

Background: Fn14 is a therapeutic target in various diseases.
Results: Anti-Fn14 antibodies activate the alternative NFκB pathway but not other Fn14-related activities induced by soluble or membrane-bound TWEAK. FcγR-bound anti-Fn14 antibodies, however, activate the full spectrum of Fn14-associated activities.
Conclusion: Anti-Fn14 antibodies elicit agonistic activities differing from those of the natural Fn14 ligand TWEAK.
Significance: These findings influence the rationale of designing Fn14-targeted therapies.
Keywords: Antibodies, Apoptosis, NF-kappa B (NF-KB), TRAF, Tumor Necrosis Factor (TNF)
Abstract
The Fn14-specific monoclonal antibodies PDL192 and P4A8, which are under consideration in clinical trials, showed no agonistic activity with respect to IL8 production and cell death induction. However, oligomerization with protein G or binding to Fcγ receptors converted both anti-Fn14 antibodies into potent agonists. TNF-like weak inducer of apoptosis (TWEAK), the ligand of Fn14, occurs naturally in two forms with partly different signaling capabilities, as a membrane-bound ligand and as a soluble trimeric molecule. Although membrane TWEAK strongly triggers all Fn14-associated pathways, soluble TWEAK predominately triggers the alternative nuclear factor κB (NFκB) pathway and enhances TNF-induced cell death but has only a poor effect on the classical NFκB pathway and chemokine production. Thus, the oligomerized and FcγR-bound anti-Fn14 mAbs mimicked the activity of membrane TWEAK. Notably, both anti-Fn14 antibodies significantly triggered p100 processing, the hallmark of the alternative NFκB pathway, and therefore resembled soluble TWEAK. In contrast to the latter, however, the anti-Fn14s showed no effect on TNF receptor 1-induced cell death and P4A8 even blocked the corresponding TWEAK response. Thus, we showed that Fn14 antibodies display an alternative NFκB pathway-specific agonistic activity but fail to phenocopy other activities of soluble TWEAK, whereas oligomerized or FcγR-bound Fn14 antibodies fully mimic the activity of membrane TWEAK. In view of the trivalent nature of the TWEAK-Fn14 interaction, this suggests that the alternative NFκB pathway is uniquely responsive already to Fn14 dimerization enabling antibodies to elicit an unnatural response pattern distinct from that of the naturally occurring Fn14 ligands.
Introduction
TNF-like weak inducer of apoptosis (TWEAK)2 is a member of the TNF ligand family that triggers cellular responses by binding to Fn14, an unusual small member of the TNF receptor family. TWEAK is initially expressed as a membrane-bound type II protein but gets readily processed in most cell types by furin proteases resulting in the release of soluble TWEAK (1). In fact, cell surface expressed TWEAK has so far only be detected on monocytes, macrophages, dendritic cells, and some breast cancer and hepatocellular cancer cell lines (1–3). Stimulation of Fn14 typically results in activation of transcription factors of the NFκB family but the stimulation of MAP kinases and the PI3K pathway as well as the induction of necrotic and apoptotic cell death have also been reported (1). Fn14 has no death domain, and in some models, its cell death-inducing activity has been indeed delineated to the NFκB-mediated induction of TNF and activation of the death receptor TNFR1 together with a concomitant depletion of TRAF2-cIAP complexes, which antagonizes TNFR1-induced cell death (4–6). Noteworthy, Fn14 differently respond to soluble and membrane-bound TWEAK. While a subset of Fn14-mediated cellular responses, e.g. activation of the alternative NFκB pathway and enhancement of TNF-induced apoptosis, is efficiently triggered by both TWEAK forms, activation of the classical NFκB pathway is primarily stimulated by membrane-bound TWEAK (7). Fn14 expression has been found on most tumor cell lines of non-lymphoid origin, but in vivo Fn14 expression is predominately expressed during development and in injured tissue (1, 8). The TWEAK/Fn14 system regulates proliferation and differentiation of mesenchymal progenitor cells, angiogenesis, but also infiltration of immune cells, cell survival, and cell death (1).
Particularly, the TWEAK-Fn14 system has been implicated in a variety of pathophysiological situations of great clinical importance. Similar to its name-giving cousin TNF, it contributes to the development of autoinflammatory diseases in various experimental models, including collagen induced arthritis (9, 10), myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis (11–13), 2,4,6-trinitrobenzenesulfonic acid-induced colitis (14, 15), and systemic lupus erythematosus-related nephritis (16) and has also been implicated in atherosclerotic plaque progression in apolipoprotein E (apoE) knock-out mice (17, 18). Furthermore, TWEAK and Fn14 play a role in detrimental inflammatory and fibrotic processes associated with the repair of injured tissue after liver damage, denervation, stroke, and renal and cerebral ischemia (19–23). In view of the tissue destruction that is inevitably associated with cancer progression, it is no surprise that high Fn14 expression is evident in most solid tumors. Notably, the high tumor-related Fn14 expression is not only evident on “activated” cells in the microenvironment but also on the malignant cells itself. Indeed, although TWEAK is cytotoxic to some tumor cell lines, in other cases, activation of Fn14 results in cell migration and survival (1, 24). Due to their disease-associated expression and function, TWEAK and Fn14 attract considerable interest as therapeutic targets. TWEAK and Fn14 can be targeted by antibodies with high selectivity, and TWEAK- and Fn14-specific antibodies are in first clinical trials for treatment of rheumatoid arthritis, lupus, and solid tumors (http://clinicaltrials.gov/). Particularly with respect to Fn14-specific antibodies different modes of action are possible in vivo. Fn14-bound antibodies might trigger destruction of the Fn14-expressing cells, e.g. by antibody-dependent cellular cytotoxicity but could also inhibit Fn14 activation by TWEAK or even act as Fn14 agonists. Dependent on the pathophysiological scenario, inhibition and activation of Fn14 by antibodies in turn can have beneficial but also exacerbating effects. The success of an anti-Fn14 antibody therapy might thus not only depends on the best possible knowledge concerning the role of TWEAK and Fn14 in the addressed application but also on the choice of an antibody that optimally modifies the activity of Fn14-expressing cells in this particular disease. We describe here that oligomerization with protein G and Fcγ receptor binding uncover a latently present high agonistic activity in a non-blocking as well as a blocking antagonistic Fn14-specific antibody. More surprisingly, we observed that both the blocking as well as the non-blocking anti-Fn14 activate selectively the alternative NFκB pathway without modulation of TNFR1-induced cell death, thus eliciting an agonistic quality distinct from those of the two naturally ligands of Fn14, soluble TWEAK and membrane TWEAK.
