

Background: Kallikreins play a pivotal role in establishing prostate cancer.
Results: In contrast to the classical Kunitz plant inhibitor SbTI, the recombinant kallikrein inhibitor (rBbKIm) led to prostate cancer cell death, whereas fibroblast viability was not affected.
Conclusion: rBbKIm shows selective cytotoxic effect and angiogenesis inhibition against prostate cancer cells.
Significance: New actions of rBbKIm may contribute to understanding the mechanisms of prostate cancer.
Keywords: Angiogenesis, Blood Coagulation Factors, Cancer Chemoprevention, Cell Adhesion, Cell Death, Cell Migration, Enzyme Inhibitors, Flow Cytometry, Kallikrein, Protein Sequence
Abstract
Prostate cancer is the most common type of cancer, and kallikreins play an important role in the establishment of this disease. rBbKIm is the recombinant Bauhinia bauhinioides kallikreins inhibitor that was modified to include the RGD/RGE motifs of the inhibitor BrTI from Bauhinia rufa. This work reports the effects of rBbKIm on DU145 and PC3 prostate cancer cell lines. rBbKIm inhibited the cell viability of DU145 and PC3 cells but did not affect the viability of fibroblasts. rBbKIm caused an arrest of the PC3 cell cycle at the G0/G1 and G2/M phases but did not affect the DU145 cell cycle, although rBbKIm triggers apoptosis and cytochrome c release into the cytosol of both cell types. The differences in caspase activation were observed because rBbKIm treatment promoted activation of caspase-3 in DU145 cells, whereas caspase-9 but not caspase-3 was activated in PC3 cells. Because angiogenesis is important to the development of a tumor, the effect of rBbKIm in this process was also analyzed, and an inhibition of 49% was observed in in vitro endothelial cell capillary-like tube network formation. In summary, we demonstrated that different properties of the protease inhibitor rBbKIm may be explored for investigating the androgen-independent prostate cancer cell lines PC3 and DU145.
Introduction
Prostate cancer is the leading cause of death in males (1). An increase in the expression of several proteases is verified in prostate cancer cells. This increase is especially present in serine proteases, which are involved in different events of tumor establishment, such as proliferation, adhesion, migration, angiogenesis, and apoptosis (2–6). The tissue serine protease kallikrein 3 (also known as prostate specific antigen) plays a relevant role in this disease; prostate cancer can be diagnosed through the quantification of prostate specific antigen levels in the serum of sick individuals (7, 8). The increase of the availability of insulin-like growth factors by tissue kallikrein proteolytic cleavage of insulin-like growth factor-binding proteins can interfere on survival and cell differentiation (9).
The ability of cells to adhere to other cells or extracellular matrix is important for growth, development, apoptosis, inflammation, migration, tumorigenesis, and immune responses (10, 11). Integrins play a central role in these processes (12–14). Integrins are heterodimeric receptors that are formed by noncovalently associated α and β subunits (10). Examples of these receptors include α2β1, α3β1, α7β1, α8β1, αvβ1, αvβ3, αvβ5, αvβ6, αvβ8, and αIIbβ3 (15). These receptors have a recognition site that binds to the extracellular matrix and consists of an arginine-glycine-aspartic acid (RGD) sequence, which is found in vitronectin, laminin, fibronectin, and osteopontin (15–19). Thus, the inhibition of ligand-receptor interactions that recognizes RGD sequence may be utilized in designing drugs (16, 20).
The progression of a normal cell to a cancer cell involves the ability of the cell to stimulate angiogenesis. When a solid tumor reaches 1–2 mm, the growth of the tumor is due to their angiogenic capacity (21). Proangiogenic factors include VEGF, basic fibroblast growth factor, PDGF, placenta growth factor, and TGF-β, among others (22, 23). Compounds that have the RGD sequence recognize αvβ3 integrin and act as antagonists that inhibit the formation of blood vessels (16, 24). These compounds can be used as antiangiogenic for the treatment of cancer.
Protease inhibitors are capable of regulating apoptosis and the cell cycle (25). Bauhinia seeds have serine and cysteine proteases inhibitors (26–28). BbKI (the Bauhinia bauhinioides kallikrein inhibitor), which is obtained from seeds of B. bauhinioides, is an 18-kDa protein that has a primary structure similar to that of other plant Kunitz-type inhibitors but is devoid of disulfide bridges (29). BbKI inhibits plasma kallikrein, plasmin, bovine trypsin, bovine chymotrypsin, porcine pancreatic kallikrein, and murine plasma kallikrein (29). BbKI recombinant protein was obtained by heterologous expression and production in Escherichia coli (30). Nakahata et al. (20) isolated an inhibitor of Bauhinia rufa named BrTI (B. rufa trypsin inhibitor) that inhibits human plasma kallikrein (Ki app = 14 nm) in addition to trypsin (Ki app = 2.9 nm). The distinctive features of the inhibitor structure are the RGD and RGE sequences responsible for the inhibition of cellular adhesion at positions 25–27 and 128–130 (26).
To evaluate the hypothesis that the presence of the RGD/RGE sequences in plant proteins is related to their defense against predators, Sumikawa et al. (31) expressed a recombinant inhibitor based on the primary sequence of BbKI. The amino acid residues around P21–28 in BbKI were replaced by those present in BrTI (V21E, S24A, H25R, H27D, and A28G) and region P127–130 (E127D and Q130E). These replacements resulted in the production of a modified inhibitor named rBbKIm (B. bauhinioides kallikrein inhibitor modified). rBbKIm inhibits trypsin (Ki app = 1.6 nm), chymotrypsin (Ki app = 7.4 nm), and human plasma kallikrein (Ki app = 3.6 nm) on the same order of magnitude as the native protein.
Because of the characteristics presented above, the use of this atypical inhibitor in a prostate cancer model is an important tool for studying the complex network involved in the proteolytic establishment of this malignancy. Therefore, this work aims to describe the action of the recombinant B. bauhinioides kallikrein inhibitor modified (rBbKIm) in DU145 and PC3 cell lines and its capacity as an antiangiogenic drug.
EXPERIMENTAL PROCEDURES
Heterologous Expression and Purification of rBbKIm
The inhibitor rBbKIm was obtained by protocols described by Sumikawa et al. (31). Briefly, the recombinant pET-29a(+) plasmid was transformed into BL21(DE3) (Novagen, Madison, WI) cells harboring pET29BbKIm and grown in LB medium (Invitrogen) supplemented with 30 μg/ml kanamycin (Invitrogen) at 37 °C. When the absorbance of the culture at 600 nm reached a value of 0.4, isopropyl β-d-thiogalactopyranoside (Invitrogen) was added at a final concentration of 0.2 or 0.5 mm, and the culture was grown for additional 3 h. Subsequently, the cells were harvested by centrifugation (4000 × g, 20 min, 4 °C) (Hitachi Himac CR21-GIII). The pellets were then resuspended in 10 ml of lysis buffer (0.05 m Tris/HCl, pH 8.0, and 0.15 m NaCl). After the bacteria were disrupted by sonication (Unique, Campinas, Brazil) and lysed in 0.5% Triton X-114 (Sigma-Aldrich) (eight cycles of ultrasound, 30 s, 40 W), the soluble protein fraction was isolated by centrifugation (4000 × g, 20 min, 4 °C). The protein was precipitated from the supernatant by adding acetone (80%, v/v) at 4 °C. The sediment was separated by centrifugation, dried at ambient temperature, dissolved in 10 mm Tris/HCl buffer, pH 8.5, and dialyzed against 50 mm Tris/HCl, pH 8.5. Subsequently, the soluble fraction was chromatographed on a DEAE-Sepharose Fast Flow (GE Healthcare) and equilibrated with 0.1 m Tris/HCl, pH 8.5. The inhibitor was eluted with 0.05 m NaCl in the same buffer. The fractions that inhibited trypsin activity were pooled and chromatographed on a Superose 12 (GE Healthcare) Äkta Purifier system for the removal of possible contaminants.
Protease Inhibition Assays
The inhibition of trypsin was used to evaluate the activity of the expressed protein as a model for enzymes with the same mechanism of action, called trypsin-type enzymes, such as the kallikrein family (32–34). The assays were performed in a 96-well plate in a final volume of 250 μl as described by Oliva et al. (35). Briefly, the inhibitor activity was determined by preincubation for 10 min at 37 °C in 0.05 m Tris/HCl, pH 8.0, containing 0.02% CaCl2 and trypsin (40 nm). Then 1.0 mm of the substrate α-benzoyl-d-l-arginine-ρ-nitroanilide was added to the reaction and incubated for 30 min at 37 °C. Ki app values were determined by adjusting the experimental points to the equation for tight binding by nonlinear regression analysis using the Grafit software (36).
