resolution” option followed by the FeatureFinder module where “charge high” and “mz tolerance” in the “isotopic pattern” section were set to 6 and 0.005 Da, respectively, and the “min rt span” in the “feature” section was set to 1/3 min. The featureXML files were exported from Proteios and aligned by using the MapAlignerPoseClustering module followed by the FeatureLinkerUnlabeled module, both run with default settings to produce a consensusXML file. Finally, the TextExporter module was run with the “consensus feature” option to convert the consensusXML into a text file that was used for further analysis.
To compute a corresponding global identity-based recall and precision for the two software solutions, the features that were identified in all files before alignment were extracted from the Proteios feature table. The feature information (m/z, charge and filename) for this set was mapped back into the resulting msInspect and OpenMS files and the corresponding cluster ids (row number for the OpenMS file) were extracted. The cluster IDs were stored in a matrix where every column represents a sample and every row a cluster. The number of true positives for each row was computed as the most frequently occurring cluster ID (one for each file). The rest were considered false negatives, because they were not aligned into the selected cluster. The most frequent cluster ID was used to represent the best cluster and false positives were subsequently extracted for every ID, computed as the number of features in the cluster that were not considered a true positive.
RESULTS AND DISCUSSION
As quality control is an important issue for label-free LC-MS analysis, we implemented a complete label-free workflow with built-in quality control metrics in the form of precision and recall, which give an estimate of the sensitivity (recall) and false discovery rate (FDR, precision = 1 - FDR) for the alignment process. The quality of the results can thus be estimated using the metrics, which are computed as described in Fig. 2 for the aligned file pairs as well as globally for the entire file batch. Two forms of precision and recall are computed during several stages of the Proteios alignment, identity-based and occurrence-based, as summarized in Fig. 4. To illustrate the feasibility of the workflow we evaluated it with two data sets consisting of mixtures of biological samples in different proportions, as proposed previously (5), using features detected by two software solutions, msInspect and OpenMS. For Fig. 4, msInspect features were used.
Fig. 4.
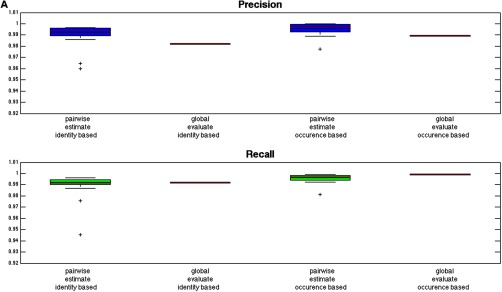
The reported precision and recall values for the Proteios alignment algorithm. The red line represents the median and the edges of the box the first and third quartile, respectively, for all the pairwise alignments in the data sets. Outliers are marked with “+ ” and deviate from the quartiles more than 1.5 times the interquartile range. Estimated values are calculated before alignment, and evaluated values are calculated afterward. Global evaluation values are reported as a red line. A, TimePoint data set (B) Clone data set. All metrics show high values. The general agreement between the pairwise and global values show that the alignment worked well even on batch scale for the parameters set during the pairwise matching. The agreement between the identity-based and occurrence-based values gives an indication of the alignment working equally well for features without identities, which is the majority, as for features with identity information. Furthermore, the generally lower values and larger interquartile range for the TimePoint data set implies a data set with larger file differences.
As seen in Fig. 4, the precision and recall values are high, as well as showing a high level of agreement between the pairwise and global values, indicating that the parameters set during the alignment resulted in the expected behavior, i.e., a high quality alignment. In general, the plot shows the global precision being slightly lower than the pairwise one and the opposite trend is seen for the global recall. This is expected and is because of the repeated matching of every file performed in the algorithm; there are as many possibilities for the feature to be aligned into the correct cluster as the number of times a file is aligned. However, the more times a file is aligned, the higher the probability of introducing false positives into the cluster. As previously mentioned and illustrated in supplemental Fig. S2, too many alignments of each file would only decrease precision at a very slight benefit for recall. Only a small decrease in the global precision compared with the pairwise ones was found, indicating that the files have not been overly aligned. It can also be noted that the TimePoint data set shows less agreement between the global and pairwise values as well as a larger spread in the estimated values, indicating a data set with larger differences and so more difficult to align. Furthermore, there is a high level of agreement between the occurrence-based and identity-based values. A good correspondence between these metrics implies that the feature matching is the same whether identity information is taken into account or not. Because most features have no sequence information, high agreement indicates that the alignment works as well for such features as for those with identity.