EXPERIMENTAL PROCEDURES
Cell Lines, Antibodies, and Reagents
The human colorectal adenocarcinoma cell line HT29, HT1080 human fibrosarcoma cells, SKOV-3 human ovarian adenocarcinoma cells, Kym-1 human rhabdomyosarcoma cells, and HEK293 human embryonic kidney cells were maintained in RPMI 1640 medium (PAA Laboratories, Pasching, Germany), containing 10% FCS (30), and WiDr human colorectal adenocarcinoma cells were cultured with minimal essential medium (Invitrogen) again containing 10% FCS. The recombinant Fn14-specific human IgG1 antibodies PDL192 and P4A8 (25–27) were generated using the published sequences of the variable domains of these antibodies (28, 29), and sequences encoding the constant region of human IgG1 (accession no. P01857) and the immunoglobulin κ chain V-J-C segment (accession no. gb AAA59000.1). Cycloheximide was obtained from Sigma, and the recombinant protein G was from Calbiochem (San Diego, CA, USA). pCMV-SPORT6 expression vectors encoding Fcγ receptors were purchased from Source Bioscience (Nottingham, UK). Production and purification of Flag-TWEAK, GpL-Flag-TNC-TWEAK, and Fc-Flag-TWEAK have already been described previously (7, 43). NIK-, IκBα-, and p-IκBα-specific antibodies were ordered from Cell Signaling (Frankfurt, Germany); PE-conjugated anti-FcγR1a (anti-CD64), anti-FcγR3a (anti-CD16), and unconjugated anti-FcγR2a (anti-CD32A) and anti-FcγR2b (anti-CD32B) were from Santa Cruz Biotechnology; and anti-tubulin was from Dunn Labortechnik (Asbach, Germany). PE-conjugated anti-TWEAK and anti-Fn14 antibodies were purchased from eBioscience (Frankfurt, Germany); anti-p100/p52 was from Upstate (Schwalbach, Germany); anti-TRAF2 was from BD Biosciences; anti-cIAP1 was from Enzo Life Sciences (Lörrach, Germany); and HRP secondary antibodies were from Dako (Hamburg, Germany).
Determination of IL8 Production
Cells (2 × 104/well) were seeded in 96-well tissue culture plates and cultured overnight. The following day, medium was exchanged to minimize background IL8 production of untreated cells. In the TWEAK inhibition experiments, cells were treated with the anti-Fn14 antibodies for 1 h before cells were stimulated with TWEAK. In the coculture experiments with Fcγ receptor expressing transfectants, target cells were added to the transiently transfected Hek293 cells, and 30 min later, the mixture of cells was challenged with the different antibodies. 18 h post stimulation, supernatants were collected and analyzed with respect to their IL8 content using a commercially available ELISA kit (BD Biosciences) according to the manufacturer's instructions.
Western Blotting Analysis
For Western blot analysis of NIK accumulation and p100 processing, whole cell lysates were prepared. Therefore, cells were harvested into ice-cold PBS and centrifuged and then directly lysed in 4× protein sample buffer (8% SDS, 0.1 m DTT, 40% glycerol, 0.2 m Tris (pH 6.8), 0.004% bromphenol blue) supplemented with both complete protease inhibitor (Roche Applied Science) and phosphatase inhibitor mixtures I and II (Sigma-Aldrich). Afterward, samples were sonificated (10 pulses). For analysis of depletion of TRAF2 and cIAP1 from the soluble compartment, cells (106) were lyzed for 20 min on ice in 100 μl of Triton X-100 buffer (30 mm Tris (pH 7.4), 10% glycerol, 125 mm NaCl, 1% Triton X-100) supplemented with complete protease inhibitor (Roche Applied Science) and phosphatase inhibitor mixtures I and II (Sigma-Aldrich). Lysates were cleared by centrifugation (14.000 rpm, 4 °C, 10 min) and mixed with 4× protein sample buffer. Total cell lysates as well as Triton X-100 lysates were boiled (95 °C, 5 min), separated by SDS-PAGE, and transferred to nitrocellulose membranes. After 1 h of blocking in Tris-buffered saline containing 0.1% Tween 20 and 5% dry milk, immunoblotting was completed with primary antibodies specific for the indicated proteins, HRP-conjugated secondary antibodies, and the commercial available ECL Western blotting detection reagents and analysis system (Amersham Biosciences, Muenchen, Germany).
Cell Death Assay
Cells (1.7 × 104/well) were seeded in 96-well tissue plates. The next day cells were treated in the presence or absence of protein G with antibodies specific for Fn14. Cell viability was determined after 16 h by crystal violet staining.
Induction of Alkaline Phosphatase Expression
The mouse myoblast cell line C2C12 (ATCC catalog no. CRL1772) was cultured in DMEM containing 10% FCS. For alkaline phosphatase induction assays, the cells were serum starved (2% FCS) and exposed to ligands for 48 h in 96-well microplates. After cell lysis, BMP2-induced alkaline phosphatase activity was measured by p-nitrophenylphosphate conversion using an ELISA reader at 405 nm.
Flow Cytometry
Cells were incubated for 20 min at 4 °C with a PE-conjugated TWEAK-specific antibody (eBioscience, Frankfurt, Germany), anti-CD16-PE, anti-CD64-PE, anti-CD32A/C, and anti CD32B were purchased from Santa Cruz Biotechnology or an appropriate isotype control. For anti-CD32A/C and anti-CD32B antibody, cells were again incubated 20 min at 4 °C with an anti-mouse PE antibody. After three cycles of washing with PBS, cells were analyzed using FACSCalibur (BD Biosciences) according to standard procedures.
RESULTS
The Anti-Fn14 mAb P4A8 Efficiently Blocks TWEAK-induced Cellular Reactions whereas the Anti-Fn14 PDL192 Barely Interferes with TWEAK Binding
We generate recombinant human IgG1 variants of the already described Fn14-specific antibodies PDL192 and P4A8 (BIIB036) (25–29). Initially, we controlled Fn14 binding of the recombinant antibodies and confirmed that they recognize different epitopes. For these purposes, we used human, murine, and cynomolgus Fn14 along with two mutants of human Fn14 (W42A and R56P) that do not interfere with ligand binding but either fail to interact with P4A8 (W42A) or with PDL192 (R56P) (28, 29). The various Fn14 variants were transiently expressed in Hek293 cells, which have only low amounts of endogenous Fn14, and antibody binding was evaluated by FACS (Fig. 1A). As expected based on the available literature (25–29), both antibodies yielded effective staining of human and cynomolgus Fn14 but reacted differently with murine Fn14 and the two mutants of human Fn14. PDL192 failed to interact with murine Fn14 and the R56P variant but significantly bound to the W42A mutant of human Fn14. In contrast, P4A8 did not interact with the latter but recognized murine Fn14 as well as R56P variant (Fig. 1A). In conclusion, this confirmed that PDL192 and P4A8 recognize two different epitopes.
FIGURE 1.

