

Background: Group A Streptococcus (GAS) translocates across the host epithelial barrier.
Results: Streptococcal pyrogenic exotoxin B (SpeB) directly cleaves junctional proteins.
Conclusion: The proteolytic efficacy of SpeB allows GAS to translocate across the epithelial barrier.
Significance: SpeB-mediated dysfunction of the epithelial barrier may have important implications for not only bacterial invasion but also dissemination of other virulence factors throughout intercellular spaces.
Keywords: Bacterial Pathogenesis, Cell Polarity, Cysteine Protease, Epithelial Cell, Junctions
Abstract
Group A Streptococcus (GAS) is an important human pathogen that possesses an ability to translocate across the epithelial barrier. In this study, culture supernatants of tested GAS strains showed proteolytic activity against human occludin and E-cadherin. Utilizing various types of protease inhibitors and amino acid sequence analysis, we identified SpeB (streptococcal pyrogenic exotoxin B) as the proteolytic factor that cleaves E-cadherin in the region neighboring the calcium-binding sites within the extracellular domain. The cleaving activities of culture supernatants from several GAS isolates were correlated with the amount of active SpeB, whereas culture supernatants from an speB mutant showed no such activities. Of note, the wild type strain efficiently translocated across the epithelial monolayer along with cleavage of occludin and E-cadherin, whereas deletion of the speB gene compromised those activities. Moreover, destabilization of the junctional proteins was apparently relieved in cells infected with the speB mutant, as compared with those infected with the wild type. Taken together, our findings indicate that the proteolytic efficacy of SpeB in junctional degradation allows GAS to invade deeper into tissues.
Introduction
Streptococcus pyogenes (group A Streptococcus; GAS)3 is well known as a human-specific pathogen responsible for numerous diseases, ranging from pharyngitis and impetigo to life-threatening invasive diseases, including necrotizing fasciitis and streptococcal toxic shock syndrome (1, 2). Serious postinfectious immune sequelae such as rheumatic fever and acute glomerulonephritis occasionally develop following repeated GAS exposure. Despite the availability of sequence information for several GAS genomes and detailed characterization of their virulence factors, a safe and effective commercial GAS vaccine has yet to be developed (3).
The pharynx and skin are thought to be the primary sites of GAS infection. Pharyngeal epithelia and keratinocytes are highly specialized physical barriers apposed tightly by tight junctions (TJs) and adherence junctions (AJs), which protect underlying sterile tissues from the external environment. Loss of cell-cell adhesion caused by bacteria has been reported to be associated with several clinical manifestations. Desmoglein 1, which plays a key role in maintaining the structure and barrier function of the epidermis, is targeted by exfoliative toxins released by Staphylococcus aureus in infectious skin diseases such as bullous impetigo and staphylococcal scalded skin syndrome (4). Development of GAS skin infections with bullous lesions also seems to be related to loss of cell-cell adhesion and inoculation of GAS into intradermal space (5).
Invasive GAS disease requires successful colonization in the pharynx or skin, followed by overcoming the host epithelial barrier together with evasion of host defense mechanisms. Multiple in vitro studies have demonstrated that GAS isolates associated with invasive diseases efficiently invade epithelial cells (6, 7). Although programmed cell death is an essential part of host defense against pathogens, it is considered that internalized GAS exploits this process to access the underlying sterile tissues (8, 9). Meanwhile, some studies that investigated the direct interactions of bacteria with epithelial junctions also elucidated the underlying mechanisms of GAS pathogenesis, with interaction of the hyaluronic acid capsule with CD44 implicated in this process (10). Furthermore, our recent study identified streptolysin S (SLS) as a novel factor that facilitates GAS translocation via degradation of intercellular junctions in concert with the host cysteine protease calpain (11). However, the precise mechanism by which GAS disrupts the epithelial barrier has yet to be completely elucidated.
During infection, GAS produces numerous secreted and cell-associated proteins, including toxins, superantigens, and proteases (12, 13). Although extracellular proteins from GAS have been extensively investigated and shown to be important for pathogenesis, its participation in epithelial barrier dysfunction is as yet unproven. Herein, we provide the first direct evidence that SpeB (streptococcal pyrogenic exotoxin B), a broad spectrum secreted cysteine protease, effectively cleaves transmembrane proteins associated with the epithelial barrier to permit bacterial penetration. Our results reveal a new mechanism to explain how GAS directly disrupts the epithelial barrier.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Culture Conditions
Invasive GAS clinical isolates, strains NIH35 (serotype M28), SSI-1 (serotype 3), SSI-9 (serotype M1), and #30 (serotype M12), were isolated from patients with streptococcal toxic shock syndrome. Other GAS clinical isolates, strains SF370 (serotype M1), TW3358 (serotype M3), TW3337 (serotype M12), TW3339 (serotype M28), NZ131 (serotype M49), and 591 (serotype M49), were used as noninvasive GAS strains. Escherichia coli XL10-Gold (Stratagene) served as a host for plasmids pAT18 and pSET4s (14, 15). GAS strains and E. coli strains were cultured at 37 °C in Todd-Hewitt broth (Becton, Dickinson and Company) supplemented with 0.2% yeast extract (Becton Dickinson) (THY medium). E. coli strains were cultured in LB medium (Sigma-Aldrich) at 37 °C with agitation. For selection and maintenance of the mutants, antibiotics were added to the media at the following concentrations: ampicillin, 100 μg/ml for E. coli; erythromycin, 150 μg/ml for E. coli and 1 μg/ml for GAS; and spectinomycin, 100 μg/ml for E. coli and GAS.
