

Background: It has not been possible to study the pumping and signaling functions of Na/K-ATPase independently in live cells.
Results: Both cell-free and cell-based assays indicate that the A420P mutation abolishes the Src regulatory function of Na/K-ATPase.
Conclusion: A420P mutant has normal pumping but not signaling function.
Significance: Identification of Src regulation-null mutants is crucial for addressing physiological role of Na/K-ATPase.
Keywords: Cell Signaling; Membrane Proteins; Mutant; Na,K-ATPase; Src; Cardiotonic Steroids
Abstract
The α1 Na/K-ATPase possesses both pumping and signaling functions. However, it has not been possible to study these functions independently in live cells. We have identified a 20-amino acid peptide (Ser-415 to Gln-434) (NaKtide) from the nucleotide binding domain of α1 Na/K-ATPase that binds and inhibits Src in vitro. The N terminus of NaKtide adapts a helical structure. In vitro kinase assays showed that replacement of residues that contain a bulky side chain in the helical structure of NaKtide by alanine abolished the inhibitory effect of the peptide on Src. Similarly, disruption of helical structure by proline replacement, either single or in combination, reduced the inhibitory potency of NaKtide on Src. To identify mutant α1 that retains normal pumping function but is defective in Src regulation, we transfected Na/K-ATPase α1 knockdown PY-17 cells with expression vectors of wild type or mutant α1 carrying Ala to Pro mutations in the region of NaKtide helical structure and generated several stable cell lines. We found that expression of either A416P or A420P or A425P mutant fully restored the α1 content and consequently the pumping capacity of cells. However, in contrast to A416P, either A420P or A425P mutant was incapable of interacting and regulating cellular Src. Consequently, expression of these two mutants caused significant inhibition of ouabain-activated signal transduction and cell growth. Thus we have identified α1 mutant that has normal pumping function but is defective in signal transduction.
Introduction
Na/K-ATPase is a ubiquitously expressed, integral membrane protein transporting Na+ and K+ across the plasma membrane by hydrolyzing ATP (1). This pumping function is essential for eukaryotic cells to maintain ionic homeostasis as well as to provide transmembrane Na+ gradients for the Na+-dependent transport of nutrients. Recent studies from different laboratories have revealed that the α1 Na/K-ATPase also has important signal transduction functions and could act as a functional receptor for cardiotonic steroids to activate protein kinase cascades (2–4), which play an important role in renal salt handling and remodeling of the heart and the kidney under pathological conditions (5). Using in vitro binding assays, we have identified two pairs of domain interactions that seem to be essential for the formation of this functional receptor. One is between the second cytoplasmic domain (CD2) of Na/K-ATPase α1 subunit and Src homology 2 (SH2) domain and the other is between the nucleotide binding domain of α1 subunit and Src kinase domain. The latter interaction keeps Src in an inactive state. Binding of cardiotonic steroids such as ouabain to the Na/K-ATPase disrupts the latter interaction, resulting in an activation of the pump-associated Src (4). Besides Src, the α1 Na/K-ATPase interacts with many other partners including phosphoinositide 3-kinase and caveolin-1 and is involved in the regulation of PI3K/Akt pathway and the formation of caveolae (6–8).
To further probe the Src-regulatory function of Na/K-ATPase, we recently mapped the structural determinant of nucleotide binding domain of the α1 subunit that is involved in the interaction with the Src kinase domain, which led to the identification of “NaKtide,” a 20-amino acid peptide located in the N terminus of the nucleotide binding domain (9). We have further engineered a cell-permeable NaKtide (pNaKtide). This peptide is a potent Src inhibitor in the in vitro and acts as a receptor antagonist by blocking the formation of functional Na/K-ATPase·Src complex when applied to cultured cells (9). Moreover, pNaKtide was effective in inducing tumor regression and inhibiting tumor growth in vivo (10). To understand the molecular basis of NaKtide-mediated Src regulation, we made several mutants of NaKtide and tested their effects on Src. These in vitro studies indicate that the N-terminal helical structure of NaKtide appears to be important for its interaction with Src. To further test this hypothesis, we made several α1 mutants and generated stable cell lines expressing these mutants. Functional studies of these stable cell lines demonstrate that the A420P mutant α1 has normal pumping function but has lost its capacity of Src regulation.
EXPERIMENTAL PROCEDURES
Materials
All the peptides of >95% purity (checked by reverse phase HPLC) were purchased from HD Biosciences (China) Co. Ltd. The polyclonal anti-Src (Tyr(P)-418) antibody, cell culture media, fetal bovine serum, trypsin, and Lipofectamine 2000 were purchased from Invitrogen. The QuikChange mutagenesis kit was obtained from Stratagene (La Jolla, CA). Image-iT FX signal enhancer, antifade kit, Alexa Fluor 488-conjugated anti-mouse IgG, and Alexa Fluor 546-conjugated anti-rabbit IgG antibodies were from Molecular Probes (Eugene, OR). Anti-Na/K-ATPase α1 polyclonal, anti-Na/K-ATPase β1 (clone C464.8) antibody, and recombinant human Src were obtained from Upstate Biotechnology (Lake Placid, NY). The monoclonal anti-α1 antibody (α6F) was from the Developmental Studies Hybridoma Bank at the University of Iowa. Anti-c-Src (B-12) monoclonal antibody, the anti-Cav1 polyclonal antibody, and all the secondary horseradish peroxidase-conjugated antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Polyclonal rat α1-specific antibody (anti-NASE) was provided by Dr. Thomas Pressley (Texas Tech University, Lubbock, TX). Radioactive 86Rb+ was from PerkinElmer Life Sciences. Protease inhibitor mixture was purchased from Sigma.
Src Kinase Assay
The activity of NaKtide and its mutant peptides was measured using in vitro Src kinase assay as described (9). Briefly, purified Src (4.5 units) was incubated with different concentrations of peptides in PBS (137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 2 mm KH2PO4, pH 7.4) for 15 min at 37 °C. Afterward 2.0 mm ATP/Mg2+ was added to induce phosphorylation. The reaction was continued for 10 min at 37 °C and was stopped by the addition of SDS sample buffer. Afterward, Western blot analysis of Src Tyr(P)-418 was conducted (9).