Characterization of the Fn14-specifc antibodies PDL192 and P4A8. A, Hek293 cells were transiently transfected with expression constructs encoding human (h), murine (m), and cynomolgus (c) Fn14 and two mutants of human Fn14 (W42A and R56P). Two days later, the two anti-Fn14 antibodies and an irrelevant IgG1 antibody were used for FACS analysis of cell surface expression of Fn14. Cells were gated to minimize the contribution of endogenous Fn14 expression and Fn14 binding was calculated as the product of percent positive cells and mean fluorescent intensity of positive cells. The average of three fully independent experiments is shown. B and C, the indicated cell lines were preincubated in triplicates in 96-well plates for 1 h with increasing concentrations of PDL192 and P4A8. Cells were then challenged with 100 ng/ml Fc-Flag-TWEAK. The next day, supernatants were collected from the HT29 and WiDr cells to determine their IL8 content by ELISA. The dotted lines indicate the level of constitutive IL8 production of non-stimulated cells. To minimize the background caused by constitutive IL8 production, cell culture medium was changed prior stimulation (B). Cell death induction in SKOV-3 and Kym-1 cells was evaluated by measuring cellular viability by staining with crystal violet (C). D, HT29 cells were again pretreated with increasing concentrations of the two anti-Fn14 antibodies and then challenged overnight with 100 ng/ml Fc-Flag-TWEAK to sensitize cells for TNF-induced apoptosis. Next day, cells were stimulated with TNF and cycloheximide to trigger TNFR1-induced cell death. After overnight incubation, cellular viability was again determined by crystal violet staining. E, C2C12 cells were pretreated with the anti-Fn14 antibodies and then alkaline phosphatase (ALP) production was induced by BMP2 (20 nm). 100 ng/ml of Fc-TWEAK was additional added to inhibit BMP2-induced alkaline phosphatase production. F, competition binding experiments with GpL-Flag-TNC-TWEAK and PDL192 and P4A8. HT29 (human) and C2C12 (murine) cells were pretreated at 37 °C for 1 h with increasing concentrations of the two anti-Fn14 antibodies. Cells were then incubated with 25 ng/ml (200 pm) of GpL-Flag-TNC-TWEAK for an additional hour, and luciferase activity of cell-bound GpL-Flag-TNC-TWEAK was determined.
Next, we analyzed the capability of the two anti-Fn14 antibodies to inhibit distinct TWEAK-induced cellular effects known to be mediated by Fn14. The Fn14-mediated induction of proinflammatory cytokines and chemokines by TWEAK has been demonstrated in a variety of cell lines. Pretreatment of HT29 and WiDr cells with PDL192 and P4A8 resulted for the latter in strong inhibition of TWEAK-induced IL8 production, whereas PDL192 showed only weak to moderate inhibition at high concentrations (Fig. 1B). The TWEAK-Fn14 system has also been recognized in some cell lines, for example SKOV3 and Kym-1, as a cell death inducer. In accordance with the inhibitory effect of P4A8 on TWEAK-induced IL8 production, we also observed complete blockade of TWEAK-induced cell death in SKOV3 and Kym-1 cells. PDL192 again showed at best a minor inhibitory effect and only at the highest concentrations (Fig. 1C). Likewise, the capability of TWEAK to enhance TNFR1-induced cell death in per se TWEAK-resistant cell lines were blocked by P4A8 but not by PDL192 (Fig. 1D). Finally, P4A8 also rescued the inhibitory effect of TWEAK on BMP2-induced production of alkaline phosphatase in the murine C2C12 model of osteoblastogenesis, whereas PDL192, which does not recognize murine Fn14, showed no effect (Fig. 1E). To evaluate the disparate inhibitory effect of the two anti-Fn14 antibodies on the interaction of TWEAK with murine and human Fn14 directly, we performed competition binding experiments with GpL-TNC-Flag-TWEAK and C2C12 and HT29 cells (Fig. 1F). Although P4A8 inhibited binding of the GpL-TWEAK fusion protein efficiently, there was practically no competition between the TWEAK variant and PDL192 for Fn14 binding (Fig. 1F).
In all functional assays described above, Fn14 was activated by help of Fc-Flag-TWEAK a highly active recombinant hexameric soluble variant of TWEAK eliciting the same cellular responses as membrane-bound TWEAK (7). Next, we tested whether P4A8 has also the capability to block the interaction of Fn14 with membrane-bound TWEAK. We thus evaluated the inhibitory effect of PDL192 and P4A8 in a coculture model where Hek293 cells transiently transfected with a non-cleavable mutant of membrane TWEAK (Fig. 2A) were used to stimulate IL8 production in HT1080 cells. In this type of assay, P4A8 showed again a strong inhibitory effect already at comparable low concentrations, whereas PDL192 again elicited at best a very moderate effect at the maximal used concentration of 10 μg/ml (Fig. 2B).
FIGURE 2.

Hek293 cells were electroporated with an expression vector encoding a non-cleavable membrane TWEAK mutant or empty vector and analyzed the next day for cell surface expression of TWEAK. A, transfectants were cocultivated with HT1080 cells in the presence and absence of the indicated concentration of PDL192 and P4A8. After overnight incubation, IL8 in the cell culture supernatants was determined by ELISA. B, an irrelevant human IgG1 antibody was included as an additional negative control.
Fn14-specific Antibodies Elicit Strong Agonistic Activity upon Oligomerization with Protein G
It has been reported for other antibodies recognizing the TNF receptor superfamily members CD95 and TRAILR2 that oligomerization can strongly enhance their agonistic activity (30–33). We therefore wondered whether PDL192 and P4A8 possess a latently present agonistic activity that can be unleashed by oligomerization. Thus, we oligomerized the two anti-Fn14 mAbs with protein G and reinvestigated their effects on Fn14-mediated induction of IL8 and cell death (Fig. 3, A and B). It turned out that protein G-mediated oligomerization converted both anti-Fn14 mAbs in highly active agonists inducing cell death in SKOV-3 cells at low concentrations and triggering a strong IL8 response in WiDr and HT29 cells (Fig. 3, A and B). In accordance with the crucial role of classical NFκB signaling for induction of IL8 gene transcription, we also observed nuclear translocation of the p65/RelA NFκB subunit, a hallmark of activation of the classical NFκB pathway (34), only with the oligomerized anti-Fn14s (Fig. 3C). Antibody concentrations up to 10 μg/ml failed to elicit significant IL8 production or cell death induction in the absence of protein G. Several other Fn14-specifc mAbs that we have not investigated here in detail also elicited agonistic activity upon protein G oligomerization (data not shown). This suggests that the sole aggregation of Fn14 molecules commonly results in receptor activation without special requirements with respect to spatial organization and receptor topology.
FIGURE 3.

