

Background: Inactivation of integrin αIIbβ3 reverses platelet aggregate formation upon coagulation.
Results and conclusion: Platelets from patient (Scott) and mouse (Capn1−/− and Ppif−/−) blood reveal a dual mechanism of αIIbβ3 inactivation: by calpain-2 cleavage of integrin-associated proteins and by cyclophilin D/TMEM16F-dependent phospholipid scrambling.
Significance: These data provide novel insight into the switch mechanisms from aggregating to procoagulant platelets.
Keywords: Calpain, Integrins, Mitochondria, Phosphatidylserine, Platelets, Cyclophilin D, Procoagulant Activity
Abstract
Aggregation of platelets via activated integrin αIIbβ3 is a prerequisite for thrombus formation. Phosphatidylserine-exposing platelets with a key role in the coagulation process disconnect from a thrombus by integrin inactivation via an unknown mechanism. Here we show that αIIbβ3 inactivation in procoagulant platelets relies on a sustained high intracellular Ca2+, stimulating intracellular cleavage of the β3 chain, talin, and Src kinase. Inhibition of calpain activity abolished protein cleavage, but only partly suppressed αIIbβ3 inactivation. Integrin αIIbβ3 inactivation was unchanged in platelets from Capn1−/− mice, suggesting a role of the calpain-2 isoform. Scott syndrome platelets, lacking the transmembrane protein TMEM16F and having low phosphatidylserine exposure, displayed reduced αIIbβ3 inactivation with the remaining activity fully dependent on calpain. In platelets from Ppif−/− mice, lacking mitochondrial permeability transition pore (mPTP) formation, agonist-induced phosphatidylserine exposure and αIIbβ3 inactivation were reduced. Treatment of human platelets with cyclosporin A gave a similar phenotype. Together, these data point to a dual mechanism of αIIbβ3 inactivation via calpain(-2) cleavage of integrin-associated proteins and via TMEM16F-dependent phospholipid scrambling with an assistant role of mPTP formation.
Introduction
Integrin αIIbβ3 plays a crucial role in platelet aggregation in response to physiological agonists. Once in its activated conformation, αIIbβ3 accomplishes platelet-platelet interactions via bridges of fibrinogen and von Willebrand factor. Under thrombotic conditions in flowing blood, αIIbβ3-dependent platelet aggregation mediates thrombus formation and, finally, occlusion of a damaged cardiac or carotid artery (1). Microscopic observations have shown that both in vivo and in parallel-plate flow chambers, the aggregated platelets in a thrombus are surrounded by patches of procoagulant platelets with quite distinct properties (2). The latter platelets characteristically are elevated in cytosolic Ca2+, have a rounded morphology with attached microparticles, and expose the negatively charged lipid, phosphatidylserine (PS).3 This contrasts to the classical pseudopod-containing structure of aggregated platelets in a thrombus, which do not expose PS (3). Time-lapse videos show that the rounded morphology arises during platelet disconnection from the thrombus core, suggesting a well controlled mechanism of platelet detachment (4). This population of PS-exposing platelets is known to bind multiple coagulation factors, greatly promoting the process of thrombin generation (3, 5). It has been argued that the platelet detachment is mediated by inactivation or closure of previously activated αIIbβ3 integrins (6). However, the regulation of such a process is not well understood.
Activation of αIIbβ3 leads to the appearance of high affinity binding sites for fibrinogen and von Willebrand factor at the platelet surface. The signaling mechanism to αIIbβ3 activation, unraveled in considerable detail, involves several pathways, i.e. via a phospholipase C and protein kinase C route, resulting in transient Ca2+ fluctuations, and via phosphoinositide 3-kinase route. The consequence is activation of a chain of regulatory proteins CalDAG-GEFI, Rap1b, and Rap1-GTP-interacting adapter molecule (RIAM) (7, 8); and these establish interaction of the cytoskeleton proteins, kindlin-3 and talin-1, with the β3 chain of the αIIbβ3 complex, with, as a result, unclasping of the αIIb and β3 chains (9, 10). Another proposed mechanism of αIIbβ3 activation is that talin-1 modulates the integrin conformation by a tilting effect on the β3 chain (11).
In response to ADP and ADP-releasing platelet agonists, the conformational change of αIIbβ3 is considered to be an intrinsically reversible process (6). The active integrin itself can evoke multiple signaling events in platelets (12). A key role in this so-called outside-in signaling pathway is provided by the protein tyrosine kinase Src, which is constitutively associated with the β3 cytoplasmic tail (13). Src, for instance, activates other tyrosine kinases such as Syk (8). In addition, a functional role of the Ca2+-dependent thiol protease, calpain, has been suggested in integrin αIIbβ3 activation and signaling, but this has remained controversial. Calpain may either activate αIIbβ3 by cleaving talin (14) or counteract the high affinity conformational state of αIIbβ3 by cleaving the cytoplasmic tail of the β3 chain (15).
Although essentially all platelet agonists cause αIIbβ3 activation, only strong agonists are capable of inducing the formation of procoagulant, PS-exposing platelets. Effective inducers of the latter response are the combination of the glycoprotein VI agonist convulxin (Cvx) with thrombin (Thr), or otherwise Ca2+ ionophores such as A23187 and ionomycin (16, 17). Common to these agonists is that they induce a high and sustained rise in cytosolic Ca2+, which is considered to trigger the Ca2+-operated membrane protein, TMEM16F, which regulates the scrambling of phospholipids and exposure of PS (18, 19). In platelets from Scott syndrome patients, who lack a functional TMEM16F, agonist-induced PS exposure is greatly diminished (20, 21). In addition, contributing to TMEM16F-mediated phospholipid scrambling is the Ca2+-dependent depolarization of the inner mitochondrial membrane and formation of a mitochondrial permeability transition pore (mPTP) (19). This is consistent with the observation that mouse platelets lacking cyclophilin D (Ppif gene), an mPTP component, are defective in PS exposure (22).
Another platelet population often described in the literature is that of coated platelets (23, 24). These are formed by dual stimulation with collagen and thrombin (but not Ca2+ ionophore) and can be characterized by high and stable surface retention of labeled fibrinogen. Fibrinogen as well as other platelet secretion products (e.g. factor V, thrombospondin, fibronectin, von Willebrand factor) bind to these platelets in an αIIbβ3-independent way via transglutaminase activity. Procoagulant and coated platelets represent two not completely overlapping populations, as not all PS-exposing platelets display high fibrinogen binding (2). In the present study, we investigated the mechanism of integrin αIIbβ3 inactivation in procoagulant platelets by determining the functional roles of calpain, Src-dependent signaling, TMEM16F, and mPTP formation.