Alignment Evaluation
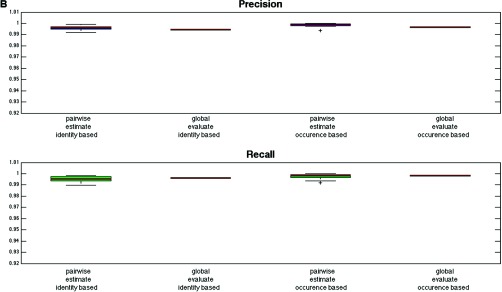
To investigate the effect of using parameter settings estimated from the data at hand as well as retention time correction aided by identity matches, the Proteios alignment was run with manually set tolerances, both when matching features and when extracting the feature set on which to base the retention time correction function. The identity-based global metrics from Fig. 4 are used for comparison in subsequent figures.
Parameter Estimation
The average mass and time tolerances used for alignment were 0.007 Da and 0.47 min, respectively, for the TimePoint data set and 0.008 Da and 0.39 min for the Clone set. Nevertheless, every file pair required its own specific settings. The mass-to-charge window spanned from 0.004 to 0.011 Da for the TimePoint data set and from 0.004 to 0.012 Da for the Clone data set. The retention time windows for the data sets were 0.15 to 0.76 min and 0.30 to 0.67 min, respectively. This shows that one tolerance is not sufficient to align sets of files and the tolerances set are even more critical if a file is aligned only once. This is of course also true for the use of a reference file, where every file will be aligned once to the reference.
The results of manually set tolerances can be seen in Fig. 5. Two sets of tolerances were chosen; one strict with a mass-to-charge and time tolerance of 0.005 Da and 0.1 min, respectively, and a wide tolerance with a mass-to-charge tolerance of 0.02 Da and time tolerance of 2 min. In general a strict tolerance will decrease the recall, but increase precision and the opposite will occur for a wide tolerance. As can be assumed, judging from the relatively wider adaptive tolerance span for the TimePoint data set, the strict tolerance affected the recall of this data set considerably more than Clone. The wide manually set tolerance increased the recall slightly but decreased the precision considerably compared with the adaptive tolerance for both data sets. To manually set a wide tolerance to make sure that as many features as possible are aligned may seem as an appropriate approach, but the inclusion of many false positives in the feature clusters can create large deviations in the feature quantities as these are summed up for the resulting peptide abundance. This in turn will lead to errors in protein quantification and unreliable results further down the workflow. To assess this, we also evaluated the effect of the parameter settings at the protein level (supplemental Fig. S3), and it was confirmed that considerable quantitative differences could be detected for the proteins in the data sets.
Fig. 5.

The deviations in identity-based recall and precision when using manually set parameters compared with adaptive for the TimePoint (A) and Clone (B) data set, respectively. As seen, the adaptive tolerance maximizes both metrics simultaneously.
Identity-aided Alignment
The effect of not using identifications as landmarks for the retention time correction function was investigated by aligning the data sets using the 10% most abundant features from every file. These features were initially matched with a mass-to-charge tolerance of 0.01 Da and retention time tolerance of 5 min to extract landmark pairs. Because no interpolation function has been estimated at this point, the time tolerance needs to be set wide enough not to miss any possible retention time drifts. Only unique pairs, i.e., those features that matched to a single other feature within the tolerance limits were selected. The similarity score was computed as the ratio of the number of matching features to the total number of the 10% set.
