

Background: ATP-dependent proteases translocate and unfold their substrates.
Results: A human virus sequence with only Gly and Ala residues causes similar dysfunctions of eukaryotic and prokaryotic protease motors: unfolding failure.
Conclusion: Sequences with amino acids of simple shape and small size impair unfolding of contiguous stable domains.
Significance: Compartmented ATP-dependent proteases of diverse origin share conserved principles of interaction between translocase/effector and substrate/recipient.
Keywords: ATP, ATP-dependent Protease, ATPases, Protease, Proteasome, Protein Degradation, Protein Turnover, Translocation, Unfolding
Abstract
ATP-dependent proteases engage, translocate, and unfold substrate proteins. A sequence with only Gly and Ala residues (glycine-alanine repeat; GAr) encoded by the Epstein-Barr virus of humans inhibits eukaryotic proteasome activity. It causes the ATPase translocase to slip on its protein track, stalling unfolding and interrupting degradation. The bacterial protease ClpXP is structurally simpler than the proteasome but has related elements: a regulatory ATPase complex (ClpX) and associated proteolytic chamber (ClpP). In this study, GAr sequences were found to impair ClpXP function much as in proteasomes. Stalling depended on interaction between a GAr and a suitably spaced and positioned folded domain resistant to mechanical unfolding. Persistent unfolding failure results in the interruption of degradation and the production of partial degradation products that include the resistant domain. The capacity of various sequences to cause unfolding failure was investigated. Among those tested, a GAr was most effective, implying that viral selection had optimized processivity failure. More generally, amino acids of simple shape and small size promoted unfolding failure. The ClpX ATPase is a homohexamer. Partial degradation products could exit the complex through transient gaps between the ClpX monomers or, alternatively, by backing out. Production of intermediates by diverse topological forms of the hexamer was shown to be similar, excluding lateral escape. In principle, a GAr could interrupt degradation because 1) the translocase thrusts forward less effectively or because 2) the translocase retains substrate less well when resetting between forward strokes. Kinetic analysis showed that the predominant effect was through the second of these mechanisms.
Introduction
ATP-dependent proteases share a common architecture (1). Proteolysis takes place in a hollow complex with narrow entry ports of sufficient diameter to admit an extended polypeptide but not native folded domains. A second complex consists of or contains an ATPase ring, which moves substrates into the degradation chamber. Because cellular proteins targeted for degradation may contain folded domains, the ATPase must act as both a translocase and unfoldase. The various elements of the protease that propel and unfold substrate have to engage in coordinated motions, grasping, unfolding, translocating, and finally clearing a path into the proteolytic chamber. Translocation and unfolding by the ATPase are thus coupled. The prevalent model of unfolding is that tugging by the ATPase ring on a region of substrate within the ring transmits a pulling force to a connected folded domain. Because the substrate constitutes a continuous connected polypeptide chain, traction is exerted on the folded domain, which, by hypothesis, is lodged at a site of constriction. Persistent traction stochastically causes unfolding and domain translocation.
In ATP-dependent proteases of bacteria, ATP binding and hydrolysis are coupled to the movement of axially positioned loops that engage and propel substrate toward proteolysis sites. Degradation is impaired by reducing ATPase activity or mutating conserved residues positioned at the tips of the axial loops that propel substrate, especially a conserved aromatic residue that presumably makes direct contact with substrate. Mutagenesis of a key aromatic residue, commonly Tyr, in the axial loops of both eukaryotic proteasomes (2) and bacterial proteases (3, 4) reduces degradation. It is likely that these mutations do so by reducing the ability of the ATPase motor to deliver a translocation force, but no experimental data provide direct comparison of force delivery by wild type versus mutated motors. Force-dependent translocation by the wild type bacterial protease ClpXP has been measured in optical trap experiments (5, 6). These studies have shown that ClpXP can act with a stall force in the range of ∼20–30 piconewtons and moves substrate in single steps of 4–8 amino acids.
Alternate but less direct means for answering questions of force transmission have been developed and applied to both bacterial proteases (7) and to eukaryotic proteasomes (8). These studies employed enzyme kinetic investigations of substrates that contain folded domains. Degrading these can require multiple rounds of ATP hydrolysis, each with a low probability of unfolding success. Using the very stable protein domain titin I27, persistent unfolding attempts were found to require hydrolysis of hundreds or thousands of ATP molecules before unfolding and subsequent substrate proteolysis is observed. Continued tugging on a mechanically stable substrate decreases the height of the energy barrier for its unfolding and eventually allows thermal fluctuations of the folded domain to cross a critical energy barrier for loss of structural integrity (9). Mutations of the conserved Tyr residue in bacterial proteases (3, 4) and of some (but not all) of the homologous residues of the six proteasome ATPases (2) increase the probability of unfolding failure or increase the time needed for unfolding.
In both bacterial proteases and the proteasome, persistent unsuccessful unfolding can be interrupted by dissociation of substrate from the motor (10, 11). This is likely to be the result of slipping of the motor on its polypeptide track. Slipping is more probable as the motor approaches stall force (6). When the motor encounters a sufficiently intractable domain, it can dissociate fully from its substrate track. Such a dissociation event is readily scored using a multidomain substrate that is degraded processively, starting strictly at one end (12). For example, if there are two domains and one has a tag at its C terminus where degradation starts and then continues, non-processive degradation events can be scored by measuring the generation of products that are missing the C-terminal domain but retain the N-terminal domain (10, 13–16). If the tag needed to initiate degradation is at the C terminus, degradation of the intermediate product cannot be reinitiated because the tag has been destroyed. Quantitative scoring of processing events that go to completion versus those that are interrupted is thus possible.
We have previously carried out studies with proteasomes to investigate whether substrate composition influences translocase activity. A Gly-Ala repeat (GAr)4 is present in Epstein-Barr virus nuclear antigen-1 (EBNA1), and its presence can inhibit proteasomal degradation of the protein (17). Previous studies have shown that a tightly folded adjacent domain collaborates with a GAr to generate degradation intermediates (15). Substrate tracts consisting only of repeated glycine and alanine residues (GArs) were found to impair the grip of the translocase. A GAr must collaborate with a tightly folded domain to generate degradation intermediates. Domains of greater mechanical stability resulted in more intermediates, and longer GAr tracts produced more intermediates than did shorter ones. For a short GAr, an optimal spacing along the polypeptide track between a folded domain and GAr was needed for intermediate production. It was concluded from these observations that a GAr is slippery and fails to adequately engage the translocase. Translocation nonetheless proceeds, unless the translocase is simultaneously presented with a GAr and the requirement to unfold a difficult load. Critical spacing between a tightly folded domain and GAr is postulated to be important because the folded cargo must be paused where it unfolds at the same time the GAr arrives in the axis of the ATPase ring.
Here these studies are extended to ClpXP. The bacterial protease is much simpler in structure than proteasomes and very distant in evolution. Using a series of substrates of systematically varied composition, the requirements for productive interaction with the ClpXP translocase were tested. Despite their great differences of taxonomy and structural complexity, proteasomes and ClpXP displayed similar enhancements of intermediate generation in response to folded substrates and a GAr. This suggests that highly conserved functional characteristics among such ATPase motors determine alternative outcomes of substrate processing.
EXPERIMENTAL PROCEDURES
Plasmid and Protein Preparation
ClpXΔN Monomer, Covalently Linked ClpX6ΔN, and ClpP
All ClpX and ClpP proteins were of Escherichia coli origin. ClpXΔN monomer (pACYC-Duet-1-ClpXΔN) and covalently linked hexamer (pACYC-Duet-1-ClpX6ΔN) plasmids were kindly provided by Dr. T. Baker (MIT) and the plasmid expressing ClpP with N-terminal His-tag (pCPX01) was a gift of Dr. H. Nakai (Georgetown University Medical Center).
Monomeric ClpX and ClpP were expressed in BL21(DE3) (Stratagene). Cells were grown at 37 °C to A600 = 0.6, expression was induced by 0.5 mm isopropyl β-d-1-thiogalactopyranoside, and cells were allowed to grow at 30 °C for 3 h before harvesting. Monomeric ClpX was purified by Ni2+-NTA chromatography as described (18), followed by mono Q ion exchange chromatography (GE Healthcare). ClpP was purified by Ni2+-NTA chromatography as described (19).