Construction of Recombinant SpeB and GAS Mutant Strains
Preparation of recombinant SpeB has been previously described (16). An in-frame speB deletion mutant, its complemented strain, and sagA-speB double mutant were constructed using pSET4s, as previously reported (11, 17). Primers speBkoF1 (5′-GCGGATCCTGTTTAATCGAAATGTTTTTTGAATGC-3′), speBkoR1 (5′-ACTTTGGTAACCGTTGAAGCCCATTTTTTTTATACCTCTTTC-3′), speBkoF2 (5′-GAAAGAGGTATAAAAAAAATGGGCTTCAACGGTTACCAAAGT-3′), and speBkoR2 (5′-AACTGCAGGTCTTAAAGGATGTACCGTATTGG-3′) were used for deletion of speB gene. For construction of EGFP-expressing GAS strains, a pAT18-EGFP vector was transformed into the GAS strains by electroporation (8).
Cell Cultures
Caco-2 cells (Riken Cell Bank) were maintained in minimum essential medium (Invitrogen) supplemented with 20% fetal bovine serum (SAFC Biosciences) and 20 μg/ml gentamicin, 17.75 mm NaHCO3 (Wako), and 15 mm HEPES (Dojindo) at pH 7.4. HaCaT cells were cultured in Dulbecco's modified Eagle's medium (Wako) supplemented with 10% fetal bovine serum (SAFC Biosciences), 20 μg/ml gentamicin. Detroit 562 cells (ATCC CCL-138; American Type Culture Collection) were maintained in minimum essential medium-α (Wako) supplemented with 10% fetal bovine serum (SAFC Biosciences) and 20 μg/ml gentamicin.
For translocation assays, Caco-2 cells were seeded at 2 × 105 cells/well onto polycarbonate Millicell culture plate inserts (12-mm diameter, 3-μm pore size; Millipore) and cultured for 5 days at 37 °C under a 5% CO2 atmosphere, as described previously (11). Transepithelial electrical resistance (TER) of the filter-grown monolayers was measured using a Millicell-ERS device (Millipore), and monolayers exhibiting TER values of 450–500 Ω·cm2 were used in the experiments.
Translocation Assay
GAS strains were grown to the exponential phase (A600 = 0.4) and centrifuged at 7000 × g for 5 min. Pelleted cells were washed with PBS and then resuspended in cell growth medium. Polarized monolayers were infected with GAS at an multiplicity of infection (MOI) of 10. The ability of GAS strains to translocate into monolayers was assessed by quantitative cultures of media obtained from the lower chambers at various times after infection as described previously (11).
Paracellular Flux of FITC-Dextrans
Caco-2 cells were grown on Millicell filters and infected with GAS at an MOI of 10 for 2 h. To remove nonadherent bacteria, the medium in the upper chamber was replaced with fresh medium at 2 h after infection. After removing nonadherent bacteria, FITC-dextran with a molecular mass of 4, 10, or 70 kDa (Sigma) was added to the apical surface of the cell monolayers. At 8 h after infection, the amount of FITC-dextran in the basolateral medium was measured using a Wallac 1420 ARVOsx fluorometer (excitation, 485 nm; emission, 535 nm; PerkinElmer Life Sciences).
Analysis of GAS-induced Cleavage of Junctional Proteins
Overnight cultures of GAS clinical isolates were centrifuged at 7000 × g for 5 min, and then the supernatants were incubated with 0.5 μg of occludin (Abnova) or E-cadherin (R&D Systems or Advanced BioMatrix) for 6 h at 37 °C. To search for a bacterial protease that cleaves E-cadherin, GAS supernatants were individually pretreated for 30 min at room temperature with the following protease inhibitors; N-ethylmaleimide (1 mm), E-64 (10 μm), chymostatin (330 μm), leupeptin (100 μm), AEBSF (1 mm), aprotinin (800 nm), benzamidine HCl (1 mm), trypsin inhibitor (100 μm), 6-aminohexanoic acid (38 mm), pepstatin (1.5 μm), phosphoramidon (10 μm), bestatin (1 mm), or EDTA (1 mm). All protease inhibitors were purchased from Sigma-Aldrich.
For infection assays, epithelial cells were seeded at 2 × 105 cells/well (35-mm diameter; Corning), cultured for 3 days, and then infected with GAS strains at an MOI of 10. At the end of the infection period, the infected cells were lysed with Laemmli gel loading buffer containing 6% 2-mercaptoethanol. Cleavage of intercellular junctions was detected by Western blot analysis (11). Western blot signals were quantified using Scion Image 4.0.3.2 software (Scion).
For identification of the cleavage site in E-cadherin, E-cadherin was incubated with DTT-activated recombinant SpeB (200 nm) for 6 h at 37 °C. Proteins were separated by SDS-PAGE and stained with Coomassie Brilliant Blue. N-terminal amino acid sequencing was performed using the Edman degradation method with an ABI protein sequencer model 491HT (Applied Biosystems).
