

Background: CXCL4L1 is a highly potent anti-angiogenic and anti-tumor chemokine, and its structural information is unknown.
Results: CXCL4L1 x-ray structure is determined, and it reveals a previously unrecognized chemokine structure adopting a novel C-terminal helix conformation.
Conclusion: The alternative helix conformation enhances the anti-angiogenic activity of CXCL4L1 by reducing the glycosaminoglycan binding ability.
Significance: Chemokine C-terminal helix orientation is critical in regulating their functions.
Keywords: Angiogenesis, Chemokines, Heparin, Platelets, X-ray Crystallography, CXCL4L1, Platelet Factor 4
Abstract
Chemokines, a subfamily of cytokines, are small, secreted proteins that mediate a variety of biological processes. Various chemokines adopt remarkable conserved tertiary structure comprising an anti-parallel β-sheet core domain followed by a C-terminal helix that packs onto the β-sheet. The conserved structural feature has been considered critical for chemokine function, including binding to cell surface receptor. The recently isolated variant, CXCL4L1, is a homologue of CXCL4 chemokine (or platelet factor 4) with potent anti-angiogenic activity and differed only in three amino acid residues of P58L, K66E, and L67H. In this study we show by x-ray structural determination that CXCL4L1 adopts a previously unrecognized structure at its C terminus. The orientation of the C-terminal helix protrudes into the aqueous space to expose the entire helix. The alternative helix orientation modifies the overall chemokine shape and surface properties. The L67H mutation is mainly responsible for the swing-out effect of the helix, whereas mutations of P58L and K66E only act secondarily. This is the first observation that reports an open conformation of the C-terminal helix in a chemokine. This change leads to a decrease of its glycosaminoglycan binding properties and to an enhancement of its anti-angiogenic and anti-tumor effects. This unique structure is recent in evolution and has allowed CXCL4L1 to gain novel functional properties.
Introduction
Chemokines are multifunctional regulators not only responsible for leukocyte recruitment but also participating in many important biological functions such as homeostasis (1), embryonic development (2), angiogenesis (2–8), and would healing and are also involved in disease (9–13). Based on the amino acid sequence, all chemokines are divided into four families including CXC, CC, C, and CX3C on the basis of their cysteine residues pattern, where C represents cysteine and X represents another residue (14). Most chemokines contain 1–3 intramolecular disulfide bonds. To date, ∼50 chemokines have been identified in human with still half of them with an unknown structure (5, 15, 16). Despite the high degree of variation in the primary structure of the amino acid sequence (14, 17, 18), a surprisingly conserved tertiary structural fold has been recognized based on the existing structures. Available chemokine structures show a highly conserved structural fold that comprises a core domain consisting of a 3-stranded anti-parallel β-sheet (β1-β3) followed by a C-terminal α helix (α1), which packs against the core β-sheet domain. A varying degree of oligomerization has been recognized for these molecules.
CXCL4 (also known as platelet factor 4 (PF4)3) is one major chemokine belonging to the CXC family. CXCL4 secreted from stimulated platelet exhibits a wide range of functions such regulation of hematopoiesis (19) and atherosclerosis (20–22), anti-angiogenesis (22–25), chemotaxis (10, 17), thrombocytopenia (26, 27), anti-microbial activity (28, 29), and inhibition of HIV-1 infection (30, 31). G-protein-coupled receptors and cell surface glycosaminoglycans (GAGs) (14, 32, 33) are implicated in the aforementioned functions. One of the major physiological roles of high affinity binding to heparin appears to be the neutralization of the anticoagulant activities on the endothelial surface of blood vessels, thereby inhibiting local antithrombin III activity and promoting coagulation at sites of vascular injury (23, 34).
CXCL4L1, a CXCL4 variant with three amino acid substitutions at P58L, K66E, and L67H in the C terminus, has recently been identified to be a potent anti-angiogenic chemokine (23, 24, 35) (Fig. 1). Interestingly, CXCL4L1 has only been identified in monkeys, chimpanzees, and humans. Because CXCL4 and CXCL4L1 genes both are localized on chromosome 4, the CXCL4L1 gene arises from recent duplication of the CXCL4 gene (24). The minor difference in their primary structure creates substantial functional differences, including enhanced anti-angiogenic activity, reduced GAG binding, and an increase in the inhibition of endothelial cell migration (24). Compared with CXCL4, CXCL4L1 is also much more effective in inhibiting FGF-2-induced chemotaxis and angiogenesis (23, 24, 36, 37). CXCL4L1 C-terminal portion containing residues 47–70 has been proved to be essential for significant anti-angiogenic and anti-tumor activity, whereas the peptide derived from the same region of CXCL4 demonstrated less effect (23, 38).
FIGURE 1.
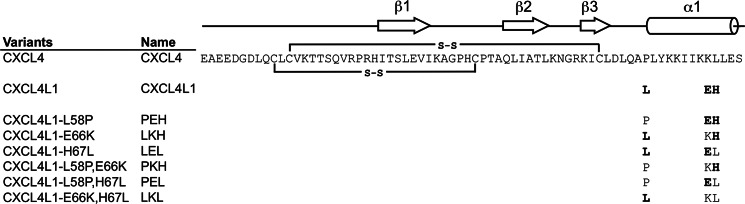
Schematic representation of the amino acid sequences of CXCL4, CXCL4L1, and the variants. The substituted residues in the mutants are highlighted in bold. The secondary structural elements determined by x-ray crystallography are indicated at the top of the sequence. Two connecting lines between Cys residues represent the disulfide connectivities of Cys-10 to Cys-36 and Cys-12 to Cys-52.
The three-dimensional structure of CXCL4 molecule (PDB codes 1RHP and 1PFM) has previously been determined by x-ray diffraction method and shown as an asymmetric tetramer (39, 40). Although the functional relevance of CXCL4 and CXCL4L1 has been under investigation for several years, structural information for CXCL4L1 is completely missing. It was suggested that the structural modifications of the C-terminal helix in CXCL4L1 are involved and that the L67H mutation plays a determinant role in mediating the functional differences between CXCL4L1 and CXCL4 (24). Solving the high resolution structure of recombinant CXCL4L1 is expected to provide the structural basis to explain these functional differences.
Here, we report the crystal structure of CXCL4L1, which consists of a β-sheet core domain very similar to other chemokines but exhibits major differences in the C terminus. Remarkably, the C-terminal helix adopts an entirely different orientation than any known chemokine and, notably, does not pack against the β-sheet core domain. This is the first observation for a chemokine in which the C-terminal helix is found in an open conformation. This modification is controlled by very few amino acid residues. Our finding provides the basis for the low binding affinity of CXCL4L1 to glycosaminoglycans and for the differences observed in biological activity.
EXPERIMENTAL PROCEDURES
Construction of CXCL4 and CXCL4L1 Plasmids
Human CXCL4 (NM_002619.3) and human CXCL4L1 (NM_002620.2) cDNA fragments were amplified by PCR followed by cloning the PCR products into pET-43.1a vector using NdeI and HindIII restriction sites. The resulting proteins contain an N-terminal methionine residue derived from the vector. The mutants used in the study were introduced by using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA), and pET-43.1a-CXCL4 or pET-43.1a-CXCL4L1 was used as the template. All constructed plasmids were verified by DNA sequencing.