Closed Circular Hexameric ClpXΔN
[T66C/T388C]·ClpX3ΔN·AviTag is a trimer of ClpX connected by a 20-residue linker, with a T66C mutation in the N-terminal ClpX copy, a T388C mutation in the C-terminal ClpX (as described in Ref. 20), and an AviTag targeting site (GLNDIFEAQKIEWHE) for enzymatic biotinylation by BirA (biotin ligase) inserted at the C terminus. The genes of BirA and ClpX trimer were constructed by PCR mutagenesis and cloned into separate multiple cloning sites in a pET-Duet vector (Novagen). The trimer and BirA protein were co-expressed in E. coli BLR (DE3) cells (Novagen) and incubated with 50 μm biotin and 0.5 mm isopropyl β-d-1-thiogalactopyranoside at room temperature overnight. Cells were sonicated in lysis buffer (25 mm potassium phosphate buffer, pH 7.0, 300 mm NaCl, 10% glycerol, and 5 mm DTT). Biotinylated ClpX trimer was affinity-purified by monomeric avidin resin (Thermo Scientific). The column was washed with 5 bed volumes of lysis buffer, and the protein was eluted with 25 mm potassium phosphate buffer, pH 7.0, 300 mm NaCl, 10% glycerol, and 2.5 mm biotin. ClpX trimer was diluted to 1 μm and incubated with 4 mm ATP, 4 mm MgCl2, and 20 μm copper phenanthroline at 4 °C overnight to induce disulfide bond formation. Oxidized protein was purified by mono Q ion exchange chromatography followed by size exclusion chromatography on Superose 6 (GE Healthcare).
GAr30 and Dihydrofolate Reductase (DHFR) Substrates
GST-I27-GAr30-GFP-ssrA and GST-I27-GAr30-GFP-ssrA/DD were assembled by sortase fusion (21, 22) from their component proteins. GST-I27-GAr30-LPETGG-His, GGG-GFP-ssrA, and GGG-GFP-ssrA/DD were expressed individually and ligated by sortase A transpeptidase from Staphylococcus aureus (SrtA). The plasmid encoding I27-GAr30-LPETGG-His was constructed by PCR mutagenesis and cloned into pGEX-4T-1. Expression and purification of GST-I27-GAr30-LPETGG-His was as for GST-I27-test sequence-GFP-ssrA. GGG-GFP-ssrA and GGG-GFP-ssrA/DD were cloned into pET28b expression vector (Novagen) and expressed in E. coli strain BL21(DE3) (Stratagene) at 30 °C overnight. GGG-GFP-ssrA and GGG-GFP-ssrA/DD were purified based on Ref. 23. Briefly, a cell pellet with expressed protein was lysed in 20 mm Tris, pH 8.0, 1 mm EDTA, and 1 mg/ml lysozyme by freeze and thaw cycles. Protein was then extracted by stepwise precipitation using triethanolamine and ammonium sulfate, followed by organic extraction. The organic chemical was removed by hydrophobic chromatography using a HighPrep PhenylFF column (GE Healthcare). The plasmid expressing SrtA with His tag was a gift from Dr. H. Ploegh (MIT, Cambridge, MA). The expression and purification of SrtA-His were performed as described (22).
Full-length GST-I27-GAr30-GFP-ssrA and GST-I27-GAr30-GFP-ssrA/DD were produced by SrtA-His sortase-mediated protein fusion. GST-I27-GAr30-LPETGG-His was incubated with GGG-GFP-ssrA or GGG-GFP-ssrA/DD in 50 mm Tris, pH 7.0, 150 mm NaCl, and 5 mm CaCl2 at 4 °C overnight. GST-I27-GAr30-LPETGG-His and SrtA-His were removed by Ni2+-NTA resin, and the full-length GST-I27-GAr30-GFP-ssrA and GST-I27-GAr30-GFP-ssrA/DD were further purified by glutathione-Sepharose (GE Healthcare) to remove unreacted GGG-GFP-ssrA. DHFR-GAr30-GFP-ssrA and DHFR-control30-GFP-ssrA were generated by the same strategy as GST-I27-GAr30-GFP-ssrA, except human DHFR was used instead of titin I27, and the GST tag was removed by PreScission protease (GE Healthcare) cleavage.
GAr and Additional Test Sequence Substrates
GFP-ssrA was cloned into the pGEX-4T-1 vector (GE Healthcare) to generate pGEX-4T-1-SalI-NcoI-GFP-ssrA. Substrates with different GAr or other test sequences and titin I27 were constructed by PCR mutagenesis and cloned into pGEX-4T-1-GFP-ssrA between the SalI and NcoI restriction sites using conventional methods. The substrates were expressed in E. coli strain BL21(DE3) (Stratagene) and induced at 30 °C overnight with isopropyl β-d-1-thiogalactopyranoside. Bacterial cells were lysed, and GST fusion proteins were purified by glutathione-Sepharose affinity chromatography (GE Healthcare) in 25 mm Tris, pH 7.5, 300 mm NaCl, 10% glycerol.
Biochemical Assay of Substrate Degradation and Quantitation of Intermediate Degradation Products
Fluorescent Labeling of Proteins
200 μg of substrate or BSA was labeled with Cy3-N-hydroxysuccinimide ester or Cy5-N-hydroxysuccinimide ester (GE Healthcare) according to the manufacturer's protocol. Briefly, the buffer was exchanged to 0.1 m NaHCO3, pH 8.3, in a 40,000 molecular weight cut-off Zeba spin desalting column (Thermo Scientific) and then incubated with Cy3- or Cy5-N-hydroxysuccinimide ester in a 20:1 molar ratio (dye/protein) at room temperature for 1 h in the dark. Labeled protein was buffer-exchanged to 20 mm Tris, pH 7.5, 150 mm NaCl, 10% glycerol by a spin desalting column to stop the reaction and remove free fluor. Protein/fluor stoichiometry of labeling was determined to be 1:1.4.
Degradation Assay
2 μm Cy3-labeled substrate was incubated with 0.3 μm ClpX hexamer and 0.7 μm ClpP tetradecamer at 30 °C in PD buffer (25 mm HEPES, pH 7.5, 100 mm KCl, 20 mm MgCl2, and 10% glycerol) with an ATP-regenerating system (2.5 mm ATP, 10 units of pyruvate kinase/l-lactate dehydrogenase (Sigma)/ml, 6 mm phosphoenolpyruvate). 1 μm Cy5-BSA was added as loading control. Time-dependent degradation was determined by collecting aliquots periodically, and the reaction was stopped by the addition of SDS loading buffer. Aliquots were fractionated by 10% SDS-PAGE, and the Cy3- and Cy5-labeled gel bands were visualized by fluorescence imaging (Fuji FLA-5100). Intensities of labeled bands corresponding to the substrate and partial degradation product were quantified using TotalLab software (Nonlinear Dynamics). Band intensities of Cy3-labeled substrate were normalized according to Cy5-labeled BSA band intensity of individual lanes. The intensity of the parent full-length substrate band at time t = 0, P0, was used as the 100% reference point for quantitation of other bands. The percentage of remaining parent band at different time points was calculated as %Pt = 100% × (Pt/P0). For quantitation of intermediate degradation products as a percentage of %Pt, the measured intermediate band intensity at the indicated time point, %It, was calculated as %It = 100% × 1/0.60 × (It − I0)/P0, where I0 is the background at time 0, and 0.60 is a factor to adjust the stoichiometric ratio of substrate and intermediate, due to their difference in fluor label intensity. This ratio was measured by comparing the band intensity of the full-length substrate containing an enterokinase cleavage site, GST-I27-DDDDKDDDDK-GFP-ssrA, and that of the GST-titin fragment after enterokinase digestion. Label intensity of the two fragments was consistent with their relative molecular masses, reflecting uniform random chemical fluor labeling of accessible reactive groups. The percentage of remaining full-length substrate (%Pt) and the percentage of intermediate generated (%It) were plotted against time to determine the rate of substrate degradation and the yield of intermediate product (intermediate fraction) as a percentage of processed full-length substrate.
Correlation of Amino Acid Properties and Intermediate Generation
The numerical values of amino acid properties selected for analysis are given in Table 2. Using the series of test sequences consisting of Ala4-X-Ala5, where X is a distinct amino acid, linear regression was performed to analyze the relationship between the percentage of intermediate generated and the various physical and chemical parameters of amino acid X. The correlation coefficients and the p value were calculated for each property. Values with p < 0.05 were considered to be statistically significant.