Circular Dichroism (CD)
These studies were done as previously described (11). Briefly, CD measurements were performed at 20 °C using Jasco J-715 and quartz flow cell with a 1-mm path length. Peptides were dissolved in PBS, pH 7.4, at a concentration of ∼0.17 mg/ml. Spectra were collected at 50 nm/min using a bandwidth of 1 nm and averaged over 10 scans, and the base line (PBS only) was subtracted from each spectrum. Percent helicity was then calculated by the equation, % helicity = [θ]222/[θ]max × 100 (where [θ]222 mean residue ellipticity and [θ]max is the maximum mean ellipticity).
Computer Modeling
Computer modeling was done using PyMOL (The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC).
Site-directed Mutagenesis
The QuikChange mutagenesis kit was used. We used the vector pRc/CMV-α1 AAC m1 as a template as described in our prior publication (3). According to the GenBankTM rat α1 sequence (NM_01254), we created A416P, A420P, A425P, and A420P/A425P mutant vectors. These four mutants were verified by DNA sequencing.
Generation of Ala to Pro Mutant Stable Cell Lines
This was done as previously described (3). Briefly, PY-17 cells were cultured in 6-well plates and transfected with the four different mutant vectors. The transfected cells were selected with 3 μm ouabain for 1 week, and the survived ouabain-resistant colonies were collected and diluted into 96-well plates to isolate a single colony. Once the colony was expanded into stable cell line, the expression of mutant rat α1 was verified by rat α1-specific antibody.
Cell Culture
Cells were cultured in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 1 μg/ml puromycin. When cells were 90–100% confluent, they were serum-starved overnight and used for experiments unless otherwise noted.
Immunoblot Analysis
After treatments, cells were washed by ice-cold PBS and lysed in ice-cold radioimmunoprecipitation buffer (0.25% sodium deoxycholate, 1% Nonidet P-40, 1 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 1 mm sodium orthovanadate, 1 mm NaF, 150 mm NaCl, 50 mm Tris-HCl, pH 7.4, and 1% protease inhibitor mixture). After the cell lysates were centrifuged at 16,000 × g for 15 min, the supernatants were collected and measured for the protein content. The same amount of protein of each sample was loaded, separated by SDS-PAGE, then transferred to an Optitran membrane and probed by indicated antibody. ImageJ 1.46 was used for analyzing the data.
Confocal Fluorescence Microscopy
Immunostaining of α1 was done as previously described (10). Briefly, cells were cultured on coverslips in 6-well plates. When cells reached 90–100% confluency, they were serum-starved overnight. After the indicated treatment, cells were washed twice with ice-cold PBS, fixed with pre-chilled (−20 °C) methanol for 15 min, and then blocked with PBS containing 1% FBS at room temperature for 30 min. The blocked cells were incubated with primary antibody overnight at 4 °C, washed, and incubated with Alexa Fluor-conjugated secondary antibody for 2 h at room temperature. The stained cells on coverslips were washed, mounted, and then visualized using a Leica DMIRE2 microscope (Wetzlar, Germany).
[3H]Ouabain Binding
To measure the surface expression of the endogenous pig Na/K-ATPase, [3H]ouabain binding assay was performed as described (8). Briefly, cells were cultured in 12-well plates until confluent and serum-starved overnight. Afterward, the cells were incubated in K+-free Krebs solution (142.4 mm NaCl, 2.8 mm CaCl2, 0.6 mm NaH2PO4, 1.2 mm MgSO4, 10 mm glucose, 15 mm Tris (pH 7.4)) for 15 min and then exposed to 200 nm [3H]ouabain for 30 min at 37 °C. After incubation, the cells were washed 3 times with ice-cold K+ free Krebs solution, solubilized in 0.1 m NaOH, 0.2% SDS, and counted in a scintillation counter for [3H]ouabain. Nonspecific binding was measured in the presence of 1 mm unlabeled ouabain and subtracted from total binding. All counts were normalized to protein amount.
Ouabain-sensitive Na/K-ATPase Activity
Cells were collected and homogenized in ice-cold buffer A (150 mm sucrose, 5 mm HEPES, 4 mm EGTA, 0.8 mm dithiothreitol) and briefly sonicated. After centrifugation (800 × g for 10 min), the post nuclear fraction was further centrifuged (45,000 × g for 45 min) to get crude membrane. The crude membrane pellet was resuspended in buffer A, and the protein content was determined. The aliquots of protein were treated with alamethicin (0.1 mg/mg of protein) for 10 min at room temperature and then added to the buffer containing 50 mm Tris (pH 7.4), 1 mm EGTA, 1 mm MgCl2, 25 mm KCl, 100 mm NaCl, 5 mm NaN3. After 15 min of preincubation at 37 °C, ATP/Mg was added to a final concentration of 2 mm to start the reaction. The reaction was continued for 45 min and stopped by adding 8% ice-cold trichloroacetic acid. Phosphate generated during the ATP hydrolysis was measured by BIOMOL GREEN Reagent (Enzo Life Science). Ouabain-sensitive Na/K-ATPase activities were calculated as the difference between the presence and absence of 1 mm ouabain. In certain experiments, the indicated vanadate or Na+ or K+ amount was added in the reaction mixture.
Cell Counting Assay
20,000 cells/well were seeded in 12-well plates and cultured in 10% FBS DMEM medium. At the indicated time, 3 wells from each cell line were trypsinized, and the number of viable cells was counted as described (8).