Protein Expression and Purification
Escherichia coli BL21(DE3) cells harboring a given plasmid were cultured in LB media or isotopically labeled minimal media with 100 μg/ml ampicillin at 37 °C. Cells were cultured until an A600 of 0.6–0.8 was reached. Cells were subsequently induced by the addition of 1 mm isopropyl-thio-β-galactopyranoside. After another 4 h of induction, we harvested the cells by centrifugation (6000 × g, 20 min, 4 °C) and stored the cells at −20 °C. The harvested cells were thawed on ice, resuspended in lysis buffer containing 50 mm Tris-HCl, pH 7.4, 100 mm NaCl with 0.5 mm freshly added phenylmethylsulfonyl fluoride and lysed by sonication. The cell lysate was separated into supernatant and pellet by high speed centrifugation (30,000 × g, 30 min, 4 °C).
Recombinant CXCL4 was isolated from the supernatant of cell lysate by affinity purification. Lysate supernatant was applied to a heparin high performance affinity column (ÄKTA FPLC system, GE Healthcare) equilibrated with 50 mm Tris-HCl, pH 7.4, 500 mm NaCl. The bound CXCL4 were eluted with a NaCl gradient (0.5–2 m). Fractions containing CXCL4 were pooled and concentrated for further purification using a Superdex 75 HR 10/300 column (ÄKTA FPLC system) in 50 mm sodium phosphate buffer, pH 7.4, 150 mm NaCl. The eluted CXCL4 was further purified to homogeneity by HPLC system (Beckman Coulter Inc.) using a C18 reverse phase column (Phenomenex Inc.). The purified CXCL4 was lyophilized and kept frozen at −80 °C. The same procedure was applied to the variant of PEL that was overexpressed in cell supernatant.
Recombinant CXCL4L1 was refolded from the pellet of cell lysate because a large portion of CXCL4L1 cannot fold properly and tends to precipitate into inclusion bodies when expressed in bacteria. Before protein refolding, we removed impurities by repeating the resuspension and centrifugation step. The pellet was resuspended in lysis buffer containing 0.8% (v/v) Triton X-100 and 0.8% sodium deoxycholate with 20 min of stirring at room temperature. Sonication followed by centrifugation was repeated. The resulted pellet was stirred again with the same buffer containing additional 10 mm EDTA at room temperature followed by centrifugation. This process was further repeated twice in the absence of Triton X-100. The final pellet with enriched CXCL4L1 was dissolved in 6 m guanidinium hydrochloride, 50 mm Tris-HCl, pH 8.0, 500 mm NaCl, and 5 mm β-mercaptoethanol. After 2 h of stirring at room temperature, the solution was dialyzed twice against chilled 0.9 m guanidinium chloride, 50 mm Tris-HCl, pH 8.0, 500 mm NaCl, 5 mm freshly added cysteine and methionine at 4 °C. The presence of cysteine and methionine enhanced the refolding of CXCL4L1, as previously reported for phospholipase (41). After 6 h of dialysis, the buffer was replaced to the same buffer without cysteine, methionine, and guanidinium chloride. The buffer exchange was repeated few times. Precipitate was removed by centrifugation (14,000 × g, 20 min, 4 °C). The protein solution containing folded CXCL4L1 was subjected to FPLC and HPLC purification using the same procedure of CXCL4 purification. Because CXCL4L1 is less stable and easily aggregates in aqueous solution, the freshly prepared CXCL4L1 was immediately used for the experiments. We applied the refolding procedure to other related mutants except PEL. To ensure the identity of proteins, mass spectrometry was used to validate the molecular weights of CXCL4, CXCL4L1, and the variants in each preparation.
Circular Dichroism (CD)
CD spectra were obtained using an AVIV 202 spectrophotometer (Aviv Biomedical Inc.). Protein concentration of 25 μm in 50 mm sodium phosphate buffer, pH 7.4, 150 mm NaCl was used for measurement in the far-UV region (190–260 nm) using a 1-mm path length cuvette at 25 °C. Data were collected every 1 nm with a 5-s averaging time. Three scans were averaged, and spectra are shown as the mean residue ellipticity (θ) in degree cm2 dmol−1. To reduce the disulfide bridges, 10 mm DTT was added to protein samples and incubated overnight at 4 °C.
Analytical Ultracentrifugation Analyses
Sedimentation equilibration was performed by Beckman XL-A Optima analytical ultracentrifuge equipped with absorbance optics and a Ti-60a titanium rotor. 500 and 25 μm protein samples were centrifuged at 45,000 rpm for 7 h at 20 °C. Concentration profiles were analyzed by program SEDFIT85.
Surface Plasmon Resonance (SPR) Analyses
The heparin binding experiments were carried out on a BIAcore 3000 instrument (GE Healthcare) by immobilizing biotinylated heparin on a streptavidin-coated surface (sensor chip SA, GE Healthcare) to a density of 37.1 resonance units. The streptavidin-coated surface without immobilized heparin was used as a reference. For kinetic binding studies, a serious concentration of chemokines was injected over the heparin surfaces with a flow rate of 40 μl/min 50 mm sodium phosphate buffer, pH 7.4, 150 mm NaCl, and 0.005% Tween 20 at 25 °C. The steady-state responses under different concentrations were measured and analyzed by Scatchard plot to determine heparin binding affinity (KD). All binding experiments were performed by a 180-s injection, then a 300-s dissociation. Surfaces were regenerated by a 30-s wash of 2.0 m NaCl. For competition assays, heparin oligosaccharides with defined lengths (from depolymerized (dp) heparin fragments, dp2-dp18) were mixed with proteins before injection onto a heparin-coated chip (42).
8-Anilino-1-naphthalenesulfonic Acid (ANS) Binding
Samples containing 10 μm chemokine were mixed with 20 μm ANS in 50 mm sodium phosphate buffer, pH 7.4, 150 mm NaCl. The ANS intensity was recorded from 400 to 600 nm with λex = 360 nm using an F-7000 fluorescence spectrofluorometer (Hitachi).
NMR HSQC Experiments
To prepare the uniformly 15N-labeled protein for NMR measurement, cells were grown on M9 minimal media with 15NH4Cl (1 g/liter) as the sole nitrogen source. HSQCs were recorded on a Bruker 500 MHz or 600 MHz spectrometer, and the acquired spectra were processed using NMRpipe (43) and analyzed with SPARKY (44).
Crystallization, Data Collection, and Processing
Crystals of CXCL4L1 were grown using the vapor diffusion method (vapor batch method). Drops containing 1 μl of protein (3.5 mg/ml) were mixed with the same volume of reservoir solution containing 0.1 m Tris-HCl, pH 8.0, 0.2 m sodium citrate, and 20% PEG 400. The 2-μl droplet was dispensed into 24-well sitting plates covered with 30 μl of a 1:1 mixture of silicone oil and paraffin oil (Hampton Research) and equilibrated against reservoir solution additionally containing 2-propanol. Single crystals of CXCL4L1 grew from the condition, and additive screening based on Hampton Research (HR2–428) was used to enhance crystal quality. Single crystals were transferred to the Fomblin Y oil (Sigma) and flash-cooled in liquid nitrogen for x-ray diffraction experiments. All x-ray diffraction data were collected at 110 K at beamline BL13B1 equipped with a CCD detector (Q315, ADSC) at the National Synchrotron Radiation Research Center in Hsinchu, Taiwan or SP44XU at SPring-8, Japan. Data sets were indexed, integrated, and scaled using the program HKL2000. The structure of CXCL4L1 was determined by molecular replacement with MOLREP (45) using the truncated structure of CXCL4 (PDB code 1F9Q) as the search model. After rigid-body refinement and simulated annealing using CNS (46), several rounds of manual model building in COOT (47) and refinement with PHENIX (48) were performed to improve the quality and completeness of the structure. Stereochemical analysis was performed with PROCHECK (49). Data collection and refinement statistics are shown in Table 1.