Anti-Fn14 antibodies strongly trigger Fn14-mediated cellular responses upon oligomerization with protein G. A and B, HT29 and WiDr cells (A) as well as SKOV-3 cells (B) were challenged in triplicates in 96-well plates with the indicated concentrations of PDL192 and P4A8 in the presence and absence of 1 μg/ml protein G (prot.G) to analyze induction of IL8 (A) and cell death induction (B). To minimize the background caused by constitutive IL8 production, cell culture medium of HT29 and WiDr cells was changed prior stimulation. IL8 content of supernatants was determined by ELISA, and cellular viability was measured by crystal violet staining. C, HT1080 cells were grown on glass coverslips and were stimulated the next day with the two anti-Fn14 antibodies (1 μg/ml) in the presence and absence of protein G (1 μg/ml) or with Flag-TWEAK (200 ng/ml) or Fc-Flag-TWEAK (200 ng/ml) and Flag-TNF (50 ng/ml) as a control for 3 h. Cells were then stained with an antibody recognizing p65. Shown are representative images (left panel) and the ratio of nuclear to cytoplasmic fluorescence intensity (right panel). To calculate the ratio of nuclear to cytoplasmic fluorescence intensity, 30 cells of each group were measured.
The Latently Present Agonistic Activity of Fn14-specific IgG1 Antibodies Is Efficiently Unleashed by Binding to Fcγ Receptors
With respect to the use of antibodies specific for members of the TNF receptor family for therapeutic applications, three major effects can be anticipated: first, receptor activation; second, inhibition of receptor activities by blockade of ligand binding; and third, stimulation of antibody-dependent immune effector mechanisms by the Fc domain of the TNF receptor-targeted antibody, which may lead to the destruction of the TNF receptor-expressing cells. The latter requires binding of antigen-bound antibodies by Fcγ receptors expressed on immune effector cells such as monocytes, macrophages, and NK cells. As oligomerization of antibodies with protein G (or secondary antibodies) is typically considered as a mean that mimics binding to Fcγ receptors, we evaluated whether binding of the various anti-Fn14 mAbs to the Fcγ receptors CD16, CD32, CD32W, and CD64 uncovers their agonistic activity observed in the protein G oligomerization experiments. For this purpose, Hek293 cells transiently expressing the aforementioned Fcγ receptors and mock-transfected control cells (Fig. 4, A and B) were cocultivated with WiDr or HT29 cells that produce high amounts of IL8 upon Fn14 stimulation, and then the cocultures were supplemented with PDL192 and P4A8. Interaction with all four types of Fcγ receptors enabled the two anti-Fn14 mAbs to trigger a strong IL8 response with ED50 values below concentrations of 100 ng/ml, whereas there was no IL8 induction with a corresponding isotype control antibody (Fig. 4, A and B). Thus, binding of anti-Fn14 IgG1 to Fcγ receptors is fully sufficient to convert these molecules in highly active agonists for Fn14 even for P4A8, which is otherwise a potent antagonist.
FIGURE 4.

Anti-Fn14 antibodies strongly trigger IL8 production upon binding to cellular Fc receptors. A and B, Hek293 cells were electroporated with expression vectors encoding CD16, CD32A, CD32B, and CD64 or empty vector and analyzed the next day for expression of the various Fc receptors. Transfectants were then cocultivated with HT29 (A) or WiDr cells (B) in the presence and absence of the indicated concentrations of PDL192 and P4A8, and after overnight incubation, IL8 in the cell culture supernatants was determined by ELISA. An irrelevant human IgG1 antibody was again included as an additional negative control.
Anti-Fn14 mAbs Act as Agonists of the Alternative NFκB Pathway without Oligomerization or FcγR Binding
Two pathways that are relevant for the majority of Fn14-mediated cellular effects are the classical and the alternative NFκB pathway. These two pathways are activated with different efficacy by membrane-bound TWEAK and soluble TWEAK trimers (7). Although the alternative NFκB pathway is strongly stimulated by both forms of TWEAK, membrane-bound TWEAK is superior to soluble TWEAK in activation of the classical NFκB pathway (7). In line with this, artificial attachment of trimeric TWEAK to cells lowers the ED50 value of IL8 induction several hundredfold but has no effect on the dose response dependence of p100 processing, the biochemical hallmark of alternative NFκB signaling (7). Noteworthy, hexameric soluble TWEAK, as used in the experiments shown in Fig. 1, and oligomerized TWEAK trimers are also highly superior to soluble TWEAK trimers with respect to classical NFκB signaling, but all three TWEAK species are comparable active with respect to p100 processing (7). Complexes of TRAF2 and cIAP1/2 inhibit the alternative NFκB pathway upstream of NIK accumulation and p100 processing and also prevent TNFR1-induced apoptosis upon recruitment to this receptor (35, 36). In accordance, it has been found that Fn14-induced enhancement of TNFR1-induced cell death, similar to alternative NFκB signaling, is efficiently triggered by all forms of TWEAK (6). In view of the results shown above, it therefore appears that oligomerized and Fcγ receptor-bound anti-Fn14 mAbs resemble membrane-bound TWEAK and potently trigger the complete set of Fn14-associated signaling pathways. P4A8 furthermore act otherwise as an antagonist of soluble and membrane-bound TWEAK (Fig. 1). For example, it blocked Fn14-mediated enhancement of TNFR1-induced cell death upon stimulation with the membrane-TWEAK mimicking Fc-TWEAK construct (Fig. 1D) but also when trimeric Flag-TWEAK was used for Fn14 stimulation (Fig. 5A). Biochemical and microscopical analysis of alternative NFκB signaling revealed, however, a more complex situation. Although P4A8 inhibited enhancement of TNF-induced cell death by trimeric soluble TWEAK efficiently, it nevertheless significantly triggered p100 processing and nuclear translocation of p52 at low to moderate concentrations (>1 μg/ml; Fig. 5, B and D). In view of the fact that soluble TWEAK trimers stimulate both p100 processing and enhancement of TNF-induced apoptosis by depletion of cytosolic TRAF2-cIAP1/2 complexes, this observation is surprising (Fig. 5, B–D). Moreover, PDL192 also triggered the alternative NFκB pathway efficiently without showing enhancement of TNFR1-induced cell death (Fig. 5, B–D). Interestingly, in accordance with the capability of the oligomerized anti-Fn14s to stimulate the classical NFκB pathway, there was an increase in the total amount of p100 and p52 in the cells stimulated with the oligomerized antibodies (Fig. 5B). A possible explanation of this at the first glance contradictory situation appears when NIK accumulation was analyzed. Stimulation of p100 processing by PDL192 and P4A8 was almost as strong as by Flag-TWEAK. The latter, however, triggered a much higher NIK accumulation (Fig. 5B). As NIK accumulation is the direct consequence of depletion of TRAF2-cIAP1/2 complexes, this observation suggests that the anti-Fn14 antibodies are far less active in TRAF2-cIAP1/2 depletion than Flag-TWEAK trimers. A reduced capability of PDL192 and P4A8 would also explain the fact that the anti-Fn14 antibodies, in contrast to Flag-TWEAK, failed to enhance TNF-induced cell death. We therefore analyzed the impact of the various Fn14 targeting reagents on TRAF2 and cIAP1. We and others previously showed that TRAF2 translocates to a Fn14/membrane-associated Triton X-100-insoluble compartment in response to TWEAK stimulation (6, 37). In confirmation of these earlier results, we observed that both TWEAK variants (soluble TWEAK and the membrane TWEAK-mimicking Fc-TWEAK) as well as the oligomerized anti-Fn14s triggered significant depletion of TRAF2 and cIAP1 from the Triton X-100 soluble fraction (Fig. 5E). However, there was no significant reduction in TRAF2 and cIAP1 levels in the Triton X-100-soluble fraction of cells challenged with non-oligomerized P4A8 and PDL192 despite robust p100 processing (Fig. 5E). The fact that minor depletion of TRAF2-cIAP1/2 and poor NIK accumulation are already sufficient to trigger considerable p100 processing without sensitizing for TNFR1-induced cell death might thus most likely reflect different thresholds of TRAF2-cIAP1/2 for these two responses. Indeed, this idea corresponds well to the molecular mode of action of the two responses. In the case of p100 processing, the moderate effect on NIK expression is potentiated as it acts catalytically and can thus activate a huge number of IKK1 molecules that in turn can trigger the processing of multiple p100 molecules. In contrast, in context of the TRAF2-cIAP1/2 complex-mediated cross-talk of Fn14 and TNFR1, there are no such amplification mechanisms, and the Fn14-induced depletion of the cytosolic TRAF2-cIAP1/2 complexes must thus be strong enough to push their concentration below a critical limit.