Cell Lines
The cell lines DU145 and PC3 were purchased from ATCC (Manassas, VA). Fibroblasts isolated from amniotic fluid (WPF5) were kindly supplied by Prof. Walter Pinto Jr. (Campinas, Brazil) and used between passages 3 and 6. Human umbilical vein endothelial cells (HUVECs)3 were kindly provided by Prof. Júlio Scharfstein. The prostate adenocarcinoma cell lines (PC3 and DU145) and HUVECs were cultured in RPMI 1640 (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), 100 μg/ml streptomycin, and 100 units/ml penicillin at 37 °C under 5.0% CO2 atmosphere. The fibroblast cell line was cultured in DMEM (Invitrogen) supplemented with 20% fetal bovine serum (Invitrogen), 100 μg/ml streptomycin, and 100 units/ml penicillin (Invitrogen) at 37 °C under 5.0% CO2 atmosphere.
Evaluation of the Degradation of the Substrate HD-Pro-Phe-Arg-ρNa by Proteins of the Conditioned Medium of DU145 and PC3
DU145 and PC3 cells (1 × 106) were seeded into plates 100 mm in diameter (TPP (Techno Plastic Products AG), Trasadingen, Switzerland) for 24 h in RPMI medium supplemented with 10% FBS. Subsequently, the medium was changed to RPMI medium without FBS for 12 h, and 50 μm rBbKIm was added. After 48 h, the supernatant was removed, centrifuged (563 × g for 10 min), and concentrated four times using an ultrafiltration membrane with an exclusion pore size of 14 kDa (Amicon, Millipore, Brazil), which resulted in the conditioned medium (CM). Total protein concentration of the CM was quantified by the micro BCA kit, according to manufacturer's instructions (Pierce).
To evaluate the degradation of the substrate HD-Pro-Phe-Arg-ρNa by proteins of the conditioned medium of DU145 and PC3, an enzymatic reaction composed of 50 mm Tris/HCl buffer, pH 8.0, 0.5 m NaCl, 50 μg of total protein present in the CM and, 0.5 mm HD-Pro-Phe-Arg-ρNa substrate was used. After 24 h at 37 °C, the absorbance was measured at 405 nm in a Packard spectrophotometer (SpectraCount model; Packard).
MTT Cell Viability Assay
Cell viability was determined by the modified colorimetric MTT (Sigma-Aldrich) assay. DU145, PC3, and fibroblast cells and HUVECs were plated in 96-well plates (TPP) at a density of 5.0 × 103 or 8.0 × 103 cells per well. Each well contained 100 μl of culture medium and 100 μl of different concentrations of rBbKIm (0–100 μm), soybean trypsin inhibitor (SbTI) (0–100 μm), and LPS (0–100 μg). After 24, 48, and 72 h of culture, MTT (0.5 mg/ml in PBS) was added to the wells (2 h, 37 °C), followed by the removal of MTT solution and the addition of 100 μl/well of Me2SO (Sigma-Aldrich) to solubilize the cells. The absorbance was measured at 540 nm using a spectrophotometer (SpectraCount model; Packard). Each experimental condition was performed in triplicate.
Cell Adhesion Assays
The cell adhesion assays were performed in triplicate according to Nakahata et al. (20). Briefly, 24-well culture plates were coated with fibronectin (Millipore), laminin (Sigma-Aldrich), and collagens I and IV (Sigma-Aldrich) (4 μg/100 μl/well) and incubated overnight at 4 °C. The wells were blocked with 1% BSA in PBS (100 μl/well) for 1 h at 37 °C and washed three times with PBS. DU145 or PC3 (5 × 104 cells/50 μl/well) cells were preincubated with rBbKIm in different concentrations for 30 min. Subsequently, the cells and inhibitor were added to the wells and incubated for 4 h at 37 °C. Nonadherent cells were washed with PBS buffer, pH 7.4, three times. The remaining adherent cells were fixed with 100% methanol for 5 min, washed three times with PBS buffer, pH 7.4, and stained with 1% (v/v) toluidine blue in 1% sodium tetraborate for 5 min. The wells were exhaustively washed with PBS, and the absorbed stain was dissolved in 1% (v/v) SDS for 30 min at 37 °C. The absorbance was measured at 540 nm using a spectrophotometer (SpectraCount model; Packard). Each experimental condition was performed in triplicate.
Wound Healing
DU145 and PC3 cells and HUVECs (2 × 105 cells/well) were seeded in 24-well culture plates (TPP) until complete confluence. With the support of a pipette tip, a slit of cells were scraped from the plate, the medium was aspirated, and the cells were treated with rBbKIm (50 and 100 μm rBbKIm in DU145 and PC3 cells, and 50 μm rBbKIm in HUVECs) and SbTI (50 and 100 μm in DU145 and PC3 cells) in 10% FBS. The wound healing was observed at 0, 15, and 23 h (DU145 and PC3) or 0 and 18 h (HUVECs). For each time of analysis, three measurements were performed from one ledge to another slit. The average distance between the edges at 15 and 23 h (DU145 and PC3) and 18 h (HUVEC) were subtracted from the average at time 0, and we then compared the mean migration of the treated cells with the control (untreated cells) (37).
Transwell Migration Chamber
To evaluate the migration of HUVEC cell in vitro, 4 × 104 cells in 250 μl of serum-free RPMI 1640 medium with or without rBbKIm were placed in the top of cell culture inserts of 8-μm pore size (Millicell; Millipore). The lower chambers were filled with 300 μl of RPMI supplemented with 2% heat-inactivated FCS (Sigma-Aldrich) to serve as a chemoattractant for cell migration. The cells were treated with 50 or 100 μm rBbKIm and the control cells with a medium containing 7 mm HEPES, pH 7.4 (vehicle). The plates were incubated for 24 h at 37 °C under 5.0% CO2 atmosphere. Transwell units were removed from 24-well plates and fixed with methanol for 30 min, the cells in the upper chamber were removed, and the cells of the lower compartment were stained with 1% toluidine blue (Sigma-Aldrich) for 15 min. Then these cells were counted under a light microscope by two independent observers.
Flow Cytometry Analysis of Cell Apoptosis
DU145 and PC3 cells (1 × 105 cells) were seeded on 6-well plates (TPP) for 24 h for complete adhesion. After this time, the wells were washed three times with medium RPMI without FBS at incubation periods of 15 min at 37 °C and 5% CO2. The cells were incubated for 24 h in RPMI without FBS to synchronize the cell cycle. Thereafter, the cells were treated with rBbKIm and SbTI (50 and 100 μm) for 24 and 48 h in RPMI without FBS at 37 °C and 5% CO2. At the end of each incubation, the cells were removed from the plate using trypsin-EDTA solution (Cultilab, Campinas, Brazil), transferred to cytometry tubes, and washed with 500 μl of binding buffer (BD PharMingen kit), centrifuged, and resuspended in 50 μl of the same buffer, 3 μl of annexin V-FITC and 5 μl of propidium iodide (PI). The reaction mixture was incubated for 30 min in the dark, and then 300 μl of the same binding buffer was added to each tube and analyzed by flow cytometry (FACSCalibur; BD Biosciences) (38, 39). The cells were fixed with 2% (v/v) paraformaldehyde for 30 min and centrifuged. One hundred microliters of PBS containing 0.01% (v/v) saponin and 0.4 mg/ml RNase A (Sigma-Aldrich) was added to each tube, and the tubes were incubated for 30 min at 37 °C. At the end of the incubation period, 5 μl of 1 mg/ml propidium iodide and 200 μl of PBS were added to each tube and analyzed by flow cytometry (FACSCalibur; BD Biosciences) (40). The results were analyzed by ModFit LT 3.2.