TABLE 1.
Crystallographic statistics of CXCL4L1
| Parameters | CXCL4L1 |
|---|---|
| Data collection | |
| Cell parameters | |
| a (Å) | 75.672 |
| b (Å) | 55.180 |
| c (Å) | 68.134 |
| Wavelength (Å) | 1.000 |
| Space group | P21212 |
| Resolution (Å) | 30.0-2.08 |
| Completeness (%)a | 99.41 (89.9) |
| Average I/σ (I) | 37.1 (214.6) |
| Number of unique reflections | 17,533 |
| Redundancy | 7.8 |
| Rmerge (%)b | 3.5 |
| Refinement | |
| Resolution range (Å) | 30.0-2.08 |
| Rfactor (%)c | 23.73 |
| Rfree (%)d | 27.09 |
| Bond length (Å)e | 0.007 |
| Bond angle (°) | 1.211 |
| Ramachandran analysis (%) | |
| Favored/allowed/generous/disallowed | 99.16/0.84/0.0/0.0 |
a Values in parentheses refer to statistics in the highest resolution shell.
b Rmerge = 〈Fobs − Fcalc〉/Fobs, where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively.
c R = Σ|Fo − Fc|/ΣFo, where Fo and Fc are the observed and calculated structure-factor amplitudes, respectively.
d Rfree was computed using 5% of the data assigned randomly.
e Root mean square deviation.
Protein Data Bank Accession Number
The coordinates of the structure of CXCL4L1 have been deposited in Protein Data Bank with the accession number 4HSV.
RESULTS
CXCL4 A-B Dimer as a Heparin Binding Unit
One of the major differences between the activities of CXCL4L1 and CXCL4 is the difference in their in cellulo diffusibility and in vivo bioavailability as a result of their difference in the GAGs binding properties (24). The heparin and heparin sulfate binding strength of CXCL4L1 are both reduced by at least 2 orders of magnitude as compared with that of CXCL4 (24). Because a distinct oligomerization state of the chemokines is known to affect both GAG and receptor binding, it is essential to define the heparin binding unit of CXCL4 before the structural characterization of CXCL4L1. Previous NMR studies on CXCL4 molecules in solution have demonstrated that CXCL4 is an asymmetric homotetramer with A, B, C, and D subunits (39). The tetramerization interfaces consist of the N terminus (A-D dimer) and the first β strand, β1 (A-B dimer). Although the atomic structure is known, it is unclear whether the tetramer is required for heparin binding.
We first obtained 1H,15N HSQC NMR spectra (Fig. 2A, boxed) to confirm that the CXCL4 molecule exists as an asymmetric homotetramer as indicated by the two sets of cross-peaks with the resonance line width corresponding to the tetrameric molecular size (40). We then carried-out experiments in the presence and absence of reducing agent, dithiothreitol (DTT), which reduces the disulfide bonds in the protein. Upon the addition of DTT, we observed mono-disperse resonances in the HSQC spectrum showing uniform intensity and significantly reduced spectral complexity as compared with that of the DTT-free CXCL4 (Fig. 2A). DTT-reduced CXCL4 is well folded, as inferred from the NMR chemical shift difference of (ΔδCα − ΔδCβ) and the profile of CD spectrum (Fig. 2, B and C). These data show that DTT-reduced CXCL4 contains the same secondary structural elements as defined by the crystal structure. Size-exclusion chromatography shows that DDT treatment of CXCL4 leads to a shift from a tetramer to a dimer (Fig. 2D). Therefore, the elimination of disulfide linkages removes the molecular asymmetry and also alters the oligomeric state of the protein.
FIGURE 2.

The structure, oligomerization state, and heparin binding properties of DTT-reduced CXCL4. A, 1H,15N HSQC spectra of CXCL4 (left) and DTT-reduced CXCL4 (right) at 600 MHz and 30 °C are shown. Two sets of resonances represent the molecular asymmetry of the tetramer (boxed, left). After reduction by the addition of 10 mm DTT, tetrameric CXCL4 with A, B, C, and D subunits was converted to an asymmetric A-B dimer. The heparin-binding sites, constituted by two C-terminal helices, are indicated by gray circles. B, shown is a secondary structure of DTT-reduced CXCL4, evaluated based on carbon chemical shifts (ΔδCα − ΔδCβ). The values of ΔδCα and ΔδCβ were, respectively, calculated from the differences between the experimental chemical shifts of 13Cα and 13Cβ and corresponding random coil values. The value of (ΔδCα − ΔδCβ) for each residue represents the average of three consecutive residues (i − 1, i, i + 1), centered at residue i. Positive values are indicative of α-helices, and negative values represent β-strands. C, shown are far-UV CD spectra of CXCL4 and DTT-reduced CXCL4. D, shown is a FPLC size-exclusion chromatogram CXCL4 and DTT-reduced CXCL4. CXCL4 elutes at the position corresponding to tetramer, and DTT-reduced CXCL4 elutes at the position of an expected dimer. The elution positions of protein standards are indicated by arrows. E, heparin binding affinities of CXCL4 and DTT-reduced CXCL4 were analyzed by a Scatchard plot derived from SPR equilibrium responses at protein concentrations of 3–55 nm. The slopes of the best-fit linear correlations in the Scatchard plot represent the binding constant (KD) values of 3 and 91 nm for the CXCL4 tetramer and dimer, respectively. F, shown are competition ratios of SPR responses of CXCL4 and DTT-reduced CXCL4 in the addition of depolymerized (dp) heparin fragments. A protein concentration of 100 nm and heparin concentration of 10 μg/ml were mixed and injected.
The secondary and tertiary structure is maintained in the DTT-reduced CXCL4 dimer, but disulfide bond linkages are required for the chemokine quaternary structure. CXCL4 contains two intramolecular disulfide bonds of Cys-10 to Cys-36 and Cys-12 to Cys-52. These connectivities anchor the N terminus to the β-sheet core domain. It is suggested that breaking down the disulfide linkages introduces flexibility in the N terminus and could potentially disrupt local interface binding. This would result in the N terminus being released into solution as either a random coil or extended loop conformation. Consistent with this notion, we observe low values of (ΔδCα − ΔδCβ) for amino acid residues located at the N terminus (Fig. 2B). Thus, the combined NMR and CD spectroscopic results indicate that CXCL4 exists as a symmetric A-B dimer under the DTT-reduced condition (Fig. 2A).
We next compared the mode and the strength of heparin binding between the DDT-reduced CXCL4 dimer and the native CXCL4 tetramer with the newly established experimental conditions. We applied Scatchard analysis to the response level of CXCL4 binding to heparin as determined by SPR. The dissociation constants (KD) for the CXCL4 dimer and tetramer were found to be about 91 and 3 nm, respectively (Fig. 2E and Table 2). By using heparin-derived fragments with different chain lengths to perform SPR competition experiment, similarity of the heparin binding mode in both the DTT-reduced CXCL4 dimer and the native CXCL4 tetramer was seen. As shown in Fig. 2F, heparin fragments longer than dodecsaccharide (dp12) significantly competed for binding to CXCL4. This suggests that the A-B dimer contains an entire heparin-binding site. The results confirm that the A-B dimer is essential for specific heparin binding (50). Thus, the CXCL4 asymmetric tetramer is assembled from two symmetric heparin-binding dimers with moderate synergy to enhance its binding strength from 91 to 3 nm.