FIGURE 5.

Anti-Fn14 antibodies efficiently trigger p100 processing at low concentrations without protein G oligomerization or binding to Fc receptors. A, HT29 cells were pretreated with increasing concentrations of the two anti-Fn14 antibodies and then challenged overnight with 100 ng/ml Flag-TWEAK to sensitize cells for TNF-induced apoptosis. On the next day, cells were challenged with TNF and cycloheximide, and after an additional overnight incubation, cellular viability was determined by crystal violet staining. B, HT29 cells were stimulated with increasing amounts of anti-Fn14 antibodies overnight with and without oligomerization by protein G (1 μg/ml) as well as with the indicated concentrations of Flag-TWEAK and Fc-Flag-TWEAK. Total cell lysates were analyzed by Western blotting with respect to the indicated proteins. C, HT29 cells were treated for 6 h in triplicates with 1 μg/ml of PDL192 and P4A8 with and without protein G (prot.-G) oligomerization and with Flag-TWEAK and Fc-Flag-TWEAK (200 ng/ml). Cells were then stimulated with TNF and cycloheximide (2.5 μg/ml) to trigger TNFR1-induced cell death. After overnight incubation, cellular viability was again determined by crystal violet staining. The dose response data for TNF alone was indicated as a dotted line in all panels for better comparison. D, HT29 cells were grown on glass coverslips and were stimulated the next day with the two anti-Fn14 antibodies (1 μg/ml) in the presence and absence of protein G (1 μg/ml)or with Flag-TWEAK (200 ng/ml) or Fc-Flag-TWEAK (200 ng/ml) overnight. Cells were then stained with an antibody recognizing p100/p52. Shown are representative images (left panel) and the ratio of nuclear to cytoplasmic fluorescence intensity (right panel). To calculate the ratio of nuclear to cytoplasmic fluorescence intensity, 30 cells of each group were measured. E, HT29 and WiDr cells were challenged with Flag-TWEAK (200 ng/ml), Fc-Flag-TWEAK (200 ng/ml), and the anti-Fn14 antibodies (2 μg/ml) with and without oligomerization by protein G (0.5 μg/ml) overnight. Triton X-100 lysates were prepared and analyzed by Western blotting with respect to the indicated proteins.
DISCUSSION
We investigated here in detail the effects of the two Fn14-specific antibodies PDL192 and P4A8 on Fn14 activity. Both antibodies showed no agonistic activity with respect to production of IL8, cell death induction, and enhancement of TNFR1-induced cell death up to concentrations of 10 μg/ml. Although PDL192 poorly affected ligand binding and only at high concentrations, P4A8 inhibited TWEAK binding and thus blocked the aforementioned Fn14-mediated responses (Figs. 1 and 2). Both anti-Fn14 mAbs, however, triggered the aforementioned Fn14-mediated effects upon oligomerization with protein G or binding to Fcγ receptors (Figs. 3 and 4).
Naturally, Fn14 is activated by membrane TWEAK or a soluble form derived thereof by proteolytic processing. Notably, binding of the two forms of TWEAK trigger qualitatively distinct states of Fn14 activity. Soluble TWEAK stimulates the alternative NFκB pathway and enhances TNFR1-induced cell death but only weakly triggers signaling via the classical NFκB pathway. In contrast, membrane TWEAK is a potent inducer of all of these pathways (6, 7). Thus, the fact that oligomerized or FcγR-bound anti-Fn14 mAbs are quite effective in the stimulation of all Fn14 responses investigated, including activation of the classical NFκB pathway, indicates that they mimic the activity of membrane TWEAK. Without oligomerization or FcγR binding, however, the effects of PDL192 and P4A8 on Fn14 activity are quite complex. On the one hand, we observed that both anti-Fn14 antibodies efficiently trigger p100 processing and nuclear translocation of p52, hallmarks of the alternative NFκB pathway (Fig. 5, B and D). Thus, in this respect, PDL192 and P4A8 resemble soluble TWEAK. On the other hand, in contrast to soluble TWEAK, both anti-Fn14 mAbs showed no effect on TNFR1-induced cell death and in the case of P4A8, this response of soluble TWEAK was actually blocked (Figs. 1D and 5A). In conclusion, this means that binding of antibodies in the absence of oligomerization or FcγR binding induces in a state of Fn14 activity distinct from those triggered by soluble and membrane TWEAK. For blocking antibodies such as PDL192, this has the quite unusual and paradoxical effect that an antibody concomitantly displays agonistic and antagonistic activities.
The unexpected and unprecedented pathway-specific agonistic activity of an otherwise antagonistic antibody that we observed for P4A8 as well as the “agonism constituting” effect of FcγR binding are not only of academic interest but of considerable relevance with respect to potential clinical applications of Fn14-specific antibodies. The TWEAK/Fn14 system is currently under clinical and preclinical investigation as a therapeutic target in tumor therapy and autoimmune diseases but also in various injuries related to traumatic tissue damage (http://clinicaltrials.gov) (1, 38). In the case of tumor therapy with anti-Fn14 mAbs, FcγR binding is typically desirable to elicit an antibody-dependent cellular cytotoxicity-related antitumoral effect. Now, our results suggest that for antibody-dependent cellular cytotoxicity resistance, this might lead to Fn14 activation and stimulation of potentially protumoral Fn14-induced effects even then when the antibody used for treatment act as an antagonist in in vitro assays. This also applies to most of the remaining potential applications of anti-Fn14 mAbs where inhibition of TWEAK/Fn14-induced signaling is aimed. The introduction of mutations or modifications in the Fc part of Fn14 antibodies that prevent their binding to Fcγ receptors could offer a solution for this problem but only as long as the anticipated therapeutic effect does not require inhibition of Fn14-induced activation of the alternative NFκB pathway. However, in situations where Fn14-driven activation of the alternative NFκB pathway contributes to the disease, this would be no solution as the alternative NFκB pathway-specific agonistic activity of otherwise antagonistic Fn14 antibodies remains unaffected and could actually exacerbate the disease. In this case, only TWEAK-neutralizing reagents and monomeric Fn14 antagonists would be useful.