A comparison of an alignment of a file pair in the Clone data set using identity-aided and tolerance-aided alignment can be seen in Fig. 6. The file pair consists of two technical replicates from the second ratio cohort run on the same day. Nevertheless, a retention time difference window between the extremes of 2 min can be seen in the identity-aided regression function. Although the width of the window remains close to constant after alignment, the difference between the bulk of the points is considerably decreased and most fall within the estimated time tolerance. The retention time window for the identity-aided alignment is marked in the corresponding tolerance-aided plots and as can be seen, larger deviations were found using a tolerance-aided alignment. This in turn led to a larger time tolerance for the tolerance-aided alignment, whereas the m/z tolerance stayed constant.
Fig. 6.
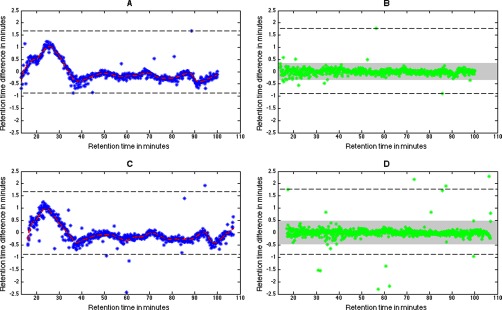
The estimated spline function between two technical replicates from the Clone data set is shown as a red dashed line where the blue markers correspond to time differences before alignment for the feature set used for interpolation. The retention time window for the identity-aided regression function is marked with a black dashed line. The right hand side figures show the time differences after applying the function to the feature set used for parameter estimation and the set time tolerance is seen in gray. The larger retention time differences seen for the tolerance-based approach (C, D), led to a larger estimated time tolerance compared with the identity-aided one (A, B). A shift in the total retention time span can also be seen in the x axis of the plots.
Fig. 7 shows the comparison of global precision and recall for the two alignment strategies. The largest differences for the two data sets can be seen in precision. This is because of the before mentioned increased time tolerance for the tolerance-aided strategy, which will have a similar effect to the wide tolerance seen in Fig. 5, albeit to less extent.
Fig. 7.
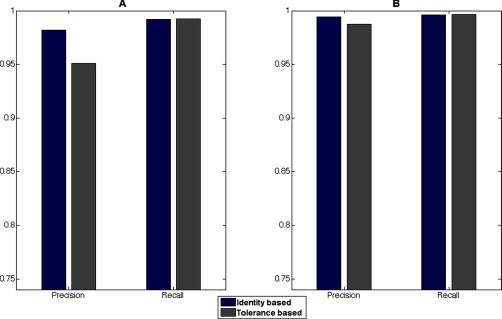
Global identity-based precision and recall for the TimePoint (A) and Clone (B) data set, respectively. There is less difference in the global precision and recall using a tolerance-aided retention time correction than using manual tolerances for the feature cluster extraction as seen in Fig. 5. However, a decrease, especially in precision, is seen because of the larger retention time differences leading to larger time tolerances set for feature cluster extraction for the tolerance-aided run. This can be compared with using only slightly too wide parameters for the same type of analysis as in Fig. 5.
It should be noted that the tolerance-aided and identity-aided approaches cover different intervals of the total retention time span as is shown in Fig. 6. The width of the span is however equivalent, ∼90 min for both of the strategies. To cover as large a time span as possible, it could be effective to combine the methods by using predominately an identity-aided alignment and add a tolerance-aided one outside of the retention time interval covered by the identifications. To maximize precision, the initial tolerances for the features to be used can be estimated from the identity-aided matches. In addition, such a combined approach can be used when there are few identifications in a file set.
Comparison to msInspect and OpenMS
The Proteios alignment was run with both msInspect and OpenMS features and the respective alignments compared using the same feature sets, i.e., msInspect alignment was compared with Proteios run on msInspect features and OpenMS alignment was compared with Proteios using OpenMS features. The alignments run in both msInspect and OpenMS use a reference-based approach.
As can be seen in Fig. 8, Proteios in general shows considerably higher precision and recall values for all sets. This can be because of a number of factors. First, the automatically selected reference files by msInspect and OpenMS were in the end point ratio cohort for both data sets. This could introduce an important amount of missed matches into the analysis using files containing very different samples as seen in (5). Furthermore, the default settings for the run OpenMS alignment module applies a linear retention time correction model, which may fail to correctly compensate for the retention time differences (36).
