

Background: Missense mutations in AHI1 result in the neurodevelopmental ciliopathy called Joubert syndrome.
Results: Mutations in AHI1 decrease cilia formation, alter its localization and stability, and change its binding to HAP1 and NPHP1.
Conclusion: Mutations in AHI1 affect ciliogenesis, AHI1 protein localization, and AHI1-protein interactions.
Significance: This study begins to describe how missense mutations in AHI1 can cause Joubert syndrome.
Keywords: Cilia, Neurodevelopment, Neurological Diseases, Neurons, Protein Complexes, AHI1, HAP1, Joubert Syndrome
Abstract
Mutations in AHI1 cause Joubert syndrome (JBTS), a neurodevelopmental ciliopathy, characterized by midbrain-hindbrain malformations and motor/cognitive deficits. Here, we show that primary cilia (PC) formation is decreased in fibroblasts from individuals with JBTS and AHI1 mutations. Most missense mutations in AHI1, causing JBTS, occur in known protein domains, however, a common V443D mutation in AHI1 is found in a region with no known protein motifs. We show that cells transfected with AHI1-V443D, or a new JBTS-causing mutation, AHI1-R351L, have aberrant localization of AHI1 at the basal bodies of PC and at cell-cell junctions, likely through decreased binding of mutant AHI1 to NPHP1 (another JBTS-causing protein). The AHI1-V443D mutation causes decreased AHI1 stability because there is a 50% reduction in AHI1-V443D protein levels compared with wild type AHI1. Huntingtin-associated protein-1 (Hap1) is a regulatory protein that binds Ahi1, and Hap1 knock-out mice have been reported to have JBTS-like phenotypes, suggesting a role for Hap1 in ciliogenesis. Fibroblasts and neurons with Hap1 deficiency form PC with normal growth factor-induced ciliary signaling, indicating that the Hap1 JBTS phenotype is likely not through effects at PC. These results also suggest that the binding of Ahi1 and Hap1 may not be critical for ciliary function. However, we show that HAP1 has decreased binding to AHI1-V443D indicating that this altered binding could be responsible for the JBTS-like phenotype through an unknown pathway. Thus, these JBTS-associated missense mutations alter their subcellular distribution and protein interactions, compromising functions of AHI1 in cell polarity and cilium-mediated signaling, thereby contributing to JBTS.
Introduction
Joubert syndrome (JBTS)3 is a recessive neurodevelopmental disorder consisting of agenesis of the cerebellar vermis, defective axonal decussation (particularly of the superior cerebellar peduncles and corticospinal tract), ataxia, hypotonia, irregular breathing, and cognitive disabilities including autism (1, 2). The diagnostic hallmark for JBTS on axial brain MRI is the molar tooth sign, a neuroanatomical malformation that results from cerebellar vermis hypoplasia, thickened and elongated superior cerebellar peduncles, and a deepened intrapeduncular fossa. In addition to the CNS phenotypes, several peripheral disturbances are often associated with JBTS, including retinal dystrophy, nephronophthisis, liver fibrosis, and polydactyly (1).
Mutations in multiple genes have been associated with JBTS with all of these genes being associated with primary cilia formation and function (3). Primary cilia are microtubule-based structures projecting from the cell surface and serve as a focused center for signal transduction (4). Given that JBTS-causing genes are involved in the formation and function of the primary cilium, and that JBTS and its related disorders display similar clinical phenotypes with other diseases resulting from dysfunctional primary cilia, JBTS is now considered a ciliopathy (5).
The Abelson-helper integration site 1 (AHI1) gene was one of the first identified genes associated with JBTS (6, 7). The cytoplasmic protein encoded by AHI1 contains an N-terminal coiled-coil domain, seven WD40 repeats, and an SH3 binding domain in its C terminus, suggesting that AHI1 could function as a scaffolding protein (8). In individuals with JBTS and AHI1 mutations, the typical genetic lesion is either a functionally null protein or a missense mutation, with the latter mutation resulting in a protein product with altered protein structure/function. Among the known missense mutations, only two, V443D (7) and E1086G (9), occur in regions of the AHI1 protein that do not contain any known specific protein domains or motifs. How these mutations affect the function of AHI1, and subsequent neuropathology associated with JBTS, is currently unknown.
Expression studies have shown that mouse Ahi1 is highly expressed in the ventral forebrain, especially in the amygdala and hypothalamus (10). At the subcellular level, Ahi1 is found in the cytoplasm and at the basal bodies of primary cilia, and inhibition of Ahi1 expression blocks the formation of primary cilia (11). Moreover, Ahi1-deficient mice display retinal degeneration resulting from defective ciliary protein trafficking (12, 13). These studies suggest a crucial role for Ahi1 in cilium formation and function. However, the mechanisms of how AHI1 mutations cause the neurological phenotypes seen in JBTS remain unclear.
Huntingtin-associated protein 1 (Hap1) and nephrocystin-1 (Nphp1) are two proteins that interact with Ahi1 (14, 15). Hap1 is a cytoplasmic protein that interacts with huntingtin (Htt), a protein known to cause Huntington disease when there is increased polyglutamine expansion in Htt (16). HAP1 is also thought to be involved in the neuropathogenesis found in Huntington disease through its aberrant binding with mutant Htt (17, 18), and may act as a modifier for Huntington disease onset (19). Unlike Htt, which is ubiquitously expressed, Hap1 is predominantly expressed in the CNS, especially in the hypothalamus, striatum, and brainstem; areas that are highly enriched in Ahi1 (10, 14). A function for Hap1 in intracellular trafficking has been demonstrated through studies showing that Hap1 associates with microtubules and membranous organelles in addition to interacting with the anterograde and retrograde molecular motors, kinesin light chain 2 and p150Glued, respectively (20).