Measurement of SpeB Activity
For quantitative assays of SpeB activity, casein hydrolysis was analyzed using FITC-labeled casein (18). Filter-sterilized supernatants from overnight GAS cultures or recombinant SpeB were activated in assay buffer (0.1 m phosphate buffer, pH 7.6, 0.01 mm EDTA) supplemented with 10 mm DTT and incubated for 30 min at 37 °C. The activated supernatants were added to an equal volume of FITC-labeled casein (Sigma-Aldrich) prepared with or without 10 μm E-64. At 6 h after incubation, the reactions were stopped by adding 5% trichloroacetic acid, and then the mixtures were stored overnight at 4 °C. Following centrifugation (15,000 × g for 5 min), the resultant supernatants were diluted with 0.5 m Tris buffer (pH 8.5), and fluorescence intensity was measured using a Wallac 1420 ARVOsx fluorometer (excitation wavelength, 485 nm; emission wavelength, 535 nm; PerkinElmer Life Sciences).
Statistical Analysis
Statistical analysis was performed using a Mann-Whitney U test. A confidence interval with a p value of < 0.05 was considered to be significant.
RESULTS
GAS Supernatant Induces Cleavage of E-cadherin
Culture supernatants from several serotypes of GAS strains recovered from invasive (strains SSI-9, SSI-1, #30, and NIH35) and noninvasive (strains SF370, TW3358, TW3337, TW3339, NZ131, and 591) diseases were incubated with the extracellular domain of human E-cadherin fused to the Fc region of human IgG1, after which cleavage was analyzed using Western blot analysis (Fig. 1). Utilizing an antibody against the extracellular domain of E-cadherin, the intact protein was detected as an ∼120-kDa band under a reducing condition. Loss of the 120-kDa band and appearance of cleavage products with apparent molecular masses of 60–100 kDa were detected in culture supernatant samples obtained from SF370, SSI-1, TW3358, #30, TW3337, NIH35, TW3339, and 591. Interestingly, cleavage was completely abrogated by pretreatment with a protease inhibitor mixture (data not shown). On the other hand, neither the culture supernatant from strain SSI-9 nor that from strain NZ131 was capable of cleaving the E-cadherin fragment. These results indicate that the culture supernatants from strains SF370, SSI-1, TW3358, #30, TW3337, NIH35, TW3339 and 591 include proteases that cleave the extracellular domain of E-cadherin. We also noted that the ability of GAS culture supernatants to cleave E-cadherin was not related to disease severity, i.e., strains from invasive as compared with those from noninvasive diseases.
FIGURE 1.

Cleavage of E-cadherin by GAS culture supernatant. Culture supernatants from several GAS clinical isolates were incubated with the recombinant extracellular domain of E-cadherin for 6 h at 37 °C. Sample proteins were separated by SDS-PAGE under a reducing condition and subjected to immunoblotting using an antibody against the extracellular domain of E-cadherin and the horseradish peroxidase-conjugated secondary antibody. The signals were developed with the peroxidase substrate tetramethylbenzidine. GAS strains recovered from invasive disease are underlined.
Identification of Bacterial Protease Responsible for Cleavage of E-cadherin
To search for bacterial proteases that contribute to the cleavage of E-cadherin, the culture supernatant of strain NIH35, a prominent degrader, was pretreated with several types of protease inhibitors before co-incubation with recombinant E-cadherin protein (Fig. 2). The cysteine protease inhibitors N-ethylmaleimide, E-64, and chymostatin completely inhibited cleavage of E-cadherin, whereas protease activity partially remained in the leupeptin-treated GAS supernatant. In contrast, protease inhibitors that target serine protease, aspartate protease, and metalloprotease had no detectable effect on E-cadherin cleavage, whereas EDTA did have an acceleratory effect. This result may reflect a chelating effect of EDTA on stabilization of the E-cadherin structure, because E-cadherin forms a homodimer in the extracellular domain in a Ca2+-dependent manner. In experiments with strain SSI-1, even though the degradation pattern of E-cadherin was slightly different from that observed with other strains (Fig. 1), the appearance of cleavage products was completely inhibited by cysteine protease inhibitors (data not shown). It has been reported that GAS secretes two major cysteine proteases, SpeB and immunoglobulin G-degrading enzyme of S. pyogenes (IdeS or Mac-1). E-64, a cysteine protease, specifically inhibits the proteolytic activity of SpeB, but not that of IdeS (19). In the present study, supernatant-induced cleavage of E-cadherin was completely inhibited by E-64 (Fig. 2). These results suggest that SpeB is a proteolytic factor for cleavage of E-cadherin. Moreover, restoration of the full-length band was observed in a leupeptin concentration-dependent manner, whereas the cleavage product did not disappear even in the presence of leupeptin at 500 μm (supplemental Fig. S1). Leupeptin is an arginine-dependent protease inhibitor. Because Cys at position 192, His at position 340, and Trp at position 357 play critical roles in the enzymatic activity of SpeB (20), leupeptin might be less effective for inhibition of SpeB activity as compared with those other cysteine proteases. Consequently, we investigated whether SpeB is involved in cleavage of E-cadherin.
FIGURE 2.

Cysteine protease contributes to cleavage of E-cadherin. Culture supernatants from strain NIH35 were pretreated with several types of protease inhibitors for 30 min at room temperature and then incubated with recombinant E-cadherin for 6 h at 37 °C. The sample proteins were separated by SDS-PAGE under a reducing condition and subjected to immunoblotting using an antibody against extracellular domain of E-cadherin and horseradish peroxidase-conjugated secondary antibody. Immunoreactive bands were detected with a peroxidase substrate tetramethylbenzidine.