Cell Surface Biotinylation of Na/K-ATPase
Biotinylation of cell surface protein of these mutant cell lines was undertaken by the protocol previously described (12). Briefly, the full confluent cells grown in 60-mm dishes were washed 3 times with ice-cold PBS followed by incubation with 2 ml of NHS-SS-biotin (1.5 mg/ml) in biotinylation buffer (10 mm triethanolamine, pH 9.0, 150 mm NaCl) for 25 min at 4 °C with very gentle horizontal motion to ensure mixing. The un-reacted biotin was quenched, and the biotinylated cells were lysed with ice-cold lysis buffer (1% Triton X-100, 150 mm NaCl, 5 mm EDTA, 50 mm Tris, 1% protease inhibitor mixture, pH 7.5). Equal amounts of protein (250 μg) were incubated with streptavidin-agarose beads (150 μl) overnight at 4 °C. The beads-bound biotinylated protein and total cell lysate (25 μg) were subjected to Western blot analysis.
Ouabain-sensitive 86Rb+ Uptake
Ouabain-sensitive 86Rb+ was measured to estimate the transport function of Na/K-ATPase as previously described (13). Briefly, cells were cultured in 12-well plates and serum-starved overnight. When the cells were >90% confluence, they were washed and incubated in DMEM with or without 1 mm ouabain for 10 min at 37 °C. 86Rb+ (1 μCi/well) was then added to the cells for 10 min, and the reaction was stopped by washing with ice-cold 0.1 m MgCl2. Trichloroacetic acid soluble 86Rb+ was counted in a Beckman scintillation counter. All counts were normalized to protein amount. In certain experiments, different concentrations of ouabain were added.
Data Analysis
Data presented are the mean ± S.E. of at least three independent experiments, statistical analysis was performed using the Student's t test, and significance was accepted at p < 0.05.
RESULTS
In Vitro Mutagenesis Analyses of NaKtide
We have shown that NaKtide binds and inhibits Src (9). Its mode of action is similar to that of purified α1 Na/K-ATPase in which it inhibits Tyr-418 phosphorylation without affecting the phosphorylation of Tyr-529 (4). The deduced structure from crystals of either the E2P (PDB ID 2ZXE) (14) or high ouabain affinity (PDB ID 3N23) (15) form indicate that NaKtide may adapt a helical structure in the N terminus (Thr-417 to Leu-427) followed by the C-terminal loop tail (Cys-428–Gln-434) (Fig. 1A). To test the importance of this helical structure in NaKtide-mediated Src inhibition, we conducted the following in vitro mutagenesis studies.
FIGURE 1.

Structure-function analysis of mutant NaKtide. Panel A, shown is the structure of NaKtide deduced from the crystal structure of Na/K-ATPase (PDB ID 2ZXE). The structure shows that side chains of Trp-418, Leu-419, and Arg-423 are exposed in the solvent. The green color represents the non-helical part, whereas the red color indicates the helical part of the peptide. The structure was generated by PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC. Panel B–D, shown is dose-dependent inhibition of Src Tyr-418 phosphorylation by NaKtide mutants. Recombinant Src (4.5 units) was incubated with different concentrations of peptides for 15 min and then assayed for Src Tyr(P)-418. Mutated residues are represented in bold and underlined. Combined data from at least four independent experiments are shown. Curve fit analysis was performed with GraphPad Prism 5. *, p < 0.05.
Based on the crystal structure (Fig. 1A), the side chains of Trp-418, Leu-419, and Arg-423 are solvent-exposed. If the helical structure is involved in the interaction with Src, these side chains may provide the contacting site and be important for Src inhibitory effect of the peptide. Indeed, replacement of these three residues with alanine significantly attenuated the inhibitory effect of peptide on Src (Fig. 1B). To further test the importance of the helical structure, we replaced Ala-420 with proline. It is known that proline replacement bends the backbone of a helical structure (16). Ala-420 resides in the middle of the helical structure. Therefore, if the helical structure is important, A420P mutation would alter the structure and attenuate the inhibitory effect of NaKtide on Src. When the dose-dependent effect of A420P mutant peptide was assessed, we found that the mutant peptide was about 10 times less effective than that of NaKtide (Fig. 1, B and C) (9). To further verify the importance of the helical structure, we also determined the effect of A425P and A420P/A425P mutant peptides on Src. As depicted in Fig. 1D, whereas A425P had a reduced effect on Src as A420P peptide, A420P/A425P double mutant exhibited no inhibition of Src.
To assess whether the helix is sufficient for the inhibition of Src, we synthesized a C-terminal-truncated peptide P3A (Ser-415 to Arg-430), removing the last four amino acid residues. As depicted in Fig. 2A, although this peptide showed an inhibitory effect on Src, both efficacy and potency were reduced. To be sure that truncation of last four amino acid residues did not reduce the helicity of the peptide, we measured the circular dichroism (CD) spectra of NaKtide and peptide P3A. As shown in Fig. 2B and Table 1, the helicity in PBS was actually increased when the C-terminal four amino acid residues were removed. In short, the above in vitro mutagenesis analyses indicate that the helical structure as well as the C-terminal tail is important for the interaction and inhibition of Src by NaKtide.
FIGURE 2.

Effect of truncation of C terminus of NaKtide on Src inhibition. Panel A, recombinant Src (4.5 units) was incubated with different concentrations of the peptide P3A for 15 min and then assayed for Tyr(P)-418 Src as described under “Experimental Procedures.” Combined data from at least four independent experiments are shown. Curve fit analysis was performed with GraphPad Prism 5. Panel B, CD spectra of NaKtide and P3A dissolved in PBS (pH 7.4) were measured as described under “Experimental Procedures.”
TABLE 1.
Calculated helicity of peptides in PBS
| Peptide | % Helicity |
|---|---|
| NaKtide | 8.4 |
| P3A | 89.4 |
Generation of Stable Cell Lines Expressing Ala to Pro Mutants
To verify the importance of the helical structure in α1 Na/K-ATPase-mediated Src regulation, we constructed the following Ala to Pro mutants based on a rat α1 cDNA expressing vector we described in our previous publications (3): A416P, A420P, A425P, and a double mutant (A420P/A425P). Based on the crystal structure, Ala-416 resides out of the helical structure. Thus, A416P mutant was selected as a control. To reduce the interference from endogenous α1 Na/K-ATPase, we rescued α1 knockdown PY-17 cells with these mutants. PY-17 cells were derived from pig LLC-PK1 cells, and the expression of endogenous α1 Na/K-ATPase was reduced >90% by the expression of α1-specific siRNA (3). Note that these cells do not express other isoforms of Na/K-ATPase (3). Wild type rat-α1-rescued PY-17 cells (AAC-19) we made previously were used as a control (3). Like wild type rat α1 (3), ouabain selection of transfected PY-17 cells resulted in numerous clones for A416P, A420P, and A425P.