Recently, a possible link for Hap1 with JBTS was highlighted in one study that showed that mice with Hap1 deletions display defects of axonal decussation at the superior cerebellar peduncles and hypoplasia of the cerebellar vermis, two key features observed in JBTS (14). Although no HAP1 mutations have been described in JBTS,4 the interaction of AHI1 and HAP1 suggests a shared pathway critical for brain formation.
NPHP1 encodes for a protein localized to cell-cell junctions and the basal body of the primary cilium. Mutations in NPHP1 are often associated with JBTS accompanied with renal dysfunction (1), and also account for a majority of cases of nephronophthisis (NPHP; a recessive renal cystic disease) (21). Although the function of NPHP1 is not well understood, the interaction of NPHP1 with other NPHP disease proteins at cell junctions and its highly regulated mRNA expression during cell polarization, suggest a role of NPHP1 in cellular organization (22, 23). An interaction of AHI1 and NPHP1 was demonstrated by yeast two-hybrid analysis in which the SH3 domain of NPHP1 bound the WD40 repeats in AHI1 (15). Moreover, the expression of AHI1 at cell-cell junctions, similar to NPHP1, supports a functional interaction of these two proteins (15). In addition to serving as a protein binding partner, AHI1 is considered a potential genetic modifier of NPHP1, because individuals with nephronophthisis and the molar tooth sign often carry mutations in NPHP1 combined with heterozygous mutations in AHI1 (24). In further support of a phenotypic connection between AHI1 and NPHP1, Nphp1−/−;Ahi1+/− mice display retinal degeneration earlier than Nphp1−/− mice, indicating that the genetic interaction of Ahi1 and Nphp1 appears to be dosage-sensitive (13).
To understand the molecular mechanisms of how AHI1 mutations cause the neurodevelopmental defects observed in JBTS, we examined the function of AHI1 through its interacting proteins to gain further insights. In this report, we determined the effects of mutant AHI1-V443D on binding to HAP1 and NPHP1 with position Val-443 serving as an important stabilizing residue for these interactions. Furthermore, we have shown that expression of AHI1-V443D and AHI1-R351L in IMCD3 cells resulted in an inability of mutant AHI1 to localize at cell-cell junctions and at the basal body of primary cilia, with a corresponding reduction in cilium formation in AHI1-V443D- and AHI1-R351L-transfected cells. These results suggest that the V443D and R351L mutations affect the distribution of AHI1, possibly due to these disrupted protein interactions. To test this, we examined the involvement of the Ahi1 and Hap1 interaction through analysis of Hap1 and Ahi1 knock-out mice. Unlike Ahi1 knock-out neurons, and fibroblasts from patients with AHI1 mutations, no defects in ciliary formation or function were observed in Hap1 knock-out neurons or fibroblasts. These experiments indicate that disrupting the binding of these two proteins is not critical for ciliary function. Because Hap1 knock-out mice were only reported to have cerebellar vermal hypoplasia and superior cerebellar peduncle defects without other JBTS-associated phenotypes (i.e. photoreceptor degeneration and cystic kidney disease) (14), this suggests that the JBTS-like phenotype in Hap1 knock-out mice may be through a defect in an unknown non-ciliary mediated mechanism or that Hap1 loss results in cerebellar developmental defects through a non-JBTS pathway.
EXPERIMENTAL PROCEDURES
Animals
The methods describing the construction of mice with a targeted deletion of Ahi1 have been reported previously (11). Hap1+/− mice were obtained from the Jackson Laboratory (Hap1tm1Xjl/J; stock number 007749) (25). Mice were maintained on a normal 12-h light-dark cycle (06:00 to 18:00) with unlimited access to food and water. All mouse procedures were performed under approval from the Institutional Animal Care and Use Committees of Rensselaer Polytechnic Institute, the Wadsworth Center, and the Albany Medical College.
Cell Culture
Human dermal fibroblasts were collected from patients using a standard skin biopsy technique after informed consent was obtained. The skin was treated with collagenase and cultured in DMEM (Invitrogen) with 10% fetal bovine serum and 1% penicillin/streptomycin. Upon reaching confluence, cells were trypsinized (0.05% trypsin/EDTA) for 5 min at 37 °C followed by trypsin inactivation in culture medium supplemented with 10% calf serum. Cell suspensions were plated on glass coverslips and were maintained at 37 °C in a humidified atmosphere (5% CO2, 95% air). To evaluate cilia formation, fibroblasts were starved in DMEM containing 0.1% fetal calf serum (FCS) for 24 h to promote robust cilia formation. Cell cultures were fixed in 4% paraformaldehyde, 4% sucrose for immunostaining. For the fibroblasts studied, the genotypes of the individuals with JBTS and AHI1 mutations were: (c.1260G>A, p.W420X and IVS8 c.1152–2a>g) and (c.2212C>T, p.R783X, and c.1976A>T, p.D659V).
Embryos from timed pregnant litters were generated by either interbreeding Ahi1+/− mice or interbreeding Hap1+/− mice. To evaluate cilium formation in neuronal cells, the hypothalamus, cortex, hippocampus, and cerebellum were harvested from embryos at day E18.5. Neuronal cultures were then prepared as previously described (10). Briefly, dissected brain regions from each embryo were incubated in 0.05% trypsin/EDTA for 10 min at 37 °C, and then were treated with 1 mg/ml of trypsin inhibitor for 5 min at room temperature (RT). Trypsin-treated tissues were dissociated by gentle trituration in Neurobasal media supplemented with B27, glucose, sodium pyruvate, glutamine, and gentamicin (Invitrogen). Cell suspensions were then cultured on poly-d-lysine (Sigma)-coated glass coverslips. Culture medium was changed twice a week. Neuronal cultures were fixed after 7 days in culture with 4% paraformaldehyde, 4% sucrose for immunostaining.