EXPERIMENTAL PROCEDURES
Materials
Horm type-I collagen was purchased from Nycomed. Cvx was purified to homogeneity from the venom of Crotalus durissus terrificus (Latoxan). Annexin A5 labeled with Alexa Fluor 647 (AF647) and Fura-2 acetoxymethyl ester were from Invitrogen. Annexin A5 labeled with fluorescein isothiocyanate (FITC) was from Pharmatarget. FITC-labeled PAC1 monoclonal antibody (mAb) against activated human integrin αIIbβ3 was from BD Bioscience. phycoerythrin (PE)-labeled JON/A mAb against activated mouse αIIbβ3 was from Emfret Analytics. Mouse anti-talin mAb and anti-v-Src were from Sigma. mAb anti-Tyr773 β3 chain (Ab38460) was from Abcam. Rabbit antibodies Ab762, Ab754, and Ab759, directed against (calpain-dependent) cleavage sites of human/mouse integrin β3, were kindly provided by Dr. X. Du (University of Illinois, Chicago) (25). Tirofiban came from MSD, human α-thrombin from Kordia, and MDL-28170 from Tocris. Prestained SDS-PAGE standards and Laemmli sample buffer were from Bio-Rad. Other reagents were purchased from Calbiochem.
Blood Collection and Platelet Preparation
Blood from healthy volunteers and a Scott syndrome patient was collected in acid-citrate-dextrose anticoagulant after full informed consent (Helsinki declaration). Experiments were approved by the local Medical Ethics Committees. Platelet-rich plasma and washed platelets were prepared as described (26). Washed platelets were suspended into Hepes buffer, pH 7.45 (10 mm Hepes, 136 mm NaCl, 2.7 mm KCl, 2 mm MgCl2, 5 mg/ml glucose, and 1 mg/ml bovine serum albumin). The final concentration of platelets in plasma or buffer medium was 1 × 108/ml, unless indicated otherwise.
Animal studies were approved by the local Animal Experimental Committees. Mice homozygous for a targeted deletion of the cyclophilin D (Ppif gene) were generated as described (22). The mice were bred on an Sv129 background and compared with Ppif+/+ animals of the same breeding. Calpain-1-deficient mice (Capn1−/−) were generated as reported previously (27, 28). These mice were bred on a C57BL/6 background and were compared with Capn1+/+ mice of the same genetic background. Mouse platelets were isolated as described before (29) and suspended in modified Hepes buffer, pH 7.45 (5 mm Hepes, 136 mm NaCl, 2.7 mm KCl, 2 mm MgCl2, 0.42 mm NaH2PO4, 5 mg/ml glucose, and 1 mg/ml bovine serum albumin). The final concentration was 1 × 108/ml, unless indicated otherwise.
Western Blot Analysis
Washed platelets in (modified) Hepes buffer, pH 7.45, were incubated with the calpain inhibitors, calpeptin (200 μm) or MDL28170 (200 μm), or vehicle for 15 min, as desired. Platelets were stimulated with Cvx (100 ng/ml), Thr (8 nm), or ionomycin (20 μm). Samples (5 × 107 platelets) were taken at the indicated time points and lysed in ice-cold 4× lysis buffer (600 mm, 10 mm Tris, 4 mm EGTA, 4 mm EDTA, 4% Nonidet P-40). Lysed samples were separated on 8% SDS-polyacrylamide gels, and proteins were transferred to PVDF blotting membranes by semidry transfer. Immunostaining was with antibodies against talin (1:500), integrin β3 (Ab762, 1:10000; Ab754, 1:1000), Tyr(P)774 β3 chain (1:500), or Src (2.5 μg/ml) for 1 h, followed by overnight incubation with horseradish peroxidase-coupled secondary antibody at 4 °C. Stained blots were visualized with an ECL system. Blot quantification was by densitometric analysis, as before (30).
Platelet Aggregation and Activation
Light transmission traces, reflecting platelet aggregation, were measured using a Chronolog aggregometer under constant stirring (37 °C). Platelets (1 × 108/ml) in Hepes buffer containing 2 mm CaCl2 were stimulated with 4 nm Thr and/or 100 ng/ml Cvx, or with 10 μm ionomycin; experiments were performed in the presence or absence of 5 μg/ml tirofiban. Aggregation of platelets was also assessed by single cell count analysis using a Coulter counter (Coulter Electronics).
For flow cytometry, washed human or mouse platelets (1 × 108/ml) were preincubated with the indicated inhibitors or dimethyl sulfoxide vehicle for 10 min and stimulated in the presence of 2 mm CaCl2 with 4 nm thrombin and/or 100 ng/ml convulxin, or with 10 μm ionomycin. In samples taken after 5–30 min, surface expression of PS was detected with AF647-labeled annexin A5. In addition, activated integrin αIIbβ3 was detected using FITC-labeled PAC1 mAb for human platelets or PE-labeled JON/A mAb for mouse platelets. Samples were analyzed with a FACScan flow cytometer (BD Accuri Cytometers).
Platelet samples were fixed and stained for transmission electron microscopy, as described before (30). Calpain activity in platelets was assessed using a calpain activity assay kit according to the manufacturer's instructions (Abcam). Results are expressed as relative fluorescence units per mg of lysate protein.
Thrombus Formation on Collagen under Flow
Collagen-induced thrombus formation was assayed as described before (20, 31). In brief, PPACK/fragmin-anticoagulated mouse blood was flowed over a coverslip coated with collagen type I in a transparent parallel-plate perfusion chamber, at shear rate of 1000 s−1 for 4 min. Thrombi formed on the collagen surface were poststained with AF647-annexin A5 and PE-JON/A mAb in modified Hepes buffer, pH 7.45, containing 2 mm CaCl2 and 1 unit/ml heparin. Phase-contrast and fluorescence images were captured for analysis of surface area coverage of adherent platelets and of platelets with active integrins or exposed PS (32). Image analysis was performed with Metamorph software Version 7.5.0.0 (MDS Analytical Technologies).
Platelet Ca2+ Responses
Cytosolic Ca2+ was measured in human platelets, preloaded with fluorescent 2.5 μm Fura-2 acetoxymethyl ester for 45 min at ambient temperature under gentle rotation (33). Washed platelets were stimulated while recording changes in fluorescence by calibrated ratio fluorometry. Calcium responses were expressed as time integrals over base line [Ca2+]i.