Fig. 8.
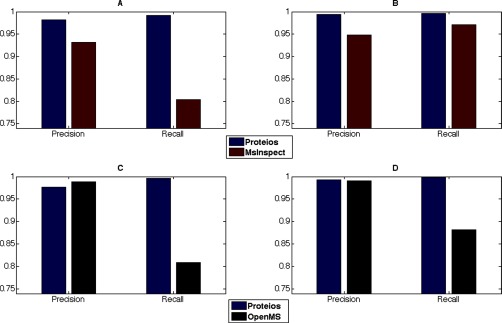
Identity-based recall and precision computed for both msInspect, OpenMS and Proteios. A, TimePoint data set (B) Clone data set for msInspect features. C, TimePoint data set and (D) Clone data set for OpenMS features. The general decrease of precision and recall for the TimePoint data set is most likely because of the timespan in which samples were run as well as one overall deviating file. The figure shows generally higher recall and precision values for the Proteios alignment, indicating a better quality analysis.
Analysis of the feature clusters used to compute precision and recall from msInspect showed that for the TimePoint data set an important amount of clusters (51%) contained 11 out of 12 possible files and 96% of those were because of systematic missing values in one file in the third ratio cohort. Corresponding analysis of the OpenMS clusters showed a similar distribution; 40% were missing one file and of those 98% were because of one specific file. This file alone is therefore responsible for introducing a considerable amount of errors in alignment and subsequently much of the decrease in true positives and associated recall seen in Fig. 8. Evidently, the computed matching order as well as repeated pairwise matching of every file in Proteios makes the software more robust against introduction of such missing values as seen by the higher recall in the figure. Furthermore, because the “–optimize” option in msInspect optimizes the number of clusters containing only one feature from each sample (25), a low number of false positives were found. Nevertheless, a handful of clusters containing more than 20 features were observed and contributed strongly to the comparative reduction in precision. For the OpenMS alignment, every cluster contained a maximum of 12 features, resulting in the high precision seen in the figure. This however, seems to have come at a cost of recall, even in the comparatively well-behaved Clone data set.
Quantification Evaluation
To assess the performance of the complete data analysis, the quantification evaluation in (5) was implemented. Two data sets were combined in linear ratio mixes as seen in Table I and Table II and the expected linear response evaluated. This method is used rather than spiked-in peptides, as using real samples makes it possible to test the analysis for the density, dynamic range and complexity seen in label-free data. The number of feature clusters with values in all files in the data set is an indicator of the sensitivity of the alignment (high recall), and the number of clusters that show the expected quantitative profile can be used to distinguish high quality clusters obtained by the algorithm (high precision). We evaluated the full Proteios workflow, as well as msInspect and OpenMS, using this strategy on both data sets. Furthermore, to evaluate the scalability of the Proteios algorithm, and its dependence on the number of files in a data set, we also analyzed the two datasets at the same time together with the other 24 files, totaling 48 files.
The two data sets were normalized by a scaling factor corresponding to the TIC of features with common identities in all files before alignment. For the Clone data set, the feature clusters containing features in all 12 files were analyzed. For the TimePoint data set, the number of feature clusters containing features in all 12 files was considerably lower for msInspect (156 clusters) and OpenMS (292 clusters) compared with Proteios (1763 and 969 clusters, with msInspect and OpenMS features, respectively). This is a result of the lower recall seen in Fig. 8 and shows Proteios' relatively larger capacity for aligning deviating samples. However, to increase the number of clusters for comparative quantification evaluation, the outlier file was disregarded and feature clusters containing features in at least 11 files were used for consecutive evaluation of the TimePoint data set. To evaluate the scaling capabilities, the two file sets were extracted from the large data set in the same manner.