Mouse embryonic fibroblasts (MEFs) were prepared by collecting the skin from Hap1−/− and Hap1+/+ embryos at day E15.5. The tissue was minced in ice-cold phosphate-buffered saline (PBS) with 10% fetal calf serum (FCS) followed by 0.05% trypsin/EDTA incubation for 10–15 min at 37 °C. Trypsin-treated tissues were then gently triturated in DMEM supplemented with 10% FCS, non-essential amino acids, and penicillin/streptomycin. Cell suspensions were then plated on gelatin-coated cell culture dishes, and cells from cell passage 6 and under were used for these experiments. To evaluate cilia instability upon growth factor stimulation, MEFs were starved in DMEM containing 0.1% FCS for 24 h prior to any experiments, thereby promoting robust cilia formation. MEFs were then cultured in 10% FCS/DMEM for various times before fixation in 4% paraformaldehyde, 4% sucrose for immunostaining.
Antibodies
Polyclonal antibodies against mouse Ahi1 were generated and described previously (10). Other antibodies used in immunolabeling and Western blot experiments included a rabbit polyclonal anti-Arl13b antibody (a gift from Dr. Caspary, Emory University), a rabbit polyclonal anti-adenylyl cyclase III antibody, a goat polyclonal anti-adenylyl cyclase III antibody, a goat polyclonal anti-MchR1 antibody, a mouse monoclonal anti-Hap1 antibody, and a goat polyclonal anti-Hap1 antibody (Santa Cruz Biotechnology), a mouse monoclonal anti-EGFP antibody, a mouse monoclonal anti-c-myc tag antibody (Clontech), a rabbit polyclonal anti-neuropeptide Y antibody, a chicken polyclonal anti-POMC antibody, a rabbit polyclonal anti-β-tubulin antibody (Abcam), a mouse polyclonal anti-HA tag antibody (Cell Signaling Technologies), a chicken anti-βIII tubulin antibody (Millipore), a mouse monoclonal anti-β-catenin antibody (BD Bioscience), a mouse monoclonal anti-γ-tubulin antibody, a mouse monoclonal anti-acetylated α-tubulin antibody (Sigma), and a mouse monoclonal anti-NeuN antibody (Chemicon).
Immunostaining
Cells were processed for immunolabeling as previously described (10, 11). Primary antibodies were diluted to the following final working concentrations: Ahi1, 1:1000 (rabbit IgG (10)), adenylyl cyclase III, 1:1000 (rabbit IgG), MchR1, 1:500 (goat IgG), Arl13b, 1:2000 (rabbit IgG (26)), NeuN, 1:1000 (mouse IgG), neuropeptide Y, 1:1000 (rabbit IgG), POMC, 1:200 (chicken IgY), β-catenin, 1:2500 (mouse IgG), acetylated α-tubulin, 1:1000 (mouse IgG), and γ-tubulin, 1:1000 (mouse IgG). Following overnight incubation in primary antibodies, cells were incubated with fluorescently conjugated secondary antibodies (1:500; Invitrogen (Molecular Probes) or Jackson ImmunoResearch Laboratories) for 1 h at RT, followed by Hoechst DNA staining (1 mg/ml). After staining, coverslips were mounted with Fluoromount-G antifade solution (Southern Biotechnology).
Microscopy
When using fluorescent secondary antibodies, images were obtained with a Zeiss AxioImager-Z1 microscope and with an AxioCam MRm camera. Images were processed with AxioVision Rel. 4.5 software and Adobe Photoshop CS2 (version 9.0.2; Adobe Systems Inc.). 3,3′-Diaminobenzidine images were acquired with an AxioCam MRc camera and processed with the software described above and MosaiX (Carl Zeiss Microimaging). Contrast and brightness were altered, as needed, through linear level adjustments to optimize the intensity range of the images.
Plasmid Constructs
For mammalian cell expression, wild type and mutant human AHI1 fragment 2 (121–580 aa, F2) and AHI1 fragment 3 (141–434 aa, F3) were constructed in pCMV-HA plasmids (Clontech). The fragment AHI1-F2-V443D was amplified by PCR and then placed in-frame into the 5′-PshAI and 3′-HindIII restriction enzyme sites (RES) in a HA-tagged and a myc-tagged wild type full-length AHI1 expressing construct to obtain full-length AHI1 with the V443D mutation (AHI1-V443D). AHI1-F3 was amplified by PCR and inserted in-frame into the 5′-EcoRI and 3′-XhoI RES of the pCMV-HA vector. The R351L mutation in AHI1 was obtained using a site-directed mutagenesis kit (Agilent Technologies) and subcloned in-frame into the 5′-SalI and 3′-HindIII RES in a myc-tagged wild type full-length AHI1 expression construct to obtain a myc-AHI1-R351L construct. Human NPHP1 cDNA clone (MGC 75120 (IMAGE: 5187280, pSPORT6-NPHP1)) was purchased from ATCC. HA-tagged-NPHP1 (HA-NPHP1) was generated by inserting the NPHP1 cDNA fragments in-frame into the 5′-NotI and 3′-EcoRI RES of the pCMV-HA vector. HA-tagged human HAP1 was purchased from GeneCopoeia. Myc-tagged human HAP1 was cloned by PCR amplification and inserted in-frame into the 5′-EcoRI and 3′-XhoI RES of the pCMV-myc vector. All DNA constructs were verified by DNA sequencing (Eurofins MWG Operon). These constructs were transfected into IMCD3 and HEK293 cells for immunocytochemical, Western blot, and co-immunoprecipitation analyses.