Statistics
Significance of differences between control and experimental groups as well as changes between groups over time were determined by one-way or two-way analysis of variance followed by a Bonferroni post hoc test. Student's t test was performed to compare paired samples. Data are expressed as means ± S.D. p values <0.05 were considered significant.
RESULTS
Closure of Activated Integrin αIIbβ3 in PS-exposing Platelets
Earlier work demonstrated the appearance of two distinct populations of platelets during thrombus formation, i.e. co-aggregated platelets with activated αIIbβ3 integrins (binding PAC1 mAb) and loosely attached platelets showing PS exposure (binding coagulation factors Va and Xa and annexin A5) (3, 20). The impaired adhesion of PS-exposing platelets seemingly contrasts to the observation that, in platelet suspensions, the Ca2+-ionophore ionomycin (causing full PS exposure) produces changes in light transmission that are suggestive of platelet aggregation (34, 35).
We reinvestigated this by first stimulating washed human platelets with 4 nm Thr or 100 ng/ml Cvx. As shown in Fig. 1A, this resulted in a rapid increase of light transmission, which was almost fully suppressed by the αIIbβ3 antagonist tirofiban (inhibition at 25 min 87 ± 3% and 72 ± 4%, respectively; mean ± S.D., n = 4). Integrin-dependent platelet aggregation with Thr or Cvx was confirmed by a major reduction in single platelet count (Fig. 1B).
FIGURE 1.

Increase in light transmission but loss of aggregating potential and inactivation of αIIbβ3 after platelet stimulation with Cvx/Thr or ionomycin. Washed human platelets were stimulated with Thr (4 nm), Cvx (100 ng/ml), Cvx/Thr or ionomycin (10 μm) ± tirofiban (5 μg/ml) in the presence of 2 mm CaCl2, as indicated. A, representative light transmission traces from five independent experiments. B, single platelet count after 30 min of stimulation. Means ± S.D. (error bars; n = 5) are shown. *, p < 0.05 versus Thr (paired t test C and D, two-color flow cytometry after stimulation at the indicated times with Cvx/Thr. Labeling was with FITC-PAC1 mAb plus AF647-annexin A5. C, representative dot plots of FL1 (FITC-PAC1 mAb) versus FL4 (AF647-annexin A5) after 5 or 25 min of activation. D, percentages of platelet populations: no staining (white bars), staining with PAC1 mAb (light gray bars), double staining (dark gray bars), and annexin A5-positive (black bars). E and F, two-color flow cytometry after stimulation with ionomycin, as for C and D.
On the other hand, the combined application of Cvx/Thr or addition of 10 μm ionomycin provoked similar rapid increases in light transmission (Fig. 1A). However, the Cvx/Thr-induced light transmission increase was only partly suppressed with tirofiban (inhibition 60 ± 7% at 25 min). In comparison, tirofiban did not affect the light transmission increase induced by ionomycin (−9 ± 8%). Platelet lysis could be excluded because single cell count analysis indicated that ionomycin treatment resulted in a platelet count that remained at 90% of the original count (Fig. 1B). Electron microscopic analysis indicated that, unlike the pseudopod-containing platelets formed by thrombin, ionomycin treatment resulted in single, rounded platelets with a translucent appearance (Fig. 2). Together, these data indicate that the pseudo-aggregation observed after Cvx/Thr or ionomycin stimulation is a consequence of the morphological change with a rounded and translucent platelet structure, reducing light transmission, rather than a consequence of integrin-dependent platelet aggregate formation.
FIGURE 2.
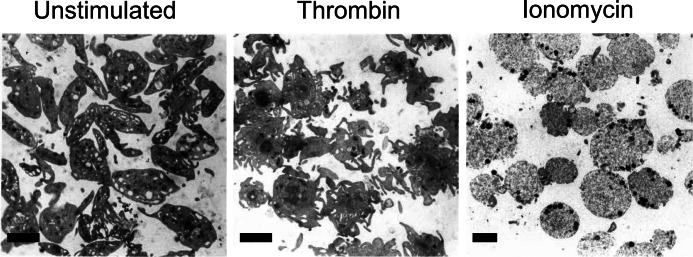
Transmission electron microscopic images of unstimulated and various types of stimulated platelets. Washed human platelets were left unstimulated or were stimulated with thrombin (4 nm) or ionomycin (10 μm) in the presence of 2 mm CaCl2 for 30 min (no stirring to prevent aggregation). Platelets were then fixed, pelleted, and prepared for transmission electron microscopic observation, as described before (30). Shown are representative images. Scale bars, 2 μm.
The absence of integrin activation with these agonists was further demonstrated by dual color flow cytometry, using FITC-PAC1 mAb and AF647-annexin A5, probing activated αIIbβ3 and PS exposure, respectively. Upon stimulation with Cvx/Thr, initially a population of platelets only binding PAC1 mAb was formed, which was gradually replaced by a population of PAC1-negative platelets which only bound annexin A5 (Fig. 1, C and D). Within the time interval of 5–30 min, 40 ± 12% (n = 4) of platelets lost the capability to bind PAC1 mAb. Similar results were found when fluorescent lactadherin was used instead of annexin A5 (data not shown). Integrin closure could not be explained by a decrease in expression levels of αIIb and β3 chains because these levels were even increased after stimulation with Thr, Cvx, or Cvx/Thr. This increase in expression levels is explained by an increase membrane surface due to granule secretion, as detected by CD62P expression (supplemental Table S1).
In response to 10 μm ionomycin, the majority of platelets bound annexin A5 but not PAC1 mAb (Fig. 1, E and F). However, at low ionomycin doses (<5 μm), transient PAC1 mAb binding was detected, accompanied by a corresponding reduction in single platelet count (data not shown). Expression levels of αIIb and β3 remained unchanged after high ionomycin stimulation (supplemental Table S1), thus indicating that this treatment precludes or antagonizes integrin activation. Increased expression levels of αIIb and β3 were not observed, which was explained by a diminished secretion after ionomycin stimulation compared with Cvx/Thr stimulation. Taken together, these results suggest that PS exposure induced by these strong agents is accompanied by either closure of activated integrin or by lack of integrin activation, which prevents the formation of large platelet aggregates.