As a first point for determining the quantification quality, a CV (coefficient of variation) cutoff was added to limit the variance allowed between technical replicates. This cutoff was set to 20%, based on the technical variability seen in (37). The data was subsequently log transformed using the natural logarithm and a least squares linear regression was performed between the different mixing ratios, to assess if the expected linear relation between the points was obtained. Linearity was estimated by the lack-of-fit sum-of-squares F-test (38). A low p value (<0.05 is used here) will lead to the rejection of the null hypothesis of a linear model, i.e., there is a systematic variation in the feature clusters that the linear model cannot account for. Quantification examples as well as their corresponding p values can be found in supplemental Figs. S4–S6. Corresponding R2 (coefficient of determination) values have also been computed for the examples for comparison and are displayed in the plots.
Fig. 9 shows the original number of clusters analyzed compared with the ones passing the CV cutoff and F-test, respectively. In Fig 9A and 9C it is seen that relatively few clusters in the TimePoint data set compared with the Clone data set passed the CV cutoff. Inspection of the data set showed that the first ratio cohort containing two recent and one older file was typically responsible for not passing the CV cutoff, this could perhaps be alleviated by a different normalization strategy.
Fig. 9.
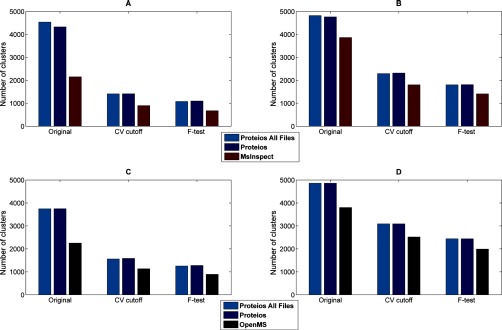
The number of feature clusters in every step of the quantification evaluation; number of clusters used for evaluation (original), number of clusters passing the CV cutoff (CV cutoff), and the number of clusters where the linear response could not be rejected at a significance level of 0. 05 (F-test). A, and (C) correspond to the TimePoint data set and (B) and (D) to the Clone data set. Features from msInspect were used in (A) and (B), whereas OpenMS features were used in (C) and (D). Proteios shows a higher number of clusters for both software comparisons, which is a result of substantially higher recall for the alignment. Overall, similar ratios of clusters are considered correctly quantified, which is an indication of the relatively lower difference in precision between the software solutions. The high agreement between Proteios runs using only the evaluated data set or the complete set of files shows the reproducibility of the analysis, irrespective of how many files are aligned at once.
The null hypothesis of linearity was rejected for 20–22% of the clusters that did pass the CV cutoff at a significance level of 0.05, irrespective of software run or data set. Only the msInspect run of the TimePoint data set had a slightly higher rejection rate of 25%. Because the alignment precision shown in Fig. 8 was high and differed relatively little between software and a shared feature cohort was used, a large difference in quantities for the features that are correctly aligned cannot be expected. The effect of the difference in alignment recall can however be seen in the lower amount of clusters compared with Proteios for both OpenMS and msInspect.
The high level of agreement between the two Proteios runs (12 files or 48 files) shows the reproducibility of the analysis when running larger sample cohorts. A slightly larger amount of feature clusters originally for the large data set can be expected as every file is aligned more often, but as can be seen, the difference disappears as the CV cut off is applied. This indicates that the number of times a file is aligned in the smaller data sets is sufficient to produce the same amount of good quality clusters.
In general, the total number of quantified clusters fulfilling the linearity and CV criteria is relatively low compared with the total number of clusters. This however does not mean that all other clusters are products of errors in alignment, but also reflect natural variation in the underlying data and artifacts introduced early in the analysis. Especially peak splitting during feature detection could contribute to increased variance and failure to pass the CV cutoff. There are furthermore difficulties with assessing quantification for low-abundance features close to the detection limit, and possibly a less strict CV cutoff should be used for these clusters. The abundance for such features may also be missing from the feature detection stage and affect the basis for quantification evaluation. The clusters containing as complete a set of features as possible are therefore the most informative in an evaluation situation and as seen above, the Proteios alignment algorithm resulted in a comparatively large as well as reproducible number of high quality clusters.