Co-immunoprecipitation
Tissue was obtained from day E18.5 pups, and brains were dissected and frozen in liquid nitrogen. Brains were homogenized as previously described (10). Brain lysates (400 μg) were incubated with 1 μg of antibody at 4 °C overnight. Protein-antibody complexes were precipitated with protein-A magnetic Dynabeads (Invitrogen) according to the manufacturer's instructions. In brief, pre-washed protein-A beads were added to samples and then mixed thoroughly. After 15 min of rotation at RT, beads were washed three times with 0.1 m sodium phosphate buffer (pH 8) before boiling in 1× SDS-PAGE sample buffer for 10 min. The supernatant was collected, separated on SDS-8% PAGE, and then processed by Western blotting.
HEK293 cells (ATCC) were co-transfected, using Lipofectamine LTX and Plus reagent, with plasmids that were indicated in the figures. Cell lysates (500 μg) were collected 24 h after transfection and incubated with 2 μg of anti-myc antibody overnight at 4 °C. The protein-antibody complexes were precipitated with Protein-G magnetic Dynabeads (Invitrogen), and the sample was rotated for 45 min at RT. The beads were washed three times with PBS containing protease inhibitors (Roche Applied Science), before boiling in 1× SDS-PAGE sample buffer. Supernatants were collected for Western blot analysis.
Western Blotting
Protein lysates were resolved by SDS-8% PAGE and electroblotted onto PVDF transfer membrane. Blots were probed with various primary antibodies as follows: Ahi1 (1:1,000 to 1:4,000 (10)), Hap1 (1:200), myc (1:1,000), HA (1:1,000), and βIII tubulin and β-tubulin (used as loading controls, 1:4,000 and 1:10,000, respectively). Primary antibodies were detected with the SuperSignal West Femto Maximum Sensitivity Substrate Chemiluminescence Kit (ThermoFisher Scientific). Signals were detected with a G:Box Chemi/Fluorescence Imager system with GeneSnap software and analyzed by GeneTool analysis software (SynGene). To quantify and compare the signal intensities between each sample, all detected signals were unsaturated and in the linear range of detection.
Analysis of AHI1-NPHP1 and AHI1-HAP1 Complexes
HEK293 cells were grown to 80% confluence and co-transfected with 1) pCMV-myc-AHI1 and -HA-HAP1; 2) pCMV-myc-AHI1-V443D and -HA-HAP1; 3) pCMV-myc-AHI and -HA-NPHP1; or 4) pCMV-myc-AHI1-V443D and -HA-NPHP1. Cells were incubated for another 24 h and lysed in RIPA buffer. The cell lysate was ultracentrifuged at 100,000 × g for 30 min and the supernatant was subjected to gel filtration chromatography in a HiLoad Superdex 200 16/60 PG column, using a buffer containing 50 mm Tris-HCl, 1 mm EDTA, 5% glycerol, 100 mm NaCl, 0.05% n-dodecyl-β-d-maltoside (pH 8.0). The high ionic strength of the buffer and the presence of the detergent prevent unspecific associations of proteins. The samples were run using a flow of 0.1 ml/min and fractions of 2 ml, containing the molecular weights between 150–750 kDa, were concentrated to 100 μl in centrifugal concentrators (Millipore). Ten μl of the concentrated fractions were analyzed by Western blot, using antibodies against the myc and HA tags. Data were analyzed by plotting the band intensities versus the fraction number. Individual protein peaks were deconvoluted, fitting the data to a multi-Gaussian function.
Quantitative Real-time PCR
HEK293 cells were transfected with myc-AHI1-WT or -V443D using Lipofectamine LTX (Invitrogen), and after 24 h, the RNA was extracted with TRIzol. cDNA was prepared from RNA using qScript cDNA Supermix (Quanta). Quantitative PCR primers were designed using Primer Express 3.0 (Applied Biosystems). Quantitative PCR primers amplified 100–200-bp fragments and were designed against a unique region of AHI1 (forward primer, 5′-AACCCAGAAACAGGAGAACAAGTAG-3′; reverse primer, 5′-ATAAGAAATGTCTCGAATGGGTGACT-3′) and against the myc tag (forward primer, 5′-TCAATGCAGAAGCTGATCTCAG-3′; reverse primer, 5′-CTCAGCTGTAGGGCTAGTCGAC-3′). cDNA was amplified using PerfeCta SYBR Green Fast Mix (Quanta) with a Step One Plus Real-time PCR System (Applied Bioscience). Relative abundance of mRNA was normalized to β-actin and calculated as 2 − (Ct gene − Ct β-actin), where Ct represents the threshold cycle for each transcript.
Protein Stability Assay
HEK293 cells were transfected with myc-tagged AHI1-WT or -V443D, and after 16 h, the cultures were treated with cycloheximide (200 μm). Lysates were obtained after 0, 1, 2, 4, 6, 12, and 24 h of cycloheximide treatment. AHI1-WT or -V443D protein levels were analyzed by Western blotting using β-tubulin as a loading control.