Interestingly, Fig. 1, C and D, points to a small population of Cvx/Thr-stimulated platelets, capable of binding both PAC1 mAb and annexin A5. Flow cytometric studies were performed to characterize this dual-labeled platelet population. It remained unchanged in size upon stimulation with Cvx/SFLLRN (thrombin receptor-activating peptide) or in the presence of transglutaminase inhibitor, cadaverine (data not shown). This population for the major part (<4%) did not exhibit high fibrinogen binding. Hence, these platelets could not be identified as “coated,” i.e. containing a transglutaminase-dependent fibrin coat. Other experiments showed that the population of dual-labeled platelets reduced in size, when incubations were performed at lower platelet count (≤1 × 108/ml), or contained a Rho kinase inhibitor, which antagonizes platelet contraction (supplemental Fig. S1). It was thus concluded that it consisted of microaggregates of perhaps contracted platelets with either active integrins or exposed PS.
Role of Calpain-mediated Protein Cleavage in Integrin αIIbβ3 Closure
Considering the described role of calpain in αIIbβ3-dependent cleavage of cytoskeletal-associated proteins (25), we examined whether relevant targets of this protease were cleaved under conditions of PS exposure. Western blot analysis was performed of gel-separated platelet lysates, using antibodies against the full-length intracellular β3 chain (Ab762) and against a calpain cleavage site in the β3 chain at residue 754 (Ab754) (15). Blots were also analyzed for the degradation of talin-1 and Src kinase, using suitable antibodies. After 5 min of stimulation with ionomycin, we noted major cleavage of the β3 chain, talin, and Src into smaller protein fragments (Fig. 3, A–D). After stimulation with Cvx/Thr, cleavage of the β3 chain, talin, and Src required ∼20 min to complete. Flow cytometry experiments, performed in parallel samples, confirmed the presence of PS exposure and absence of integrin activation with ionomycin and a gradual PS exposure and integrin closure with Cvx/Thr (compare Fig. 1). Together, these results suggested that phospholipid scrambling is one of the mechanisms contributing to intracellular integrin cleavage and inability to activation.
FIGURE 3.

Intracellular cleavage of β3 chain, talin, and Src after platelet stimulation with Cvx/Thr or ionomycin. Platelets were left unstimulated or stimulated with Cvx/Thr or ionomycin, as described for Fig. 1. Samples (5 × 107 platelets) were lysed after 5 or 30 min and subjected to electrophoresis and Western blotting. Blots were stained for full-length β3 chain with Ab762 (A), calpain-cleaved β3 chain with Ab754 (B), anti-talin mAb (C), or anti-Src mAb (D). Shown are representative blots and bar graphs of densitometric analysis of stained bands, corrected for loading control (anti-tubulin mAb). Graphs indicate means ± S.D. (error bars; n = 3–4). *, p < 0.05 (one-way analysis of variance, Bonferroni correction).
Considering that calpain may regulate tyrosine phosphorylation of the β3 chain (27), we checked how platelet stimulation with Cvx/Thr or ionomycin influenced the β3 phosphorylation at Tyr733, i.e. a phosphorylation site indicative for αIIbβ3 outside-in signaling (36). Whereas platelet stimulation with Thr alone resulted in persistent Tyr773 phosphorylation, stimulation with Cvx/Thr resulted in a loss of phosphorylation at 30 min, whereas ionomycin did not give any phosphorylation at all (supplemental Fig. S2). Hence, this phosphorylation site appears to be lost in platelets stimulated with the PS-exposing agonists.
To investigate a functional role of calpain activity in this protein degradation, two structurally different pharmacological inhibitors were used, i.e. calpeptin and MDL-28170, both of which are established inhibitors of Ca2+-dependent proteases (28). Either compound fully inhibited the degradation of β3 chain, talin, and Src in platelets that were stimulated with Cvx/Thr or ionomycin (Fig. 4, A–D).
FIGURE 4.

Role of calpain in cleavage of β3 chain, talin, and Src induced by Cvx/Thr or ionomycin. Platelets were stimulated as described for Fig. 2 but preincubated for 10 min with either vehicle or calpain inhibitors, calpeptin (200 μm) or MDL-28170 (200 μm). Western blots from protein-separated lysates were stained for full-length β3 chain with Ab762 (A), calpain-cleaved β3 chain with Ab754 (B), anti-talin mAb (C), or anti-Src mAb (D). Shown are representative blots and bar graphs of densitometric analysis of stained protein bands, corrected for loading control (anti-tubulin mAb). Graphs indicate means ± S.D. (error bars; n = 4). *, p < 0.05 (one-way analysis of variance, Bonferroni correction).
We then determined effects of these inhibitors on the process of integrin inactivation. In the time frame of 5–30 min, calpeptin as well as MDL-28170 significantly but incompletely affected the integrin closure in response to Cvx/Thr (Fig. 5, A and B). Measurement of calpain activity indicated that both calpeptin and MDL-28170 nearly completely blocked this proteolytic activity in the stimulated platelets (Fig. 5C). Neither of the inhibitors affected initial PS exposure (78 ± 13% and 107 ± 12% of control, respectively). In Fura-2-loaded platelets, neither inhibitor changed the Cvx/Thr-induced intracellular Ca2+ rises (Fig. 5D).
FIGURE 5.

Partial role of calpain in agonist-induced αIIbβ3 closure. Platelets (1 × 108/ml) were pretreated with vehicle, calpeptin (200 μm), or MDL-28170 (200 μm) for 10 min and then stimulated with Cvx/Thr for 5–30 min. A and B, samples were analyzed by flow cytometry in the presence of FITC-PAC1 mAb. Shown are percentages of PAC1-binding platelets (A) and extent of integrin closure between 5 and 30 min (B). C, calpain activity was assessed after 30 min of stimulation. Data are expressed as relative fluorescence units (RFU). D, calcium responses to Cvx/Thr in Fura-2-loaded platelets were assessed. Shown are changes in [Ca2+]i time integrals (nm × 10 min) relative to vehicle control. Graphs indicate means ± S.D. (error bars; n = 4). *, p < 0.05 (one-way analysis of variance, Bonferroni correction).