Interestingly, cells transfected with the double mutation (A420P/A425P) cDNA did not produce any viable clone in the presence of ouabain. To explore whether the expressed A420P/A425P double mutant could function as a pump, we conducted a transient transfection assay, made crude membrane preparations, and measured ouabain-sensitive ATPase activity. Like wild type rat α1, transient transfection of PY-17 cells with A420P/A425P double mutant produced a comparable increase in ouabain-sensitive ATPase activity as the wild type rat α1 (Table 2). Thus, it is unlikely that the failure of generating viable clones from A420P/A425P mutant α1 is because of defects in the pumping capacity of this mutant.
TABLE 2.
Ouabain-sensitive ATPase activity
| Transient transfection (plasmid) | Rat α1 | A420P/A425P |
|---|---|---|
| Ouabain-sensitive ATPase activity (%), n ≥3 | 100 | 115.3 ± 9.8 |
Western blot analyses using rat α1-specific antibody showed that every viable clone tested expressed rat α1 mutant and that the expression level of mutant α1 varied among different clones (supplemental Fig. 1). After this initial Western blot screening, we picked clone 4 from A416P-transfected (named as LL-A416P-4, abbreviated as A416P), clone 20 from A420P-transfected (named as LL-A420P-20, abbreviated as A420P), and clone 9 from A425P-transfected (named as LL-A425P-9, abbreviated as A425P) cells. As depicted in Fig. 3A, the expression of mutant α1 in these cell lines was comparable with that of wild type rat α1 in AAC-19 cells. Because rat α1-specific antibody was used, no detectable signal of rat α1 was observed in the PY-17 cell lysates, derived from pig LLC-PK1 cells (Fig. 3A). However, when the blot was analyzed by a generic α1-specific monoclonal antibody, a weak signal was detected in samples from PY-17 cells as we previously reported (3). Moreover, the overall α1 expression level in the rescued cell lines was comparable with each other. This is further verified by immunostaining of these cell lines. Strong and comparable signals were detected in the plasma membrane area in all of the rescued cell lines but not the parental PY-17 cells (Fig. 3B). Finally, biotinylation analysis indicates that the ratio of biotinylated surface α1 to total α1 in the cell lysates was similar among AAC-19, A416P mutant, and A420P and A425P cells (Fig. 3C).
FIGURE 3.

Expression of Na/K-ATPase. Panel A, total cell lysates from different cell lines were separated by SDS-PAGE and analyzed by Western blot for the expression of the rat α1 Na/K-ATPase using polyclonal rat α1-specific antibody (anti-NASE) and the total α1 using monoclonal anti-α1 antibody (α6F). A representative Western blot is shown, and quantitative data (mean ± S.E.) of total α1subunit were calculated from at least three separate experiments. **, p < 0.01 versus AAC-19 cells. Panel B, cells were immunostained with anti-α1 primary and Alexa Fluor-conjugated secondary antibody as described under “Experimental Procedures.” Representative image of three separate experiments is shown for each cell line. Panel C, cells were biotinylated and processed as described under “Experimental Procedures.” An aliquot of 25 μg of cell lysate (T) and biotinylated membrane protein (M) from 250 μg of total cell lysates was subjected to SDS-PAGE and probed with α6F antibody. Representative Western blots are shown, and quantitative data are calculated based on at least three independent experiments as relative ratio of membrane α1 to total α1. Values are the mean ± S.E. Panel D, total cell lysates were analyzed by Western blot using anti-Na/K-ATPase β1 antibody. A representative Western blot of at least three independent experiments is shown.
Na/K-ATPase Activity in Mutant-rescued Cells
To characterize the pumping function of mutant Na/K-ATPase, we first checked the expression of β1 subunit. Knockdown of α1 subunit reduced the expression and glycosylation of β1 subunit (12). As shown in Fig. 3D, all three α1 mutants were able to rescue the expression and glycosylation of β1 as did wild type α1. These findings indicate that the expressed mutant α1 is fully capable of assembling with the β1 subunit into functional Na/K-ATPase, which is consistent with the findings depicted in Fig. 3, B and C.
To assess the pumping capability of these mutants, we measured ouabain-sensitive 86Rb+ uptake in these cell lines (Fig. 4A). No difference in pump activity was detected among different cell lines, indicating that the Ala to Pro mutation made in this particular area of nucleotide binding domain of α1 subunit did not cause an apparent defect in pumping function of the Na/K-ATPase. This notion is further supported by the fact that the expressed mutants showed the similar sensitivity to vanadate (Fig. 4B) and ouabain (Fig. 4C).
FIGURE 4.

Pumping activity in mutant-rescued cells. Panel A, ouabain-sensitive 86Rb+ uptake was measured as described under “Experimental Procedures.” Values are normalized to per protein amount and then calculated as % of AAC-19 cells (mean ± S.E.). Panel B, vanadate sensitivity analysis is shown. Crude membrane fractions were prepared from AAC-19, A416P, A420P, and A425P cells and Na/K-ATPase activity was assayed as described under “Experimental Procedures” in the presence of different concentrations of vanadate. The data points are shown as the percentage of the Na/K-ATPase activity in the absence of vanadate (mean ± S.E.). Panel C, ouabain dose-response curves are shown. AAC-19, A416P, A420P, and A425P cells were cultured in 12-well plates and serum-starved overnight. After reaching 100% confluency, cells were pretreated with different concentrations of ouabain as indicated for 10 min and assayed for 86Rb+ uptake. Data are calculated from at least three repeats and shown as % of respective control (mean ± S.E.). Curve fit analysis was performed by GraphPad Prism 5. Panel D, measurement of Na+ Km is shown. Crude membrane preparations were made from A416P and A420P cells and measured for the Na/K-ATPase activity as a function of Na+ concentration as described under “Experimental Procedures.” Choline chloride was used to substitute NaCl so that ionic strength was kept constant. The combined data from at least three repeats were shown, and Km values (mean ± S.E.) were calculated using GraphPad Prism 5. Panel E, measurement of K+ Km is shown. Crude membrane preparations were made from A416P and A420P cells and measured for the Na/K-ATPase activity as a function of K+ concentration as described under “Experimental Procedures.” Choline chloride was used to substitute KCl so that ionic strength was kept constant. The combined data from at least three repeats were shown, and Km values (mean ± S.E.) were calculated using GraphPad Prism 5. Panel F, [3H]ouabain binding was performed as described under “Experimental Procedures.” Cells were incubated with 200 nm ouabain for 30 min and then assayed for ouabain binding. *, p < 0.05; **, p < 0.01 versus PY-17 cells.