TABLE 2.
Numerical values of selected amino acid properties
| Amino acid | Properties |
||||
|---|---|---|---|---|---|
| Molecular weight | Shapea | Flexibilityb | Hydrophobicityc | Isoelectric pointd | |
| Ala | 89 | 0 | 0 | 1.8 | 6.00 |
| Arg | 174 | 5 | 5 | −4.5 | 11.15 |
| Asp | 133 | 2 | 2 | −3.5 | 2.77 |
| Glu | 147 | 3 | 3 | −3.5 | 3.22 |
| Gly | 75 | 0 | 0 | −0.4 | 5.97 |
| His | 155 | 2 | 2 | −3.2 | 7.47 |
| Lys | 146 | 0 | 4 | −3.9 | 9.59 |
| Phe | 165 | 2 | 2 | 2.8 | 5.48 |
| Ser | 105 | 0 | 1 | −0.8 | 5.68 |
| Trp | 204 | 2 | 2 | −0.9 | 5.89 |
| Tyr | 181 | 2 | 2 | −1.3 | 5.66 |
| Val | 117 | 1 | 1 | 4.2 | 5.96 |
Determination of Rate Constants for Resolution of the Complex of ClpXP with Partial Degradation Products
Single-turnover reaction conditions were used that favored the formation of ClpXP engaged with partially degraded substrates (Enz·I27-frag). 1 μm Cy3-labeled GST-I27-GAr10-GFP-ssrA or GST-I27-control10-GFP-ssrA substrate was incubated with 2 μm ClpX hexamer and 4 μm ClpP tetradecamer at 30 °C in PD with an ATP regeneration system. 5 μm GFP-ssrA was added immediately before t = 4 min as a competitive inhibitor to assure that unprocessed GAr10 or control10 substrates did not subsequently enter the Enz·I27-frag pool. 1 μm Cy5-labeled BSA was included as a loading control. Time-dependent degradation of substrate and accumulation of intermediates was followed as described under “Degradation Assay.”
To calculate the rate constants that characterize resumption of processive degradation of the I27-frag by Enz·I27-frag, the data were analyzed as follows. Degradation of associated I27-frag by the complex Enz·I27-frag was regarded as beginning at t = 4 min., immediately after the addition of the competitive inhibitor, a time that coincides with the peak of intermediate abundance. At that time, intermediates constitute two pools: those I27-frag molecules in association with ClpXP and those that had previously been released from the enzyme. The decline in intermediate abundance after t = 4 min is due to degradation by Enz·I27-frag. Determining the first order rate constant of this degradative process (kproc) requires estimating the pool of intermediate present at t = 4 min plus that which exits the enzyme without further processing during the subsequent reaction period. That pool size can be regarded as the intermediate pool present near the end of the reaction, period, int(end). int(end) was determined as the mean of intermediate abundance present late in the reaction course, t = 20, 25, and 30 min. The half-life (t½) and other kinetic parameters for processive degradation by Enz·I27-frag were determined as follows. From each data point describing the total amount of intermediate present, inttotal(t), early in the time course (t = 4–11 min), inttotal(t) − int(end) was calculated to determine intproc(t), the amount of I27-frag not yet degraded by Enz·I27-frag at time t. These data were normalized to a scale of 0.0–1.0 by calculating the equation, (intproc(t) − int(end))/(intproc(0) − int(end)) = normintproc(t).
The log10 values of normintproc(t) were determined, and their time slope was calculated by linear regression. In this way, values were obtained (for both GAr10 and control10 substrates) of the half-life t½, the mean lifetime τ = t½/ln2, and λ = 1/τ, where λ is the decay constant (N/No = e−λt), which here constitutes the first order rate constant, kproc, for degradation of the I27-frag by the Enz·I27-frag complex.
kout, the rate constant for dissociation of I27-frag from Enz·I27-frag, was calculated from the relationship, intermediate fraction = kout/(kout + kproc).
RESULTS
GAr Must Collaborate with a Tightly Folded Domain to Generate Degradation Intermediates
ClpXP and other proteases that contain an AAA+ ATPase can initiate degradation at either end of a polypeptide substrate or at its interior (24). In proteasomes, the interaction between a folded domain and a GAr that generates intermediates requires specific spacing and orientation of those two elements. The GAr must engage the ATPase ring translocase first while the trailing folded domain is paused, awaiting unfolding and subsequent passage through the translocase (15). To test the relevance of this paradigm to ClpXP, we designed substrates (Fig. 1) for which degradation initiates at the C-terminal ssrA tag and then continues through the adjacent GFP domain, with subsequent translocation next proceeding through a GAr (or alternate test sequence) and then through a folded domain. The C-terminal SsrA peptide is an 11-residue peptide tag that interacts with ClpX. Substrates of both proteasomes and ClpXP require in addition to an association domain (such as ssrA) a second structural element, one that initiates invasion of the protease (8, 11, 16). Processive degradation begins at such an initiator element, which has a minimum chain length and is weakly structured or unstructured (16, 25). The substrates used in the experiments described here are designed to bind and initiate degradation strictly at their C terminus. In particular, they lack adventitious large unstructured regions that could provide alternate degradation initiation sites. If degradation is strictly unidirectional one can test the following hypothesis. In a substrate with a GAr and folded domain positioned so that the GAr arrives at the ClpX translocase at the same time that a trailing folded domain arrives at a site of constriction where its unfolding takes place, failure to unfold depends on both the structural stability of the folded domain and the ability of the GAr to impede translocation, which drives unfolding. In short, is a GAr more slippery for the translocase compared with other sequences, as measured by failure to unfold a hard-to-unfold domain?
FIGURE 1.
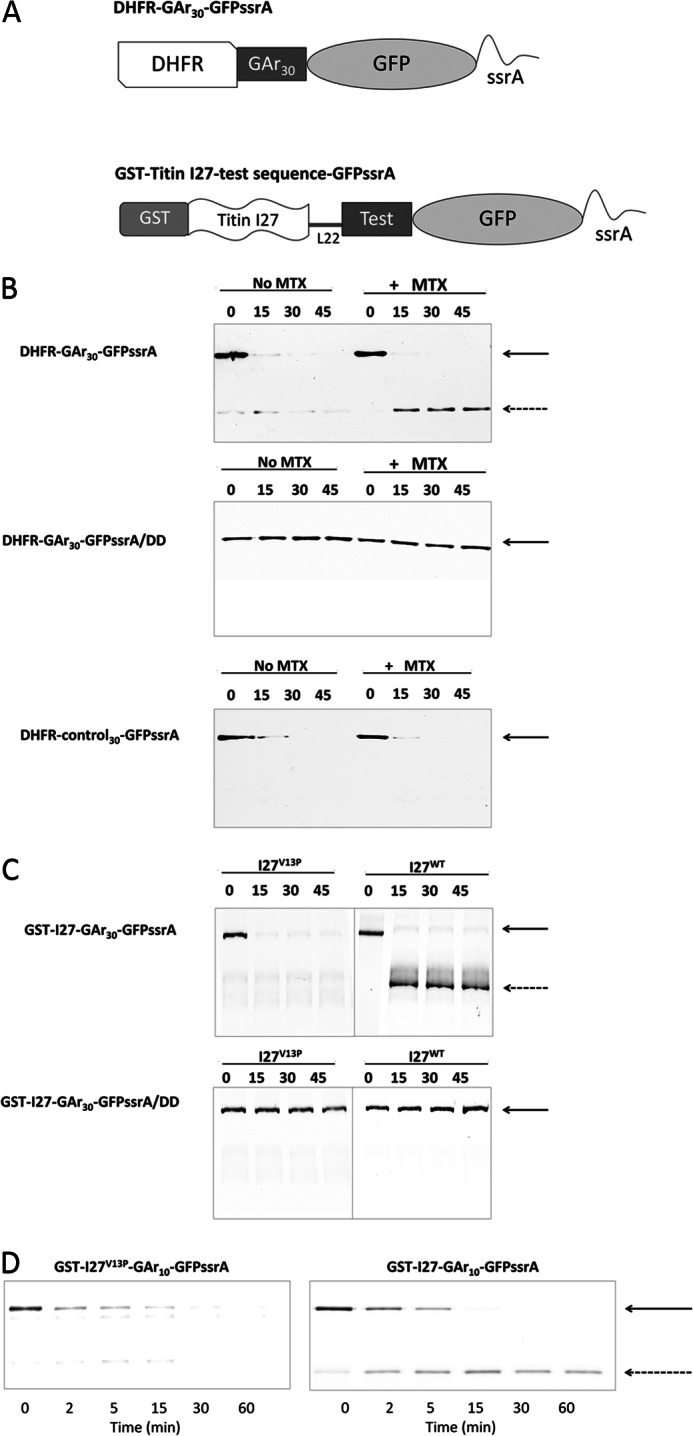
The accumulation of degradation intermediates requires collaboration between a GAr and a stable folded domain. A, schematic representation of substrate proteins used in this paper. A human DHFR or titin I27 immunoglobulin coding sequence was included to provide a domain requiring active unfolding by the ClpX translocase/unfoldase. Sequences of varying length and amino acid composition (Test), some of which consist of Gly-Ala repeat sequences (GAr) were placed between DHFR or I27 and GFP-ssrA to determine their effects on the production of intermediate products of stalled degradation. L22, a linker of 22 amino acids. B, top, degradation of DHFR-GAr30-GFP-ssrA in the presence (+MTX) or absence (No MTX) of 20 μm methotrexate. Bands corresponding to full-length parent substrate (solid arrow) or degradation intermediates (dashed arrow) were detected by Western blotting using anti-DHFR. Middle, as in the top, but the substrate contains a terminal dipeptide mutation of ssrA that cannot be recognized by ClpX. Bottom, as in the top, but DHFR-control30-GFP-ssrA contains a 30-mer diverse amino acid sequence instead of GAr30. C, top, I27V13P-GAr30-GFP-ssrA and I27-GAr30-GFP-ssrA substrates were subjected to degradation by ClpXP. Bottom, the same proteins (GST-I27-GAr30-GFP-ssrA/DD) but with a terminal dipeptide mutation of ssrA that cannot be recognized by ClpX. D, degradation of GST-I27-GAr10-GFP-ssrA. The titin I27 domain contains a destabilizing V13P point mutation (left) or is wild type (right).