Subsequent experiments were performed with mice lacking the major Ca2+-dependent cysteine protease, calpain-1 (μ-calpain, Capn1−/− gene), that accounts for ∼80% of the calpain activity in mouse and human platelets (37). Assessment of collagen-dependent thrombus formation indicated that the Capn1−/− blood formed enlarged thrombi, which displayed increased integrin αIIbβ3 activation (JON/A mAb binding) but decreased PS exposure (AF647-annexin A5 binding) compared with wild type Capn1+/+ blood (Fig. 6A). This suggests a negative role of calpain-1 in murine αIIbβ3 activation under these conditions. In platelet suspensions stimulated with Cvx/Thr, Capn1−/− and Capn1+/+ platelets displayed similar αIIbβ3 activation and closure, while PS exposure was comparable (Fig. 6, B and C). Treatment of Capn1−/− and Capn1+/+ platelets with MDL-28170 partly suppressed integrin closure but did not result in a significant decrease in PS exposure (Fig. 6, D and E).
FIGURE 6.

Unchanged agonist-induced PS exposure and αIIbβ3 closure in calpain-1-deficient platelets. A, blood from Capn1+/+ or Capn1−/− mice was perfused over collagen for 4 min at 1000 s−1. Thrombi were labeled with PE-JON/A mAb and AF647-annexin A5. Shown are representative brightfield and fluorescence images (scale bars, 20 μm), as well as quantitative analyses of (fluorescent) platelet deposition. B–E, washed mouse platelets were stimulated with Cvx/Thr for 5–30 mi, and analyzed by flow cytometry. Shown are percentages of platelets binding PE-JON/A (B) or AF647-annexin A5 (C). Platelets were pretreated with vehicle or MDL-28170 (200 μm), as indicated. Treatment effects are shown of time-dependent changes in αIIbβ3 closure (D) or PS exposure (E). Graphs indicate means ± S.D. (error bars; n = 3–4). *, p < 0.05 versus vehicle (two-way analysis of variance, Bonferroni correction).
After stimulation with Cvx/Thr or ionomycin, the knock-out and wild type platelets gave similar cleavage patterns of the integrin β3 chain (supplemental Fig. S3). Furthermore, total calpain inhibition with MDL-28170 fully blocked the cleavage of β3 chain in Capn1+/+ and Capn1−/− platelets (supplemental Fig. S4). In conclusion, whereas the pharmacological data suggest a partial role for calpain activity in the process of integrin αIIbβ3 closure of PS-exposing mouse and human platelets, calpain-1 (at least in mice) does not appear to be the main β3 chain/talin-degrading protease or target of the calpain inhibitors.
Role of Phospholipid Scrambling in Integrin αIIbβ3 Closure
Inactivation of integrin αIIbβ3 under conditions of PS exposure may point to a causal link between αIIbβ3 closure and scrambling of membrane phospholipids. This possibility was investigated using platelets from a Scott syndrome patient lacking the transmembrane protein TMEM16F, whose platelets are impaired in phospholipid scrambling in response to Cvx/Thr or ionomycin (38). Defective PS exposure in response to Cvx/Thr in the patient's platelets was accompanied by a more persistent integrin αIIbβ3 activation (Fig. 7, A and B). In the patient, ionomycin caused partial and transient αIIbβ3 activation, indicating that a mechanism of integrin closure was still operating. Interestingly, treatment of the Scott platelets with MDL-28170 fully blocked the time-dependent closure of integrin αIIbβ3, in contrast to the partial blockage observed in control platelets (Fig. 7C). Western blotting indicated that the cleavage pattern of the β3 chain was not affected in Scott platelets after stimulation with ionomycin (Fig. 8) or Cvx/Thr (data not shown). Furthermore, the cleavage was fully inhibited by MDL-21870. Together, these results suggest a dual mechanism of integrin αIIbβ3 closure in procoagulant platelets partly via TMEM16F-dependent scrambling of the plasma membrane phospholipids and partly via calpain-dependent cleavage of intracellular proteins including the integrin β3 chain.
FIGURE 7.

Partial role of TMEM16F in agonist-induced PS exposure and αIIbβ3 closure. Platelets from control subjects or a Scott patient (deficient in TMEM16F) were stimulated with Cvx/Thr (100 ng/ml, 4 nm) or ionomycin (19 μm) for 5–30 min, as in Fig. 1. Samples were analyzed by flow cytometry for PS exposure (AF647-annexin A5 binding) or αIIbβ3 activation (FITC-PAC1 binding), as indicated. A, percentages of PS-exposing platelets and increase in PS exposure between 5 and 30 min. B, percentages of PAC1-binding platelets and percentages of αIIbβ3 closure between 5 and 30 min. C, effect of platelet treatment with MDL-28170 (200 μm) on PAC1 binding and αIIbβ3 closure. Graphs indicate means ± S.D. (error bars; n = 4). *, p < 0.05 versus vehicle (two-way analysis of variance, Bonferroni correction).
FIGURE 8.
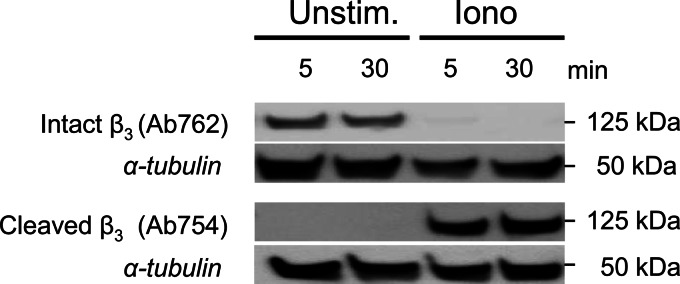
Unchanged agonist-induced cleavage of β3 chain in the absence of TMEM16F. Platelets from the Scott patient (deficient in TMEM16F) were stimulated with ionomycin for 5–30 min, as in Fig. 1. Western blots from samples were stained for full-length β3 chain (Ab762) or calpain-cleaved β3 chain (Ab754). Blots were reprobed with anti-tubulin mAb as loading control. Shown are representative blots.
Involvement of Mitochondrial Transition Pore Formation in αIIbβ3 Closure
Considering that mPTP formation plays an important role in Cvx/Thr-stimulated PS exposure in platelets (22), we investigated whether this mitochondrial process is also involved in integrin αIIbβ3 closure. Experiments were performed with platelets harvested from mice deficient in cyclophilin D (Ppif gene), which forms part of the mPTP. Hence, Ppif−/− mouse platelets allow evaluating integrin closure in a second model, next to Scott platelets, in which PS exposure is blunted. Perfusion of Ppif−/− blood over collagen resulted in decreased platelet deposition and PS exposure (AF647-annexin A5 binding) compared with wild type Ppif+/+ blood (Fig. 9A). Also, in suspensions of Ppif−/− platelets stimulated with Cvx/Thr, PS exposure was markedly reduced, whereas αIIbβ3 activation and closure were suppressed in a similar way (Fig. 9B). Treatment of wild type Ppif+/+ platelets with cyclosporin A, a compound known to block mPTP formation (39), suppressed PS exposure and integrin closure to the level observed in the knock-out platelets, whereas cyclosporin A treatment of Ppif−/− platelets was without any effect. Intracellular Ca2+ rises were not affected in Ppif−/− platelets compared with controls.4
FIGURE 9.