TABLE 2.
Summary of binding affinities of CXCL4, CXCL4L1, and the variants to heparin
| CXCL4 variants | K1aa | NaCl concentration to elute from heparin-agarose |
|---|---|---|
| nm | m | |
| CXCL4 | 3.71 | 1.5 |
| PEH (K66E, L67H) | 255.04 | 0.9 |
| LKH (P58L, L67H) | 107.53 | 1.1 |
| LEL (P58L, K66E) | 4.08 | 1.3 |
| PKH (L67H) | 101.01 | 1.1 |
| PEL (K66E) | 4.28 | 1.4 |
| LKL (P58L) | 4.14 | 1.5 |
| CXCL4L1 | 251.13 | 1.0 |
| CXCL4/DTT | 91.4 | 1.0 |
a Values from the slope of the linear regression of Scatchard-plot analyses of SPR measurements.
Deficient Heparin Binding of CXCL4L1 Tetramer in Solution
CXCL4L1 is a nonallelic variant of CXCL4 containing three mutations in the C-terminal helix (Fig. 1) that leads to significantly altered biological properties. To assess the molecular basis of the altered function of CXCL4L1, we first compared the biophysical properties and the heparin binding of CXCL4L1 in solution in comparison to CXCL4 and then determined its crystal structure. Both chemokines are shown by size-exclusion chromatography to be tetramers in solution with no detectable dimer or monomer populations during the purification processes (Fig. 3A). However, the apparent size of CXCL4L1 is slightly bigger as evidenced by analytical ultracentrifugation experiments (Fig. 3B) as well as FPLC size-exclusion analysis (Fig. 3A) despite the same number of amino acids. The apparent molecular sizes determined by analytical ultracentrifugation are 28 and 32 kDa for CXCL4 and CXCL4L1, respectively. This implies that the molecular packing of the CXCL4L1 tetramer is less globular and likely more elongated than CXCL4 tetramer. The comparison of CD spectra demonstrated a slightly more helical content for CXCL4L1 than for CXCL4 (Fig. 3C). The result indicates that both the secondary and tertiary structures of the two chemokines are different in aqueous solution.
FIGURE 3.

Comparison of biophysical properties and heparin binding of CXCL4L1 and CXCL4. A, FPLC size-exclusion elution profiles are shown. The elution positions of protein standards are indicated by arrows. CXCL4 and CXCL4L1 both are eluted at the position corresponding to a tetramer. B, analytical ultracentrifugation (AUC) profiles are shown. The estimated apparent molecular masses are 28 and 32 kDa for CXCL4 and CXCL4L1, respectively. C, far-UV CD spectra for CXCL4 and CXCL4L1 are shown. D, shown are fluorescent emission spectra of ANS fluorescent dye after binding to the hydrophobic surface of CXCL4 and CXCL4L1. The stronger fluorescent intensity represents the larger portion of the hydrophobic surface exposed to the solvent. E, SPR sensograms of heparin binding are shown. Left, sensograms were obtained by injecting different concentrations of CXCL4 or CXCL4L1 (3–55 nm) over a heparin-coating chip. The inset shows the injections of CXCL4L1 with higher concentrations (0.3–1.5 μm), which were carried out due to very low affinity of CXCL4L1 for heparin. Right, sensograms were derived from injections of the same concentration (55 nm) of CXCL4, CXCL4L1, and the variants of LKL, PEL, PKH.
The packing difference between the CXCL4L1 and CXCL4 molecule can also be observed in binding studies using the ANS hydrophobic probe. ANS emits an intensive fluorescence signal at 480 nm when binding occurs in a hydrophobic environment. As shown in Fig. 3D, the fluorescence emission spectra for the CXCL4L1-ANS complex is significantly greater than that for the CXCL4-ANS complex. This indicates that CXCL4L1 contains a larger exposed hydrophobic surface.
Finally, we determined the heparin binding of CXCL4L1 together with a set of mutants by using SPR (Fig. 3E, supplemental Fig. S1, and Table 2). Previous work by using glutathione S-transferase fusion chimeric protein has reported that the heparin binding affinity is 100-fold reduced in CXCL4L1 (24). In this study we confirm the reduction of the heparin binding activity of the intrinsic CXCL4L1 tetramer and show that only moderate responses can be detected after injecting a 10× excess of CXCL4L1 (Fig. 3E, left). A comparative study on a series of CXCL4 mutants (Fig. 3E, right) also indicates that the L67H substitution rather than K66E substitution resulted in the greatest reduction in heparin binding affinity, and the P58L substitution had no measurable effect. Introducing a K66E mutation into CXCL4 (termed PEL) preserved heparin binding affinity, whereas L67H (termed LKH) caused a more significant decrease in SPR response. The K66E/L67H double mutant (PEH in supplemental Fig. S1) abolished heparin binding, suggesting that L67H is the key mutation.
Crystal Structure of CXCL4L1
To gain deeper insights into the structural modifications responsible for the altered heparin binding, we determined CXCL4L1 x-ray structure. The crystal structure was solved at 2.0 Å resolution (Fig. 4A and Table 1). In line with the spectroscopic data obtained in solution, CXCL4L1 also packed as a tetramer in the crystalline state, comprising two nearly identical dimers. The two dimers are asymmetrically arranged to form a tetrameric structure composed of core β-sheet domains flanked by four “open” C-terminal α-helices. Unlike other chemokine structures, the C-terminal helices of CXCL4L1 do not pack against the core β-sheet and, therefore, have a less globular shape as compared with CXCL4.
FIGURE 4.
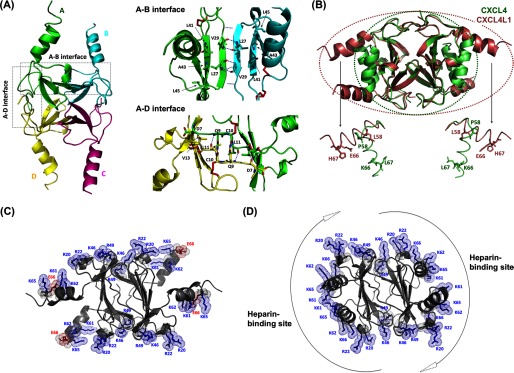
Crystal structure of CXCL4L1. A, shown is a schematic representation of CXCL4L1 tetramer (left) and the A-B and A-D interface where A/B/C/D monomer subunits are depicted by different colors. The hydrogen bonds for monomer association in A-B and A-D dimer interfaces are indicated by dotted lines. The residues constituting the exposed hydrophobic surface in A-B interface are shown as well as the residues responsible for association of N terminus in A-D interface. B, shown is the superposition of CXCL4 (PDB code 1F9Q) and CXCL4L1. The β-sheet core domain is almost identical, but helix α1 is rotated by ∼87° in CXCL4L1 relative to CXCL4, thus altering the overall shape of the molecule. The positions of the three mutations are indicated in two of the subunits by sticks. C, the distribution of positively charged residues and Glu-66 in CXCL4L1 is shown. D, the distribution of positively charged residues in CXCL4 is shown. The surrounding positive region is purposed to be able to recruit two individual anionic polysaccharides.