Directed Yeast Two-hybrid Analyses
Full-length human AHI1 (1–1196 aa) and four individual fragments of AHI1 (fragment 1 (F1, coiled-coil domain: 1–140 aa), fragment 2 (F2, no putative protein domains: 121–580 aa), fragment 4 (F4, WD40 repeats: 551–940 aa) and fragment 5 (F5, SH3 domain: 911–1196 aa)) were fused in-frame with the coding sequence of the DNA-binding domain of LexA in the pBTM116 vector to generate bait plasmids (pBTM-AHI1-F1, -F2, -F4, and -F5). In addition, a sequence of AHI1-F2 containing: 1) a control mutation (D358N), 2) a mutation producing neurological alterations in nephronophthisis (D330G), or 3) a JBTS-causing missense mutation (V443D) was also fused in-frame with the coding sequence of the DNA-binding domain of LexA in the pBTM116 vector (pBTM-AHI1-F2-D358N, -F2-D330G, and -F2-V443D). Full-length human HAP1 and two individual fragments (fragment 1 (F1, 1–320 aa) and fragment 2 (F2, 287–671 aa)) were subcloned into pVP16 and fused in-frame with the LexA activation domain to create pVP16-HAP1-F1 and pVP16-HAP1-F2 as prey plasmids. The pVP16 HAP1 plasmids, separately with each individual bait plasmid, were co-transformed into the recipient L40 yeast strain, which contains a chromosomal copy of each reporter gene HIS3 and LacZ, whose activity is driven by a synthetic LexA-dependent promoter. Double transformants were selected on plates Yc/–Ura/–Trp/–Leu. Transformants expressing the HIS3 reporter were selected on high stringency plates (on Yc/–Ura/–Trp/–Leu/–His/–Lys plus 5 mm 3-amino-1,2,4-triazole). Direct protein interactions were confirmed by a β-galactosidase colony-lift filter assay (4 mg/ml of X-Gal in Z buffer).
Histological Procedures and Immunostaining
To obtain tissue for immunohistochemistry, Ahi1+/+ and Ahi1−/− P19 animals were overdosed with sodium pentobarbital and transcardially perfused with 0.01 m PBS, followed by 4% paraformaldehyde made in PBS. Brains were removed and post-fixed for 3 days in 4% paraformaldehyde, and then cryoprotected in 30% sucrose/PBS. Brains were sectioned in the sagittal plane at a thickness of 20 μm using a Microm cryostat (Richard-Allan Scientific), mounted on Superfrost Plus microscope slides (ThermoFisher Scientific), and allowed to air dry at RT. Tissue sections were permeabilized with 0.04% Triton X-100/PBS and endogenous peroxidases were quenched via 0.03% H2O2/methanol (30 min). Sections were washed thoroughly with Triton X-100/PBS and blocked for 1 h in 10% normal horse serum, followed by overnight incubation with anti-Hap1 antibodies. For primary antibody detection, a Vector Elite ABC kit and a 3,3′-diaminobenzidine kit (Vector Laboratories) were used according to the manufacturer's instructions.
Hap1 Stability Upon AHI1 Expression
PC12 cells were transfected with myc-AHI1-WT (0.6 and 2.4 μg) using Lipofectamine LTX with Plus reagent. For controls without myc-AHI1-WT (0 μg), only Lipofectamine and Plus reagent were added to the cells. After 24 h, cell lysates were collected and 40 μg of protein were analyzed by Western blotting. Hap1 levels were determined using β-tubulin as a loading control.
RESULTS
Human Dermal Fibroblasts from Individuals with JBTS Carrying AHI1 Mutations Have Reduced Primary Cilia Formation and Protein Distribution
To determine whether loss of AHI1 results in an impairment in primary cilia formation and protein distribution, human dermal fibroblasts from unaffected, and individuals with JBTS and AHI1 mutations, were cultured in serum-deprived conditions to promote cilia formation. Fibroblasts from individuals with JBTS showed an ∼50% decrease in primary cilia formation (p < 0.005; Fig. 1, A and B). Primary cilia protein distribution was also evaluated in human dermal fibroblasts by ARL13B immunolabeling. JBTS fibroblasts displayed a significant reduction in ARL13B ciliary localization in their remaining primary cilia as compared with ARL13B ciliary localization in normal fibroblasts (% of unaffected cells with ciliary localized ARL13B, 84.76 ± 0.24; % of JBTS cells with ciliary localized ARL13B, 39.07 ± 3.28; p < 0.05) (Fig. 1, A and C). These results confirm previous data from Ahi1 knock-out mice that showed decreases in ciliogenesis (11), and further support a role of AHI1 in ciliogenesis and primary cilia function in both rodents and humans. Importantly, these ARL13B data indicate that the remaining primary cilia on fibroblasts from individuals with JBTS are likely not entirely functional, because they are missing an important ciliary protein, ARL13B.
FIGURE 1.

Primary cilia formation and localization of ciliary proteins in the remaining cilia are significantly decreased in human dermal fibroblasts from individuals with JBTS and having AHI1 mutations. A, representative images of primary cultured human dermal fibroblasts from unaffected controls (top) and individuals with JBTS having mutations in AHI1 (bottom) labeled with the ciliary marker, acetylated α-tubulin (green), and with a ciliary-localizing protein necessary for proper primary cilium function, ARL13B (red). Arrowheads point to primary cilia. Scale bar = 5 μm. B, JBTS dermal fibroblasts have significantly diminished primary cilia formation compared with unaffected dermal fibroblasts (p < 0.005, Mann-Whitney U test). Primary cilia formation was evaluated by counting the number of cells having acetylated α-tubulin-positive cilia (≥100 cells per case). The individual points represent the mean for each individual fibroblast line. Asterisk denotes significance from unaffected individuals. C, primary cilia on dermal fibroblasts from individuals with JBST having AHI1 mutations showed a significantly decreased localization of ARL13B to acetylated α-tubulin-positive cilia as compared with unaffected individuals (p < 0.05, Mann-Whitney U test). The number of cells having primary cilia labeled with both acetylated α-tubulin and ARL13B were counted in unaffected and JBTS fibroblasts (≥ 100 cells per case). Results are expressed as mean ± S.E. Asterisk denotes the significance from unaffected individuals. DNA was visualized with Hoechst 33258 (blue).