SpeB Cleaves Intercellular Junctional Proteins
To examine whether SpeB is responsible for GAS supernatant-induced cleavage of intercellular junctions, an in-frame speB deletion mutant and its complemented strain were constructed. Equivalent growth rates of wild type and mutant strains were observed in conventional liquid medium (data not shown). GAS culture supernatant-induced cleavage of E-cadherin was nearly abolished by mutagenesis of the speB gene, whereas it was completely restored by the complementation (Fig. 3A). A similar phenomenon was observed with strains #30, TW3337, and TW3339 (supplemental Fig. S2), indicating that SpeB-dependent protease activity toward E-cadherin is conserved among clinical isolates with no relation to disease severity. These observations prompted us to compare the SpeB-specific protease activities in the culture supernatants of the previously examined GAS strains (Fig. 3B). High caseinolytic activities were detected in culture supernatants from strains #30, TW3337, NIH35, TW3339, and 591, which are findings consistent with its ability to cleave recombinant E-cadherin (Fig. 1). In contrast, lower levels of SpeB activity were observed in supernatants from strains SSI-9, SF370, SSI-1, TW3358, and NZ131. Together, these results indicate that SpeB-associated cysteine protease activity is correlated with the cleavage of intercellular junctions.
FIGURE 3.

SpeB cleaves intercellular junctional proteins. A, recombinant E-cadherin was treated with culture supernatants from strain NIH35, an speB deletion mutant, and a complemented strain for 6 h at 37 °C. Cleavage of E-cadherin was detected by Western blotting. B, SpeB activities present in the culture supernatants from several GAS clinical isolates were analyzed using FITC-labeled casein. For quantitative evaluation, the casein hydrolysis activity of recombinant SpeB was used to prepare a standard curve. To ensure the specificity of SpeB, the cysteine protease inhibitor E-64 was routinely added to selected samples. All of the experiments were performed six times with three technical repeats. The data are shown as the means ± S.D. of six samples from representative findings obtained in three independent experiments. C, recombinant E-cadherin was incubated with various concentrations of recombinant SpeB for 6 h at 37 °C. Cleavage of E-cadherin was detected by Western blotting. Five N-terminal residues in the cleavage fragments of E-cadherin were determined by peptide sequence analysis. The scheme represents estimated cleavage fragments of E-cadherin. D, full-length recombinant occludin was incubated with various concentrations of recombinant SpeB for 6 h at 37 °C.
SpeB is secreted as a 42-kDa zymogen and autocatalyzed into an active 28-kDa cysteine protease (18, 20), and its substrate specificity is similar to that of the papain family of proteases (21). Active SpeB cleaves the host extracellular matrix (22), immunoglobulins (23, 24), and complement components (16). In addition to its ability to cleave host proteins, SpeB exerts proteolytic activity toward streptococcal surface proteins (25–27). Numerous investigations have reported that this proteolytic activity contributes to bacterial evasion from the host defense system and systemic dissemination (reviewed in Refs. 28 and 29). Therefore, we postulated that SpeB directly cleaves intercellular junctional proteins, and GAS translocates through the opening of paracellular junctions.
To examine whether SpeB directly cleaves transmembrane junctions, recombinant E-cadherin was treated with various concentrations of recombinant SpeB (Fig. 3C). An SpeB-dependent loss of the full-length band and appearance of several cleaved products, which reacted with an antibody against the extracellular domain of E-cadherin, were detected. Thus, five N-terminal residues in the cleavage products of E-cadherin in the presence of 200 nm SpeB were determined by peptide sequence analysis, which identified the N-terminal sequence of fragments with molecular masses of ∼80 and 65 kDa as DWVIP, which is identical to the N terminus of the EC1 domain, i.e., the N terminus of the recombinant protein. SpeB has been reported to cleave IgG in the hinge region (23). Because recombinant E-cadherin is the extracellular domain fused to the IgG Fc region at the C terminus, it is likely that an 80-kDa band represents a fragment with intact extracellular E-cadherin and a truncated Fc fragment. Deduced from this size, the 65-kDa band represents a fragment cleaved within the EC domains, possibly between EC4 and EC5. Meanwhile, the sequence of cleavage fragments with molecular masses of ∼55 and 35 kDa revealed VTDTN, which corresponds to the neighboring sequence of the calcium-binding site between the EC2 and EC3 domains. Speculating from these sizes, the C terminus of the 55- and 35-kDa fragments would be within the Fc region and within the region between EC4 and EC5, respectively. Because each EC domain of E-cadherin contains DXD or DXNDN motifs responsible for mediating calcium-dependent adhesion, SpeB may cleave the regions neighboring these motifs between each EC domain. Furthermore, SpeB-mediated cleavage of occludin, the most prominent member of TJ, was examined using an antibody against the C terminus of occludin (Fig. 3D). Cleavage of the full-length form of occludin, which was recognized as an 85-kDa band, and distinct cleavage products were detected in an SpeB concentration-dependent manner. These findings suggest that SpeB directly cleaves occludin at several sites.
SpeB Is a Bacterial Determinant for GAS-induced Cleavage of Intercellular Junctions
To investigate destabilization of the intercellular junctions in GAS-infected epithelial Caco-2 cells, a typical epithelial in vitro model, cells were infected with GAS strains, and the cleavage of junctional proteins was detected using Western blot analysis (Fig. 4). Marked cleavage of E-cadherin and occludin was detected in Caco-2 cells infected with the wild type strain, whereas that was significantly repressed by mutagenesis of the speB gene and restored by complementation. Similar results were observed in keratinocyte HaCaT and pharyngeal epithelial Detroit 562 cells, which are human cell lines derived from primary anatomical sites of GAS infection. On the other hand, the cleavage of JAM-1, a member of the group of TJ proteins, was not detected in cells infected with any of the strains. These findings indicate that SpeB contributes to cleavage of intercellular junctions in GAS-infected epithelial cells.