Cytochrome c Release Assay
DU145 and PC3 cells were seeded (5 × 104 cells/well) on 13-mm glass coverslips, placed in 24-well plates and incubated for 24 h at 37 °C and 5% CO2 for full compliance. After this period, the cells were incubated in RPMI medium without FBS for 24 h. Subsequently, the cells were treated with 50 and 100 μm rBbKIm in RPMI medium without FBS and incubated at 37 °C and 5% CO2 for 24 h. At the end of the treatment, the cells were washed with PBS, and the mitochondria were labeled with Mitotracker Deep Red 633 (1:500) (Molecular Probes) for 20 min in the dark and fixed with 2% (v/v) paraformaldehyde in PBS for 15 min. The cells were washed again three times with PBS containing 0.01 m glycine and then permeabilized with 0.01% saponin for 15 min. After this time, the cells were stained with primary antibody, mouse anti-cytochrome c (1:400) (R&D Systems), for 2 h. Afterward, the cells were stained with secondary antibody, anti-mouse IgG Alexa 488 (1:500) (Invitrogen), for 40 min. Next, the cell nuclei were labeled with the fluorophore DAPI (1:3000) (Invitrogen) for 30 min and then fixed to slides with 7 μl of fluoromount G (Electron Microscopy Sciences). The visualization and measurement of the fluorescence slides were performed in a confocal scanning microscope Carl Zeiss LSM780 laser model, 63× objective, using oil immersion and a numerical aperture of 1.43. The program used for image acquisition was Zen 2010.
Determination of Caspase-3 Activation by Flow Cytometry
DU145 and PC3 cells (1 × 105 cells) were seeded in each well of a 6-well plate (TPP) containing RPMI medium supplemented with 10% fetal bovine serum and 100 IU/ml penicillin/streptomycin solution for adhesion. The cells were then washed three times with RPMI medium without FBS and incubated for 24 h in RPMI without FBS. Twenty-four hours later, the medium was replaced with two different concentrations (50 and 100 μm) of rBbKIm and SbTI for 48 h at 37 °C and 5% CO2. At the end of the treatment, the cells were removed from the plate using a trypsin-EDTA solution (Cultilab), transferred to cytometry tubes, and fixed with 100 μl of 2% paraformaldehyde (v/v) for 30 min at room temperature. The cells were resuspended in 200 μl of 0.01% glycine in PBS and incubated for 15 min at room temperature. The cells were then resuspended in 200 μl of 0.01% saponin in PBS and incubated for 15 min at room temperature. Finally, the cells were incubated with 10 μl of anti-cleaved caspase-3 conjugated with Alexa Fluor 488 (BD Biosciences) for 40 min. The results were analyzed by flow cytometry (FACSCalibur; BD Biosciences).
Determination of Caspase-9 Activation
DU145 and PC3 cells (1 × 105 cells) were seeded into plates 100 mm in diameter (TPP) containing RPMI medium supplemented with 10% fetal bovine serum and 100 IU/ml penicillin/streptomycin solution for 24 h. The cells were then washed three times with RPMI medium without FBS and incubated for 12 h in the same medium. After 12 h, the medium was replaced with RPMI medium without FBS, and the cells were treated with 50 μm rBbKIm for 48 h. After 48 h, the cells were washed twice with cold PBS, scraped, and centrifuged. The pellet was frozen at −80 °C. For cell lysis, a lysis buffer containing 25 mm HEPES buffer, pH 7.4 plus 0.1% Chaps (Sigma-Aldrich), 1 mm EDTA, 2 mm MgCl2, 2 mm DTT, and mixture protease inhibitors (1:10) (Roche Applied Science) was used. The pellet was resuspended in 50 μl of lysis buffer and incubated on ice for 20 min. The cell pellets were frozen in dry ice, thawed at 37 °C, and vortexed; this cycle was repeated 10 times. At the end, the cells were centrifuged at 15,771 × g for 10 min at 4 °C. The supernatant with the cytoplasmic proteins was quantified by the micro BCA method (Pierce). Thirty micrograms of total protein was incubated in buffer (25 mm HEPES buffer, pH 7.4 containing 10% sucrose, 0.1% Chaps, and 1 mm DTT) with 20 mm Ac-Leu-Glu-His-Asp-AFC substrate (Calbiochem) at 37 °C, and readings were performed every 15 min in a fluorometer (Spectramax Gemini EM, Molecular Devices Corporation) using λex = 400 nm and λem = 505 nm.
Measurement of Ca2+ Mobilization in Cells
Ca2+ was measured spectrophotometrically using the fluorescent probe fura-2/AM (Molecular Probes). DU145 (3 × 104 cell/well) and PC3 (3.5 × 104 cell/well) cells were seeded in 6-well plates (TPP) for 24 h for complete adhesion. After this time, the cells were suspended in Hanks' balanced salt solution and treated with rBbKIm (100 and 200 μm) for 1 h at 37 °C and 5% CO2. Subsequently, the cells were loaded with the probe (2 μm) for 40 min and transferred to a fluorometer (SPEX FluoroLog-2, AR-CM System; PerkinElmer Life Sciences) using λexc = 340 e 380 nm and λem = 505 nm. The cells were either stimulated with thrombin (0.5 UI/ml), an agonist for PARs receptors, or not stimulated.
In Vitro Angiogenesis Assay on Matrigel
To evaluate angiogenic structure formation, HUVECs were used. This assay was performed as described by Paschoalin et al. (41). MatrigelTM (BD Biosciences) was added to cold 96-well plates (TPP) and incubated at 37 °C for 1 h for their polymerization. HUVECs supplemented with 0.2% FBS were seeded on MatrigelTM at a density of 5 × 103 cells/well in the presence and absence of 50 and 100 μm of rBbKIm at 37 °C and 5% CO2. After 24 h, the tubular structures that were formed were observed under a microscope light and photographed for subsequent counting. The control sample (untreated) was considered to be 100%, and the formation of tubular structures of each treated sample was calculated in relation to control.
Statistical Analysis
The data were analyzed as the means ± S.D., and statistical significance was determined using ANOVA and t tests (SigmaPlot 10.0). p < 0.05 (*) was considered to be significant.
RESULTS
Action of rBbKIm and SbTI on Cell Viability and Cell Death
The effect of rBbKIm on the viability of DU145 and PC3 cells was analyzed by the MTT assay at different time points (24, 48, and 72 h) (Fig. 1). The results of our studies have shown that rBbKIm in DU145 cells were more subtle because the inhibitor was not effective at low concentrations (less than 50 μm). However, after 48 h, rBbKIm effectively inhibited cell viability at concentrations in the range of 50–100 μm (reduction by 33 and 56%, respectively), and a slight increase in this effect was observed after 72 h of incubation (Fig. 1A). The action of rBbKIm in the early phase of proliferation (24 h) was effective in PC3 cells, and 25 and 50 μm of rBbKIm inhibited the cell viability by 29 and 49%, respectively. The effectiveness of inhibition was better observed in PC3 cells after 48 h of incubation with 25 and 50 μm rBbKIm in which a decrease in cell viability of 58 and 73% was observed, respectively. Compared with 48 h, no significant modification in this activity was observed after 72 h of incubation (Fig. 1B). We should note that the reduction of fibroblast viability was less at concentrations above 25 μm (Fig. 1C). Comparing the effects of rBbKIm with that of SbTI, at incubation times and concentrations in which the rBbKIm action was more significant, one can notice that SbTI only slightly decreased PC3 cell viability (100 μm, 48 h), and also this inhibitor did not interfere on DU145 cell viability (Fig. 1, D and E). Indeed, to directly address whether components from the bacterial preparations could be responsible for the rBbKIm effects, the bacterial endotoxin LPS was also investigated on DU145 and PC3 (Fig. 1F). The results showed that, if present, this bacterial component did not interfere with cell viability. Therefore, these results suggest that rBbKIm has a more specific effect on prostate cancer cells.
FIGURE 1.

The effect of rBbKIm, SbTI, and LPS on cell viability. A–C, DU145 (A), PC3 (B), and fibroblast (C) cells were treated with increasing concentrations (0–100 μm) of rBbKIm for 24, 48, and 72 h. D and E, DU145 (D) and PC3 (E) cells were treated with increasing concentrations (0–100 μm) of SbTI for 24 and 48 h. F, DU145 and PC3 cells were treated with increasing concentrations (0–100 μm) of LPS for 24 h. Cell viability was measured using the MTT assay. The control cells were treated with a medium containing 7 mm HEPES, pH 7.4 (vehicle). The calculation of the percentage of inhibition was performed for the control cells (untreated) at each incubation time. The values are expressed as the means ± standard deviation of the representative experiment. Significant differences versus controls are presented (ANOVA; *, p < 0.05).