The V443D Mutation in AHI1 Disrupts the Localization of AHI1 at the Cell-Cell Junction and Basal Body
Most of the missense mutations in AHI1 occur in protein binding motifs or the SH3 domain of AHI1, with such mutations affecting protein-protein interactions. However, AHI1 does contain a large region that does not contain any known protein motifs. Therefore, we were interested in exploring how mutations in this region could possibly affect AHI1 function. Given that the V443D mutation in AHI1 results in JBTS and is in this region of AHI1 devoid of known protein motifs, we were interested in how the V443D mutation affected the function of AHI1 at the primary cilium. Because AHI1 is a basal body localizing protein (11, 15), we first examined whether AHI1-V443D had the same distribution pattern as wild type AHI1. We evaluated the co-localization of γ-tubulin labeled centrioles (one which is the basal body) with wild type AHI1 or AHI1-V443D in IMCD3 cells by immunolabeling. These results showed that ∼80% of the cells expressing the wild type AHI1 (AHI1-WT) protein had AHI1 at the basal body of the primary cilium, whereas less than 5% of the cells expressing AHI1-V443D had co-localization with γ-tubulin at the basal body (p < 0.0001; Fig. 2, A and D). This drastic shift was also observed in the distribution of AHI1 at cell-cell junctions (Fig. 2, C and D). That is, AHI1-V443D was rarely co-localized to cell junctions labeled by Arl13b, a marker for primary cilia and cell junctions (Fig. 2, B and C), whereas more than 60% of the cells expressing AHI1-WT had AHI1 localized to the cell junctions of IMCD3 cells (p < 0.0001; Fig. 2, C and D). These results indicate that the V443D mutation impacts the native distribution of AHI1 in the cell, specifically the localization at the basal body and cell-cell junctions. Because the V443D mutation in AHI1 results in the abnormalities described in JBTS, the distribution of AHI1 at cell-cell junctions and the basal body appears to be essential for the proper function of AHI1.
FIGURE 2.

Loss of AHI1 distribution at the basal body and cell-cell junctions with the JBTS-associated AHI1-V443D mutation. A, myc-tagged full-length AHI1-WT or AHI1-V443D was transiently expressed in IMCD3 cells and the co-localization of transfected proteins (myc, red) and the centrosome (γ-tub, green) was evaluated by immunostaining. The arrowheads point to the co-localization of γ-tubulin and AHI1 in AHI1-WT cells at the basal body (top), with no co-localization of AHI1 and γ-tubulin in AHI1-V443D cells (bottom). B, Arl13b staining (green) is co-localized with β-catenin (β-ctnn; red) at cell junctions (arrowhead) indicating that Arl13b is a good marker for cell-cell junctions. C, transfected cells were cultured in serum-free medium to examine the distribution of transfected proteins (myc, red) at cell junctions and the basal body of primary cilia (Arl13b, green). Co-localization of transfected AHI1 and Arl13b was found at the basal body (white arrowhead) and cell junctions (open arrowhead) in AHI1-WT cells, but not in AHI1-V443D cells. Scale bars = 5 μm. D, the graph represents the percentage of transfected AHI1 protein, localized at the basal body and cell junctions, as well as the incidence of cilia formation in these transfected cells. Results were collected from ≥3 independent transfection assays and more than 100 transfected cells were counted from each coverslip. The error bar represents the S.E. Asterisks denote the significance from AHI1-WT transfected cells (basal body co-localization, p < 0.0001; cell junction distribution, p < 0.0001; cilium formation, p < 0.02).
Given that AHI1 is required for cilium formation, disrupting the localization of AHI1 at the basal body could impair ciliogenesis. Therefore, the percentage of ciliated cells from AHI1-WT- and AHI1-V443D-transfected IMCD3 cells were assessed by Arl13b immunolabeling. A slight but significant reduction in cilium formation was seen in cells expressing AHI1-V443D as compared with AHI1-WT (Fig. 2D, AHI-WT, 76.16 ± 2.38%; AHI1-V443D, 59.22 ± 4.37%; p < 0.02). A likely reason for AHI1-V443D expressing cells not having a more significant blockade of cilium formation (as seen in Ahi1-deficient cells (11)) is that these cells still express endogenous Ahi1. Furthermore, this result suggests that AHI1-V443D is not functioning as a complete dominant negative in cilium formation in the presence of wild type Ahi1.
The V443D Mutation Reduces the Interaction of AHI1 and NPHP1
To determine the mechanism of how AHI1-V443D results in its inability to localize to the basal body and cell junctions, we examined another AHI1-binding partner, NPHP1, which has been shown to co-localize with Ahi1 (15). Given our results showing that AHI1-V443D localization is disrupted at cell-cell junctions, we assessed whether the interaction of AHI1 and NPHP1 could be disrupted by the V443D mutation. To test this hypothesis, we performed co-immunoprecipitation experiments in which HA-tagged NPHP1, and either myc-tagged AHI1-WT or AHI1-V443D, were co-expressed. As expected, myc-AHI1-WT binds HA-NPHP1 (Fig. 3A). However, HA-NPHP1 was still pulled down by AHI1-V443D (Fig. 3A).