To further confirm that these mutations do not alter the Na/K-ATPase pumping properties, we compared the kinetic properties of A416P and A420P mutants. The crude membranes were prepared from A416P and A420P cells, and ATPase activity was measured in the presence of different concentrations of Na+ and K+. As depicted in Fig. 4, D and E, the Km values of Na+ and K+ were comparable between A416P and A420P mutants. These values are similar to those reported in the literature for α1 Na/K-ATPase (17).
To assess the level of endogenous pig α1 expression in the mutant-rescued cells, we conducted [3H]ouabain binding analyses. Because ouabain dissociates from rat α1 much faster than from the endogenous pig α1, this binding assay allows us to assess the surface expression of endogenous α1 in the presence of highly expressed rat α1 in the rescued cells. The parental PY-17 cells were used as control. As depicted in Fig. 4F, expression of endogenous α1 in either AAC-19 cells or mutant-rescued cells was further reduced compared with that in PY-17 cells.
Taken together, the above findings indicate that the expression of mutants restored total cellular α1 Na/K-ATPase and consequently the pumping capacity in A416P, A420P, and A425P cells to the level comparable to that in AAC-19 cells. Moreover, the level of endogenous α1 in the rescued cells was lower than that of PY-17 cells, amounts less than 10% that of total α1 Na/K-ATPase.
The Expressed Mutants Have Different Effects on Caveolin-1 Expression
We showed that knockdown of α1 Na/K-ATPase increased endocytosis and degradation of caveolin-1 in PY-17 cells. As reported (18), this defect could be rescued by the expression of rat α1 as shown in Fig. 5. When caveolin-1 was measured in mutant-rescued cells, we found that expression of A416P or A420P, was sufficient to restore the expression of caveolin-1 similar to that in AAC-19 cells. However, A425P failed to restore the expression of caveolin-1 (Fig. 5).
FIGURE 5.
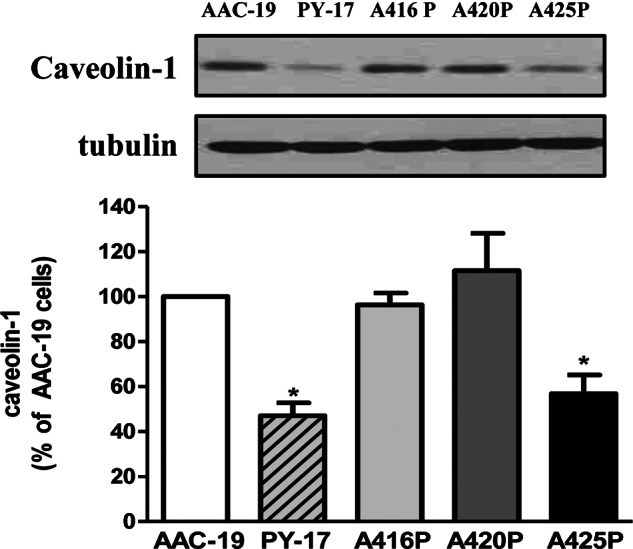
Caveolin-1 expression level in the mutant cells. Total cell lysates from AAC-19, A416P, A420P, and A425P cells were separated by SDS-PAGE and analyzed by Western blot for caveolin-1. A representative Western blot is shown, and quantitative data (mean ± S.E.) were calculated from at least three separate experiments. * p < 0.05 versus control AAC-19 cells.
Regulation of Src by Mutant α1
To assess whether the Src regulatory function of α1 Na/K-ATPase was altered in the mutant-rescued cells, we first determined the basal Src activity in these cells. We previously reported that knockdown of Na/K-ATPase increased basal Src activity in PY-17 cells and that rescuing the knockdown cells with rat α1 reduced the elevated basal Src activity in AAC-19 cells (3). This is indeed the case as shown in Fig. 6 by Western blot analysis of active Src as indicated by Tyr-418 phosphorylation. Interestingly, only the expression of A416P, but not A420P and A425P mutant, showed the same effect as wild type α1. The basal Src activity in both A420P and A425P cells was as high as that in PY-17 cells. These data suggest that bending the helical structure of intact α1 subunit, as in NaKtide (Fig. 1), may reduce the capability of α1 Na/K-ATPase to interact and regulate Src. Taken together, the above experiments indicate that A416P mutant works like wild type α1, whereas expression of A420P mutant restores the expression of caveolin-1 but fails to inhibit Src. The expression of A425Pmutant, on the other hand, could not restore either Src regulation or caveolin-1 expression.
FIGURE 6.

Regulation of Src by mutant α1. Total cell lysates from AAC-19, A420P, and A425P cells were separated by SDS-PAGE and analyzed by Western blot for Tyr(P)-418 (pY418) Src and total Src. Quantitative data are the mean ± S.E. from at least three independent experiments. *, p < 0.05 versus control AAC-19 cells.