TWEAK is a trimer that interacts with three Fn14 molecules, whereas the IgG1 antibodies used in this study have only two Fn14 binding sides. This suggests that the alternative NFκB pathway is unique among the Fn14-associated signaling pathways as it is already highly responsive to Fn14 dimerization allowing antibodies to trigger an unusual response pattern distinct from that of the naturally occurring Fn14 ligands. Based on the established trimeric structure of TRAF2 (39, 40) and what is known about the role of TRAF2 and cIAPs in NFκB signaling (34, 36), we propose the following model (Fig. 6) of antibody, soluble TWEAK, and membrane TWEAK-induced Fn14 activation.
FIGURE 6.
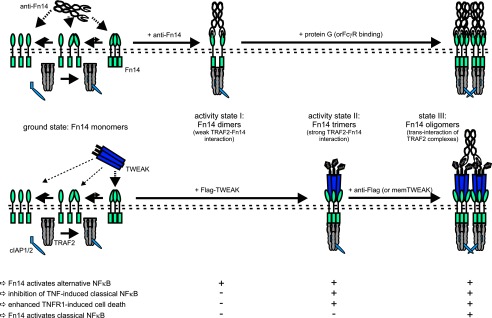
Model of distinct states of Fn14 activity induced by Fn14-specific antibodies and soluble and membrane-bound TWEAK.
In non-stimulated cells, Fn14 predominately occurs as monomers that do not interact with TRAF2-cIAP complexes or only undergo short-lived instable interactions with one of the three TRAF2 protomers contained in a TRAF2-cIAP complex. In any case, there is no relevant activation of Fn14-related signaling pathways (Fig. 6, ground state). If bivalent Fn14-specific antibodies are applied, Fn14 become trapped in complexes containing two adjacent Fn14 molecules (αFn14-Fn142). Now, two Fn14 molecules of such a complex concomitantly interact with two of the three protomers of a TRAF2 trimer, resulting in enhanced but still transient and submaximal interaction between Fn14 molecules and TRAF2-cIAP complexes. The Fn14 interaction with TRAF2-cIAP complexes is now strong enough to result in a minor depletion of cytosolic TRAF2-cIAP complexes or the displacement of the latter from NIK degradation-triggering TRAF2-cIAP-TRAF3-NIK complexes. The resulting very weak accumulation of NIK, however, is relevant enough to trigger p100 processing because this involves two catalytic amplification steps, NIK-mediated phosphorylation and activation of IKK1 and phosphorylation of p100 by the latter resulting in p100 processing (Fig. 6, activity state I). TRAF2-cIAP complexes also inhibit caspase-8 activation in context of TNFR1 signaling and thus protects against TNF-induced apoptosis (41). Here, the TRAF2-cIAP complexes act in a stoichiometric manner without amplificatory mechanisms. Thus, the minor reduction in the availability of TRAF2-cIAP complexes related to the weak depletion caused by antibody stimulation of anti-Fn14 is too weak to get apparent. With respect to the activation of the classical NFκB pathway, trans-interaction of two IAPs seems to be necessary. As a TRAF2 trimer interacts with only a single cIAP molecule (42), this cannot occur in the anti-Fn14-induced complexes of two Fn14 molecules with a single TRAF2 trimer.
For soluble TWEAK, a ligand trimer interacts with three Fn14 molecules (TWEAK-Fn143) resulting in a complex that can undergo concomitant interaction all protomers of a TRAF2 trimer resulting in stronger durable recruitment of TRAF2-cIAP complexes than for the anti-Fn14 induced receptor dimers. As a consequence, there is significant depletion of cytosolic TRAF2-cIAP complexes. Now, the latter is strong enough to lead to enhancement of TNF-induced apoptosis and also to much stronger NIK accumulation (Fig. 6, activity state II). Due to the two amplificatory catalytic steps connecting NIK accumulation and p100 processing, however, the alternative pathway was already maximally triggered by low NIK levels so that the further increase in NIK accumulation has no chance to translate in a corresponding enhanced p100 processing activity. There is still no or only weak classical NFκB signaling as there is still no trans-interaction of two TRAF2-IAP complexes due to the lack of secondary interaction of two TWEAK-Fn143 complexes.
For stimulation of membrane TWEAK, oligomerized soluble TWEAK trimers or hexameric Fc-TWEAK, the TWEAK-Fn143 complexes become oligomerized resulting in close neighborhood of two or more TRAF2-cIAP complexes and trans-activation of two IAPs and thus additional activation of the classical NFκB pathway (Fig. 6, activity state III). Likewise, protein G oligomerization and FcγR binding lead to supramolecular clustering of several αFn14-Fn142 complexes, resulting in tighter binding of TRAF2-cIAP complexes and again in transactivation of the IAP component of these complexes and stimulation of the classical NFκB pathway (Fig. 6, activity state III).
This work was supported by the Deutsche Forschungsgemeinschaft (DFG Wa 1025/19-2, SFB 487, TP B7), Deutsche Krebshilfe (Project 109922), and Argen-X BVBA. H.W. is a consultant of Argen-X BVBA. Because of a Research Services Agreement between the University Hospital Würzburg and Argen-X BVBA, antibodies not described in this publication have been analyzed for Argen-X BVBA.
- TWEAK
- TNF-like weak inducer of apoptosis
- cIAP1/2
- cellular inhibitor of apoptosis 1/2
- PE
- phycoerythrin
- Fcγ receptor
- FcγR
- Fn14
- fibroblast growth factor-inducible 14
- NIK
- NFκB-inducing kinase
- TRAF2
- TNF receptor associated factor 2
- TNFR
- TNF receptor.