FIGURE 4.
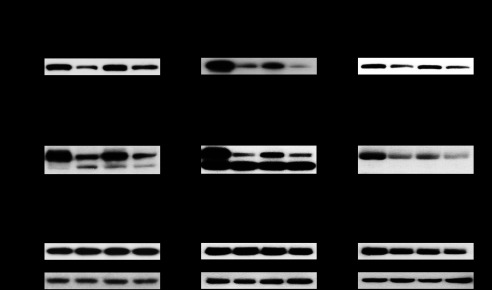
SpeB is related to GAS-induced destabilization of intercellular junctions. Caco-2, HaCaT, and Detroit 562 cells were infected with strain NIH35, an speB deletion mutant, or a complemented strain at an MOI of 10 for 6 h (Caco-2 and Detroit 562) or 8 h (HaCaT). Cleavage of E-cadherin, occludin, and JAM-1 was detected in whole cell lysates by Western blot analysis, with β-actin used as a loading control. The graphs below the representative blots show fold changes of band intensities of E-cadherin and occludin normalized to that of β-actin. The data are shown as the means ± S.D. of three independent experiments. *, p < 0.05.
SpeB Contributes to Translocation of GAS across Epithelial Barrier
Destabilization of key components involved in maintenance of the epithelial barrier is expected to increase bacterial translocation via a paracellular route. In most cases of GAS infections, skin and pharyngeal epithelium are thought to be the initial sites of bacterial colonization. Pharyngeal epithelial Detroit 562 cells and keratinocyte HaCaT cells were grown in a Millicell filter system under conditions that induced both physiological polarization of the epithelial cells and formation of intercellular junctions. However, the unstable junctional integrity of Detroit 562 and HaCaT cells, which was confirmed by TER, did not allow for assessing GAS translocation. Hence, polarized Caco-2 cells, which express typical epithelial markers of differentiation and are widely used as an in vitro model of the epithelial barrier (30), were utilized for the translocation assay (Fig. 5A). The apical surfaces of Caco-2 monolayers were infected with strain NIH35 or the speB deletion mutant, and then the ability of GAS to translocate across the monolayers was assessed for up to 8 h at 2-h intervals. The wild type strain was detected in basolateral media, indicating migration of bacteria through the monolayers. Although deletion of the speB gene had no effect on the production of capsule and SLS (data not shown), translocation of the speB mutant was significantly decreased. The translocation phenotype of the complemented strain was restored to the wild type level. To further investigate the generality of the role of SpeB in the translocation process, we also constructed speB deletion mutants with a background of #30, TW3337, TW3339, and 591, which showed a relatively high level of SpeB activity (Fig. 3B). For all of the tested strains except strain 591, the translocation ability of their speB deletion mutants was decreased as compared with the wild type (Fig. 5B). An antibiotic protection assay confirmed that strain 591 tends to internalize in an intracellular niche (data not shown). These results suggest that SpeB-dependent translocation is widely conserved among a majority of clinical isolates.
FIGURE 5.

SpeB is involved in the process of GAS translocation. Caco-2 cells were grown on Millicell filters and then infected with GAS strains at an MOI of 10 for 2 h. After removing nonadhered bacteria, the ability of the GAS strains to translocate across epithelial cells was assessed by examining medium samples obtained from the lower chambers at the indicated time of infection. Shown are the results of translocation rates of strain NIH35 (A) and clinical isolates (B). Three experiments were performed, and the data are presented as the means ± S.D. of six samples from representative findings obtained in three independent experiments. *, p < 0.01.
It is generally accepted that loss of the barrier function in monolayers is reflected by a decrease in TER (31, 32). Although the values for TER in Caco-2 monolayers infected with GAS strains were decreased by ∼40% as compared with those in noninfected cells, no comparable difference in decrease of TER values between the wild type and speB deletion mutant was observed (data not shown). Our previous study reported that SLS, which is encoded by the sagA gene, facilitates GAS translocation via degradation of epithelial intercellular junctions, accompanied by a decrease in TER. Therefore, decreased TER value in the Caco-2 cell monolayers infected with the speB mutant was attributed to SLS-induced loss of intercellular junction integrity. Furthermore, the bacterial translocation rate of the sagA mutant was considerably lower than that of the speB mutant (supplemental Fig. S3A). To clarify the relative contribution of SpeB in bacterial translocation, we constructed a double deletion mutant lacking the sagA and speB genes. Unexpectedly, no significant difference in the ability of translocation across Caco-2 monolayers between sagA mutant and the sagA-speB double mutant was observed. We next assessed the effects of SpeB on junctional integrity using a biochemical assay that measured transepithelial transport of FITC-labeled dextrans (supplemental Fig. S3B). Interestingly, only passive diffusion of dextran with a molecular mass of 4 kDa across the cell monolayers was decreased in cells infected with the sagA-speB double mutant, as compared with those infected with the sagA mutant. These results suggest that the proteolytic activity of SpeB contributes to dysfunction of the paracellular barrier in concert with SLS through a different mechanism. Additionally, we emphasize that single deletion of the speB gene compromised the ability of bacteria to translocate across the epithelial monolayer (Fig. 5). Therefore, we argue that SpeB is one of the determinants for the bacterial translocation across epithelial barrier.