As in the CXCL4 structure (PDB code 1F9Q), we observed two intramolecular disulfide linkages in one subunit: Cys-10 to Cys-36 and Cys-12 to Cys-52. The β-sheet core domain forms the interface of the tetramer, and the domain structure is identical to the corresponding region in CXCL4. Superposition of the two structures shows similar core domains with an root mean square deviation of ∼0.4 Å for Cα atoms of residues between Cys-10 and Cys-52 (Fig. 4B). The hydrogen bonds between two β1 strands stabilize the A-B (and C-D) interface, and the β1-β3 antiparallel β-sheet extends to another subunit to form a 6-stranded β-sheet (Fig. 4A, A-B interface). The N-terminal region, which forms an antiparallel β-sheet, provides additional interactions to stabilize the A-D (and B-C) interface (Fig. 4A). The structural symmetry is distorted by the in-equivalent interactions at the N termini, as the N-terminal residues of the A-D interface provide more interactions than at the B-C interface. Overall, there are no notable structural differences between the core domains of CXCL4 and CXCL4L1.
The CXCL4L1 structure is unique in the conformation adopted by the C-terminal helix. A careful comparison of the CXCL4 structure to all known chemokine structures shows the C-terminal helix of CXCL4L1 to be oriented orthogonal to the C-terminal helices of known structures. The helices, rotating ∼87 degrees, protrude perpendicularly from the protein surfaces and show less contact with the rest of the tetrameric complex. In contrast to the conformation defined in CXCL4, the helices in CXCL4L1 are spatially isolated and exclusively linked to the β-sheet core domain through the protein backbone (Fig. 4A). It is also of note that the two C-terminal helices in an individual A-B dimer unit are separated by a large distance of 12–13 Å. The open helical conformation exposes the hydrophobic surface constituted by one-side residues of the 6-stranded β-sheet, i.e. Leu-27, Val-29, Leu-41, Ala-43, and Leu-45 (Fig. 4A, A-B interface), which is consistent with the ANS binding data (Fig. 3D).
We need emphasis that the structural difference between the two chemokines is not derived from the different sample preparation procedures. CXCL4L1 was refolded from inclusion body, whereas CXCL4 was isolated from the supernatant of cell lysate. We have isolated trace amounts of correctly folded CXCL4L1 from the cell lysate. The CXCL4L1 demonstrated the same CD spectrum, HPLC elution profile, and heparin binding in SPR assay as that previously identified in the refolded CXCL4L1. There was no detectable difference between the two proteins derived from different preparation methods. Similarly, refolded CXCL4 from inclusion body also reproducibly gave the same structure and biophysical properties in experiments.
We have shown that the A-B dimer unit is adequate for the heparin binding in CXCL4 (Fig. 2). The open conformation of the C-terminal helix in CXCL4L1 disrupts the continuity of the positively charged protein surface, rendering the protein surface unfavorable for binding. It has also been shown that the positively charged cluster in the C-terminal helix (Lys-61, Lys-62, Lys-65, and Lys-66) constitute the most noteworthy part of the heparin-binding site and other cationic residues located in the middle of sequence (i.e. Arg-20, Arg-22, Lys-46, Arg-49) are synergistically involved (Fig. 4D) (50). The protrusion of the helices and the additional exposed hydrophobic region in CXCL4L1 creates a steric barrier to GAG binding (Fig. 4C).
Structure of the C-terminal Helix
The orientation of the C-terminal helix in each CXCL4L1 monomer is not identical when the four monomers of the tetramer are superimposed (Fig. 5A). There is an ∼5º rotation between the helices of monomers A/D relative to monomers B/C. Without packing constraint from the core region, the swing-out helix is expected to generate a certain degree of flexibility in the CXCL4L1 molecule.
FIGURE 5.

Orientations of helix α1 in CXCL4 and CXCL4L1. A, the two helical orientations (A/D and B/C) were identified in CXCL4L1 monomers, and only one orientation (A/B/C/D) was found in CXCL4 monomers. B, shown is a secondary structure for CXCL4 and CXCL4L1. Two disulfide bonds of Cys-10 to Cys-36 and Cys-12 to Cys-52 are shown as open curved bars. The hydrogen bonds of the backbone are indicated by arrows pointed from hydrogen bond donor to acceptor. The helix α1 contains residues Leu-59—Ser-70 in CXCL4 and residues Asp-54–His-67 in CXCL4L1, and the helical wheel of CXCL4 α1 is depicted to show the amphipathic property.
We also compared the topology and hydrogen bond patterns of CXCL4 and CXCL4L1 (Fig. 5B). As expected, the hydrogen bond pattern in the β-sheet domain is identical, but there is a significant difference at the connecting loop from residue 54 to 58 between β3 and α1. Proline is a well known helix breaker. Therefore, Pro-58 plays a role in rotating α1 by 90º to allow the helix to pack against the surface of β-strands in CXCL4 molecule. The helical element immediately starts from the next residue, Leu-59, and extends to the end of C-terminal residue, Ser-70. The P58L mutation in CXCL4L1 allows the helix α1 formation starting immediately after β3 at residue Asp-54. Therefore, its helix could then start from residue Asp-54 and extend to the end of C terminus. This also explains why a slightly larger helical content of CXCL4L1 is detected in CD (Fig. 3C).
Helix α1 in CXCL4 is amphipathic and facilitates hydrophobic interactions between the side chains of Leu-67 (α1) and Leu-27 (β1). As a result of the substitution Leu-67 with His-67 in CXCL4L1, this hydrophobic packing is expected to be disrupted and thus allows the release of the α1 helix from protein surface (Fig. 5B). The L67H mutation also creates a change in the local helicity of helix α1 probably due to its multiple H-bond forming ability. The regular helical structure in CXCL4L1 actually only reaches to residue His-67 with a more loosely helical formation afterward.
The K66E mutation is located in the middle of the hydrophilic side of helix α1. CXCL4 K66 is exposed to solution and does not play any role in mediating intramolecular interaction. Instead, the replacement of a positively charged residue with a negatively charged one might result in decreased ability for ligand binding as Lys-66 is located within the positively charged cluster region. We expect that K66E might simply modulate the interaction of the protein with anionic GAGs rather than altering the chemokine structure. The conformational difference in helix α1 orientations is likely controlled by the two substitutions, Pro-58 to Leu and Leu-67 to His.
Effect of Single Mutations on Chemokine Folding and Stability
Because we could obtain excellent NMR spectra for DTT-reduced CXCL4, we used the same method to determine the structural modifications produced by single mutations. We obtained HSQCs of CXCL4L1 and all related mutants in the presence of DTT (Fig. 6 and supplemental Fig. S2). Similar to CXCL4, CXCL4L1 also adopts a dimer formation under reducing conditions. However, severe resonance broadening, including missing and weak resonances, was observed in the 1H,15N HSQC spectrum of CXCL4L1 in the presence of DTT (Fig. 6). This is not due to protein aggregation as the NMR experiment was carefully controlled under low protein concentration. In contrast, it suggests that the molecule is not well folded into a tight ensemble of conformations and behaves more like a molten globule or partially denatured structure. In fact, a number of intense cross-peaks appear between 1H chemical shift 7.5 and 9.0 ppm corresponding to the “random coil” region. Therefore, some portion of DTT-reduced CXCL4L1 molecule is likely to be unstructured. We conclude that DTT-reduced CXCL4L1 is not stable upon reduction of the disulfide linkages. Compared with the result for CXCL4, the helix α1 orientation may become important in maintaining chemokine fold in the reduced condition.