Moreover, the experimental data on the cell cycle phases showed a distinct effect of the inhibitor, because DU145 rBbKIm treatment (50 and 100 μm, 24 and 48 h) did not significantly alter the cell cycle compared with the control (untreated cells) (Fig. 2A). Whereas rBbKIm did not affect the PC3 cell cycle in an early phase (24 h), after 48 h 100 μm rBbKIm induced cell cycle arrest in the G0/G1 and G2/M phases (Fig. 2B). Moreover, SbTI did not lead to DU145 and PC3 cell cycle arrest (Fig. 2, C and D).
FIGURE 2.

rBbKIm induces apoptosis and cell arrest in prostate cancer lineages after 48 h of treatment. The cells were exposed to 50 and 100 μm of rBbKIm and SbTI for 24 and 48 h. A–D, subsequently, the cell cycle was evaluated using the PI label, G0/G1 (black columns), S (white columns), and G2/M (gray columns). E–H, cell death was assessed by flow cytometry using the annexin V-FITC and PI label, annexin V−PI− (black columns), annexin V+PI− (light gray columns), annexin V+PI+ (dark gray columns), and annexin V−PI+ (white columns). A and B, rBbKIm promotes cell cycle arrest after 48 h of treatment in PC3 cells (B) but not in DU145 cells (A). C and D, SbTI had no effect on cell cycle in both cells. E and F, low levels of cell death at 24 h of rBbKIm treatment in DU145 (E) and PC3 (F) cells was observed; however, after 48 h, ∼50% of the cells died by apoptosis (annexin V+PI−). G and H, SbTI did not induced DU145 (G) and PC3 (H) apoptosis. The experiment is representative of two independent experiments. The values are expressed as the means ± standard deviation. Significant differences versus controls are presented (ANOVA; *, p < 0.05).
In addition, DU145 and PC3 cells were treated with 50 and 100 μm rBbKIm for 24 and 48 h to evaluate cell death by apoptosis using annexin V and PI. When incubating DU145 cells with 50 μm and 100 μm rBbKIm for 24 h, 45% and 60% of DU145 cells were annexin V+PI−, respectively (Fig. 2E). After 48 h of treatment with 50 or 100 μm rBbKIm, 64 and 68% of DU145 cells were annexin V+PI−, respectively (Fig. 2E). Furthermore, after a 24 h treatment with 50 and 100 μm rBbKIm, PC3 cells showed a slight increase in apoptotic cell death in comparison to control without inhibitor (Fig. 2F). And, after 48 h of treatment with 50 or 100 μm rBbKIm, 48 and 44% of PC3 cells were annexin V+PI−, respectively (Fig. 2F). Together, these results suggest that apoptotic cell death was induced by rBbKIm. In contrast, SbTI, at 24 and 48 h, did not induce apoptotic cell death in both cell lines (Fig. 2, G and H).
To corroborate whether cell death induced by rBbKIm occurs by apoptosis, cytochrome c release and caspase activation analyses were performed in DU145 and PC3 cells. Previous to rBbKIm treatment, we observed a colocalization of MitoTracker Red and cytochrome c in both cell lines, indicating that cytochrome c is present in the mitochondria. Cytochrome c did not colocalize with the MitoTracker after rBbKIm (50 or 100 μm) treatment, indicating the release of cytochrome c from the mitochondria to the cytoplasm of DU145 and PC3 cells (Fig. 3).
FIGURE 3.

Mitochondrial cytochrome c is released by DU145 and PC3 cells. Scanning confocal microscopy of DU145 (A) and PC3 (B) cells was performed after treatment with 50 and 100 μm rBbKIm for 24 h. The mitochondria were stained with MitoTracker Red (red), cytochrome c was labeled using anti-cytochrome c, and the secondary antibody, mouse anti-IgG, was conjugated with Alexa Fluor 488 (green). The nuclei were stained with DAPI (blue). It is possible to observe that both treatments released cytochrome c from the mitochondria. The images were acquired using the confocal scanning microscope Carl Zeiss LSM780. Bars, 10 nm.
The apoptotic process involves the activation of caspases, which are essential to unraveling the mechanism of the action of cytotoxic drugs. Flow cytometric analysis showed an activation of caspase-3 in the DU145 cells treated with rBbKIm (50 and 100 μm) for 48 h (Fig. 4A), but not with SbTI (Fig. 4C). Similarly, caspase-3 was not activated in PC3 cells treated with rBbKIm (Fig. 4B) and SbTI (Fig. 4D). The activation of caspase-9 was evaluated with the fluorogenic substrate LQHD-AFC, and the cleavage of the AFC fluorophore was monitored. Only PC3 showed a significant activation of caspase-9; DU145 did not present any caspase-9 activity (Fig. 4E).
FIGURE 4.

Analysis of the activation of caspase-3 and caspase-9. The caspase activity and activation were investigated by a fluorescence assay and flow cytometry. A–D, flow cytometric analysis of the endogenous levels of activated caspase-3 in DU145 and PC3 cells treated with rBbKIm (A and B) and SbTI (C and D) 50 and 100 μm. The filled and open histograms represent unstimulated and stimulated samples, respectively. E, caspase-9 activation assay was performed by incubation of the substrate LQHD-AFC 20 mm with cellular lysate at 37 °C for 4 h (λexc = 400 nm and λem = 505 nm). The values are expressed as the means ± standard deviation of a representative experiment performed in triplicate. Significant differences versus controls are presented (t test; *, p < 0.05).
Hydrolytic Activity of Proteases in the Conditioned Medium of DU145 and PC3 Cells
To evaluate the effect of rBbKIm on DU145 and PC3 extracellular proteases, the culture medium supernatant of DU145 and PC3 cells (CM) previously treated with rBbKIm for 48 h (RPMI medium without FBS) was submitted to an enzymatic reaction in the presence of HD-Pro-Phe-Arg-ρNa, a specific substrate for plasma and tissue kallikreins. As depicted in Fig. 5, in which the DU145-CM and PC3-CM were incubated at 37 °C for 24 h, there was a 95% inhibition of substrate hydrolysis, demonstrating the effectiveness of the inhibitor for blocking serine protease release by both prostate cancer cells.
FIGURE 5.
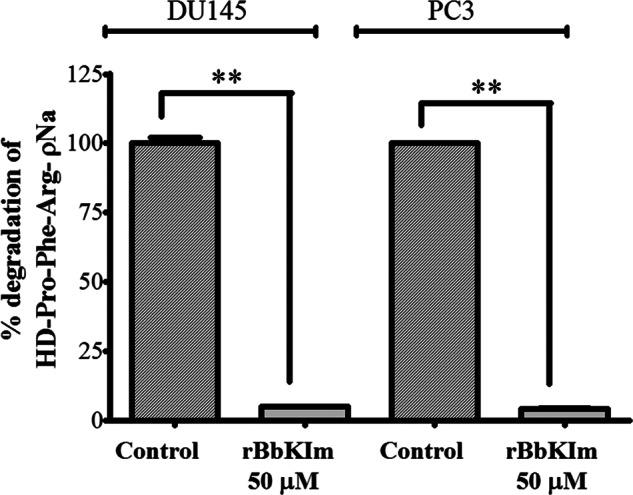
Evaluation of the degradation of the substrate HD-Pro-Phe-Arg-ρNa proteins by DU145 and PC3-CM cells pretreated with rBbKIm. CM-DU145 and CM-PC3 were collected from untreated cells (control) and cells treated with rBbKIm for 48 h. CM-DU145 and CM-PC3 (50 μg) were incubated in Tris/HCl 50 mm, pH 8.0, containing 0.5 m NaCl and the substrate HD-Pro-Phe-Arg-ρNa (0.5 mm) at 37 °C for 24 h. The values are expressed as the means ± standard deviation of a representative experiment performed in triplicate. Significant differences versus controls are presented (t test; **, p < 0.0001).
Action of rBbKIm on DU145 and PC3 Cell Adhesion
The effect of DU145 and PC3 cell adhesion on extracellular matrix proteins such as fibronectin, laminin, and collagen I and IV in the presence of rBbKIm was evaluated. The results show that rBbKIm (3.125–100 μm) did not inhibit DU145 cell adhesion to fibronectin, laminin, or collagen I and IV (Fig. 6, A and B). The effect of rBbKIm on PC3 cell adhesion was very similar to DU145 cell adhesion, and there was no inhibition of PC3 cell adhesion to matrix proteins (Fig. 6, C and D). Conversely, rBbKIm increased the adherence of DU145 cells to collagen IV at concentrations up to 12.5 μm (Fig. 6B) and of PC3 cells to laminin and collagen I and V (Fig. 6, C and D).