Partial role of mPTP formation in agonist-induced PS exposure and αIIbβ3 closure. A, blood from Ppif+/+ or Ppif−/− mice was perfused over collagen for 4 min at 1000 s−1. Thrombi were labeled with PE-JON/A mAb and AF647-annexin A5. Shown are representative brightfield and fluorescence images (scale bars, 20 μm), as well as quantitative analyses of (fluorescent) platelet deposition. B and C, washed mouse platelets were pretreated with vehicle (0.2% dimethyl sulfoxide) or cyclosporin A (4 μm), stimulated for 5–30 min with Cvx/Thr, and analyzed by flow cytometry. Bars indicate percentages of platelets binding PE-JON/A (B) or AF647-annexin A5 (C). Graphs indicate means ± S.D. (error bars; n = 3–4). *, p < 0.05 (two-way analysis of variance, Bonferroni correction). D and E, human platelets, preincubated with vehicle (0.2% dimethyl sulfoxide) or cyclosporin A (4 μm), were stimulated for 5–30 min with Cvx/Thr. Shown are time-dependent changes in percentages of platelets binding FITC-PAC1 (D) or AF647-annexin A5 (E). Graphs indicate means ± S.D. (error bars; n = 4). *, p < 0.05 (one-way analysis of variance, Bonferroni correction).
Similar experiments were performed with human platelets treated with cyclosporin A. Again, cyclosporin A suppressed PS exposure as well as integrin closure in response to Cvx/Thr (Fig. 9C). In Fura-2-loaded platelets, cyclosporin A did not affect Cvx/Thr-induced intracellular Ca2+ rises (data not shown). In addition, Src cleavage observed after prolonged Cvx/Thr stimulation was prevented by cyclosporin A treatment (data not shown). Together, these observations point to a central role of mPTP formation in PS exposure and integrin closure.
DISCUSSION
In this work, we demonstrate a dual mechanism responsible for integrin αIIbβ3 closure (inactivation) in PS-exposing platelets being dependent on Ca2+ and calpain activity (but not calpain-1) and relying on Ca2+-dependent phospholipid scrambling mediated by the transmembrane protein TMEM16F. The calpain-dependent proteolysis under conditions of PS exposure involves intracellular cleavage of the integrin β3 chain, talin, and Src kinase. These findings implicate that PS-exposing, procoagulant platelets lose not only their capability of integrin-dependent adhesion and aggregate formation, but also the competence for integrin-dependent outside-in signaling and downstream responses including formation of tight platelet-platelet contacts. This model provides an explanation for the loose association of PS-exposing platelets in a thrombus and begins to outline a basis for bound coagulation factors and thrombin generation. The recognized mechanisms of integrin closure or inability to activation thus explain the earlier recognized heterogeneity of platelets in thrombus formation (5, 20).
High and sustained elevation in cytosolic Ca2+, a requirement for PS exposure (40), appears to be required for achieving optimal integrin closure, most likely because of the Ca2+ dependence of calpain activity and TMEM16F. Mouse platelets deficient in calpain-1 showed a moderate increase in integrin activation and reduced PS exposure upon collagen-dependent thrombus formation, which is consistent with a moderate reduction in αIIbβ3 closure under these mild activation conditions. This result seemingly contrasts with earlier findings that Capn1−/− platelets show reduced aggregation at low thrombin concentrations (27), but stipulates that a direct role of calpain-1 in integrin activation remains subtle. In contrast, we found that the Capn1−/− platelets show nearly normal integrin closure as well as cleavage of β3, talin, and Src, when stimulated with strong agonists such as Cvx/Thr. Given the complete abolishment of protein cleavage with two calpain inhibitors (calpeptin and MDL-28170), we concluded that another cysteine protease is involved under conditions of high Ca2+ concentration. It is likely that this protease is calpain-2, which accounts for ∼20% of the platelet calpain activity and is known to require millimolar Ca2+ concentrations for activation (41). Given the heterogeneity in intracellular Ca2+ concentrations these high levels can be reached locally in procoagulant platelets. Unfortunately, a role for calpain-2 could not be tested directly in platelets because calpain-2 deficiency in mice is lethal (42). The mechanistic reason for this lethality is still unknown, but a general role for this calpain isoform in cell proliferation and cell cycle progression has been suggested.
Regarding the integrin closure of Cvx/Thr-stimulated platelets, inhibitor studies demonstrated clear, but partial, inhibitory effects of MDL-28170 and calpeptin. In suspensions of platelets where microaggregate formation was prevented, both compounds suppressed integrin closure of control platelets by ∼50% (under conditions where the cleavage of β3 chain, talin, and Src was blocked), whereas it fully abrogated the partly impaired and delayed integrin closure in Scott syndrome platelets, which do not show PS exposure. This clearly supports a model of dual mechanism of integrin closure that is in part calpain-dependent (likely through the degradation of proteins implicated in αIIbβ3 activation) and in part mediated by TMEM16F (via Ca2+-mediated phospholipid scrambling). The latter pathway is absent in the Scott syndrome platelets. It is conceivable that the profound intramembrane changes caused by phospholipid scrambling affect interactions of the integrin αIIb and β3 chains or of integrin-associated proteins even in the absence of calpain cleavage. However, the precise mechanism remains to be investigated.
Although the calpain-mediated cleavage of specific proteins in PS-exposing platelets has been reported before (4), we now show that the protease responsible is not calpain-1, but likely calpain-2. Moreover, this cleavage occurs at a much faster and extensive scale than anticipated, including complete degradation of several proteins that are known to play a key role in integrin activation and signaling, i.e. the β3 chain, talin, and Src kinase. The eventual result will be that these PS-exposing platelets (with a prominent role in coagulation) are no longer capable of carrying out the Src/Syk- and talin-dependent responses of aggregated platelets using active integrins, including clot retraction and tight platelet-platelet contact formation.