FIGURE 6.

Effect of mutations on chemokine folding and stability. 1H,15N HSQC spectra of DTT-reduced CXCL4, CXCL4L1, and PKH, LKL, and PEL mutants are compared (0.2 mm protein concentration, 10 mm DTT, 25 °C, and 600 MHz).
To dissect the role of each mutation, we obtained NMR spectra for combinations of the point mutation-based CXCL4 sequence (Fig. 6). The CXCL4 variant PEL with one substitution of E66 shows similar HSQC character as that in CXCL4, indicating a minor effect on CXCL4 structure. The CXCL4 variant LKL with one substitution of Leu-56 also exhibited a widely dispersed cross-peak. However, there appear to be non-uniform intensities for the resonances within the spectrum. Because some of the relatively strong cross-peaks also appear in the central region of spectrum, the fold of the LKL mutant may mainly adopt the CXCL4 fold but with certain structural instability. Finally, the CXCL4 variant PKH with one substitution of His-67 has prominently broadened and overlapping resonances. The spectrum differs from those obtained for CXCL4 and CXCL4L1 samples, indicating a collapsed structure with significant conformational exchange. This observation is consistent with the notion that L67H plays a determinant role in the structural stability, possibly disrupting the packing between helix α1 and the β-sheet core domain. The additional incorporation of substitution Leu-58 (mutant termed LKH) converted structural property to be more similar to CXCL4L1 (supplemental Fig. S2), indicating that the substitutions at residues 58 and 67 have a synergistic effect for converting the molecule into a CXCL4L1 structure.
DISCUSSION
Alternate C-terminal Helical Orientation and Conformation Regulate Functions
Although chemokine CXCL4 and CXCL4L1 both act on endothelial cell proliferation and migration, CXCL4L1 appears to be of much more potent anti-angiogenic activity than CXCL4. CXCL4L1 differs from CXCL4 by only three amino acids located in the C-terminal helix α1. Our current understanding of the structural and functional relationship of chemokines is mainly based on the canonic chemokine structure including the C-terminal helix packing onto the β-sheet core domain and different oligomerization states. However, the structure of CXCL4L1 that we elucidated in this article is very different from any known chemokine structure and leads to novel insights in the structure-function relationships of chemokines. The C-terminal helix in CXCL4L1 adopts an opened solvent-accessible conformation with certain degree of flexibility, which is controlled by a few amino acids. This conformational change leads to dramatic changes in the biological functions of this chemokine.
The C-terminal helix greatly influences the biological properties of CXCL4s (23). CXCL4 is capable of binding to heparin and heparan sulfates and, thus, neutralizing their biological activities. The interaction is also responsible for the strong matrix binding, rapid clearance of CXCL4 from the circulation, and its internalization by endothelial cells (24). The loss of heparin recognition in CXCL4L1 is primarily due to the conformational change induced by residue His-67. The charge replacement at residue 66 has a secondary role in this interaction. This results in a low heparin binding ability for CXCL4L1 and together completely eliminates the binding to chondroitin sulfate (24). This also translates in a modification of the bioavailability of CXCL4L1 by increasing its half-life and diffusibility in circulation (24). The orientation of helix α1 critically impacts on the heparin binding capability of these chemokines. An open conformation of the C terminus favors lower heparin binding capability as seen in CXCL4L1.
CXCL4L1 inhibits angiogenesis and tumor growth more potently than CXCL4. His-67 is the most critical residue for these activities (24). Considering the weak heparin binding in CXCL4L1, this implies that the recognition to cell surface GAGs and anti-angiostatic properties are less related. It is likely that the lower GAG binding of CXCL4L1 makes the chemokine more available to interact with cell surface receptors such as CXCR3 (23). The C terminus of CXCL4L1 should cover all or part of receptor binding site. The N terminus together with its N-loop between the first two cysteines and the 310 helix adjunct to β1 has usually been proposed to act as the key domain responsible for receptor binding (14). The opened C-terminal helix α1 in CXCL4L1, as revealed in this study, seems to provide an alternative or additional domain for cell surface receptor interaction. In addition, the open conformation may also impact on the structure of the N-terminal domain, allowing a better interaction with cell surface receptors.
Implications to Other Chemokine Families
Among the three mutants, P58L and L67H substitutions together modulate the helix orientation, whereas the K66E substitution has no effect. Leu-58 replaces the helix breaker, Pro-58, to allow extended helix α1 formation along the N-terminal direction. His-67 in CXCL4 reduces the helical hydrophobicity to allow the helix to swing out from the protein surface and also affects protein stability during the folding process. This implies a need for a modification of the protein secretion pathway for His-67-containing chemokine. Indeed, the protein secretion pathway and the regulation of expression of both chemokines have been shown to be significantly different (51). We also noticed that the K66E mutation is only found in human, whereas the other two mutations, L67H and P58L, are both found in chimpanzee and orangutan. It will be interesting to investigate how a change in this charged residue within a heavily concentrated positively charged cluster affects its binding to GAGs and how this influences the function of the human protein. In other chemokines such as in human CXC16 and CXC17, a hydrophilic residue of Asn or His at the corresponding position of His-67 in CXCL4L1 is also found. Therefore, it will be interesting to investigate the orientation of their C-terminal helix and also the folding and secretion pathway of these chemokines.
Chemokine Heteromerization
The core domain of anti-parallel β-sheet is stable even in the absence of contact with helix α1. Hence, in the presence of disulfide linkages, the C-terminal helix seems not required for maintaining the fold of anti-parallel β-sheet domain. The observation is actually in agreement with the previous assumption that the positively charged C-terminal helices in the CXCL4 A-B (and C-D) dimer induce thermo-instability for the β-sheet core domain (52). This is due to the repulsive force between helices to repel each other. By eliminating the repulsive force, the open C-terminal helices in CXCL4L1 contain less positive potential and a more stable oligomerization state. This indicates that the CXCL4L1 tetramer is more stable than the CXCL4 tetramer and that the C-terminal helix might regulate the stability of the oligomerized chemokine. The idea is supported by the observation that CXCL4 fails to dissociate the CXCL4L1 tetramer to constitute a CXCL4-CXCL4L1 heteromer. Therefore, CXCL4 and CXCL4L1 do not heterodimerize and may function independently. However, this needs to be confirmed when modifying the experimental conditions and concentrations.
There is much recent interest in the study of heteromerization between CXC and CC-type chemokines (52) This provides an additional regulatory mechanism for chemokine functions. Indeed, CXCL4 has been shown to be able to associate with CCL5 and CXCL8 chemokines (52–55). To form a heteromer, the CXCL4 tetramer needs to be dissociated when encountering other chemokines. Because of the higher stability of the tetramer, CXCL4L1 may have less ability to form heteromers with other chemokines. Thus, our study also sheds some light to a better understanding of the structural basis of hetero- and/or homo-oligomerization of chemokines.
Taken together, we describe for the first time a novel and unique structure for a CXC chemokine that is due to only a few mutations among which only one has the principal role. The previously unrecognized helix α1 orientation has significant consequences for its physicochemical and in vitro and in vivo biological properties. This work also constitutes fertile ground for further investigations on the structure-function relationships of this family of chemokines.
Acknowledgments
We acknowledge the NMR facility at National Tsing Hua University and x-ray beamlines of BL13B1 and BL13C1 at the National Synchrotron Radiation Research Center (Taiwan) and SP44XU at SPring-8 (Japan). We thank Dr. G. Bhabha for valuable suggestions and careful reading of this paper.