In conclusion, we have presented an integrated workflow for label-free LC-MS which utilizes quality-control to optimize parameter settings, and also to evaluate the analysis outcome. No spiked-in or specific sets of samples are necessary to overview the data analysis as we introduce data set-independent quality metrics to monitor the performance of the system. Using this setup we integrated a novel adaptive alignment algorithm into the Proteios Software Environment. The parameter-free environment guarantees a reproducible analysis for a sample cohort as well as user-friendliness. Incorporation of real time quality control in combination with a computed matching order in the alignment algorithm minimizes possible errors in alignment. This ensures the reliable peptide and subsequent protein quantification necessary for robust statistical analysis in differential expression proteomics studies.
Supplementary Material
Footnotes
* This work was supported by the Swedish Foundation for Strategic Research (RBb08-0006) and the Swedish Research Council (project 2007-5188). Support by BILS (Bioinformatics Infrastructure for Life Sciences) is gratefully acknowledged.
 This article contains supplemental Figs. S1 to S6 and Tables S1 to S3.
This article contains supplemental Figs. S1 to S6 and Tables S1 to S3.
1 The abbreviations used are:
- LC
- liquid chromatography
- CV
- coefficient of variation
- FDR
- false discovery rate
- LOWESS
- locally weighted scatterplot smoothing
- LTQ
- linear trap quadrupole
- OMSSA
- open mass spectrometry search algorithm
- R2
- coefficient of determination
- TIC
- total ion current.
REFERENCES
- 1. Neilson K. A., Ali N. A., Muralidharan S., Mirzaei M., Mariani M., Assadourian G., Lee A., van Sluyter S. C., Haynes P. A. (2011) Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics 11, 535–553 [DOI] [PubMed] [Google Scholar]
- 2. America A. H., Cordewener J. H. (2008) Comparative LC-MS: a landscape of peaks and valleys. Proteomics 8, 731–749 [DOI] [PubMed] [Google Scholar]
- 3. Zhu W. H., Smith J. W., Huang C. M. (2010) Mass Spectrometry-Based Label-Free Quantitative Proteomics. J. Biomed. Biotechnol. doi:10.1155/2010/840518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Listgarten J., Emili A. (2005) Statistical and computational methods for comparative proteomic profiling using liquid chromatography-tandem mass spectrometry. Mol. Cell. Proteomics 4, 419–434 [DOI] [PubMed] [Google Scholar]
- 5. Sandin M., Krogh M., Hansson K., Levander F. (2011) Generic workflow for quality assessment of quantitative label-free LC-MS analysis. Proteomics 11, 1114–1124 [DOI] [PubMed] [Google Scholar]
- 6. Hoekman B., Breitling R., Suits F., Bischoff R., Horvatovich P. (2012) msCompare: A Framework for Quantitative Analysis of Label-free LC-MS Data for Comparative Candidate Biomarker Studies. Mol. Cell. Proteomics 11, M111.015974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Colaert N., Van Huele C., Degroeve S., Staes A., Vandekerckhove J., Gevaert K., Martens L. (2011) Combining quantitative proteomics data processing workflows for greater sensitivity. Nat. Methods 8, 481–483 [DOI] [PubMed] [Google Scholar]
- 8. Old W. M., Meyer-Arendt K., Aveline-Wolf L., Pierce K. G., Mendoza A., Sevinsky J. R., Resing K. A., Ahn N. G. (2005) Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 4, 1487–1502 [DOI] [PubMed] [Google Scholar]
- 9. Li Z., Adams R. M., Chourey K., Hurst G. B., Hettich R. L., Pan C. L. (2012) Systematic Comparison of Label-Free, Metabolic Labeling, and Isobaric Chemical Labeling for Quantitative Proteomics on LTQ Orbitrap Velos. J. Proteome Res. 11, 1582–1590 [DOI] [PubMed] [Google Scholar]
- 10. Michalski A., Cox J., Mann M. (2011) More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J. Proteome Res. 10, 1785–1793 [DOI] [PubMed] [Google Scholar]