FIGURE 3.

The V443D mutation in AHI1 alters the binding between AHI1 and NPHP1. A, co-immunoprecipitation of HA-tagged NPHP1 (HA-NPHP1) and myc-tagged AHI1 (WT or V443D) was performed from lysates of transiently transfected HEK293 cells. Proteins were precipitated with anti-myc antibodies and analyzed by Western blotting (representative experiment, n = 3). B, Western blots and plots showing the quantified band intensities against the fraction number of AHI1-WT/NPHP1 soluble proteins derived from HEK293 cells transiently expressing full-length HA-NPHP1 and myc-AHI1-WT and subjected to gel filtration chromatography. Fractions 18–29 (750–150 kDa) were analyzed by Western blotting (representative blots from triplicate experiments). The data were fitted to a multi-Gaussian function. The intensity distribution pattern for HA-NPHP1 and myc-AHI1-WT indicates that they co-migrate at two different peaks of 430 and 210 kDa, corresponding to a heterotetramer and a heterodimer, respectively. C, Western blots and plots showing the quantified band intensities against the fraction number of AHI1-V443D/NPHP1 soluble proteins derived from HEK293 cells transiently expressing full-length HA-NPHP1 and myc-AHI1-V443D and subjected to gel filtration chromatography. Fractions 18–29 (750–150 kDa) were analyzed by Western blotting (representative blots from triplicate experiments). The data were fitted to a multi-Gaussian function. The intensity distribution curve denotes that myc-AHI1-V443D and HA-NPHP1 co-migrate at a peak of 190 kDa corresponding to a heterodimer. The V443D mutation in AHI1 results in the loss of the AHI1-NPHP1 heterotetramer resulting in two peaks of AHI1 and a peak of NPHP1 of different molecular weights. Asterisks denote that this is the mutant AHI1-V443D in this subfigure. IB, immunoblot.
Given the non-quantitative nature of co-immunoprecipitation assays, and to study in more detail the binding between AHI1-V443D with NPHP1, we analyzed the interactions of AHI1 and NPHP1 in HEK293 cells. This system has several advantages, such as a high transfection efficiency in these cells, and because HEK293 cells are a human cell line, they have the full complement of chaperones and other proteins to ensure that the recombinant proteins are correctly folded. After co-transfection of AHI1 and NPHP1 or AHI1-V443D and NPHP1, the cells were lysed, and the soluble fractions, containing the overexpressed proteins, were separated from cell membranes and other organelles by differential centrifugation. The ability of the different proteins to associate with each other was evaluated by separation of fractions containing different molecular weights by gel filtration. The buffer used in the chromatography had a high ionic strength, which guarantees that the associations are specific, and had a relatively low amount of non-ionic detergent, β-d-dodecyl maltoside, to prevent nonspecific hydrophobic interactions. The mild detergent dodecyl maltoside has been used extensively in the characterization of protein complexes from different types of membranes, whereas serving as an effective agent for solubilization and extraction of most membrane proteins (27, 28). Moreover, it does not interfere with high affinity associations (27, 28).
In these types of experiments, two different behaviors can be anticipated. For two proteins that do not interact, it would be expected that they would migrate in single peaks representing monomers, but for proteins that interact with each other, it is expected that one or more complexes are formed, which migrate at high molecular weight masses than the monomeric species. Fig. 3, B and C, shows the distribution of AHI1 and NPHP1 in the different fractions of soluble proteins from cell lysates of cells co-transfected with NPHP1 and AHI1-WT, or NPHP1 and AHI1-V443D. AHI1-WT and NPHP1 are enriched in the same fractions 21–27 (Fig. 3B, bottom). The plots of the intensity versus the fraction number (Fig. 3B, top) revealed a series of peaks, corresponding to different complexes of AHI1 and NPHP1, which were different between AHI1-WT and AHI1-V443D (compare Fig. 3, B and C). AHI1-WT and NPHP1 co-migrated in two different size complexes, corresponding to molecular masses of 430 and 210 kDa, which possibly represent a heterotetramer (AHI12-NPHP12) and a heterodimer (AHI1-NPHP1) (Fig. 3B). Conversely, the V443D mutation in AHI1 greatly disrupted the association of AHI1-V443D and NPHP1. Although a heterodimer can still be observed, the heterotetramer is lost, although other complexes where found, which probably represent self-associations of AHI1 and NPHP1, such as the tetramer of NPHP1 and the tetramers and dimers of AHI1, with molecular masses of 300, 500, and 250 kDa, respectively (Fig. 3C). Overall, our results indicate that the V443D mutation in AHI1 specifically alters the formation of a tetramer, which according to our data is one of the two functional forms of these proteins. The reduced binding ability of AHI1-V443D to NPHP1 may result in the phenotype of mislocalization observed in AHI1-V443D-transfected cells.
The V443D Mutation Results in an Increased Rate of AHI1 Protein Decay
In addition to possibly altering protein function, the JBTS-associated missense mutation, V443D in AHI1, resulting in an amino acid substitution from a hydrophobic to hydrophilic amino acid, could also perturb protein structure stability and lead to faster degradation. Although effects of the V443D mutation in AHI1 on its subcellular distribution and protein interactions were determined previously, it was also important to elucidate whether this mutant AHI1 could be expressed with a normal half-life in cells. We first determined the mRNA expression levels for the AHI1-WT and AHI1-mutant (V443D). Equal amounts of plasmid DNA expressing myc-tagged AHI-WT or -mutant (V443D) were transfected in HEK293 cells. RNA was extracted and cDNA was synthesized and amplified by real-time PCR. The gene expression levels of both constructs were not significantly different using two separate primer sets (Fig. 4A).