DISCUSSION
Epithelial barrier integrity is maintained by physical interactions of cell-cell junctional complexes, including TJs and AJs (33). TJs seal intercellular space between adjacent epithelial cells to protect subepithelial tissue against microbial invaders, whereas AJs are required for integrity of the TJs. Several human pathogens have evolved strategies to target intercellular junctions and their components (34, 35). GAS also possesses some tools, such as a hyaluronic acid capsule (10) and SLS (11), to trigger signals that disrupt intercellular junctions and permit bacterial penetration via a paracellular route. In this study, we sought to define the role played by potential proteolytic activity of SpeB in destabilization of epithelial cell-cell junctional complexes. Here, we report for the first time that SpeB directly cleaves intercellular junctions and that its proteolytic activity contributes to GAS translocation across the epithelial barrier.
E-cadherin is a constituent of AJs and spans paracellular space through five extracellular domains repeated in tandem, EC1–EC5 (36). The extracellular portion of E-cadherin contains the conserved motifs DXD and DXNDN, which bind Ca2+ and serve as templates for homophilic binding with E-cadherin molecules on the surface of neighboring cells. It has been reported that single mutations introduced into calcium-binding motifs resulted in abrogated calcium binding activity and cell adhesion potential (37). Notably, we revealed in the present study that SpeB cleaves the ectodomain of E-cadherin at a site close to the region of calcium-binding sites. Together, these results suggest that proteolytic activity of SpeB induces destabilization of the epithelial barrier.
Unexpectedly, no significant difference in TER values between Caco-2 cells infected with the wild type strain and those infected with the speB mutant strain was observed. SLS also induces cleavage of E-cadherin and occludin via activation of the host protease calpain, accompanied by a decrease in TER values in those infected cells (11). Because of SLS activity in the speB mutant strain, TER values may not be altered by the mutation, whereas the cleavage of E-cadherin and occludin was inhibited by mutagenesis of the speB gene. SLS-activated calpain cleaves E-cadherin at the cytoplasmic domain. Because the cytoplasmic domain of E-cadherin and occludin is linked to the actin cytoskeleton and other signaling elements, truncation of that portion rapidly breaches the integrity of the epithelial barrier. Therefore, SLS-induced cleavage of E-cadherin and occludin may strongly reflect the reduction in TER values, as compared with SpeB-induced cleavage. Indeed, passive diffusion of FITC-dextran, which has a molecular mass of 4 kDa, but not 10 or 70 kDa, across cell monolayers after infection with the sagA-speB double mutant was decreased, as compared with infection with the sagA mutant. Our results suggest that SpeB-induced destabilization of the epithelial barrier allows for permeation of bacterial secretory molecules with a low molecular mass in concert with SLS.
In the present study, we found a positive correlation between the amount of active SpeB and cleavage of E-cadherin, whereas its proteolysis activity was not associated with the severity of disease related to GAS isolates derived from invasive infections. In clinical epidemiology surveillance, the association of SpeB expression with disease severity in GAS infections remains controversial. Although the cysteine protease activities of strains SSI-9 and SSI-1, isolates from invasive diseases, were relatively low in our in vitro experiments, we previously demonstrated that SpeB is a key factor for survival of SSI-9 in a mouse model (16). Therefore, it is likely that the protease activities of these strains are increased in vivo. Because we also noted that several non-invasive isolates possess a potential ability to destabilize intercellular junctions, disruption of cell-cell adhesion is likely related to a variety of clinical symptoms involving the skin and soft tissue. Consequently, the proteolysis action of SpeB may be involved in the development of skin and purulent infections.
It was recently reported that naturally occurring single amino acid replacement in ropB (38), a critical positive regulator of the speB gene, may have a role in expression of active SpeB in in vivo conditions. Furthermore, it has been suggested that spontaneous mutations within genes encoding the CovRS two-component system affect transcriptional activity of several virulence factors including SpeB during dissemination in vivo (27, 39, 40). A dynamic shift of SpeB expression in vivo caused by these mutations or sensing the surrounding environment would affect the translocation phenotype of GAS, especially in the early stages of infection.
In summary, the present findings provide evidence that SpeB can cleave intercellular junctions, thereby allowing bacterial translocation across the epithelial barrier. We speculate that SpeB-induced opening of paracellular junctions also permits penetration of several virulence factors, including toxins and superantigens, and leads to exacerbation of clinical manifestations. Following bacterial translocation across the epithelial barrier, evasion of the host immune system is essential for development of invasive infections. Accordingly, a part of colonized bacteria in superficial epithelial cells of the pharynx or skin might preferentially invade through a paracellular route to avoid being killed in intracellular space. However, a universal issue with the in vitro model of the epithelial barrier used in this study is differences in epithelial stratification between the polarized cell line and in vivo conditions. Both the pharynx and skin, primary sites of GAS infection, are guarded by multilayered stratified epithelium, whereas the cells used in this study differentiate into a polarized monolayer. Therefore, further investigations such as in vivo animal studies are necessary for proper interpretation of our in vitro findings. Although the precise role of SpeB in the pathogenesis of invasive GAS disease remains controversial, the present results support the notion that SpeB acts as a multifunctional element in the initial stages of infection.