FIGURE 6.

The effect of rBbKIm on cell adhesion. A and B, DU145 cell adhesion. C and D, PC3 cell adhesion. The control cells were treated with a medium containing 7 mm HEPES, pH 7.4 (vehicle). BSA promotes the inhibition of cell adhesion to the plastic. The experiment was performed in triplicate (ANOVA; *, p < 0.05).
Action of rBbKIm and SbTI on DU145 and PC3 Cell Migration
The action of the inhibitors on the cell motility of DU145 cells (Fig. 7, A and B) indicates that 100 μm rBbKIm did not inhibit DU145 cell migration when incubated for 15 and 23 h of treatment, and 50 and 100 μm rBbKIm inhibited 23 and 38% of PC3 migration, respectively (Fig. 7, C and D), whereas SbTI inhibitor did not interfere on DU145 and PC3 cells migration (Fig. 7, E–H).
FIGURE 7.

DU145 and PC3 cell migration assay. The graph represents the percentage of cell migration after 15 and 23 h. A and B, DU145 cell migration with or without rBbKIm. C and D, PC3 cell migration with or without rBbKIm. E and F, DU145 cell migration with or without SbTI. G and H, PC3 cell migration with or without SbTI. The values are expressed as the means ± standard deviation of a representative experiment performed in triplicate. Significant differences versus controls are presented (ANOVA; *, p < 0.05).
Action of rBbKIm on Ca2+ Mobilization
The ability of rBbKIm to stimulate calcium mobilization was evaluated and compared with thrombin, a canonical endogenous agonist for PARs. Thrombin greatly increases Ca2+ released from internal storages. rBbKIm itself was not capable of inducing calcium mobilization, but in the presence of thrombin, an enzyme in which the proteolysis activity is not affected by the inhibitor, an important increase of calcium mobilization was observed (∼71%) in both lineages DU145 (Fig. 8A) and PC3 (Fig. 8B). These results suggest that the effects of rBbKIm appear to be dependent on integrin activation, and the interaction of rBbKIm with RGD-binding integrins facilitates the increase of intracellular calcium (Fig. 8).
FIGURE 8.
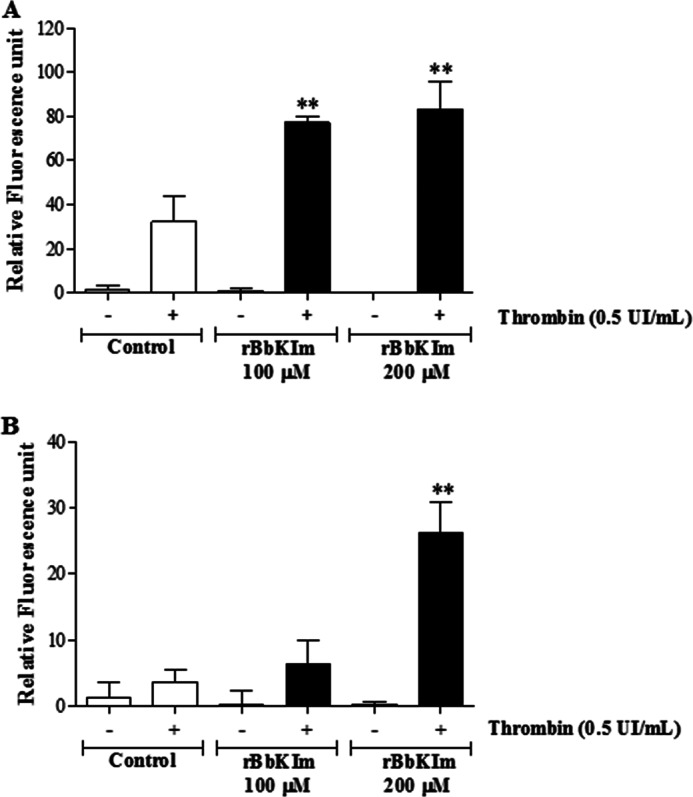
Cytosolic calcium mobilization. DU145 (A) and PC3 (B) cells were loaded with Ca2+ indicator Fura-2/AM, and fluorescence and intensity measurements were performed with a fluorometer. The cells were untreated (control) or pretreated with 100 or 200 μm rBbKIm. The values are expressed as the means ± standard deviation of a representative experiment performed in triplicate (ANOVA; **, p < 0.0001).
Action of rBbKIm on Angiogenesis
Numerous characteristics are being explored for the development of antitumorogenic agents. Among these agents are angiogenesis inhibitor drugs that are capable of modulating endothelial cell viability and migration. Thus, our study evaluated the effect of rBbKIm on cell viability, cell migration, and the formation of angiogenic structures in human endothelial cells (HUVECs). After 24 h of treatment, rBbKIm reduced HUVEC viability by 28 and 27% when concentrations of 50 and 100 μm were used, respectively (Fig. 9A). The effect of rBbKIm on the migration of HUVECs was measured by evaluating the percentage of cells that migrated through the slit after scraping the cells and the Transwell migration chamber. In the wound healing assay, rBbKIm (50 μm) inhibited migration by 22% after 18 h of treatment (Fig. 9B), similar results were obtained by Transwell assay (20 and 30% of inhibition with 50 and 100 μm rBbKIm, respectively, after 24 h of treatment) (Fig. 9C). Angiogenesis was tested in Matrigel using HUVECs, and an inhibition of angiogenic structures was observed when cells were treated with 50 and 100 μm of rBbKIm. In the presence of rBbKIm 50 μm, there was a 49% decrease in the formation of angiogenic structures. A 70% inhibition was observed when the cells were treated with 100 μm rBbKIm (Fig. 9, D and E). These results show that the inhibitor rBbKIm, in addition to acting on DU145 and PC3 prostate cancer cells, can also be a potent inhibitor of angiogenesis.
FIGURE 9.

Antiangiogenic activity of rBbKIm in vitro. A, the effect of rBbKI on HUVEC viability was determined by the MTT assay. HUVECs were incubated for 24 h with rBbKIm at different concentrations (0–100 μm). B, the HUVEC wound healing assay in the presence of rBbKIm (50 μm). C, the HUVEC Transwell migration assay in the presence of rBbKIm (50 and 100 μm). D, typical images of untreated HUVECs (control) and cells treated with rBbKIm (50 and 100 μm) were captured in an inverted microscope with a 5× objective glass. The arrows indicate the formation of angiogenic structures. E, angiogenic structure formation measurement. The values are expressed as the means ± standard deviation of two independent experiments. Significant differences versus controls are presented (ANOVA; *, p < 0.05).
DISCUSSION
The action of protease inhibitors has been studied in different models. Specially in cancer, protease inhibitors from Bowman-Birk and Kunitz families are the most studied (14, 42–45) not only because of the inhibition of the net activation of proteases but also because of the presence of atypical structures that invoke mechanisms that suppress pathways fundamental to the establishment of tumors. In this study, we used the chimeric inhibitor, rBbKIm. In addition to being a potent inhibitor of kallikreins, rBbKIm has a very atypical peptide sequence from another Bauhinia inhibitor that is known to be responsible for the inhibition of cellular adhesion (20). These features attracted our attention, and we selected prostate cancer as a model for our studies because of the importance of tissue kallikrein activation in the development of this disease (9, 46). The conserved activity of the purified cloned inhibitor was confirmed by its ability to block trypsin activity in vitro.
The importance of the inhibitor selectivity for trypsin-like activity secreted by these cells was demonstrated to be important and may reflect the cellular signaling events triggered by proteolysis, such as the activation of protease-activated receptors (PARs). Studies have shown that expression of PAR1 is clearly increased in advanced prostate cancer (47–49). The activation of PARs promotes proliferation, invasion, and release of angiogenic factors by cancer cells (50, 51), and kallikreins are enzymes that are known to cleave this receptor with a specificity similar to thrombin (52, 53). Thus, it is possible to infer that rBbKIm led to the impairment of these prostate cellular events by its direct inhibitory action.