- 11. Khan Z., Bloom J. S., Garcia B. A., Singh M., Kruglyak L. (2009) Protein quantification across hundreds of experimental conditions. Proc. Natl. Acad. Sci. U.S.A. 106, 15544–15548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang H., Alvarez S., Hicks L. M. (2012) Comprehensive comparison of iTRAQ and label-free LC-based quantitative proteomics approaches using two Chlamydomonas reinhardtii strains of interest for biofuels engineering. J. Proteome Res. 11, 487–501 [DOI] [PubMed] [Google Scholar]
- 13. Schilling B., Rardin M. J., MacLean B. X., Zawadzka A. M., Frewen B. E., Cusack M. P., Sorensen D. J., Bereman M. S., Jing E., Wu C. C., Verdin E., Kahn C. R., Maccoss M. J., Gibson B. W. (2012) Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: application to protein acetylation and phosphorylation. Mol. Cell. Proteomics 11, 202–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lange E., Gropl C., Reinert K., Kohlbacher O., Hildebrandt A. (2006) High-accuracy peak picking of proteomics data using wavelet techniques. Pac. Symp. Biocomput. 243–254 [PubMed] [Google Scholar]
- 15. Zhang J., Gonzalez E., Hestilow T., Haskins W., Huang Y. (2009) Review of Peak Detection Algorithms in Liquid-Chromatography-Mass Spectrometry. Curr. Genomics 10, 388–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vandenbogaert M., Li-Thiao-Té S., Kaltenbach H. M., Zhang R., Aittokallio T., Schwikowski B. (2008) Alignment of LC-MS images, with applications to biomarker discovery and protein identification. Proteomics 8, 650–672 [DOI] [PubMed] [Google Scholar]
- 17. Zhang R., Barton A., Brittenden J., Huang J., Crowther D. (2010) Evaluation for computational platforms of LC-MS based label-free quantitative proteomics: a global view. J. Proteomics Bioinform. 3, 260–265 [Google Scholar]
- 18. Valot B., Langella O., Nano E., Zivy M. (2011) MassChroQ: a versatile tool for mass spectrometry quantification. Proteomics 11, 3572–3577 [DOI] [PubMed] [Google Scholar]
- 19. Ballardini R., Benevento M., Arrigoni G., Pattini L., Roda A. (2011) MassUntangler: a novel alignment tool for label-free liquid chromatography-mass spectrometry proteomic data. J Chromatogr A 1218, 8859–8868 [DOI] [PubMed] [Google Scholar]
- 20. Pluskal T., Castillo S., Villar-Briones A., Oresic M. (2010) MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Voss B., Hanselmann M., Renard B. Y., Lindner M. S., Köthe U., Kirchner M., Hamprecht F. A. (2011) SIMA: simultaneous multiple alignment of LC/MS peak lists. Bioinformatics 27, 987–993 [DOI] [PubMed] [Google Scholar]
- 22. Tautenhahn R., Böttcher C., Neumann S. (2008) Highly sensitive feature detection for high resolution LC/MS. BMC Bioinformatics 9, 504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lange E., Tautenhahn R., Neumann S., Gröpl C. (2008) Critical assessment of alignment procedures for LC-MS proteomics and metabolomics measurements. BMC Bioinformatics 9, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Häkkinen J., Vincic G., Mansson O., Wårell K., Levander F. (2009) The proteios software environment: an extensible multiuser platform for management and analysis of proteomics data. J. Proteome Res. 8, 3037–3043 [DOI] [PubMed] [Google Scholar]
- 25. Bellew M., Coram M., Fitzgibbon M., Igra M., Randolph T., Wang P., May D., Eng J., Fang R., Lin C., Chen J., Goodlett D., Whiteaker J., Paulovich A., McIntosh M. (2006) A suite of algorithms for the comprehensive analysis of complex protein mixtures using high-resolution LC-MS. Bioinformatics 22, 1902–1909 [DOI] [PubMed] [Google Scholar]