Acknowledgments
We thank H. Watanabe and T. Murai for providing the GAS strains. We gratefully acknowledge T. Sekizaki and D. Takamatsu for the pSET4s plasmid. We thank Y. Fujinaga for offering helpful advice.
This work was supported in part by a Grant-in-aid for Scientific Research (B) 20390465 from the Japan Society for the Promotion of Science and a Grant-in-aid for Scientific Research on Priority Areas 18073011; a Grant-in-aid for Scientific Research for Young Scientists (A) 2189048; and Grants-in-aid for Scientific Research for Young Scientists (B) 20791336 and 21791786 from the Ministry of Education, Culture, Sports, Science and Technology.

This article contains supplemental Figs. S1–S3.
- GAS
- group A Streptococcus
- TJ
- tight junction
- AJ
- adherence junction
- TER
- transepithelial electrical resistance
- MOI
- multiplicity of infection
- SLS
- streptolysin S.
REFERENCES
- 1. Carapetis J. R., Steer A. C., Mulholland E. K., Weber M. (2005) The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5, 685–694 [DOI] [PubMed] [Google Scholar]
- 2. Tart A. H., Walker M. J., Musser J. M. (2007) New understanding of the group A Streptococcus pathogenesis cycle. Trends Microbiol. 15, 318–325 [DOI] [PubMed] [Google Scholar]
- 3. Cole J. N., Henningham A., Gillen C. M., Ramachandran V., Walker M. J. (2008) Human pathogenic streptococcal proteomics and vaccine development. Proteomics Clin. Appl. 2, 387–410 [DOI] [PubMed] [Google Scholar]
- 4. Amagai M. (2003) Desmoglein as a target in autoimmunity and infection. J. Am. Acad. Dermatol. 48, 244–252 [DOI] [PubMed] [Google Scholar]
- 5. Ferrieri P., Dajani A. S., Wannamaker L. W., Chapman S. S. (1972) Natural history of impetigo. I. Site sequence of acquisition and familial patterns of spread of cutaneous streptococci. J. Clin. Invest. 51, 2851–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Terao Y., Kawabata S., Kunitomo E., Murakami J., Nakagawa I., Hamada S. (2001) Fba, a novel fibronectin-binding protein from Streptococcus pyogenes, promotes bacterial entry into epithelial cells, and the fba gene is positively transcribed under the Mga regulator. Mol. Microbiol. 42, 75–86 [DOI] [PubMed] [Google Scholar]
- 7. Bisno A. L., Brito M. O., Collins C. M. (2003) Molecular basis of group A streptococcal virulence. Lancet Infect. Dis. 3, 191–200 [DOI] [PubMed] [Google Scholar]
- 8. Nakagawa I., Nakata M., Kawabata S., Hamada S. (2001) Cytochrome c-mediated caspase-9 activation triggers apoptosis in Streptococcus pyogenes-infected epithelial cells. Cell. Microbiol. 3, 395–405 [DOI] [PubMed] [Google Scholar]
- 9. Cywes Bentley C., Hakansson A., Christianson J., Wessels M. R. (2005) Extracellular group A Streptococcus induces keratinocyte apoptosis by dysregulating calcium signaling. Cell. Microbiol. 7, 945–955 [DOI] [PubMed] [Google Scholar]
- 10. Cywes C., Wessels M. R. (2001) Group A Streptococcus tissue invasion by CD44-mediated cell signaling. Nature 414, 648–652 [DOI] [PubMed] [Google Scholar]
- 11. Sumitomo T., Nakata M., Higashino M., Jin Y., Terao Y., Fujinaga Y., Kawabata S. (2011) Streptolysin S contributes to group A streptococcal translocation across an epithelial barrier. J. Biol. Chem. 286, 2750–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cunningham M. W. (2000) Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 13, 470–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Olsen R. J., Shelburne S. A., Musser J. M. (2009) Molecular mechanisms underlying group A streptococcal pathogenesis. Cell. Microbiol. 11, 1–12 [DOI] [PubMed] [Google Scholar]
- 14. Trieu-Cuot P., Carlier C., Poyart-Salmeron C., Courvalin P. (1991) Shuttle vectors containing a multiple cloning site and a lacZ α gene for conjugal transfer of DNA from Escherichia coli to Gram-positive bacteria. Gene 102, 99–104 [DOI] [PubMed] [Google Scholar]
- 15. Takamatsu D., Osaki M., Sekizaki T. (2001) Thermosensitive suicide vectors for gene replacement in Streptococcus suis. Plasmid 46, 140–148 [DOI] [PubMed] [Google Scholar]
- 16. Terao Y., Mori Y., Yamaguchi M., Shimizu Y., Ooe K., Hamada S., Kawabata S. (2008) Group A streptococcal cysteine protease degrades C3 (C3b) and contributes to evasion of innate immunity. J. Biol. Chem. 283, 6253–6260 [DOI] [PubMed] [Google Scholar]
- 17. Nakata M., Kimura K. R., Sumitomo T., Wada S., Sugauchi A., Oiki E., Higashino M., Kreikemeyer B., Podbielski A., Okahashi N., Hamada S., Isoda R., Terao Y., Kawabata S. (2011) Assembly mechanism of FCT region type 1 pili in serotype M6 Streptococcus pyogenes. J. Biol. Chem. 286, 37566–37577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hauser A. R., Schlievert P. M. (1990) Nucleotide sequence of the streptococcal pyrogenic exotoxin type B gene and relationship between the toxin and the streptococcal proteinase precursor. J. Bacteriol. 172, 4536–4542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. von Pawel-Rammingen U., Johansson B. P., Björck L. (2002) IdeS, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin G. EMBO J. 21, 1607–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carroll R. K., Musser J. M. (2011) From transcription to activation. How group A streptococcus, the flesh-eating pathogen, regulates SpeB cysteine protease production. Mol. Microbiol. 81, 588–601 [DOI] [PubMed] [Google Scholar]