The contribution of the motif sequences RGD/RGE in the suppression of cell adhesion differed from previous reports, which highlights the importance of the recognition by integrins. This recognition regulates the invasive and metastatic potential of tumor cells (16), and peptides bearing this motif have been utilized in cell attachment inhibition (54–57). In particular, PC3 and DU145 cells express high levels of α5β1 and αvβ3 integrins that recognize the RGD sequence in cell adhesion proteins (54). The activation of PAR1 decreases PC3 cell adhesion to collagen I and IV and laminin (58), which leads to an enhancement of metastasis through improved DU145 and PC3 cell motility, and apoptosis is prevented (59). This information helped us understand the effect of rBbKIm: the RGD domain promoted the adhesion of DU145 and PC3 cells to collagen IV and PC3 cells to laminin and collagen I. An increase of 37 and 71% in adhesion was observed when DU145 cells were treated with 100 μm rBbKIm and seeded on either laminin or collagen IV, respectively. In PC3 cell adhesion, an increase of 43% on laminin and 51% on collagen I was observed. A slight increase of 23% in cell adhesion was also observed when PC3 cells were seeded on collagen IV. Our results suggest that these effects may be a consequence of the inhibition of extracellular proteases capable of activating the PAR1 receptor localized in the cell membrane. The ineffectiveness of rBbKIm on DU145 and PC3 adhesion to fibronectin may be related to the fact that the adhesion to fibronectin is poorly affected by the activation of PAR1, which is mainly due to the α5 subunit expression being lower compared with that of collagen and laminin receptors (58). Cell adherence on the stromal prostate is important to prevent the entry of cancer cells into the bloodstream, thus preventing metastasis in distant tissues (56).
Although the binding of rBbKIm to the cell surface did not impair cell adhesion, the deleterious effect on DU145 and PC3 viability suggests that this macromolecule provides signals that interfere with cell survival. Integrins are unable to bind to their receptors in an inactive state (60). Extracellular matrix-integrin binding evokes molecular signaling cascades. By using fluorescence assays, we were able to demonstrate that rBbKIm potentiates increase of cytoplasmic calcium concentration by thrombin. This effect may be the consequence of the amplification of RGD/RGE binding recognition by the receptors that are more exposed after PAR activation (61). Thus, the rBbKIm binding signaling pathway is dependent of integrin activation. Our experimental procedure demonstrated that thrombin induces PAR activation and is a protease that is not affected by rBbKIm. Therefore, in cell culture, integrin activation is stretched by proteolysis produced by enzymes inert to rBbKIm action. Thus, this inhibitory action impaired the action of the PAR and delayed integrin activation. However, this inhibitory action may also contribute to calcium increase and subsequently trigger apoptosis signaling pathways (62).
The effect of rBbKIm was compared with the classical Kunitz plant inhibitor, SbTI, and in contrast to the inert effect of SbTI, rBbKIm was selective, causing prostate cancer cell death. The induction of apoptosis in DU145 is not related to cell cycle arrest. Studies have shown that the DU145 cell line has a mutation in the RB tumor suppressor gene (63) caused by a mutation in exon 21 (64), which encodes a truncated form of the Rb protein, promoting resistance to cell cycle arrest (65). Apoptosis in these cells suggests that an activation of caspase-3 occurs. However, we could not detect an activation of caspase-9, although the route has been activated via the mitochondrial release of cytochrome c.
Treatment of PC3 with rBbKIm suggests an induction of apoptosis via mitochondrial cytochrome c release and caspase-3 activation. This result was similar to previous results obtained (66) showing that the treatment of neuroblastoma cells with TNF-α induces cytochrome c release and caspase-9 activation but not caspase-3 activation. The PC3 cell death that occurred with rBbKIm treatment (100 μm/48 h) may be related to G0/G1 and G2/M cell cycle arrest as similarly demonstrated by Agarwal et al. (67) in which CR9–7 human fibroblasts were treated with tetracycline.
Another event implicated in the activation of PAR1 in PC3 is the increase in migratory ability, which is important in the process of invasion and metastasis (68, 69). rBbKIm inhibits cell migration of PC3 cells; however, this effect is not observed in DU145 cells. The distinct effect on PC3 cells can be understood if we consider the different proteases involved in the mechanism of cell migration in the cells that are affected by the inhibitor.
Cytotoxic drugs generally do not differentiate normal from metastatic tissues. Thus, it is important to develop new drugs that target proteins that are differentially expressed in cancer cells compared with normal adult tissues (70). This reasoning is why the action of rBbKIm on the viability of the amniotic fluid human fibroblast was analyzed. This drug did not inhibit the viability of these cells, which indicates its selectivity for tumor cells.
Angiogenesis occurs when the tumor is large in size and requires the diffusion of nutrients and oxygen (71, 72). The impairment of HUVEC cell migration, promoted by rBbKIm, may be a consequence of its effect on cell viability, leading to angiogenesis inhibition. These characteristics may be relevant to the establishment of antiangiogenic properties because the inhibitor reduced the formation of angiogenic structures in Matrigel, suggesting an ability to interfere with tumor metastasis. In summary, our findings provide a basis for using a distinct inhibitor to investigate the participation of proteases and activation of signaling pathways in different cell lines.
Acknowledgment
We thank Lucimeire Aparecida Santana (Instituto Nacional de Farmacologia/Universidade Federal de São Paulo, Brazil) for technical assistance.
This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico, by Fundação de Amparo à Pesquisa do Estado de São Paulo Grant 2009/53766-5, and by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior.
- HUVEC
- human umbilical vein endothelial cell
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- CM
- conditioned medium
- SbTI
- soybean trypsin inhibitor
- PI
- propidium iodide
- ANOVA
- analysis of variance
- PAR
- protease-activated receptor.
REFERENCES
- 1. Siegel R., Ward E., Brawley O., Jemal A. (2011) Cancer statistics, 2011. The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J. Clin. 61, 212–236 [DOI] [PubMed] [Google Scholar]
- 2. Chang C., Werb Z. (2001) The many faces of metalloproteases. Cell growth, invasion, angiogenesis and metastasis. Trends Cell Biol. 11, S37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bok R. A., Hansell E. J., Nguyen T. P., Greenberg N. M., McKerrow J. H., Shuman M. A. (2003) Patterns of protease production during prostate cancer progression. Proteomic evidence for cascades in a transgenic model. Prostate Cancer Prostatic Dis. 6, 272–280 [DOI] [PubMed] [Google Scholar]
- 4. Darmoul D., Gratio V., Devaud H., Lehy T., Laburthe M. (2003) Aberrant expression and activation of the thrombin receptor protease-activated receptor-1 induces cell proliferation and motility in human colon cancer cells. Am. J. Pathol. 162, 1503–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Podgorski I., Linebaugh B. E., Sameni M., Jedeszko C., Bhagat S., Cher M. L., Sloane B. F. (2005) Bone microenvironment modulates expression and activity of cathepsin B in prostate cancer. Neoplasia 7, 207–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. López-Otín C., Matrisian L. M. (2007) Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 7, 800–808 [DOI] [PubMed] [Google Scholar]