Initial experiments were performed using substrates that tested the interaction between mammalian DHFR as the folded domain and a GAr 30 residues in length (GAr30). These were of the form DHFR-GAr30-GFP-ssrA and are schematically represented in Fig. 1A. Substrate and reaction products were resolved by SDS-PAGE and imaged by Western blotting with anti-DHFR antiserum. DHFR-GAr30-GFP-ssrA was rapidly degraded by ClpXP; only a small fraction of the substrate initially present remained intact after a 15-min incubation using a ∼7-fold molar excess of DHFR-GAr30-GFP-ssrA over ClpXP complex (Fig. 1B, top left). The reaction produced a trace amount of a fragment with the approximate size of DHFR, an intermediate remnant resulting from ClpXP degradative processing that destroys GFP-ssrA. The amount of that intermediate product was too small for accurate measurement. The ligand methotrexate (MTX) increases the mechanical stability of DHFR (26). In contrast to the reaction without MTX, the reaction in the presence of MTX yielded a marked increase in production of the DHFR intermediate (Fig. 1B, top right). Under these conditions, the yield of intermediate was about half of the starting substrate. The same substrate, but with a terminal DD in place of the AA of ssrA, a mutation that renders ssrA inactive as a degradation tag, resulted in no degradation of substrate regardless of the presence or absence of MTX (Fig. 1B, middle). When a 30-mer control30 sequence (QDDGTLPMSCAQESGMDRHPAACASARINV) was used in place of the GAr30, no intermediate was detected in the presence or absence of MTX (Fig. 1B, bottom).
These data demonstrate that the GAr sequence results in more intermediates than a nonspecific control sequence, that greater domain stability augments production of intermediates, and that these processing events depend on the integrity of ssrA function. A substrate of similar structure, containing DHFR followed by a GAr30 sequence, has previously been analyzed in yeast cells (15) and produced similar findings; the yield of intermediate products of proteasome processing was increased by the addition of MTX to cells and was reduced by point mutations of DHFR that destabilized its structure.
To determine whether these results hold true for the interaction between the GAr30 and a different folded domain, we used titin I27. I27 is a tightly folded β sandwich (27), one of the immunoglobin fold repeat elements of the giant titin protein (28). To examine the effect of domain stability, we compared I27 and its V13P mutation, a single residue change that destabilizes I27 (29). We generated two substrates, I27V13P-GAr30-GFP-ssrA and I27-GAr30-GFP-ssrA. With the protein containing the wild type form of I27, essentially all of the products of degradation were intermediates, as suggested by visual inspection of band density (Fig. 1C, top right) and quantitative analysis of band intensity (data not shown). Degradation was stalled almost completely by the combination of I27 and GAr30. In strong contrast, no interruption of degradation was observed using an otherwise identical substrate but with I27 containing the destabilizing V13P single amino acid mutation (Fig. 1C, top left). The terminal DD mutation of ssrA prevented degradation of both substrates (Fig. 1C, bottom) These data demonstrate that for these substrates, the wild I27 domain collaborates with GAr30 to interrupt ClpXP degradation, but the destabilized I27 almost never does so.
The Inhibitory Effect of Gly-Ala Repeats Is Dependent on Its Length
In its native biological context, the EBNA1 protein of Epstein-Barr virus, a GAr can consist of up to ∼300 contiguous Gly and Ala residues (30). To determine the dependence of intermediate generation on GAr length, we constructed substrates of the form GST-I27-GArN-GFP-ssrA, containing GAr sequences with n = 7, 8, 9, 10, or 15 amino acids. The GST tag was included to facilitate affinity purification. Substrate was similarly degraded in all cases (Fig. 2, A and B), as expected from events that begin at GFP-ssrA and are independent of the remainder of the protein in their initial processing trajectory. However, the amount of intermediate increased with longer GAr sequences (Fig. 2, A and B) ranging from 33 up to 77% (Fig. 2C). No significant increment was found when the length was increased from 10 to 15 residues. A positive correlation between GAr length and intermediate accumulation was previously found in yeast and human proteasomes (13, 15, 31). We also tested the effect of domain stability with a GAr10. In GST-I27-GAr10-GFP-ssrA, introducing the V13P destabilizing mutation reduced the yield of intermediates from 75% to an undetectable level (Fig. 1D), an effect qualitatively similar to that observed using the longer GAr30 (Fig. 1C).
FIGURE 2.

Production of degradation intermediate increases with increasing length of a GAr. A, substrates (Cy3-labeled) of the form GST-I27-GArN-GFP-ssrA, containing GAr sequences with n = 7, 8, 9, 10, or 15 amino acids were degraded by ClpXP for 60 min, and samples were periodically analyzed. Samples were separated by SDS-PAGE and scanned by fluorescence imaging, using fluor-specific excitation and emission wavelengths. Cy5-labeled BSA included in each sample was fractionated on the same gel and analyzed as a loading control (lower portion of each panel). The position of full-length substrate (solid arrows) or degradation intermediate (dashed arrows) is indicated. B, the abundance of the full-length substrate (Parent) and degradation intermediate in A were quantified, normalized to the intensity of the Cy5-BSA bands in the same lane. The values for substrate with GAr of indicated length and the corresponding degradation intermediates are expressed as a percentage of the total of the full-length parent protein present at t = 0. C, the molar yield of intermediate as a percentage of the decrease in parent substrate was calculated from the mean of 15, 30, and 60 min time points, as described under “Experimental Procedures.” Means ± S.D. (error bars) of three independent experiments are plotted.