- 21. Nomizu M., Pietrzynski G., Kato T., Lachance P., Menard R., Ziomek E. (2001) Substrate specificity of the streptococcal cysteine protease. J. Biol. Chem. 276, 44551–44556 [DOI] [PubMed] [Google Scholar]
- 22. Kapur V., Topouzis S., Majesky M. W., Li L. L., Hamrick M. R., Hamill R. J., Patti J. M., Musser J. M. (1993) A conserved Streptococcus pyogenes extracellular cysteine protease cleaves human fibronectin and degrades vitronectin. Microb. Pathog. 15, 327–346 [DOI] [PubMed] [Google Scholar]
- 23. Collin M., Olsén A. (2001) Effect of SpeB and EndoS from Streptococcus pyogenes on human immunoglobulins. Infect. Immun. 69, 7187–7189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eriksson A., Norgren M. (2003) Cleavage of antigen-bound immunoglobulin G by SpeB contributes to streptococcal persistence in opsonizing blood. Infect. Immun. 71, 211–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Berge A., Björck L. (1995) Streptococcal cysteine proteinase releases biologically active fragments of streptococcal surface proteins. J. Biol. Chem. 270, 9862–9867 [DOI] [PubMed] [Google Scholar]
- 26. Chaussee M. S., Cole R. L., van Putten J. P. (2000) Streptococcal erythrogenic toxin B abrogates fibronectin-dependent internalization of Streptococcus pyogenes by cultured mammalian cells. Infect. Immun. 68, 3226–3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walker M. J., Hollands A., Sanderson-Smith M. L., Cole J. N., Kirk J. K., Henningham A., McArthur J. D., Dinkla K., Aziz R. K., Kansal R. G., Simpson A. J., Buchanan J. T., Chhatwal G. S., Kotb M., Nizet V. (2007) DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat. Med. 13, 981–985 [DOI] [PubMed] [Google Scholar]
- 28. Chiang-Ni C., Wu J. J. (2008) Effects of streptococcal pyrogenic exotoxin B on pathogenesis of Streptococcus pyogenes. J. Formos. Med. Assoc. 107, 677–685 [DOI] [PubMed] [Google Scholar]
- 29. Nelson D. C., Garbe J., Collin M. (2011) Cysteine proteinase SpeB from Streptococcus pyogenes. A potent modifier of immunologically important host and bacterial proteins. Biol. Chem. 392, 1077–1088 [DOI] [PubMed] [Google Scholar]
- 30. Sun H., Chow E. C., Liu S., Du Y., Pang K. S. (2008) The Caco-2 cell monolayer. Usefulness and limitations. Expert Opin. Drug Metab. Toxicol. 4, 395–411 [DOI] [PubMed] [Google Scholar]
- 31. González-Mariscal L., Betanzos A., Nava P., Jaramillo B. E. (2003) Tight junction proteins. Prog. Biophys. Mol. Biol. 81, 1–44 [DOI] [PubMed] [Google Scholar]
- 32. Schneeberger E. E., Lynch R. D. (2004) The tight junction. A multifunctional complex. Am. J. Physiol. Cell. Physiol. 286, C1213–C1228 [DOI] [PubMed] [Google Scholar]
- 33. Hartsock A., Nelson W. J. (2008) Adherens and tight junctions. Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 1778, 660–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Balkovetz D. F., Katz J. (2003) Bacterial invasion by a paracellular route. Divide and conquer. Microbes Infect. 5, 613–619 [DOI] [PubMed] [Google Scholar]
- 35. Sousa S., Lecuit M., Cossart P. (2005) Microbial strategies to target, cross or disrupt epithelia. Curr. Opin. Cell. Biol. 17, 489–498 [DOI] [PubMed] [Google Scholar]
- 36. Bryant D. M., Stow J. L. (2004) The ins and outs of E-cadherin trafficking. Trends Cell. Biol. 14, 427–434 [DOI] [PubMed] [Google Scholar]
- 37. Ozawa M., Engel J., Kemler R. (1990) Single amino acid substitutions in one Ca2+ binding site of uvomorulin abolish the adhesive function. Cell. 63, 1033–1038 [DOI] [PubMed] [Google Scholar]
- 38. Carroll R. K., Shelburne S. A., 3rd, Olsen R. J., Suber B., Sahasrabhojane P., Kumaraswami M., Beres S. B., Shea P. R., Flores A. R., Musser J. M. (2011) Naturally occurring single amino acid replacements in a regulatory protein alter streptococcal gene expression and virulence in mice. J. Clin. Invest. 121, 1956–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Engleberg N. C., Heath A., Miller A., Rivera C., DiRita V. J. (2001) Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenes result in enhanced virulence in a murine model of skin and soft tissue infection. J. Infect. Dis. 183, 1043–1054 [DOI] [PubMed] [Google Scholar]
- 40. Sumby P., Whitney A. R., Graviss E. A., DeLeo F. R., Musser J. M. (2006) Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog. 2, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]