- 26. Sturm M., Bertsch A., Gröpl C., Hildebrandt A., Hussong R., Lange E., Pfeifer N., Schulz-Trieglaff O., Zerck A., Reinert K., Kohlbacher O. (2008) OpenMS - an open-source software framework for mass spectrometry. BMC Bioinformatics 9, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Levander F., Krogh M., Warell K., Gärden P., James P., Häkkinen J. (2007) Automated reporting from gel-based proteomics experiments using the open source Proteios database application. Proteomics 7, 668–674 [DOI] [PubMed] [Google Scholar]
- 28. MacLean B., Eng J. K., Beavis R. C., McIntosh M. (2006) General framework for developing and evaluating database scoring algorithms using the TANDEM search engine. Bioinformatics 22, 2830–2832 [DOI] [PubMed] [Google Scholar]
- 29. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 30. de Groot J. C., Fiers M. W., van Ham R. C., America A. H. (2008) Post alignment clustering procedure for comparative quantitative proteomics LC-MS data. Proteomics 8, 32–36 [DOI] [PubMed] [Google Scholar]
- 31. Cleveland W. S. (1979) Robust locally weighted regression and smoothing scatterplots. J. Am. Stat. Assoc. 74, 829–836 [Google Scholar]
- 32. Ali A., Moushib L. I., Lenman M., Levander F., Olsson K., Carlson-Nilson U., Zoteyeva N., Liljeroth E., Andreasson E. (2012) Paranoid potato: Phytophthora-resistant genotype shows constitutively activated defense. Plant Signal Behav 7, 400–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martens L., Chambers M., Sturm M., Kessner D., Levander F., Shofstahl J., Tang W. H., Römpp A., Neumann S., Pizarro A. D., Montecchi-Palazzi L., Tasman N., Coleman M., Reisinger F., Souda P., Hermjakob H., Binz P. A., Deutsch E. W. (2011) mzML–a community standard for mass spectrometry data. Mol. Cell. Proteomics 10, R110 000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kessner D., Chambers M., Burke R., Agus D., Mallick P. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu X., Pan S., Cheng S., Zhang B., Mu D., Ni P., Zhang G., Yang S., Li R., Wang J., Orjeda G., Guzman F., Torres M., Lozano R., Ponce O., Martinez D., De la Cruz G., Chakrabarti S. K., Patil V. U., Skryabin K. G., Kuznetsov B. B., Ravin N. V., Kolganova T. V., Beletsky A. V., Mardanov A. V., Di Genova A., Bolser D. M., Martin D. M., Li G., Yang Y., Kuang H., Hu Q., Xiong X., Bishop G. J., Sagredo B., Mejia N., Zagorski W., Gromadka R., Gawor J., Szczesny P., Huang S., Zhang Z., Liang C., He J., Li Y., He Y., Xu J., Zhang Y., Xie B., Du Y., Qu D., Bonierbale M., Ghislain M., Herrera Mdel R., Giuliano G., Pietrella M., Perrotta G., Facella P., O'Brien K., Feingold S. E., Barreiro L. E., Massa G. A., Diambra L., Whitty B. R., Vaillancourt B., Lin H., Massa A. N., Geoffroy M., Lundback S., DellaPenna D., Buell C. R., Sharma S. K., Marshall D. F., Waugh R., Bryan G. J., Destefanis M., Nagy I., Milbourne D., Thomson S. J., Fiers M., Jacobs J. M., Nielsen K. L., Sonderkaer M., Iovene M., Torres G. A., Jiang J., Veilleux R. E., Bachem C. W., de Boer J., Borm T., Kloosterman B., van Eck H., Datema E., Hekkert B. L., Goverse A., van Ham R. C., Visser R. G. (2011) Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195 [DOI] [PubMed] [Google Scholar]
- 36. Podwojski K., Fritsch A., Chamrad D. C., Paul W., Sitek B., Stühler K., Mutzel P., Stephan C., Meyer H. E., Urfer W., Ickstadt K., Rahnenfuhrer J. (2009) Retention time alignment algorithms for LC/MS data must consider non-linear shifts. Bioinformatics 25, 758–764 [DOI] [PubMed] [Google Scholar]
- 37. Teleman J., Karlsson C., Waldemarson S., Hansson K., James P., Malmström J., Levander F. (2012) Automated selected reaction monitoring software for accurate label-free protein quantification. J. Proteome Res. 11, 3766–3773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Montgomery D. C. (2005) Design and Analysis of Experiments, 6th Ed., John Wiley & Sons Inc., Hoboken, NJ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.