Stalling Requires Optimal Spacing between a Folded Domain and GAr
Comparison of GST-I27-GArN-GFP-ssrA substrates with n = 10 or 15 showed that extending the C-terminal boundary of the GAr in GST-I27-GAr10-GFP-ssrA by an additional five residues did not significantly augment intermediate generation. This may be because GAr tracts of 10 or 15 residues are equipotent or because the spacing between the two significant elements, I27 and GAr, is critical and the additional five residues are too distant from I27 to influence their interaction. To test the sensitivity of intermediate production to the spacing between I27 and GAr, we moved GAr10 closer to the folded domain. In the GST-I27-GArN-GFP-ssrA series of substrates (Fig. 1A), there is an adventitious 22-residue polypeptide (derived from prior (10, 32) and present molecular manipulations of substrate modules), which separates the C terminus of the I27 β sandwich from the initial (N-terminal) residue of GArN. Deletions were made in GST-I27-GAr10-GFP-ssrA to remove either 11 residues of that intervening sequence or all 22 residues. The effect of these deletions is to move GAr10 closer to I27. Positioning at a distance of 22, 11, or 0 residues caused a diminution of intermediate accumulation from 75 to 56 to 47%, respectively (Fig. 3A). Distance makes a difference, but no sharp optimum was observed. This may be because the position within ClpX where I27 pauses is not geometrically well defined or because it is dynamically modulated by changes of the ATPase conformation.
FIGURE 3.

Effects of GAr spacing and composition on production of intermediates. A, GST-I27-GAr10-GFP-ssrA substrate with different length spacers between I27 and the GAr10 motif was degraded by ClpXP. The lengths of the spacers were 0, 11, or 22 amino acids. The position of full-length substrate (solid arrows) or degradation intermediate (dashed arrows) is indicated. B, the production of intermediates from substrates I27-test-GFP-ssrA with test sequences Gly10 and Ala10 were compared with GAr10 and with a 10-mer control10 of diverse composition (sequence shown). Data were generated and analyzed as in Fig. 2. Error bars, S.D.
The Role of Ala and Gly Residues in the Gly-Ala Repeats
To determine the role of amino acid composition in a GAr sequence, we compared homogeneous oligomers, Ala10 and Gly10 tracts, as test sequences to GAr10 in the context of GST-I27–10-mer-GFP-ssrA. A control10 10-mer of diverse composition (IEGRGIEGRG) was also used in this comparison. Ala10 (56%) and Gly10 sequences (40%) produced less intermediate than GAr10 (75%), whereas the control10 sequence yielded still less (34%) (Fig. 3B). Compared with GAr10, both Ala10 and Gly10 caused a decrease in intermediate production. The small amino acid side chains present in Gly and Ala cannot therefore be the sole relevant property of a GAr.
Next, we further investigated the relationship between amino acid composition and intermediate formation, introducing various single amino acid substitutions in Ala10. When the second Ala was replaced by a Gly (forming AGA8), more intermediate was detected compared with Ala10 (Fig. 4). When the Gly substitution was at a more central position (A4GA5), the inhibitory effect remained similar to that of Ala10, showing that the Gly position plays a role in the inhibitory effect. When the second Ala was instead replaced by the negatively charged residue Glu, to form AEA8, the inhibitory effect was decreased significantly. Substituting Glu for a central Ala (A4EA5) further decreased the inhibitory effect. A similar result was obtained for A4DA5. In A4RA5, a central Ala was replaced by the positively charged residue Arg. In this case, the accumulation of intermediate was reduced to the lowest level among all of the 10-mer sequences tested. However, substitution of a central Ala by another positively charged residue, Lys (A4KA5), produced a more modest reduction. When a central Ala was substituted by Trp (A4WA5) or Phe (A4FA5), the inhibitory effect of Ala10 was strongly diminished. However, a substitution by the aromatic residue Tyr or His produced a more modest decrease in intermediate accumulation. Val caused little decrease in intermediate accumulation compared with Gly.
FIGURE 4.

Effect on intermediate production of single amino acid substitutions within Ala10. Test sequences consisted of a homologous set of single amino acid substitutions (X) for a central Ala in Ala10 (Ala4-X-Ala5). Data were generated and analyzed as in Fig. 2. The numerical values for intermediate generation are summarized in Table 1.
Table 1 summarizes the degradation properties of the homologous set of substitutions of a central Ala in Ala10, displayed in descending rank order of efficacy for intermediate generation. Substituents producing the most intermediates have small hydrophobic side chains. To more systemically analyze the relationship between side chain chemical and physical properties and intermediate generation, we plotted selected physical parameters (Table 2) of the substituent amino acids: molecular mass, shape (position of branch in a side chain), flexibility (number of side chain torsional angles), hydrophobicity, and isoelectric point (Fig. 5). Linear regression analysis showed that the mass, shape, and flexibility of amino acid were significantly correlated with intermediate generation (p < 0.05). However, there were no significant correlations with hydrophobicity or isoelectric point. Among the tested amino acid properties, shape shows the highest single-property correlation (R2 = 0.71); molecular weight and flexibility gave coefficients of determination R2 of 0.47 and 0.52, respectively. This analysis shows that certain physical properties of a single amino acid substituted within the Ala10 motif can affect its capacity to generate intermediates.
TABLE 1.
Percentage of intermediate production by single residue X substitution in Ala4-X-Ala5
The substrate consisted of GST-I27–10-mer-GFP-ssrA. Test 10-mers are listed in descending order of potency for producing intermediates.
| Test sequence (10-mer) | Percentage of intermediate |
|---|---|
| % | |
| A4GA5 | 56 ± 7 |
| A10 | 56 ± 6 |
| A4VA5 | 51 ± 7 |
| A4YA5 | 49 ± 6 |
| A4KA5 | 48 ± 7 |
| A4HA5 | 47 ± 5 |
| A4SA5 | 44 ± 5 |
| A4DA5 | 38 ± 4 |
| A4FA5 | 35 ± 4 |
| A4EA5 | 32 ± 4 |
| A4WA5 | 32 ± 5 |
| A4RA5 | 22 ± 4 |
FIGURE 5.

The coefficient of determination (R2) and regression line for percentage of intermediate generation (Table 1) versus selected properties (Table 2) of single amino acid residue substituents in Ala10. A, residue molecular mass; B, shape; C, flexibility; D, hydrophobicity; E, isoelectric point. The R2 and the p values are shown in each plot.
Route of Escape of Intermediate Degradation Products from ClpXP
There are limited data on the influence of substrate interactions on the stability of assembly of AAA+ protease complexes. Multiple rounds of degradation do not cause disassembly of the ClpAP complex (33). Biochemical data support the conclusion that intermediate products of degradation by proteasomes (15) or bacterial ATP-dependent proteases (10) do not remain associated with the cleavage machinery. Competition assays were performed to determine whether intermediates remain associated with ClpXP and thus impair its degradation activity. The degradation of GFP-ssrA was measured 5 min after preincubation with I27-GAr10-GFP-ssrA or after a 60-min preincubation with that substrate. Degradation of GFP-ssrA was competitively impaired by the presence of the I27-GAr10-GFP-ssrA substrate, which remains present after early addition (5 min) of GFP-ssrA, but GFP-ssrA degradation is not impaired after its late addition (60 min), at which time I27-GAr10-GFP-ssrA has been exhausted and confers no residual inhibitory effect (Fig. 6). Intermediate generation therefore does not clog the enzyme.
FIGURE 6.

Neither GAr10 substrate nor its intermediate degradation product is trapped by ClpX. Shown is competition between I27-GAr10-GFP-ssrA and GFP-ssrA for degradation by ClpXP. Each substrate was added at ∼13-fold molar excess to ClpXP complex. After a 5- or 60-min preincubation of ClpXP (0.3 μm ClpX hexamer and 0.7 μm ClpP tetradecamer) with Cy3-labeled I27-GAr10-GFP-ssrA substrate (2 μm), Cy5-labeled GFP-ssrA (2 μm) was added. The time course of degradation of GFP-ssrA was monitored (left). Time-dependent GFP-ssrA degradation was quantified and plotted (right). ClpXP degradation of labeled GFP-ssrA was determined after early (5 min) or late (60 min) addition or (as control) with no addition of GAr10 substrate. Error bars, S.D.
A substrate that has been partially cleaved and released has been stymied in its advancement by the ATPase translocase. It retains a folded domain plus some remnant of a contiguous region. The size of that remnant is determined by the distance between the sites of proteolysis (here in the interior of ClpP14) and the position (with respect to ClpX6) at which the folded domain pauses during futile unfolding attempts. In principle, there are two exit routes. The remnant extension can back out in the opposite direction from which it entered. Alternatively, because stability of the ClpX6 ring depends on non-covalent associations between its adjacent monomers, a transient gap may appear that allows sideways exit. If the second mode is the dominant escape route, covalently connecting the monomers should cut off escape. Because this would clog ClpX6 with intermediates that cannot be cleared, the expected results are a lower rate of degradation of substrate and reduced generation of intermediates.