Expression of A420P Mutant Inhibits Ouabain-induced Activation of the Src Pathway
We have shown that ouabain is a specific agonist of α1 Na/K-ATPase·Src receptor complex (4). Binding of ouabain to the α1 Na/K-ATPase stimulated the Src pathway within minutes and increased the expression of α1 Na/K-ATPase in hours (2, 4). Indeed, these changes in response to ouabain (10 to100 μm) were observed in the control AAC-19 and A416P cells (Fig. 7, A and B). If the A420P mutant could not interact with Src as evidenced by the findings in Figs. 1 and 6, this mutant would not be able to constitute a functional receptor for cardiotonic steroids such as ouabain to activate Src. This was the case as shown in Fig. 7, A and B. Ouabain was able to stimulate Src in A416P but not in A420P cells. Furthermore, we reported that ouabain could increase the expression of α1 in LLC-PK1 and rat α1-rescued AAC-19 cells (8). It failed to do so in A420P cells. To seek further evidence that the helical structure is important for the formation of a functional Na/K-ATPase·Src complex, we repeated these studies in A425P cells. As shown in Fig. 7, A and B, A425P mutant, like A420P mutant, lost its ability to allow ouabain to stimulate Src pathway. It is important to note that AAC-19 cells express ouabain-insensitive rat α1 Na/K-ATPase. Therefore, μm instead of nm concentrations of ouabain were used in these experiments (3).
FIGURE 7.

Expression of mutant α1 inhibits ouabain-induced signal transduction. Panel A, confluent cells were treated with different concentrations of ouabain for 10 min and then harvested, and cell lysates were analyzed by Western blot for Src Tyr(P)-418 (pTyr 418). Panel B, confluent cells were treated with different concentrations of ouabain for 24 h, then harvested and analyzed for Na/K-ATPase α1 expression by Western blot. A representative Western blot is shown, and quantitative data are presented as the mean ± S.E. of at least three independent experiments. * p < 0.05 versus 0 mm ouabain.
Expression of A420P and A425P Mutants Inhibits Cell Proliferation
Src-mediated pathways are known to play an important role in cell proliferation. We have shown that alteration in Na/K-ATPase-mediated Src regulation affects cell growth (26). However, these previous studies were done in cell lines where both pumping and signaling functions of Na/K-ATPase were altered. To further demonstrate a role of α1 Na/K-ATPase-mediated Src regulation in control of cell growth, we cultured control and mutant cells in full medium and counted the number of cells at different time points. As shown in Fig. 8, A416P grew similarly as AAC-19 cells. However, cell growth was significantly reduced in A420P and A425P cells.
FIGURE 8.

Effects of mutant α1 expression on cell growth. AAC-19, A416P, A420P, and A425P cells were plated in 12-well plates (20,000 cells/well), cultured for different times, and then collected and counted. The values are the mean ± S.E. from four independent experiments. **, p < 0.01 versus AAC-19 cells.
DISCUSSION
We report here the identification of α1 mutants that retain normal pumping activity but fail to interact and regulate Src and Src-mediated signaling pathways in cell cultures. These findings reaffirm the importance of the helical structure of NaKtide in binding and regulating Src. Moreover, we suggest that the newly identified α1 mutants would allow us to fully assess the importance of Src-regulatory function of α1 Na/K-ATPase as well as the CTS3-induced signaling in animal physiology, pathology, and pharmacology.
Identification of the A420P Mutant α1 as a Src Regulation-null Pump
Based on the crystal structure (20), the NaKtide sequence can adapt a helical structure at the N terminus followed by a C-terminal loop (Fig. 1). Kinase activity assays indicate that replacement of either Ala-420 or Ala-425 by proline was sufficient to reduce the peptide-induced inhibition of Src (Fig. 1). Moreover, alanine replacement of residues that contain a bulky side chain in the helical structure (i.e. Trp-418, Leu-419, and Arg-423) also resulted in a loss of the inhibitory effect of the peptide on Src. These new findings suggest an important role of the helical structure and amino acid residue Trp-418, Leu-419, and Arg-423 in the binding and regulation of Src. Because pNaKtide acts as an effective antagonist of receptor Na/K-ATPase·Src complex in vitro and in vivo (9, 10), the new findings provide important structural information for directing the development of second generation of pNaKtide derivatives.
PY-17 cells were derived from LLC-PK1 cells stably transfected with plasmids expressing α1-specific siRNA (3). The expression of α1-specific siRNA produced >90% down-regulation of endogenous α1 and consequently abolished the signaling function of Na/K-ATPase. As such, these cells have been used by us to study the signaling function of α1 Na/K-ATPase and the structure/function relationships of the pump in signal transduction after the cells were rescued by either wild type rat α1 or α1 mutants (3). The intrinsic limitation of these early studies is the fact that we could not separate the pumping function of Na/K-ATPase from its signaling function. For instance, knockdown of α1 Na/K-ATPase abolished ouabain-induced activation of protein kinases in PY-17 cells. However, we could not rule out the influence of reduced transport activity across the cell membrane and/or the consequent changes in intracellular ion concentrations on protein kinase activity (12). Furthermore, we could not attribute the effects of either endogenous or exogenous ouabain on cellular functions solely to the changes in protein kinase activity as ouabain also inhibits the pumping function of Na/K-ATPase. Thus, it would be more desirable to have a mutant Na/K-ATPase that can pump but not signal. We believe that A420P mutant α1 is such a mutant pump. First of all, expression of A420P not only rescued cellular α1 but also restored the expression and glycosylation of the β1 subunit, indicating that the mutant α1 is fully capable of assembling with the β1 subunit into functional pump in the plasma membrane. This is consistent with the findings presented in Fig. 3 showing that the mutant α1 was expressed in the plasma membrane as revealed by both immunostaining and biotinylation. Functionally, A420P mutant exhibited comparable pumping activity to that of wild type rat α1 and A416P mutant as measured by ouabain-sensitive 86Rb+ uptake. Moreover, the expressed mutant showed the same ouabain sensitivity as that of wild type α1. This is in sharp contrast to many reported mutants of α1 Na/K-ATPase in the literature (21). For example, the identified mutants defective in E1/E2 transitions exhibit reduced pump activity and altered ouabain sensitivity (22). Our recent rescuing studies of I279A and F286A mutants in PY-17 cells confirm these early findings (26). Second, unlike the wild type α1 or A416P mutant, expression of A420P mutant failed to restore the basal Src activity in the rescued cells. Most importantly, although ouabain was able to inhibit the pumping function, it failed to stimulate Src and Src effectors in the rescued A420P cells (Fig. 6). Third, it is of interest to compare A420P and A425P mutants. Although both of these mutants showed a defect in Src regulation, expression of A420P, but not A425P, was able to, like wild type α1, rescue the expression of caveolin-1. This is important because caveolae play a critical role in cell signal transduction in general (23, 24), and use of A425P in the study would further complicate the signaling pathways.