- 7. Clements J. A., Willemsen N. M., Myers S. A., Dong Y. (2004) The tissue kallikrein family of serine proteases. Functional roles in human disease and potential as clinical biomarkers. Crit. Rev. Clin. Lab. Sci. 41, 265–312 [DOI] [PubMed] [Google Scholar]
- 8. Thompson I. M., Pauler-Ankerst D., Chi C., Goodman P. J., Tangen C. M., Lippman S. M., Lucia M. S., Parnes H. L., Coltman C. A., Jr. (2007) Prediction of prostate cancer for patients receiving finasteride. Results from the Prostate Cancer Prevention Trial. J. Clin. Oncol. 25, 3076–3081 [DOI] [PubMed] [Google Scholar]
- 9. Pampalakis G., Sotiropoulou G. (2007) Tissue kallikrein proteolytic cascade pathways in normal physiology and cancer. Biochim. Biophys. Acta 1776, 22–31 [DOI] [PubMed] [Google Scholar]
- 10. Hynes R. O. (2002) Integrins. Bidirectional, allosteric signaling machines. Cell 110, 673–687 [DOI] [PubMed] [Google Scholar]
- 11. Legate K. R., Montañez E., Kudlacek O., Fässler R. (2006) ILK, PINCH and parvin. The tIPP of integrin signalling. Nat. Rev. Mol. Cell Biol. 7, 20–31 [DOI] [PubMed] [Google Scholar]
- 12. Arya M., Bott S. R., Shergill I. S., Ahmed H. U., Williamson M., Patel H. R. (2006) The metastatic cascade in prostate cancer. Surg. Oncol. 15, 117–128 [DOI] [PubMed] [Google Scholar]
- 13. Whitbread A. K., Veveris-Lowe T. L., Lawrence M. G., Nicol D. L., Clements J. A. (2006) The role of kallikrein-related peptidases in prostate cancer. Potential involvement in an epithelial to mesenchymal transition. Biol. Chem. 387, 707–714 [DOI] [PubMed] [Google Scholar]
- 14. de Paula C. A., Coulson-Thomas V. J., Ferreira J. G., Maza P. K., Suzuki E., Nakahata A. M., Nader H. B., Sampaio M. U., Oliva M. L. (2012) Enterolobium contortisiliquum trypsin inhibitor (EcTI), a plant proteinase inhibitor, decreases in vitro cell adhesion and invasion by inhibition of Src protein-focal adhesion kinase (FAK) signaling pathways. J. Biol. Chem. 287, 170–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Desgrosellier J. S., Cheresh D. A. (2010) Integrins in cancer. Biological implications and therapeutic opportunities. Nat. Rev. Cancer 10, 9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Trabocchi A., Menchi G., Cini N., Bianchini F., Raspanti S., Bottoncetti A., Pupi A., Calorini L., Guarna A. (2010) Click-chemistry-derived triazole ligands of arginine-glycine-aspartate (RGD) integrins with a broad capacity to inhibit adhesion of melanoma cells and both in vitro and in vivo angiogenesis. J. Med. Chem. 53, 7119–7128 [DOI] [PubMed] [Google Scholar]
- 17. Yamada K. M., Kennedy D. W. (1984) Dualistic nature of adhesive protein function. Fibronectin and its biologically active peptide fragments can autoinhibit fibronectin function. J. Cell Biol. 99, 29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pytela R., Pierschbacher M. D., Ruoslahti E. (1985) A 125/115-kDa cell surface receptor specific for vitronectin interacts with the arginine-glycine-aspartic acid adhesion sequence derived from fibronectin. Proc. Natl. Acad. Sci. U.S.A. 82, 5766–5770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Humphries J. D., Byron A., Humphries M. J. (2006) Integrin ligands at a glance. J. Cell Sci. 119, 3901–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nakahata A. M., Bueno N. R., Rocha H. A., Franco C. R., Chammas R., Nakaie C. R., Jasiulionis M. G., Nader H. B., Santana L. A., Sampaio M. U., Oliva M. L. (2006) Structural and inhibitory properties of a plant proteinase inhibitor containing the RGD motif. Int. J. Biol. Macromol. 40, 22–29 [DOI] [PubMed] [Google Scholar]
- 21. Folkman J. (1990) What is the evidence that tumors are angiogenesis dependent? J. Natl. Cancer Inst. 82, 4–6 [DOI] [PubMed] [Google Scholar]
- 22. Ferrara N. (1999) Molecular and biological properties of vascular endothelial growth factor. J. Mol. Med. 77, 527–543 [DOI] [PubMed] [Google Scholar]
- 23. Chau C. H., Figg W. D. (2010) Drug Management of Prostate Cancer: Principles of Antiangiogenic Therapy, Springer, New York [Google Scholar]
- 24. Giannis A., Rübsam F. (1997) Integrin antagonists and other low molecular weight compounds as inhibitors of angiogenesis: new drugs in cancer therapy. Angew. Chem. Int. Ed. Engl. 36, 588–590 [Google Scholar]
- 25. Kennedy A. R. (1998) Chemopreventive agents. Protease inhibitors. Pharmacol. Ther. 78, 167–209 [DOI] [PubMed] [Google Scholar]
- 26. Oliva M. L., Sampaio U. M. (2008) Bauhinia Kunitz-type proteinase inhibitors. Structural characteristics and biological properties. Biol. Chem. 389, 1007–1013 [DOI] [PubMed] [Google Scholar]
- 27. Oliva M. L., Silva M. C., Sallai R. C., Brito M. V., Sampaio M. U. (2010) A novel subclassification for Kunitz proteinase inhibitors from leguminous seeds. Biochimie 92, 1667–1673 [DOI] [PubMed] [Google Scholar]
- 28. Oliva M. L., da Silva-Ferreira R., Ferreira J. G., de Paula C. A., Salas C. E., Sampaio M. U. (2011) Structural and functional properties of kunitz proteinase inhibitors from leguminosae. A mini review. Curr. Protein Pept. Sci. 12, 348–357 [DOI] [PubMed] [Google Scholar]
- 29. Oliva M. L., Andrade S. A., Batista I. F., Sampaio M. U., Juliano M., Fritz H., Auerswald E. A., Sampaio C. A., (1999) Human plasma kallikrein and tissue kallikrein binding to a substrate based on the reactive site of a factor Xa inhibitor isolated from Bauhinia ungulata seeds. Immunopharmacology 45, 145–149 [DOI] [PubMed] [Google Scholar]
- 30. Araújo A. P., Hansen D., Vieira D. F., Oliveira C., Santana L. A., Beltramini L. M., Sampaio C. A., Sampaio M. U., Oliva M. L. (2005) Kunitz-type Bauhinia bauhinioides inhibitors devoid of disulfide bridges. Isolation of the cDNAs, heterologous expression and structural studies. Biol. Chem. 386, 561–568 [DOI] [PubMed] [Google Scholar]
- 31. Sumikawa J. T., Brito M. V., Macedo M. L., Uchoa A. F., Miranda A., Araujo A. P., Silva-Lucca R. A., Sampaio M. U., Oliva M. L. (2010) The defensive functions of plant inhibitors are not restricted to insect enzyme inhibition. Phytochemistry 71, 214–220 [DOI] [PubMed] [Google Scholar]
- 32. Dufton M. J. (1990) Could domain movements be involved in the mechanism of trypsin-like serine proteases? FEBS Lett. 271, 9–13 [DOI] [PubMed] [Google Scholar]
- 33. Krem M. M., Rose T., Di Cera E., (1999) The C-terminal sequence encodes function in serine proteases. J. Biol. Chem. 274, 28063–28066 [DOI] [PubMed] [Google Scholar]
- 34. Andrade D., Assis D. M., Lima A. R., Oliveira J. R., Araujo M. S., Blaber S. I., Blaber M., Juliano M. A., Juliano L. (2010) Substrate specificity and inhibition of human kallikrein-related peptidase 3 (KLK3 or PSA) activated with sodium citrate and glycosaminoglycans. Arch. Biochem. Biophys. 498, 74–82 [DOI] [PubMed] [Google Scholar]
- 35. Oliva M. L., Santomauro-Vaz E. M., Andrade S. A., Juliano M. A., Pott V. J., Sampaio M. U., Sampaio C. A. (2001) Synthetic peptides and fluorogenic substrates related to the reactive site sequence of Kunitz-type inhibitors isolated from Bauhinia. Interaction with human plasma kallikrein. Biol. Chem. 382, 109–113 [DOI] [PubMed] [Google Scholar]
- 36. Knight C. G. (1986) In Proteinase Inhibitors (Barret A.J., Salvesen G., eds) pp. 23–51, Elsevier, Amsterdam [Google Scholar]
- 37. Rodriguez L. G., Wu X., Guan J. L. (2005) Wound-healing assay. Methods Mol. Biol. 294, 23–29 [DOI] [PubMed] [Google Scholar]
- 38. Andree H. A., Reutelingsperger C. P., Hauptmann R., Hemker H. C., Hermens W. T., Willems G. M. (1990) Binding of vascular anticoagulant α (VACα) to planar phospholipid bilayers. J. Biol. Chem. 265, 4923–4928 [PubMed] [Google Scholar]