ClpX hexamers have been described that consist of six copies of the monomer connected by peptide linkers (L-ClpX6) (18). These are approximately as active as the cognate non-covalent hexamer in ATPase activity and in their capacity to collaborate with ClpP in degradation. Whereas the non-covalent ClpX6 has six potential sites for lateral escape, the linear covalent hexamer has but one. Furthermore, covalently closed circular forms of ClpX6 (C-ClpX6) have been reported (20) in which there remain no junctures that can permit lateral escape. (C-ClpX6 used in this study was made by a modification of the method described in Ref. 20; a pair of ClpX covalent trimers were circularized by sealing the two junctions between ClpX trimers with disulfide bonds.) Each of these three topological forms of ClpX6 was prepared to test the lateral escape hypothesis (Fig. 7A). We examined the degradation of GST-I27-GAr10-GFP-ssrA by these distinct forms of ClpXP. The rate of degradation of substrate and the rate and extent of appearance of the intermediate degradation product did not differ significantly among the three (Fig. 7B). C-ClpX6 was found to retain its stabilizing disulfides throughout the reaction period (data not shown). Lateral escape of intermediates through interstices between pairs of ClpX monomers therefore cannot be the dominant mode of their dissociation.
FIGURE 7.

Degradation rate and intermediate production by various topological forms of ClpX. A, molecular mobility of ClpX covalent linear (L-ClpX6) and closed circular (C-ClpX6) hexamers. Proteins were fractionated by SDS-PAGE in the absence of dithiothreitol (−DTT) or the presence of dithiothreitol (+DTT), conditions that maintain or reduce disulfide bonds, respectively. L-ClpX6 and C-ClpX6 were generated and purified as described under “Experimental Procedures.” C-ClpX6 is a dimer of linear covalent trimers, in which the two trimer junctions are connected by disulfide bonds. The C-ClpX6 protein consists of ∼85% closed circular hexamer (two disulfide bonds) and ∼15% linear hexamer (one disulfide bond). Disulfide bonds in C-ClpX6 were broken in the presence of 10 mm DTT; thus, the ClpX trimer (ClpX3) was the sole species present under +DTT reducing conditions. B, monomeric ClpX (ClpX1), L-ClpX6, and C-ClpX6 were compared in assays of I27-GAr10-GFP-ssrA degradation. i, time-dependent substrate degradation and production of intermediates. ii, quantitation of data in i. Data were generated and analyzed as in Fig. 2.
Effect of Sequence on the Rate Constants That Determine Intermediate Generation
A substrate like GST-I27-test-GFP-ssrA pauses while the I27 domain is undergoing unfolding attempts by ClpX. That pause in proteolysis takes place within a transient complex that consists of ClpXP and the partially degraded substrate, containing I27 plus some C-terminal extension, an as yet undigested remnant fragment derived from test sequence plus GFP. This complex will be designated Enz·I27-frag. The complex of enzyme and partially degraded protein can resolve in two ways (Fig. 8A). I27-frag can exit by backing out. Alternatively, it can resume degradation, completing processive proteolysis. Only the first of these alternatives results in the production of a stable durable intermediate. The yield of intermediates can thus be formulated as the probability of the first of these outcomes, backing out. Each of these events is associated with a rate constant, kout for exit and kproc for resumption of processive degradation. Consequently, intermediate yield = kout/(kout + kproc).
FIGURE 8.

Determination of kinetic constants associated with partition between exit and degradation of a proteolytic fragment in complex with ClpXP. A, schematic representation of the degradation of substrate. After substrate association with ClpXP, the GFP-ssrA domain of the I27-test-GFP-ssrA substrate is degraded by ClpXP and degradation pauses, forming the Enz·I27-frag complex. Enz·I27-frag either continues to degrade I27-frag (determined by rate constant kproc), or I27-frag dissociates from the enzyme (determined by rate constant kout). B, intermediate as a percentage of substrate at initial time with test sequence GAr10 or control10. Data were generated and analyzed as in Fig. 3, except an excess of enzyme (2:1 molar ratio of ClpXP/substrate) was used. The quantity of intermediate reached a maximum at t = 4 min. Data shown are representative of three independent experiments. C, the time-dependent decay of I27-frag derived from substrates with test sequences consisting of GAr10 or control10 is plotted against time. Data were analyzed as described under “Experimental Procedures.” The time axis is transformed so that t = 0 corresponds to the 4 min point in B, the time of maximum intermediate accumulation. Error bars, S.D. of three independent experiments.
When a test sequence, such as a GAr, increases the intermediate yield, it could in principle do so in two ways: by increasing kout or by reducing kproc. To resolve that question, these kinetic parameters were determined for the substrates GST-I27-GAr10-GFP-ssrA and GST-I27-control10-GFP-ssrA, the first of which produces a higher intermediate yield. We determined kproc directly by observing the time-dependent destruction of I27-frag by Enz·I27-frag. By using a 2-fold molar excess of ClpXP (in contrast to a molar excess of substrate in the experiments described previously), a significant pool of Enz·I27-frag could be generated, and its subsequent means of resolution could be followed, as shown schematically in Fig. 8A. Under these specific conditions of enzyme excess, the amount of intermediates was maximal at 4 min after the initiation of degradation (Fig. 8B). At this time, some portion of the I27-frag intermediates is enzyme-associated, whereas a distinct pool has already dissociated. Subsequent decline from this peak of intermediate abundance is due to resumption or continuation of processive degradation by Enz·I27-frag. Adding an excess of a GFP-ssrA as a competitive inhibitor immediately before the 4 min time point ensured that any unprocessed substrate remaining in solution could not subsequently enter the Enz·I27-frag pool. The Enz·I27-frag complex can resolve in two ways, by releasing I27-frag or further degrading it, but only degradation, not release, reduces the amount of intermediate present. The first order rate constant kproc can therefore be calculated by measuring the rate of decline of intermediate abundance after background subtraction of intermediates present at time points late in the reaction course, those released by the enzyme rather than further processed. For GST-I27-GAr10-GFP-ssrA and GST-I27-control10-GFP-ssrA, kproc was 0.063 and 0.091 min−1, respectively (Fig. 8C). The intermediate fractions that accumulate from GST-I27-GAr10-GFP-ssrA and GST-I27-control10-GFP-ssrA are 0.75 and 0.15, respectively, of input substrate at late time points (Fig. 8B); the relationship intermediate fraction = kout/(kout + kproc) allows direct calculation of kout for the two substrates, yielding the following: for GAr10, kout = 0.146 min−1 and kproc = 0.063 min−1; for control10, kout = 0.016 min−1 and kproc = 0.091 min−1.
For the GAr substrate compared with the control substrate, the rate of escape is about 9.2-fold higher. The rate of moving forward processively is less markedly changed, about 1.4-fold lower. Therefore, a GAr increases the generation of intermediates mostly by promoting their escape.
DISCUSSION
ATP-dependent proteases convert the chemical energy of ATP binding and hydrolysis to mechanical work, which moves substrates and concomitantly unfolds them. There is a general consensus that this energy transmission proceeds through an interaction between axial moving loops of the ATPase, which act as propulsive paddles, and an extended substrate polypeptide chain positioned in the ATPase channel (3, 4). There is less agreement as to whether and, if so, in what way the amino acid composition of the substrate polypeptide influences the efficiency of energy transmission. Because translocation and unfolding are coupled, the effects of varying coupling efficiency are most readily elicited using substrate with domains that require significant energy for unfolding. Mutations of key residues of the propulsive loops have been found to impair unfolding of substrates that present a high mechanical load (3, 4). We show here that for ClpXP substrate sequence also influences unfolding. In particular, we find that a virus-derived sequence containing only Gly and Ala residues is especially effective in frustrating processivity.