Earlier studies have shown a critical role of the mPTP and cyclophilin D in Cvx/Thr-induced but not ionomycin-induced PS externalization (22). Using platelets from Ppif−/− mice, this was confirmed in the present study. In whole blood flow assay over collagen, the cyclophilin D-deficient platelets showed a consistent reduction in thrombus formation and PS exposure. Moreover, the reduced PS exposure was accompanied by a similar reduction in integrin closure, thus suggesting a common, mitochondrial-dependent denominator for the impairment of PS exposure and αIIbβ3 inactivation. Taken together, our findings reveal a novel, dual mechanism of αIIbβ3 inactivation via calpain (not calpain-1) dependent cleavage of integrin-associated proteins and via TMEM16F-dependent phospholipid scrambling, with an assistant role of mPTP formation.
This work was supported by the Cardiovascular Centre, Maastricht, the Center for Translational Molecular Medicine Innovative Coagulation Diagnostics (INCOAG), and the Netherlands Heart Foundation grant 2011T6 (to J. M. E. M. C.) and National Institute of Health grant HL089519 (to A. H. C.).

This article contains supplemental Table S1 and Figs. S1–S4.
S. M. Jobe, unpublished observation.
- PS
- phosphatidylserine
- AF647
- Alexa Fluor 647
- Cvx
- convulxin
- mPTP
- mitochondrial permeability transition pore
- PE
- phycoerythrin
- Thr
- thrombin.
REFERENCES
- 1. Jackson S. P. (2011) Arterial thrombosis: insidious, unpredictable and deadly. Nat. Med. 17, 1423–1436 [DOI] [PubMed] [Google Scholar]
- 2. Heemskerk J. W., Mattheij N. J., Cosemans J. M. (2013) Platelet-based coagulation: different populations, different functions. J. Thromb. Haemost. 11, 2–16 [DOI] [PubMed] [Google Scholar]
- 3. Munnix I. C., Kuijpers M. J., Auger J., Thomassen C. M., Panizzi P., van Zandvoort M. A., Rosing J., Bock P. E., Watson S. P., Heemskerk J. W. (2007) Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation: regulation by transient integrin activation. Arterioscler. Thromb. Vasc. Biol. 27, 2484–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kulkarni S., Jackson S. P. (2004) Platelet factor XIII and calpain negatively regulate integrin αIIbβ3 adhesive function and thrombus growth. J. Biol. Chem. 279, 30697–30706 [DOI] [PubMed] [Google Scholar]
- 5. Berny M. A., Munnix I. C., Auger J. M., Schols S. E., Cosemans J. M., Panizzi P., Bock P. E., Watson S. P., McCarty O. J., Heemskerk J. W. (2010) Spatial distribution of factor Xa, thrombin, and fibrin(ogen) on thrombi at venous shear. Plos One 5, e10415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cosemans J. M., Iserbyt B. F., Deckmyn H., Heemskerk J. W. (2008) Multiple ways to switch platelet integrins on and off. J. Thromb. Haemost. 6, 1253–1261 [DOI] [PubMed] [Google Scholar]
- 7. Shattil S. J., Kim C., Ginsberg M. H. (2010) The final steps of integrin activation: the end game. Nat. Rev. Mol. Cell Biol. 11, 288–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Watson S. P., Auger J. M., McCarty O. J., Pearce A. C. (2005) GPVI and integrin αIIbβ3 signaling in platelets. J. Thromb. Haemost. 3, 1752–1762 [DOI] [PubMed] [Google Scholar]
- 9. Ma Y. Q., Qin J., Plow E. F. (2007) Platelet integrin αIIbβ3: activation mechanisms. J. Thromb. Haemost. 5, 1345–1352 [DOI] [PubMed] [Google Scholar]
- 10. Nieswandt B., Varga-Szabo D., Elvers M. (2009) Integrins in platelet activation. J. Thromb. Haemost. 7, 206–209 [DOI] [PubMed] [Google Scholar]
- 11. Kim C., Ye F., Hu X., Ginsberg M. H. (2012) Talin activates integrins by altering the topology of the β transmembrane domain. J. Cell Biol. 197, 605–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shattil S. J., Newman P. J. (2004) Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood 104, 1606–1615 [DOI] [PubMed] [Google Scholar]
- 13. Arias-Salgado E. G., Lizano S., Sarkar S., Brugge J. S., Ginsberg M. H., Shattil S. J. (2003) Src kinase activation by direct interaction with the integrin β cytoplasmic domain. Proc. Natl. Acad. Sci. U.S.A. 100, 13298–13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yan B., Calderwood D. A., Yaspan B., Ginsberg M. H. (2001) Calpain cleavage promotes talin binding to the β3 integrin cytoplasmic domain. J. Biol. Chem. 276, 28164–28170 [DOI] [PubMed] [Google Scholar]
- 15. Pfaff M., Du X., Ginsberg M. H. (1999) Calpain cleavage of integrin β cytoplasmic domains. FEBS Lett. 460, 17–22 [DOI] [PubMed] [Google Scholar]
- 16. Arachiche A., Kerbiriou-Nabias D., Garcin I., Letellier T., Dachary-Prigent J. (2009) Rapid procoagulant phosphatidylserine exposure relies on high cytosolic calcium rather than on mitochondrial depolarization. Arterioscler. Thromb. Vasc. Biol. 29, 1883–1889 [DOI] [PubMed] [Google Scholar]
- 17. Siljander P. R., Munnix I. C., Smethurst P. A., Deckmyn H., Lindhout T., Ouwehand W. H., Farndale R. W., Heemskerk J. W. (2004) Platelet receptor interplay regulates collagen-induced thrombus formation in flowing human blood. Blood 103, 1333–1341 [DOI] [PubMed] [Google Scholar]
- 18. Suzuki J., Umeda M., Sims P. J., Nagata S. (2010) Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838 [DOI] [PubMed] [Google Scholar]
- 19. van Kruchten R., Mattheij N. J., Saunders C., Feijge M. A., Swieringa F., Wolfs J. L., Collins P. W., Heemskerk J. W., Bevers E. M. (2013) Both TMEM16F-dependent and TMEM16F-independent pathways contribute to phosphatidylserine exposure in platelet apoptosis and platelet activation. Blood 121, 1850–1857 [DOI] [PubMed] [Google Scholar]
- 20. Munnix I. C., Cosemans J. M., Auger J. M., Heemskerk J. W. (2009) Platelet response heterogeneity in thrombus formation. Thromb. Haemost. 102, 1149–1156 [DOI] [PubMed] [Google Scholar]