- 39. Raynal P., Pollard H. B. (1994) Annexins. The problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins. Biochim. Biophys. Acta 1197, 63–93 [DOI] [PubMed] [Google Scholar]
- 40. Darzynkiewicz Z., Juan G., Li X., Gorczyca W., Murakami T., Traganos F. (1997) Cytometry in cell necrobiology. Analysis of apoptosis and accidental cell death (necrosis). Cytometry 27, 1–20 [PubMed] [Google Scholar]
- 41. Paschoalin T., Carmona A. K., Rodrigues E. G., Oliveira V., Monteiro H. P., Juliano M. A., Juliano L., Travassos L. R. (2007) Characterization of thimet oligopeptidase and neurolysin activities in B16F10-Nex2 tumor cells and their involvement in angiogenesis and tumor growth. Mol. Cancer 6, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kennedy A. R. (1994) Prevention of carcinogenesis by protease inhibitors. Cancer Res. 54, 1999s–2005s [PubMed] [Google Scholar]
- 43. Birk Y. (2003) Plant Protease Inhibitors: Significance in Nutrition, Plant Protection, Cancer Prevention and Genetic Engineering, pp. 1–126, Springer-Verlag, Berlin [Google Scholar]
- 44. Goettig P., Magdolen V., Brandstetter H. (2010) Natural and synthetic inhibitors of kallikrein-related peptidases (KLKs). Biochimie 92, 1546–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nakahata A. M., Mayer B., Ries C., de Paula C. A., Karow M., Neth P., Sampaio M. U., Jochum M., Oliva M. L. (2011) The effects of a plant proteinase inhibitor from Enterolobium contortisiliquum on human tumor cell lines. Biol. Chem. 392, 327–336 [DOI] [PubMed] [Google Scholar]
- 46. Wilkinson R., Woods K., D'Rozario R., Prue R., Vari F., Hardy M. Y., Dong Y., Clements J. A., Hart D. N., Radford K. J. (2012) Human kallikrein 4 signal peptide induces cytotoxic T cell responses in healthy donors and prostate cancer patients. Cancer Immunol. Immunother. 61, 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu J., Bastian M., Kohlschein P., Schuff-Werner P., Steiner M. (2003) Expression of functional protease-activated receptor 1 in human prostate cancer cell lines. Urol. Res. 31, 163–168 [DOI] [PubMed] [Google Scholar]
- 48. Kaushal V., Kohli M., Dennis R. A., Siegel E. R., Chiles W. W., Mukunyadzi P. (2006) Thrombin receptor expression is upregulated in prostate cancer. Prostate 66, 273–282 [DOI] [PubMed] [Google Scholar]
- 49. Black P. C., Mize G. J., Karlin P., Greenberg D. L., Hawley S. J., True L. D., Vessella R. L., Takayama T. K. (2007) Overexpression of protease-activated receptors-1,-2, and-4 (PAR-1, -2, and -4) in prostate cancer. Prostate 67, 743–756 [DOI] [PubMed] [Google Scholar]
- 50. Macfarlane S. R., Seatter M. J., Kanke T., Hunter G. D., Plevin R. (2001) Proteinase-activated receptors. Pharmacol. Rev. 53, 245–282 [PubMed] [Google Scholar]
- 51. Ossovskaya V. S., Bunnett N. W. (2004) Protease-activated receptors. Contribution to physiology and disease. Physiol. Rev. 84, 579–621 [DOI] [PubMed] [Google Scholar]
- 52. Obiezu C. V., Diamandis E. P. (2005) Human tissue kallikrein gene family. Applications in cancer. Cancer Lett. 224, 1–22 [DOI] [PubMed] [Google Scholar]
- 53. Abdallah R. T., Keum J. S., El-Shewy H. M., Lee M. H., Wang B., Gooz M., Luttrell D. K., Luttrell L. M., Jaffa A. A. (2010) Plasma kallikrein promotes epidermal growth factor receptor transactivation and signaling in vascular smooth muscle through direct activation of protease-activated receptors. J. Biol. Chem. 285, 35206–35215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cooper C. R., Chay C. H., Pienta K. J. (2002) The role of αvβ3 in prostate cancer progression. Neoplasia 4, 191–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dardik R., Kaufmann Y., Savion N., Rosenberg N., Shenkman B., Varon D. (1997) Platelets mediate tumor cell adhesion to the subendothelium under flow conditions. Involvement of platelet GPIIb-IIIa and tumor cell αv integrins. Int. J. Cancer 70, 201–207 [DOI] [PubMed] [Google Scholar]
- 56. Tantivejkul K., Kalikin L. M., Pienta K. J. (2004) Dynamic process of prostate cancer metastasis to bone. J. Cell Biochem. 91, 706–717 [DOI] [PubMed] [Google Scholar]
- 57. Metcalf D. G., Moore D. T., Wu Y., Kielec J. M., Molnar K., Valentine K. G., Wand A. J., Bennett J. S., DeGrado W. F. (2010) NMR analysis of the αIIbβ3 cytoplasmic interaction suggests a mechanism for integrin regulation. Proc. Natl. Acad. Sci. U.S.A. 107, 22481–22486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Loberg R. D., Tantivejkul K., Craig M., Neeley C. K., Pienta K. J. (2007) PAR1-mediated RhoA activation facilitates CCL2-induced chemotaxis in PC-3 cells. J. Cell Biochem. 101, 1292–1300 [DOI] [PubMed] [Google Scholar]
- 59. Shi X., Gangadharan B., Brass L. F., Ruf W., Mueller B. M. (2004) Protease-activated receptors (PAR1 and PAR2) contribute to tumor cell motility and metastasis. Mol. Cancer Res. 2, 395–402 [PubMed] [Google Scholar]
- 60. Anthis N. J., Campbell I. D. (2011) The tail of integrin activation. Trends Biochem. Sci. 36, 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Meoli D. F., White R. J. (2009) Thrombin induces fibronectin-specific migration of pulmonary microvascular endothelial cells. Requirement of calcium/calmodulin-dependent protein kinase II. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L706–L714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Prevarskaya N., Skryma R., Shuba Y. (2011) Calcium in tumour metastasis. New roles for known actors. Nat. Rev. Cancer 11, 609–618 [DOI] [PubMed] [Google Scholar]
- 63. Bookstein R., Shew J. Y., Chen P. L., Scully P., Lee W. H. (1990) Suppression of tumorigenicity of human prostate carcinoma cells by replacing a mutated RB gene. Science 247, 712–715 [DOI] [PubMed] [Google Scholar]
- 64. MacGrogan D., Bookstein R. (1997) Tumour suppressor genes in prostate cancer. Semin. Cancer Biol. 8, 11–19 [DOI] [PubMed] [Google Scholar]
- 65. Noonan E. J., Place R. F., Basak S., Pookot D., Li L. C. (2010) miR-449a causes Rb-dependent cell cycle arrest and senescence in prostate cancer cells. Oncotarget 1, 349–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Alvarez S., Blanco A., Fresno M., Muñoz-Fernández M. A. (2011) TNF-α contributes to caspase-3 independent apoptosis in neuroblastoma cells. Role of NFAT. PLoS One 6, e16100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Agarwal M. L., Agarwal A., Taylor W. R., Stark G. R. (1995) p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 92, 8493–8497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cohen I., Maoz M., Turm H., Grisaru-Granovsky S., Maly B., Uziely B., Weiss E., Abramovitch R., Gross E., Barzilay O., Qiu Y., Bar-Shavit R. (2010) Etk/Bmx regulates proteinase-activated-receptor1 (PAR1) in breast cancer invasion. Signaling partners, hierarchy and physiological significance. PLoS One 5, e11135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chieng-Yane P., Bocquet A., Létienne R., Bourbon T., Sablayrolles S., Perez M., Hatem S. N., Lompré A. M., Le Grand B., David-Dufilho M. (2011) Protease-activated receptor-1 antagonist F 16618 reduces arterial restenosis by down-regulation of tumor necrosis factor α and matrix metalloproteinase 7 expression, migration, and proliferation of vascular smooth muscle cells. J. Pharmacol. Exp. Ther. 336, 643–651 [DOI] [PubMed] [Google Scholar]
- 70. Katayama H., Sen S. (2010) Aurora kinase inhibitors as anticancer molecules. Biochim. Biophys. Acta 1799, 829–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Carmeliet P., Jain R. K. (2000) Angiogenesis in cancer and other diseases. Nature 407, 249–257 [DOI] [PubMed] [Google Scholar]
- 72. Veiseh O., Kievit F. M., Ellenbogen R. G., Zhang M. (2011) Cancer cell invasion. Treatment and monitoring opportunities in nanomedicine. Adv. Drug Deliv. Rev. 63, 582–596 [DOI] [PMC free article] [PubMed] [Google Scholar]