The Epstein-Barr virus of humans causes acute infections but can also establish a dormant state in which B cells harboring the virus undergo malignant transformation. Under these conditions, expression of a single viral protein is required (34). The viral EBNA1 protein contains a tract consisting solely of Gly and Ala residues. The length of such GAr tracts varies among individual viral isolates but can be as long as 300 amino acids (30). There must be strong evolutionary constraints that exclude the presence within a GAr of the 18 other native amino acids. A GAr has two known cis-acting functions; it impairs translation (35) and proteasomal degradation (36). Both of these effects have the capacity to impede host immune surveillance, especially so because proteasome degradation provides a source of MHC class I immunopeptides (37). We have previously examined the cis-inhibitory role of GAr sequences in degradation by proteasomes (13, 15). Extending these studies to the bacterial protease ClpXP is important and informative for several reasons. First, it provides a test of functional generality; do proteases that are architecturally similar (1) but very divergent in evolution behave according to similar rules of translocase-substrate interaction? Second, the regulatory complex of ClpXP, the ClpX homohexameric ATPase, is structurally much simpler than the corresponding 19 S regulatory complex of proteasomes, in which all six ATPases are different (but homologous) and more than a dozen other proteins are present (38). The greater structural complexity of the proteasome makes it harder to conclude that any sequence-dependent differences of substrate behavior are the direct results of differential interactions with the ATPase translocase rather than with some other component of the regulatory complex. Third, the greater simplicity of performing biochemical assays with the bacterial protease compared with proteasomes facilitates systematic structure/function analysis of substrate processing and the mechanism of partitioning between alternate processing outcomes.
In these studies, we used a method of analysis previously employed with proteasomes to test the capacity of a specific amino acid tract to impair unfolding and degradation of a juxtaposed folded domain (15). We found that for ClpXP, as with proteasomes, a GAr tract positioned near a folded domain prolongs or prevents its unfolding in such a way that a partial degradation product is produced and dissociates from the protease. Mechanical stability of the domain is important to that outcome; more stable domains result in a larger fraction of intermediate products. The position of the GAr with respect to the folded domain is also important for proteasomes (15) and, as demonstrated here, ClpXP. This dependence on spacing is readily rationalized, because the distance between folded domain and polypeptide region undergoing translocation provides a molecular ruler marking the distance between the site of propulsion and a second site where unfolding is paused. The simultaneous arrival of each substrate component at its respective site within the protease is a precondition for unfolding failure.
We systematically tested various sequences for their ability to impair unfolding. We scored the fraction of degradation events that produce intermediates. Among the sequences tested here, the virus-related GAr was the most effective in generating intermediates. Strong effects of polypeptide composition were observed in this assay. The effects of amino acid sequence and composition on translocation by protease ATPases have been investigated previously. Using a fluor-quencher pair separated by various tracts of natural amino acids (or non-native polymers), only modest differences of translocation rate by ClpXP were observed (39), leading to the conclusion that a broad range of chemically diverse linear polymers could be accommodated. However, these various substrates were not tested under conditions of significant mechanical load. The assay of translocase function used here is specific to conditions of high mechanical load. The present data are therefore not in conflict with those reported previously (39), which measured the sequence dependence of substrate translocation velocity by ClpXP under minimal load conditions. In another study with proteasomes and variants of native proteins that generate processing intermediates, it was concluded that interrupting degradation processivity depends on the presence of a tightly folded domain plus a sequence of special character. Sequences that impaired processivity were found to be “simple” (i.e. of low compositional complexity); amino acid identity or chemistry was deemed not to be relevant (40). However, a limited number of sequences were tested, and these bore no systematic relationship to each other. In the present experiments with ClpXP, no correlation was observed between compositional complexity of test sequence and outcomes.
If the translocase is viewed as a friction drive acting on an extended polypeptide chain, then asperity, the roughness of its amino acid side chains, can naively be expected to promote interaction and the opposite to impair it. The series of test sequences consisting of a run of 10 Ala residues substituted by a single non-Ala amino acid at position 5 provided a uniform context for testing the effects of diverse side chains on function (Fig. 4). Plotting intermediate yield versus physical and chemical parameters of the individual amino acid substituent (Fig. 5) revealed the properties that did and did not influence outcomes. Hydrophobicity and isoelectric point were essentially irrelevant. In contrast, measures of bulk or “bumpiness” gave better correlations. Residue molecular mass showed a modest but significant correlation (R2 = 0.47, higher molecular weight associated with more intermediates), and a parameter that assesses the number and position of side chain branches showed an even better correlation (R2 = 0.71, greater branching associated with more intermediates).
We found that changing the topology of the ClpX hexamer to seal junctures between the individual monomers had little or no effect on the generation of intermediates. We interpret these data to demonstrate that dissociation of intermediates from the enzyme takes place by backing out, not by lateral escape through transient gaps between ClpX monomers. This finding emphasizes the importance of the balance between substrate retention and escape in determining the ability of the translocase to act processively.
GAr sequences with a length of 7–15 amino acids promoted progressively more intermediate production, but GAr15 was only modestly more effective than GAr10. This length dependence can be regarded from two perspectives. The first relates to the power stroke length of the ClpX translocase. Its smallest translocation steps have been reported to be 5–8 amino acids in length, with longer 10–13-residue increments corresponding to sequential but unresolved steps (5). A GAr may therefore be more effective as its length approaches and exceeds the length of the power stroke. From a second prospective, a GAr of greater length is more likely to promote stochastic escape, because a longer GAr may promote larger or more prolonged Brownian excursions within the translocase tunnel.
The extent of intermediate formation as a fraction of all degradation events is governed by two kinetic parameters, kout and kproc. These determine the resolution of a complex consisting of a partially degraded substrate that remains associated with ClpXP and is paused in the process of unfolding a protein domain. By comparing a GAr and a control sequence, the predominant effect of sequence on intermediate generation was shown to arise from its influence on the rate of escape, an effect on kout rather than kproc. One can imagine the translocating loops of ClpX acting as paddles on substrate, to deliver force to move substrate forward (power stroke) and also retain substrate in the axial pore (return stroke or pause). If such a cycle is to avoid futility, the forward and return strokes must follow different trajectories. Molecular dynamic simulations (41) of a related ATPase motor support such a paddling mechanism and substantiate the effect of substrate sequence on paddle grip. Additionally, the axial pore of the ATPase is a dynamic system in which the geometry of the enzyme (20) and path of the substrate are likely to be undergoing cycle-associated changes. The interactions between translocase/effector and substrate/recipient can therefore change in multiple ways during different phases of the ATP-driven mechanical cycle. The data described here point to substrate sequence as important for events that take place between power strokes, rather than during the power stroke. As unfolding persists, a GAr causes a 9.2-fold failure of retention but a mere 1.4-fold impairment to force delivery.
This work was supported, in whole or in part, by National Institutes of Health Grants GM45335 (to P. C.) and GM097974 (to Sanford Simon, Rockefeller University).
- GAr
- glycine-alanine repeat
- ClpX6
- ClpX hexamer
- C-ClpX6
- closed circular ClpX hexamer
- L-ClpX6
- linear ClpX hexamer
- DHFR
- dihydrofolate reductase
- EBNA1
- Epstein-Barr virus nuclear antigen-1
- MTX
- methotrexate.