Src is known to play an important role in the regulation of cell growth (25). The α1 Na/K-ATPase exists in two major conformations, namely E1 and E2. We have found that expression of α1 Na/K-ATPase mutants that are defective in either E1 or E2 conformational transition alter the dynamic nature of Src regulation, resulting in cell growth inhibition (26). These early findings suggest that the α1 Na/K-ATPase is a key regulator of cellular Src activity and consequently cell growth. However, because E1 and E2 mutants are also defective in pumping (26), it is conceivable that changes in cellular pumping activity could at least be part of altered Src regulation and cell growth. To this end, the new findings as depicted in Fig. 8 are very important because they clearly demonstrate that alteration in the Src regulatory capacity of α1 Na/K-ATPase is sufficient to inhibit cell growth. Therefore, we believe that the α1 Na/K-ATPase provides cells a critical ability of dynamic Src regulation.
Uncertainties and Implications
Recent studies have identified both ouabain and Marinobufagenin (MBG) as endogenous steroids whose production and secretion are regulated by stimuli including angiotensin II and adrenocorticotropic hormone (27, 28). Although their pathophysiological significance has been a subject of debate for many years (29), several gene replacement studies from Lingrel and co-workers (30, 31) have unequivocally demonstrated an important role of endogenous CTS in regulation of renal Na+ excretion and blood pressure. In addition, we and others showed that CTS not only induced hypertension in rats but also caused significant cardiovascular remodeling independent of their effect on blood pressure (5, 20, 31, 32). Moreover, CTS may play an important role in embryonic development (19). However, the question remains of whether the signaling function of α1 Na/K-ATPase is involved in the aforementioned physiological and pathological processes. To this end, we know that the Src pathway is activated by high salt intake and by infusion of physiologically relevant amount of CTS (5). Thus, α1 gene replacement using the newly identified A420P mutant would allow us to conduct experiments similar to those carried out by Lingrel and co-workers (30, 31) to assess specifically the Src regulatory function of Na/K-ATPase in renal salt handling, organ remodeling, and embryonic development.
Although our new findings suggest that the helical structure of NaKtide is important for regulating Src and the formation of receptor Na/K-ATPase·Src complex, more studies are required to reveal the detailed structural determinants. For example, it remains to be determined whether mutation of either Trp-418 or Leu-419 or Arg-423 is sufficient for abolishing the inhibitory effect of NaKtide on Src. It will also be of interest to test whether alanine replacement of these residues could produce another mutant α1 that retains the full pumping capacity but no Src regulatory function. Finally, we do not know why expression of the double mutant A420P/A425P α1 failed to produce viable clones in the presence of ouabain. Transient transfection assays indicate that the expressed mutant apparently had a normal ATPase activity as other mutants. However, it is possible that the double mutation alters ouabain sensitivity or makes the mutant pump interact with key growth-related pathways other than Src.
In short, this work taken together with our prior reports reveals that the α1 Na/K-ATPase is an important Src regulator. Mutations that affect the interaction between the NaKtide region of α1 subunit and Src appear to have a specific effect on the α1 Na/K-ATPase-mediated Src regulation without compromising the normal pumping function.
Acknowledgment
We thank Tillekeratne Manoranjini for technical support.
This work was supported, in whole or in part, by National Institutes of Health Grant HL-109015 (NHLBI).

This article contains supplemental Fig. 1.
- CTS
- cardiotonic steroid.
REFERENCES
- 1. Lingrel J. B., Kuntzweiler T. (1994) Na+,K(+)-ATPase. J. Biol. Chem. 269, 19659–19662 [PubMed] [Google Scholar]
- 2. Haas M., Askari A., Xie Z. (2000) Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J. Biol. Chem. 275, 27832–27837 [DOI] [PubMed] [Google Scholar]
- 3. Liang M., Cai T., Tian J., Qu W., Xie Z. J. (2006) Functional characterization of Src-interacting Na/K-ATPase using RNA interference assay. J. Biol. Chem. 281, 19709–19719 [DOI] [PubMed] [Google Scholar]
- 4. Tian J., Cai T., Yuan Z., Wang H., Liu L., Haas M., Maksimova E., Huang X. Y., Xie Z. J. (2006) Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol. Cell 17, 317–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu J., Yan Y., Liu L., Xie Z., Malhotra D., Joe B., Shapiro J. I. (2011) Impairment of Na/K-ATPase signaling in renal proximal tubule contributes to Dahl salt-sensitive hypertension. J. Biol. Chem. 286, 22806–22813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu J., Kesiry R., Periyasamy S. M., Malhotra D., Xie Z., Shapiro J. I. (2004) Ouabain induces endocytosis of plasmalemmal Na/K-ATPase in LLC-PK1 cells by a clathrin-dependent mechanism. Kidney Int. 66, 227–241 [DOI] [PubMed] [Google Scholar]