- 21. Castoldi E., Collins P. W., Williamson P. L., Bevers E. M. (2011) Compound heterozygosity for two novel TMEM16F mutations in a patient with Scott syndrome. Blood 117, 4399–4400 [DOI] [PubMed] [Google Scholar]
- 22. Jobe S. M., Wilson K. M., Leo L., Raimondi A., Molkentin J. D., Lentz S. R., Di Paola J. (2008) Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 111, 1257–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dale G. L. (2005) Coated-platelets: an emergic component of the procoagulant response. J. Thromb. Haemost. 3, 2185–2192 [DOI] [PubMed] [Google Scholar]
- 24. Jobe S. M., Leo L., Eastvold J. S., Dickneite G., Ratliff T. L., Lentz S. R., Di Paola J. (2005) Role of FcRγ and factor XIIIa in coated platelet formation. Blood 106, 4146–4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Du X., Saido T. C., Tsubuki S., Indig F. E., Williams M. J., Ginsberg M. H. (1995) Calpain cleavage of the cytoplasmic domain of the integrin β3 subunit. J. Biol. Chem. 270, 26146–26151 [DOI] [PubMed] [Google Scholar]
- 26. van der Meijden P. E., Schoenwaelder S. M., Feijge M. A., Cosemans J. M., Munnix I. C., Wetzker R., Heller R., Jackson S. P., Heemskerk J. W. (2008) Dual P2Y12 receptor signaling in thrombin-stimulated platelets: involvement of phosphoinositide 3-kinase β but not γ isoform in Ca2+ mobilization and procoagulant activity. FEBS J. 275, 371–385 [DOI] [PubMed] [Google Scholar]
- 27. Kuchay S. M., Kim N., Grunz E. A., Fay W. P., Chishti A. H. (2007) Double knockouts reveal that protein tyrosine phosphatase 1B is a physiological target of calpain-1 in platelets. Mol. Cell. Biol. 27, 6038–6052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuchay S. M., Wieschhaus A. J., Marinkovic M., Herman I. M., Chishti A. H. (2012) Targeted gene inactivation reveals a functional role of calpain-1 in platelet spreading. J. Thromb. Haemost. 10, 1120–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gilio K., van Kruchten R., Braun A., Berna-Erro A., Feijge M. A., Stegner D., van der Meijden P. E., Kuijpers M. J., Varga-Szabo D., Heemskerk J. W., Nieswandt B. (2010) Roles of platelet STIM1 and Orai1 in glycoprotein VI- and thrombin-dependent procoagulant activity and thrombus formation. J. Biol. Chem. 285, 23629–23638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cosemans J. M., Van Kruchten R., Olieslagers S., Schurgers L. J., Verheyen F. K., Munnix I. C., Waltenberger J., Angelillo-Scherrer A., Hoylaerts M. F., Carmeliet P., Heemskerk J. W. (2010) Potentiating role of Gas6 and Tyro3, Axl and Mer (TAM) receptors in human and murine platelet activation and thrombus stabilization. J. Thromb. Haemost. 8, 1797–1808 [DOI] [PubMed] [Google Scholar]
- 31. Gilio K., Harper M. T., Cosemans J. M., Konopatskaya O., Munnix I. C., Prinzen L., Leitges M., Liu Q., Molkentin J. D., Heemskerk J. W., Poole A. W. (2010) Functional divergence of platelet protein kinase C (PKC) isoforms in thrombus formation on collagen. J. Biol. Chem. 285, 23410–23419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cosemans J. M., Schols S. E., Stefanini L., de Witt S., Feijge M. A., Hamulyák K., Deckmyn H., Bergmeier W., Heemskerk J. W. (2011) Key role of glycoprotein Ib/V/IX and von Willebrand factor in platelet activation-dependent fibrin formation at low shear flow. Blood 117, 651–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heemskerk J. W., Feijge M. A., Henneman L., Rosing J., Hemker H. C. (1997) The Ca2+-mobilizing potency of a-thrombin and thrombin-receptor-activating peptide on human platelets: concentration and time effects of thrombin-induced Ca2+ signaling. Eur. J. Biochem. 249, 547–555 [DOI] [PubMed] [Google Scholar]
- 34. Wolfs J. L., Comfurius P., Rasmussen J. T., Keuren J. F., Lindhout T., Zwaal R. F., Bevers E. M. (2005) Activated scramblase and inhibited aminophospholipid translocase cause phosphatidylserine exposure in a distinct platelet fraction. Cell. Mol. Life Sci. 62, 1514–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fuse I., Higuchi W., Uesugi Y., Aizawa Y. (2001) Pathogenetic analysis of three cases with a bleeding disorder characterized by defective platelet aggregation induced by Ca2+ ionophores. Br. J. Haematol. 112, 603–608 [DOI] [PubMed] [Google Scholar]
- 36. Naik M. U., Stalker T. J., Brass L. F., Naik U. P. (2012) JAM-A protects from thrombosis by suppressing integrin αIIbβ3-dependent outside-in signaling in platelets. Blood 119, 3352–3360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuchay S. M., Chishti A. H. (2007) Calpain-mediated regulation of platelet signaling pathways. Curr. Opin. Hematol. 14, 249–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Munnix I. C., Harmsma M., Giddings J. C., Collins P. W., Feijge M. A., Comfurius P., Heemskerk J. W., Bevers E. M. (2003) Store-mediated calcium entry in the regulation of phosphatidylserine exposure in blood cells from Scott patients. Thromb. Haemost. 89, 687–695 [PubMed] [Google Scholar]
- 39. Leytin V., Allen D. J., Mutlu A., Gyulkhandanyan A. V., Mykhaylov S., Freedman J. (2009) Mitochondrial control of platelet apoptosis: effect of cyclosporin A, an inhibitor of the mitochondrial permeability transition pore. Lab. Invest. 89, 374–384 [DOI] [PubMed] [Google Scholar]
- 40. Heemskerk J. W., Bevers E. M., Lindhout T. (2002) Platelet activation and blood coagulation. Thromb. Haemost. 88, 186–193 [PubMed] [Google Scholar]
- 41. Goll D. E., Thompson V. F., Li H., Wei W., Cong J. (2003) The calpain system. Physiol. Rev. 83, 731–801 [DOI] [PubMed] [Google Scholar]
- 42. Dutt P., Croall D. E., Arthur J. S., Veyra T. D., Williams K., Elce J. S., Greer P. A. (2006) m-Calpain is required for preimplantation embryonic development in mice. BMC Dev. Biol. 6, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]