This work was supported by Taiwan National Science Council grants (to W.-g. W. and S.-C. S.), a National Tsing Hua University and Chang Gung Memorial Hospital joint research grant (to S.-C. S.), an Agence National pour le Recherche grant, a National Cancer Institute (INCA) grant, and by Ligue National du Cancer grants (to A. B.).

This article contains supplemental Figs. S1 and S2.
The atomic coordinates and structure factors (code 4HSV) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PF4
- platelet factor 4
- GAG
- glycosaminoglycan
- SPR
- Surface Plasmon Resonance
- dp
- depolymerized
- ANS
- 8-anilino-1-naphthalenesulfonic acid
- HSQC
- heteronuclear single quantum correlation.
REFERENCES
- 1. Raman D., Sobolik-Delmaire T., Richmond A. (2011) Chemokines in health and disease. Exp. Cell Res. 317, 575–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Keeley E. C., Mehrad B., Strieter R. M. (2011) Chemokines as mediators of tumor angiogenesis and neovascularization. Exp. Cell Res. 317, 685–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mukaida N., Baba T. (2012) Chemokines in tumor development and progression. Exp. Cell Res. 318, 95–102 [DOI] [PubMed] [Google Scholar]
- 4. Bikfalvi A. (2012) Angiogenesis and invasion in cancer. Handb. Clin. Neurol. 104, 35–43 [DOI] [PubMed] [Google Scholar]
- 5. Balkwill F. R. (2012) The chemokine system and cancer. J. Pathol. 226, 148–157 [DOI] [PubMed] [Google Scholar]
- 6. Kiefer F., Siekmann A. F. (2011) The role of chemokines and their receptors in angiogenesis. Cell. Mol. Life Sci. 68, 2811–2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bikfalvi A., Moenner M., Javerzat S., North S., Hagedorn M. (2011) Inhibition of angiogenesis and the angiogenesis/invasion shift. Biochem. Soc. Trans. 39, 1560–1564 [DOI] [PubMed] [Google Scholar]
- 8. Bikfalvi A. (2009) Tumoral angiogenesis. Models, targets, and inhibition. J. Soc. Biol. 203, 167–170 [DOI] [PubMed] [Google Scholar]
- 9. Scholten D. J., Canals M., Maussang D., Roumen L., Smit M. J., Wijtmans M., de Graaf C., Vischer H. F., Leurs R. (2012) Pharmacological modulation of chemokine receptor function. Br. J. Pharmacol. 165, 1617–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koelink P. J., Overbeek S. A., Braber S., de Kruijf P., Folkerts G., Smit M. J., Kraneveld A. D. (2012) Targeting chemokine receptors in chronic inflammatory diseases. An extensive review. Pharmacol. Ther. 133, 1–18 [DOI] [PubMed] [Google Scholar]
- 11. Van Sweringen H. L., Sakai N., Tevar A. D., Burns J. M., Edwards M. J., Lentsch A. B. (2011) CXC chemokine signaling in the liver. Impact on repair and regeneration. Hepatology 54, 1445–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Flad H. D., Brandt E. (2010) Platelet-derived chemokines. Pathophysiology and therapeutic aspects. Cell. Mol. Life Sci. 67, 2363–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gleissner C. A., von Hundelshausen P., Ley K. (2008) Platelet chemokines in vascular disease. Arterioscler. Thromb. Vasc. Biol. 28, 1920–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Salanga C. L., Handel T. M. (2011) Chemokine oligomerization and interactions with receptors and glycosaminoglycans. The role of structural dynamics in function. Exp. Cell Res. 317, 590–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zlotnik A., Yoshie O. (2012) The chemokine superfamily revisited. Immunity 36, 705–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nomiyama H., Osada N., Yoshie O. (2010) The evolution of mammalian chemokine genes. Cytokine Growth Factor Rev. 21, 253–262 [DOI] [PubMed] [Google Scholar]
- 17. Balestrieri M. L., Balestrieri A., Mancini F. P., Napoli C. (2008) Understanding the immunoangiostatic CXC chemokine network. Cardiovasc. Res. 78, 250–256 [DOI] [PubMed] [Google Scholar]
- 18. Allen S. J., Crown S. E., Handel T. M. (2007) Chemokine. Receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 25, 787–820 [DOI] [PubMed] [Google Scholar]
- 19. Kowalska M. A., Rauova L., Poncz M. (2010) Role of the platelet chemokine platelet factor 4 (PF4) in hemostasis and thrombosis. Thromb. Res. 125, 292–296 [DOI] [PubMed] [Google Scholar]
- 20. Gleissner C. A. (2012) Platelet-derived chemokines in atherogenesis. What's new? Curr. Vasc. Pharmacol. 10, 563–569 [DOI] [PubMed] [Google Scholar]
- 21. Koenen R. R., Weber C. (2011) Chemokines. established and novel targets in atherosclerosis. EMBO Mol. Med. 3, 713–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aidoudi S., Bikfalvi A. (2010) Interaction of PF4 (CXCL4) with the vasculature. A role in atherosclerosis and angiogenesis. Thromb. Haemost 104, 941–948 [DOI] [PubMed] [Google Scholar]
- 23. Vandercappellen J., Van Damme J., Struyf S. (2011) The role of the CXC chemokines platelet factor-4 (CXCL4/PF-4) and its variant (CXCL4L1/PF-4var) in inflammation, angiogenesis, and cancer. Cytokine Growth Factor Rev. 22, 1–18 [DOI] [PubMed] [Google Scholar]
- 24. Dubrac A., Quemener C., Lacazette E., Lopez F., Zanibellato C., Wu W. G., Bikfalvi A., Prats H. (2010) Functional divergence between 2 chemokines is conferred by single amino acid change. Blood 116, 4703–4711 [DOI] [PubMed] [Google Scholar]
- 25. Aidoudi S., Bujakowska K., Kieffer N., Bikfalvi A. (2008) The CXC-chemokine CXCL4 interacts with integrins implicated in angiogenesis. PLoS One 3, e2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reilly M. P., Taylor S. M., Hartman N. K., Arepally G. M., Sachais B. S., Cines D. B., Poncz M., McKenzie S. E. (2001) Heparin-induced thrombocytopenia/thrombosis in a transgenic mouse model requires human platelet factor 4 and platelet activation through FcγRIIA. Blood 98, 2442–2447 [DOI] [PubMed] [Google Scholar]
- 27. Pouplard C., May M. A., Iochmann S., Amiral J., Vissac A. M., Marchand M., Gruel Y. (1999) Antibodies to platelet factor 4-heparin after cardiopulmonary bypass in patients anticoagulated with unfractionated heparin or a low-molecular-weight heparin. clinical implications for heparin-induced thrombocytopenia. Circulation 99, 2530–2536 [DOI] [PubMed] [Google Scholar]
- 28. Yeaman M. R., Yount N. Y., Waring A. J., Gank K. D., Kupferwasser D., Wiese R., Bayer A. S., Welch W. H. (2007) Modular determinants of antimicrobial activity in platelet factor-4 family kinocidins. Biochim. Biophys. Acta 1768, 609–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cole A. M., Ganz T., Liese A. M., Burdick M. D., Liu L., Strieter R. M. (2001) Cutting edge. IFN-inducible ELR- CXC chemokines display defensin-like antimicrobial activity. J. Immunol. 167, 623–627 [DOI] [PubMed] [Google Scholar]