- 7. Cai T., Wang H., Chen Y., Liu L., Gunning W. T., Quintas L. E., Xie Z. J. (2008) Regulation of caveolin-1 membrane trafficking by the Na/K-ATPase. J. Cell Biol. 182, 1153–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tian J., Li X., Liang M., Liu L., Xie J. X., Ye Q., Kometiani P., Tillekeratne M., Jin R., Xie Z. (2009) Changes in sodium pump expression dictate the effects of ouabain on cell growth. J. Biol. Chem. 284, 14921–14929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Z., Cai T., Tian J., Xie J. X., Zhao X., Liu L., Shapiro J. I., Xie Z. (2009) NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J. Biol. Chem. 284, 21066–21076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li Z., Zhang Z., Xie J. X., Li X., Tian J., Cai T., Cui H., Ding H., Shapiro J. I., Xie Z. (2011) Na/K-ATPase mimetic pNaKtide peptide inhibits the growth of human cancer cells. J. Biol. Chem. 286, 32394–32403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kawamoto S. A., Coleska A., Ran X., Yi H., Yang C. Y., Wang S. (2012) Design of triazole-stapled BCL9 α-helical peptides to target the β-catenin/B-cell CLL/lymphoma 9 (BCL9) protein-protein interaction. J. Med. Chem. 55, 1137–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liang M., Tian J., Liu L., Pierre S., Liu J., Shapiro J., Xie Z. J. (2007) Identification of a pool of non-pumping Na/K-ATPase. J. Biol. Chem. 282, 10585–10593 [DOI] [PubMed] [Google Scholar]
- 13. Liu J., Periyasamy S. M., Gunning W., Fedorova O. V., Bagrov A. Y., Malhotra D., Xie Z., Shapiro J. I. (2002) Effects of cardiac glycosides on sodium pump expression and function in LLC-PK1 and MDCK cells. Kidney Int. 62, 2118–2125 [DOI] [PubMed] [Google Scholar]
- 14. Shinoda T., Ogawa H., Cornelius F., Toyoshima C. (2009) Crystal structure of the sodium-potassium pump at 2.4 A resolution. Nature 459, 446–450 [DOI] [PubMed] [Google Scholar]
- 15. Yatime L., Laursen M., Morth J. P., Esmann M., Nissen P., Fedosova N. U. (2011) Structural insights into the high affinity binding of cardiotonic steroids to the Na+,K+-ATPase. J. Struct. Biol. 174, 296–306 [DOI] [PubMed] [Google Scholar]
- 16. Barlow D. J., Thornton J. M. (1988) Helix geometry in proteins. J. Mol. Biol. 201, 601–619 [DOI] [PubMed] [Google Scholar]
- 17. Blanco G. (2005) Na,K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. Semin. Nephrol. 25, 292–303 [DOI] [PubMed] [Google Scholar]
- 18. Chen Y., Cai T., Wang H., Li Z., Loreaux E., Lingrel J. B., Xie Z. (2009) Regulation of intracellular cholesterol distribution by Na/K-ATPase. J. Biol. Chem. 284, 14881–14890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li J., Khodus G. R., Kruusmägi M., Kamali-Zare P., Liu X. L., Eklöf A. C., Zelenin S., Brismar H., Aperia A. (2010) Ouabain protects against adverse developmental programming of the kidney. Nat. Commun. 1, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ogawa H., Shinoda T., Cornelius F., Toyoshima C. (2009) Crystal structure of the sodium-potassium pump (Na+,K+-ATPase) with bound potassium and ouabain. Proc. Natl. Acad. Sci. U.S.A. 106, 13742–13747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boxenbaum N., Daly S. E., Javaid Z. Z., Lane L. K., Blostein R. (1998) Changes in steady-state conformational equilibrium resulting from cytoplasmic mutations of the Na,K-ATPase α-subunit. J. Biol. Chem. 273, 23086–23092 [DOI] [PubMed] [Google Scholar]
- 22. Toustrup-Jensen M., Vilsen B. (2003) Functional consequences of alterations to Ile-279, Ile-283, Glu-284, His-285, Phe-286, and His-288 in the NH2-terminal part of transmembrane helix M3 of the Na+,K+-ATPase. J. Biol. Chem. 278, 38653–38664 [DOI] [PubMed] [Google Scholar]
- 23. Parton R. G., Simons K. (2007) The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 8, 185–194 [DOI] [PubMed] [Google Scholar]
- 24. Salanueva I. J., Cerezo A., Guadamillas M. C., del Pozo M. A. (2007) Integrin regulation of caveolin function. J. Cell. Mol. Med. 11, 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parsons S. J., Parsons J. T. (2004) Src family kinases, key regulators of signal transduction. Oncogene 23, 7906–7909 [DOI] [PubMed] [Google Scholar]
- 26. Ye Q., Lai F., Banerjee M., Duan Q., Li Z., Si S., Xie Z. (2013) Expression of mutant α1 Na/K-ATPase defective in conformational transition attenuates Src-mediated signal transduction. J. Biol. Chem. 288, 5803–5814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bagrov A. Y., Shapiro J. I., Fedorova O. V. (2009) Endogenous cardiotonic steroids. Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 61, 9–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hamlyn J. M., Ringel R., Schaeffer J., Levinson P. D., Hamilton B. P., Kowarski A. A., Blaustein M. P. (1982) A circulating inhibitor of (Na+ + K+)ATPase associated with essential hypertension. Nature 300, 650–652 [DOI] [PubMed] [Google Scholar]
- 29. Kelly R. A., Smith T. W. (1989) The search for the endogenous digitalis. An alternative hypothesis. Am. J. Physiol. 256, C937–C950 [DOI] [PubMed] [Google Scholar]
- 30. Dostanic-Larson I., Van Huysse J. W., Lorenz J. N., Lingrel J. B. (2005) The highly conserved cardiac glycoside binding site of Na,K-ATPase plays a role in blood pressure regulation. Proc. Natl. Acad. Sci. U.S.A. 102, 15845–15850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Loreaux E. L., Kaul B., Lorenz J. N., Lingrel J. B. (2008) Ouabain-sensitive α1 Na,K-ATPase enhances natriuretic response to saline load. J. Am. Soc. Nephrol. 19, 1947–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kennedy D. J., Vetteth S., Periyasamy S. M., Kanj M., Fedorova L., Khouri S., Kahaleh M. B., Xie Z., Malhotra D., Kolodkin N. I., Lakatta E. G., Fedorova O. V., Bagrov A. Y., Shapiro J. I. (2006) Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 47, 488–495 [DOI] [PubMed] [Google Scholar]