REFERENCES
- 1. Winkles J. A. (2008) The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat. Rev. Drug Discov. 7, 411–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kawakita T., Shiraki K., Yamanaka Y., Yamaguchi Y., Saitou Y., Enokimura N., Yamamoto N., Okano H., Sugimoto K., Murata K., Nakano T. (2004) Functional expression of TWEAK in human hepatocellular carcinoma: possible implication in cell proliferation and tumor angiogenesis. Biochem. Biophys. Res. Commun. 318, 726–733 [DOI] [PubMed] [Google Scholar]
- 3. Willis A. L., Tran N. L., Chatigny J. M., Charlton N., Vu H., Brown S. A., Black M. A., McDonough W. S., Fortin S. P., Niska J. R., Winkles J. A., Cunliffe H. E. (2008) The fibroblast growth factor-inducible 14 receptor is highly expressed in HER2-positive breast tumors and regulates breast cancer cell invasive capacity. Mol. Cancer Res. 6, 725–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ikner A., Ashkenazi A. (2011) TWEAK induces apoptosis through a death-signaling complex comprising receptor-interacting protein 1 (RIP1), Fas-associated death domain (FADD), and caspase-8. J. Biol. Chem. 286, 21546–21554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schneider P., Schwenzer R., Haas E., Mühlenbeck F., Schubert G., Scheurich P., Tschopp J., Wajant H. (1999) TWEAK can induce cell death via endogenous TNF and TNF receptor 1. Eur. J. Immunol. 29, 1785–1792 [DOI] [PubMed] [Google Scholar]
- 6. Wicovsky A., Salzmann S., Roos C., Ehrenschwender M., Rosenthal T., Siegmund D., Henkler F., Gohlke F., Kneitz C., Wajant H. (2009) TNF-like weak inducer of apoptosis inhibits proinflammatory TNF receptor-1 signaling. Cell Death Differ. 16, 1445–1459 [DOI] [PubMed] [Google Scholar]
- 7. Roos C., Wicovsky A., Müller N., Salzmann S., Rosenthal T., Kalthoff H., Trauzold A., Seher A., Henkler F., Kneitz C., Wajant H. (2010) Soluble and transmembrane TNF-like weak inducer of apoptosis differentially activate the classical and noncanonical NF-κB pathway. J. Immunol. 185, 1593–1605 [DOI] [PubMed] [Google Scholar]
- 8. Meighan-Mantha R. L., Hsu D. K., Guo Y., Brown S. A., Feng S. L., Peifley K. A., Alberts G. F., Copeland N. G., Gilbert D. J., Jenkins N. A., Richards C. M., Winkles J. A. (1999) The mitogen-inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J. Biol. Chem. 274, 33166–33176 [DOI] [PubMed] [Google Scholar]
- 9. Kamata K., Kamijo S., Nakajima A., Koyanagi A., Kurosawa H., Yagita H., Okumura K. (2006) Involvement of TNF-like weak inducer of apoptosis in the pathogenesis of collagen-induced arthritis. J. Immunol. 177, 6433–6439 [DOI] [PubMed] [Google Scholar]
- 10. Perper S. J., Browning B., Burkly L. C., Weng S., Gao C., Giza K., Su L., Tarilonte L., Crowell T., Rajman L., Runkel L., Scott M., Atkins G. J., Findlay D. M., Zheng T. S., Hess H. (2006) TWEAK is a novel arthritogenic mediator. J. Immunol. 177, 2610–2620 [DOI] [PubMed] [Google Scholar]
- 11. Desplat-Jégo S., Creidy R., Varriale S., Allaire N., Luo Y., Bernard D., Hahm K., Burkly L., Boucraut J. (2005) Anti-TWEAK monoclonal antibodies reduce immune cell infiltration in the central nervous system and severity of experimental autoimmune encephalomyelitis. Clin. Immunol. 117, 15–23 [DOI] [PubMed] [Google Scholar]
- 12. Desplat-Jégo S., Varriale S., Creidy R., Terra R., Bernard D., Khrestchatisky M., Izui S., Chicheportiche Y., Boucraut J. (2002) TWEAK is expressed by glial cells, induces astrocyte proliferation and increases EAE severity. J. Neuroimmunol. 133, 116–123 [DOI] [PubMed] [Google Scholar]
- 13. Mueller A. M., Pedré X., Kleiter I., Hornberg M., Steinbrecher A., Giegerich G. (2005) Targeting fibroblast growth factor-inducible-14 signaling protects from chronic relapsing experimental autoimmune encephalomyelitis. J. Neuroimmunol. 159, 55–65 [DOI] [PubMed] [Google Scholar]
- 14. Dohi T., Borodovsky A., Wu P., Shearstone J. R., Kawashima R., Runkel L., Rajman L., Dong X., Scott M. L., Michaelson J. S., Jakubowski A., Burkly L. C. (2009) TWEAK/Fn14 pathway: a nonredundant role in intestinal damage in mice through a TWEAK/intestinal epithelial cell axis. Gastroenterology 136, 912–923 [DOI] [PubMed] [Google Scholar]
- 15. Kawashima R., Kawamura Y. I., Oshio T., Son A., Yamazaki M., Hagiwara T., Okada T., Inagaki-Ohara K., Wu P., Szak S., Kawamura Y. J., Konishi F., Miyake O., Yano H., Saito Y., Burkly L. C., Dohi T. (2011) Interleukin-13 damages intestinal mucosa via TWEAK and Fn14 in mice-a pathway associated with ulcerative colitis. Gastroenterology 141, 2119–2129.e8 [DOI] [PubMed] [Google Scholar]
- 16. Zhao Z., Burkly L. C., Campbell S., Schwartz N., Molano A., Choudhury A., Eisenberg R. A., Michaelson J. S., Putterman C. (2007) TWEAK/Fn14 interactions are instrumental in the pathogenesis of nephritis in the chronic graft-versus-host model of systemic lupus erythematosus. J. Immunol. 179, 7949–7958 [DOI] [PubMed] [Google Scholar]
- 17. Muñoz-García B., Moreno J. A., López-Franco O., Sanz A. B., Martín-Ventura J. L., Blanco J., Jakubowski A., Burkly L. C., Ortiz A., Egido J., Blanco-Colio L. M. (2009) Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) enhances vascular and renal damage induced by hyperlipidemic diet in ApoE-knockout mice. Arterioscler. Thromb. Vasc. Biol. 29, 2061–2068 [DOI] [PubMed] [Google Scholar]
- 18. Schapira K., Burkly L. C., Zheng T. S., Wu P., Groeneweg M., Rousch M., Kockx M. M., Daemen M. J., Heeneman S. (2009) Fn14-Fc fusion protein regulates atherosclerosis in ApoE-/- mice and inhibits macrophage lipid uptake in vitro. Arterioscler. Thromb. Vasc. Biol. 29, 2021–2027 [DOI] [PubMed] [Google Scholar]
- 19. Hotta K., Sho M., Yamato I., Shimada K., Harada H., Akahori T., Nakamura S., Konishi N., Yagita H., Nonomura K., Nakajima Y. (2011) Direct targeting of fibroblast growth factor-inducible 14 protein protects against renal ischemia reperfusion injury. Kidney Int. 79, 179–188 [DOI] [PubMed] [Google Scholar]