- 30. Auerbach D. J., Lin Y., Miao H., Cimbro R., Difiore M. J., Gianolini M. E., Furci L., Biswas P., Fauci A. S., Lusso P. (2012) Identification of the platelet-derived chemokine CXCL4/PF-4 as a broad-spectrum HIV-1 inhibitor. Proc. Natl. Acad. Sci. U.S.A. 109, 9569–9574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwartzkopff F., Grimm T. A., Lankford C. S., Fields K., Wang J., Brandt E., Clouse K. A. (2009) Platelet factor 4 (CXCL4) facilitates human macrophage infection with HIV-1 and potentiates virus replication. Innate Immun. 15, 368–379 [DOI] [PubMed] [Google Scholar]
- 32. Struyf S., Salogni L., Burdick M. D., Vandercappellen J., Gouwy M., Noppen S., Proost P., Opdenakker G., Parmentier M., Gerard C., Sozzani S., Strieter R. M., Van Damme J. (2011) Angiostatic and chemotactic activities of the CXC chemokine CXCL4L1 (platelet factor-4 variant) are mediated by CXCR3. Blood 117, 480–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de Jong E. K., de Haas A. H., Brouwer N., van Weering H. R., Hensens M., Bechmann I., Pratley P., Wesseling E., Boddeke H. W., Biber K. (2008) Expression of CXCL4 in microglia in vitro and in vivo and its possible signaling through CXCR3. J. Neurochem. 105, 1726–1736 [DOI] [PubMed] [Google Scholar]
- 34. Fiore M. M., Mackie I. J. (2009) Dual effect of platelet factor 4 on the activities of factor Xa. Biochem. Biophys. Res. Commun. 379, 1072–1075 [DOI] [PubMed] [Google Scholar]
- 35. Sarabi A., Kramp B. K., Drechsler M., Hackeng T. M., Soehnlein O., Weber C., Koenen R. R., Von Hundelshausen P. (2011) CXCL4L1 inhibits angiogenesis and induces undirected endothelial cell migration without affecting endothelial cell proliferation and monocyte recruitment. J. Thromb. Haemost. 9, 209–219 . [DOI] [PubMed] [Google Scholar]
- 36. Struyf S., Burdick M. D., Peeters E., Van den Broeck K., Dillen C., Proost P., Van Damme J., Strieter R. M. (2007) Platelet factor-4 variant chemokine CXCL4L1 inhibits melanoma and lung carcinoma growth and metastasis by preventing angiogenesis. Cancer Res. 67, 5940–5948 [DOI] [PubMed] [Google Scholar]
- 37. Struyf S., Burdick M. D., Proost P., Van Damme J., Strieter R. M. (2004) Platelets release CXCL4L1, a nonallelic variant of the chemokine platelet factor-4/CXCL4 and potent inhibitor of angiogenesis. Circ. Res. 95, 855–857 [DOI] [PubMed] [Google Scholar]
- 38. Vandercappellen J., Liekens S., Bronckaers A., Noppen S., Ronsse I., Dillen C., Belleri M., Mitola S., Proost P., Presta M., Struyf S., Van Damme J. (2010) The COOH-terminal peptide of platelet factor-4 variant (CXCL4L1/PF-4var47–70) strongly inhibits angiogenesis and suppresses B16 melanoma growth in vivo. Mol. Cancer Res. 8, 322–334 [DOI] [PubMed] [Google Scholar]
- 39. Mayo K. H., Roongta V., Ilyina E., Milius R., Barker S., Quinlan C., La Rosa G., Daly T. J. (1995) NMR solution structure of the 32-kDa platelet factor 4 ELR-motif N-terminal chimera. A symmetric tetramer. Biochemistry 34, 11399–11409 [DOI] [PubMed] [Google Scholar]
- 40. Zhang X., Chen L., Bancroft D. P., Lai C. K., Maione T. E. (1994) Crystal structure of recombinant human platelet factor 4. Biochemistry 33, 8361–8366 [DOI] [PubMed] [Google Scholar]
- 41. Bezzine S., Koduri R. S., Valentin E., Murakami M., Kudo I., Ghomashchi F., Sadilek M., Lambeau G., Gelb M. H. (2000) Exogenously added human group X secreted phospholipase A2 but not the group IB, IIA, and V enzymes efficiently release arachidonic acid from adherent mammalian cells. J. Biol. Chem. 275, 3179–3191 [DOI] [PubMed] [Google Scholar]
- 42. Wang Y. L., Kuo J. H., Lee S. C., Liu J. S., Hsieh Y. C., Shih Y. T., Chen C. J., Chiu J. J., Wu W. G. (2010) Cobra CRISP functions as an inflammatory modulator via a novel Zn2+- and heparan sulfate-dependent transcriptional regulation of endothelial cell adhesion molecules. J. Biol. Chem. 285, 37872–37883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe. A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 44. Goddard T. D., Kneller D. G. SPARKY 3. University of California, San Francisco [Google Scholar]
- 45. Collaborative Computational Project, Number 4 (1994) The CCP4 suite. programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 46. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography and NMR system. A new software suite for macromolecular structure determination. Acta Crystallogr. D. Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 47. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 48. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Laskowski R. A. (2001) PDBsum. Summaries and analyses of PDB structures. Nucleic Acids Res. 29, 221–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lortat-Jacob H., Grosdidier A., Imberty A. (2002) Structural diversity of heparan sulfate binding domains in chemokines. Proc. Natl. Acad. Sci. U.S.A. 99, 1229–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lasagni L., Grepin R., Mazzinghi B., Lazzeri E., Meini C., Sagrinati C., Liotta F., Frosali F., Ronconi E., Alain-Courtois N., Ballerini L., Netti G. S., Maggi E., Annunziato F., Serio M., Romagnani S., Bikfalvi A., Romagnani P. (2007) PF-4/CXCL4 and CXCL4L1 exhibit distinct subcellular localization and a differentially regulated mechanism of secretion. Blood 109, 4127–4134 [DOI] [PubMed] [Google Scholar]
- 52. Nesmelova I. V., Sham Y., Gao J., Mayo K. H. (2008) CXC and CC chemokines form mixed heterodimers. Association free energies from molecular dynamics simulations and experimental correlations. J. Biol. Chem. 283, 24155–24166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Koenen R. R., von Hundelshausen P., Nesmelova I. V., Zernecke A., Liehn E. A., Sarabi A., Kramp B. K., Piccinini A. M., Paludan S. R., Kowalska M. A., Kungl A. J., Hackeng T. M., Mayo K. H., Weber C. (2009) Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat. Med. 15, 97–103 [DOI] [PubMed] [Google Scholar]
- 54. Gouwy M., Struyf S., Noppen S., Schutyser E., Springael J. Y., Parmentier M., Proost P., Van Damme J. (2008) Synergy between coproduced CC and CXC chemokines in monocyte chemotaxis through receptor-mediated events. Mol. Pharmacol. 74, 485–495 [DOI] [PubMed] [Google Scholar]
- 55. Nesmelova I. V., Sham Y., Dudek A. Z., van Eijk L. I., Wu G., Slungaard A., Mortari F., Griffioen A. W., Mayo K. H. (2005) Platelet factor 4 and interleukin-8 CXC chemokine heterodimer formation modulates function at the quaternary structural level. J. Biol. Chem. 280, 4948–4958 [DOI] [PubMed] [Google Scholar]