REFERENCES
- 1. Pickart C. M., Cohen R. E. (2004) Proteasomes and their kin. Proteases in the machine age. Nat. Rev. 5, 177–187 [DOI] [PubMed] [Google Scholar]
- 2. Erales J., Hoyt M. A., Troll F., Coffino P. (2012) Functional asymmetries of proteasome translocase pore. J. Biol. Chem. 287, 18535–18543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hinnerwisch J., Fenton W. A., Furtak K. J., Farr G. W., Horwich A. L. (2005) Loops in the central channel of ClpA chaperone mediate protein binding, unfolding, and translocation. Cell 121, 1029–1041 [DOI] [PubMed] [Google Scholar]
- 4. Martin A., Baker T. A., Sauer R. T. (2008) Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding. Nat. Struct. Mol. Biol. 15, 1147–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aubin-Tam M.-E., Olivares A. O., Sauer R. T., Baker T. A., Lang M. J. (2011) Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell 145, 257–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maillard R. A., Chistol G., Sen M., Righini M., Tan J., Kaiser C. M., Hodges C., Martin A., Bustamante C. (2011) ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell. 145, 459–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kenniston J. A., Baker T. A., Fernandez J. M., Sauer R. T. (2003) Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell 114, 511–520 [DOI] [PubMed] [Google Scholar]
- 8. Henderson A., Erales J., Hoyt M. A., Coffino P. (2011) Dependence of proteasome processing rate on substrate unfolding. J. Biol. Chem. 286, 17495–17502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bustamante C., Chemla Y. R., Forde N. R., Izhaky D. (2004) Mechanical processes in biochemistry. Annu. Rev. Biochem. 73, 705–748 [DOI] [PubMed] [Google Scholar]
- 10. Kenniston J. A., Baker T. A., Sauer R. T. (2005) Partitioning between unfolding and release of native domains during ClpXP degradation determines substrate selectivity and partial processing. Proc. Natl. Acad. Sci. U.S.A. 102, 1390–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takeuchi J., Chen H., Coffino P. (2007) Proteasome substrate degradation requires association plus extended peptide. EMBO J. 26, 123–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fan C. M., Maniatis T. (1991) Generation of p50 subunit of NF-κB by processing of p105 through an ATP-dependent pathway. Nature 354, 395–398 [DOI] [PubMed] [Google Scholar]
- 13. Zhang M., Coffino P. (2004) Repeat sequence of Epstein-Barr virus-encoded nuclear antigen 1 protein interrupts proteasome substrate processing. J. Biol. Chem. 279, 8635–8641 [DOI] [PubMed] [Google Scholar]
- 14. Zhang M., MacDonald A. I., Hoyt M. A., Coffino P. (2004) Proteasomes begin ornithine decarboxylase digestion at the C terminus. J. Biol. Chem. 279, 20959–20965 [DOI] [PubMed] [Google Scholar]
- 15. Hoyt M. A., Zich J., Takeuchi J., Zhang M., Govaerts C., Coffino P. (2006) Glycine-alanine repeats impair proper substrate unfolding by the proteasome. EMBO J. 25, 1720–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Prakash S., Tian L., Ratliff K. S., Lehotzky R. E., Matouschek A. (2004) An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat. Struct. Mol. Biol. 11, 830–837 [DOI] [PubMed] [Google Scholar]
- 17. Levitskaya J., Coram M., Levitsky V., Imreh S., Steigerwald-Mullen P. M., Klein G., Kurilla M. G., Masucci M. G. (1995) Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature 375, 685–688 [DOI] [PubMed] [Google Scholar]
- 18. Martin A., Baker T. A., Sauer R. T. (2005) Rebuilt AAA+ motors reveal operating principles for ATP-fuelled machines. Nature 437, 1115–1120 [DOI] [PubMed] [Google Scholar]
- 19. Jones J. M., Welty D. J., Nakai H. (1998) Versatile action of Escherichia coli ClpXP as protease or molecular chaperone for bacteriophage Mu transposition. J. Biol. Chem. 273, 459–465 [DOI] [PubMed] [Google Scholar]
- 20. Glynn S. E., Nager A. R., Baker T. A., Sauer R. T. (2012) Dynamic and static components power unfolding in topologically closed rings of a AAA+ proteolytic machine. Nat. Struct. Mol. Biol. 19, 616–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Popp M. W., Antos J. M., Grotenbreg G. M., Spooner E., Ploegh H. L. (2007) Sortagging. A versatile method for protein labeling. Nat. Chem. Biol. 3, 707–708 [DOI] [PubMed] [Google Scholar]
- 22. Popp M. W., Antos J. M., Ploegh H. L. (2009) Site-specific protein labeling via sortase-mediated transpeptidation. Curr. Protoc. Protein Sci., Chapter 15, Unit 15.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yakhnin A. V., Vinokurov L. M., Surin A. K., Alakhov Y. B. (1998) Green fluorescent protein purification by organic extraction. Protein Expr. Purif. 14, 382–386 [DOI] [PubMed] [Google Scholar]
- 24. Hoskins J. R., Yanagihara K., Mizuuchi K., Wickner S. (2002) ClpAP and ClpXP degrade proteins with tags located in the interior of the primary sequence. Proc. Natl. Acad. Sci. U.S.A. 99, 11037–11042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gottesman S., Roche E., Zhou Y., Sauer R. T. (1998) The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev. 12, 1338–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eilers M., Schatz G. (1986) Binding of a specific ligand inhibits import of a purified precursor protein into mitochondria. Nature 322, 228–232 [DOI] [PubMed] [Google Scholar]
- 27. Improta S., Politou A. S., Pastore A. (1996) Immunoglobulin-like modules from titin I-band. Extensible components of muscle elasticity. Structure 4, 323–337 [DOI] [PubMed] [Google Scholar]
- 28. Tskhovrebova L., Trinick J. (2003) Titin. Properties and family relationships. Nat. Rev. 4, 679–689 [DOI] [PubMed] [Google Scholar]
- 29. Li H., Carrion-Vazquez M., Oberhauser A. F., Marszalek P. E., Fernandez J. M. (2000) Point mutations alter the mechanical stability of immunoglobulin modules. Nat. Struct. Biol. 7, 1117–1120 [DOI] [PubMed] [Google Scholar]
- 30. Falk K., Gratama J. W., Rowe M., Zou J. Z., Khanim F., Young L. S., Oosterveer M. A., Ernberg I. (1995) The role of repetitive DNA sequences in the size variation of Epstein-Barr virus (EBV) nuclear antigens and the identification of different EBV isolates using RFLP and PCR analysis. J. Gen. Virol. 76, 779–790 [DOI] [PubMed] [Google Scholar]
- 31. Sharipo A., Imreh M., Leonchiks A., Imreh S., Masucci M. G. (1998) A minimal glycine-alanine repeat prevents the interaction of ubiquitinated I κB α with the proteasome. A new mechanism for selective inhibition of proteolysis. Nat. Med. 4, 939–944 [DOI] [PubMed] [Google Scholar]
- 32. Carrion-Vazquez M., Oberhauser A. F., Fowler S. B., Marszalek P. E., Broedel S. E., Clarke J., Fernandez J. M. (1999) Mechanical and chemical unfolding of a single protein. A comparison. Proc. Natl. Acad. Sci. U.S.A. 96, 3694–3699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Singh S. K., Guo F., Maurizi M. R. (1999) ClpA and ClpP remain associated during multiple rounds of ATP-dependent protein degradation by ClpAP protease. Biochemistry 38, 14906–14915 [DOI] [PubMed] [Google Scholar]
- 34. Leight E. R., Sugden B. (2000) EBNA-1. A protein pivotal to latent infection by Epstein-Barr virus. Rev. Med. Virol. 10, 83–100 [DOI] [PubMed] [Google Scholar]
- 35. Yin Y., Manoury B., Fåhraeus R. (2003) Self-inhibition of synthesis and antigen presentation by Epstein-Barr virus-encoded EBNA1. Science 301, 1371–1374 [DOI] [PubMed] [Google Scholar]
- 36. Sharipo A., Imreh M., Leonchiks A., Brändén C., Masucci M. G. (2001) cis-Inhibition of proteasomal degradation by viral repeats. Impact of length and amino acid composition. FEBS Lett. 499, 137–142 [DOI] [PubMed] [Google Scholar]
- 37. Kloetzel P. M., Ossendorp F. (2004) Proteasome and peptidase function in MHC-class-I-mediated antigen presentation. Curr. Opin. Immunol. 16, 76–81 [DOI] [PubMed] [Google Scholar]
- 38. Finley D. (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Barkow S. R., Levchenko I., Baker T. A., Sauer R. T. (2009) Polypeptide translocation by the AAA+ ClpXP protease machine. Chem. Biol. 16, 605–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tian L., Holmgren R. A., Matouschek A. (2005) A conserved processing mechanism regulates the activity of transcription factors Cubitus interruptus and NF-κB. Nat. Struct. Mol. Biol. 12, 1045–1053 [DOI] [PubMed] [Google Scholar]
- 41. Koga N., Kameda T., Okazaki K., Takada S. (2009) Paddling mechanism for the substrate translocation by AAA+ motor revealed by multiscale molecular simulations. Proc. Natl. Acad. Sci. U.S.A. 106, 18237–18242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. van Gunsteren W. F., Mark A. E. (1992) Prediction of the activity and stability effects of site-directed mutagenesis on a protein core. J. Mol. Biol. 227, 389–395 [DOI] [PubMed] [Google Scholar]
- 43. Kyte J., Doolittle R. F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 44. Zimmerman J. M., Eliezer N., Simha R. (1968) The characterization of amino acid sequences in proteins by statistical methods. J. Theor. Biol. 21, 170–201 [DOI] [PubMed] [Google Scholar]