- 20. Jakubowski A., Ambrose C., Parr M., Lincecum J. M., Wang M. Z., Zheng T. S., Browning B., Michaelson J. S., Baetscher M., Baestcher M., Wang B., Bissell D. M., Burkly L. C. (2005) TWEAK induces liver progenitor cell proliferation. J. Clin. Invest. 115, 2330–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mittal A., Bhatnagar S., Kumar A., Lach-Trifilieff E., Wauters S., Li H., Makonchuk D. Y., Glass D. J. (2010) The TWEAK-Fn14 system is a critical regulator of denervation-induced skeletal muscle atrophy in mice. J. Cell Biol. 188, 833–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Potrovita I., Zhang W., Burkly L., Hahm K., Lincecum J., Wang M. Z., Maurer M. H., Rossner M., Schneider A., Schwaninger M. (2004) Tumor necrosis factor-like weak inducer of apoptosis-induced neurodegeneration. J. Neurosci. 24, 8237–8244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yepes M., Brown S. A., Moore E. G., Smith E. P., Lawrence D. A., Winkles J. A. (2005) A soluble Fn14-Fc decoy receptor reduces infarct volume in a murine model of cerebral ischemia. Am. J. Pathol. 166, 511–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Michaelson J. S., Burkly L. C. (2009) Therapeutic targeting of TWEAK/Fnl4 in cancer: exploiting the intrinsic tumor cell killing capacity of the pathway. Results Probl. Cell Differ. 49, 145–160 [DOI] [PubMed] [Google Scholar]
- 25. Culp P. A., Choi D., Zhang Y., Yin J., Seto P., Ybarra S. E., Su M., Sho M., Steinle R., Wong M. H., Evangelista F., Grove J., Cardenas M., James M., Hsi E. D., Chao D. T., Powers D. B., Ramakrishnan V., Dubridge R. (2010) Antibodies to TWEAK receptor inhibit human tumor growth through dual mechanisms. Clin. Cancer Res. 16, 497–508 [DOI] [PubMed] [Google Scholar]
- 26. Michaelson J. S., Amatucci A., Kelly R., Su L., Garber E., Day E. S., Berquist L., Cho S., Li Y., Parr M., Wille L., Schneider P., Wortham K., Burkly L. C., Hsu Y. M., Joseph I. B. (2011) Development of an Fn14 agonistic antibody as an anti-tumor agent. MAbs 3, 362–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tran N. L., McDonough W. S., Savitch B. A., Fortin S. P., Winkles J. A., Symons M., Nakada M., Cunliffe H. E., Hostetter G., Hoelzinger D. B., Rennert J. L., Michaelson J. S., Burkly L. C., Lipinski C. A., Loftus J. C., Mariani L., Berens M. E. (2006) Increased fibroblast growth factor-inducible 14 expression levels promote glioma cell invasion via Rac1 and nuclear factor-κB and correlate with poor patient outcome. Cancer Res. 66, 9535–9542 [DOI] [PubMed] [Google Scholar]
- 28. Culp P. (2009) Therapeutic use of anti-TWEAK antibodies. WO2009020933A2 [Google Scholar]
- 29. Garber E., Burkly L., Michealson J., Lugovskoy A., Hsu Y.-M., Hanf K. (2009) Anti-Fn14 antibodies and uses thereof. WO2009140177A2 [Google Scholar]
- 30. Adams C., Totpal K., Lawrence D., Marsters S., Pitti R., Yee S., Ross S., Deforge L., Koeppen H., Sagolla M., Compaan D., Lowman H., Hymowitz S., Ashkenazi A. (2008) Structural and functional analysis of the interaction between the agonistic monoclonal antibody Apomab and the proapoptotic receptor DR5. Cell Death Differ. 15, 751–761 [DOI] [PubMed] [Google Scholar]
- 31. Dhein J., Daniel P. T., Trauth B. C., Oehm A., Möller P., Krammer P. H. (1992) Induction of apoptosis by monoclonal antibody anti-APO-1 class switch variants is dependent on cross-linking of APO-1 cell surface antigens. J. Immunol. 149, 3166–3173 [PubMed] [Google Scholar]
- 32. Natoni A., MacFarlane M., Inoue S., Walewska R., Majid A., Knee D., Stover D. R., Dyer M. J., Cohen G. M. (2007) TRAIL signals to apoptosis in chronic lymphocytic leukaemia cells primarily through TRAIL-R1 whereas cross-linked agonistic TRAIL-R2 antibodies facilitate signalling via TRAIL-R2. Br. J. Haematol. 139, 568–577 [DOI] [PubMed] [Google Scholar]
- 33. Wilson N. S., Yang B., Yang A., Loeser S., Marsters S., Lawrence D., Li Y., Pitti R., Totpal K., Yee S., Ross S., Vernes J. M., Lu Y., Adams C., Offringa R., Kelley B., Hymowitz S., Daniel D., Meng G., Ashkenazi A. (2011) An Fcγ receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell 19, 101–113 [DOI] [PubMed] [Google Scholar]
- 34. Kanarek N., Ben-Neriah Y. (2012) Regulation of NF-κB by ubiquitination and degradation of the IκBs. Immunol. Rev. 246, 77–94 [DOI] [PubMed] [Google Scholar]
- 35. O'Donnell M. A., Ting A. T. (2011) RIP1 comes back to life as a cell death regulator in TNFR1 signaling. FEBS J 278, 877–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Razani B., Reichardt A. D., Cheng G. (2011) Non-canonical NF-κB signaling activation and regulation: principles and perspectives. Immunol. Rev. 244, 44–54 [DOI] [PubMed] [Google Scholar]
- 37. Vince J. E., Chau D., Callus B., Wong W. W., Hawkins C. J., Schneider P., McKinlay M., Benetatos C. A., Condon S. M., Chunduru S. K., Yeoh G., Brink R., Vaux D. L., Silke J. (2008) TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFα. J. Cell Biol. 182, 171–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ortiz A., Sanz A. B., Muñoz García B., Moreno J. A., Sánchez Niño M. D., Martín-Ventura J. L., Egido J., Blanco-Colio L. M. (2009) Considering TWEAK as a target for therapy in renal and vascular injury. Cytokine. Growth Factor. Rev. 20, 251–258 [DOI] [PubMed] [Google Scholar]
- 39. Park Y. C., Burkitt V., Villa A. R., Tong L., Wu H. (1999) Structural basis for self-association and receptor recognition of human TRAF2. Nature 398, 533–538 [DOI] [PubMed] [Google Scholar]
- 40. Ye H., Park Y. C., Kreishman M., Kieff E., Wu H. (1999) The structural basis for the recognition of diverse receptor sequences by TRAF2. Mol. Cell 4, 321–330 [DOI] [PubMed] [Google Scholar]
- 41. Wang C. Y., Mayo M. W., Korneluk R. G., Goeddel D. V., Baldwin A. S., Jr. (1998) NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 281, 1680–1683 [DOI] [PubMed] [Google Scholar]
- 42. Zheng C., Kabaleeswaran V., Wang Y., Cheng G., Wu H. (2010) Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: affinity, specificity, and regulation. Mol. Cell 38, 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fick A., Lang I., Schäfer V., Seher A., Trebing J., Weisenberger D., Wajant H. (2012) Studies of binding of tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK) to fibroblast growth factor inducible 14 (Fn14). J. Biol. Chem. 287, 484–495 [DOI] [PMC free article] [PubMed] [Google Scholar]