FIGURE 4.

AHI1 protein levels are decreased with the human JBTS AHI1 mutation (V443D). A, AHI1-WT or AHI1-V443D mRNA levels from HEK293 cells transiently expressing myc-tagged AHI1-WT or AHI1-V443D cDNA. RNA was extracted and cDNA was synthetized and amplified by quantitative PCR. mRNA relative abundance was normalized to β-actin levels. Results were expressed as mean ± S.E. from three independent transfection experiments. AHI1-WT or AHI1-V443D mRNA levels are not significantly different using either an internal primer set or using primers for myc. B, protein stability of the AHI1-V443D is reduced. HEK293 cells were transfected with myc-AHI1-WT or -V443D and then were treated with cycloheximide (CHX) at several time points followed by cell lysis. Protein levels were analyzed by Western blotting. C, graphical display of the expression level of myc-AHI1-WT or -V443D from each lysate that was normalized to levels of β-tubulin (loading control). To compare AHI1-WT or AHI1-V443D protein levels at different time points, t0 values were set as 1. Results were expressed as mean ± S.E. using three independent transfections experiments. The asterisk indicates significance from AHI1-WT (p < 0.05).
To test for a possible reduction in AHI1-V443D stability at the protein level, we followed the decay of AHI1-WT and AHI1-V443D levels in the presence of the protein synthesis inhibitor, cycloheximide. The levels of transfected proteins were then evaluated by Western blot analysis (Fig. 4B). Levels of myc-tagged AHI1 proteins were normalized by β-tubulin protein levels (Fig. 4C). We found a significant reduction in AHI1-V443D protein levels that was 50% of that found for AHI1-WT levels (p < 0.05), suggesting that the V443D mutation results in an unstable AHI1 protein that subsequently can be degraded at a higher rate than wild type AHI1.
A New AHI1 Mutation, R351L, Also Disrupts the Localization of AHI1 at the Cell-Cell Junction and the Basal Body
Here we present a new mutation in AHI1, which causes JBTS with this mutation, also occurring in the region of AHI1 that is devoid of any known protein domains. The homozygous c.1052G>T, p.R351L variant was identified in the daughter of first-cousin, Middle Eastern parents within a 40.5 MB region of homozygosity encompassing AHI1. This missense mutation was not seen in 11,974 exome chromosomes (Exome Variant Server, NHLBI GO Exome Sequencing Project). The patient had developmental delay, ataxia, seizures, and classic brain imaging findings for JBTS, but no other features. Arg-351 is conserved throughout vertebrates and is predicted to be damaging by PolyPhen2 (HumDiv score 1.0, HumVar score 0.998).
To determine whether this JBTS-causative mutation in AHI1, in the same protein domain lacking region as V443D, can have similar phenotypes, we examined the AHI1-R351L mutation, using the same methodologies as described for the V443D mutation. Transfection of AHI1-R351L in cells resulted in a slight, but significant decrease in cilia formation compared with AHI1-WT using the ciliary marker, acetylated-α-tubulin (Fig. 5C). The lack of a more drastic effect on ciliogenesis is likely because endogenous Ahi1 is still expressed in the cells, suggesting that AHI1-R351L is also a weak dominant negative. Similar to the V443D mutation, the R351L mutation in AHI1 also affects its cellular localization. We found that only 17% of the cells expressing AHI1-R351L had AHI1-R351L co-localized with γ-tubulin at the basal body, whereas 63% of the cells expressing AHI1-WT had AHI1-WT and γ-tubulin co-localized at the basal body (Fig. 5, A and C). We also explored the distribution of AHI1-R351L at cell junctions and found a significant reduction in the number of cells expressing AHI1-R351L at cell junctions compared with the number of cells expressing AHI1-WT at junctions (Fig. 5, B and C). Taken together, the JBTS-associated mutations, V443D and R351L in AHI1, alter the distribution of AHI1 at cell-cell junctions and the basal body, which could further disrupt the function of AHI1 in the cell.
FIGURE 5.

Loss of AHI1 distribution at the basal body and cell-cell junctions with the JBTS-associated AHI1-R351L mutation. A, IMCD3 cells were transiently transfected with myc-AHI1-WT or myc-AHI1-R351L, fixed, and immunostained with myc (red) and γ-tubulin (green) to evaluate co-localization of transfected proteins (myc) and the centriole (basal body) marker (γ-tubulin). Arrowheads point to the co-localization of transfected AHI1 proteins and γ-tubulin in AHI1-WT cells (top), but an absence of co-localization in AHI1-R351L cells (bottom). Scale bar = 5 μm. B, IMCD3 cells were transiently transfected with myc-AHI1-WT or myc-AHI1-R351L, fixed, and immunostained with myc (red) and β-catenin (β-ctnn)(green) to evaluate co-localization of transfected proteins (myc) and a cell junction marker (β-ctnn). Arrowheads point to the co-localization of transfected AHI1 proteins and β-ctnn in AHI1-WT cells (top), but an absence of co-localization at cell junctions in AHI1-R351L cells (bottom). Scale bar = 5 μm. C, the graph represents the percentage of transfected AHI1 protein, localized at the basal body and cell junctions, as well as the incidence of cilia formation in these transfected cells. Cilia formation was quantified by evaluating the presence of cilia in IMCD3 cells transiently transfected with myc-AHI1-WT or myc-AHI1-R351L, and serum starved for 24 h to induce robust cilia formation, fixed, and immunostained with myc and acetylated-α-tubulin. Co-localization of proteins and cilia formation was evaluated on three different transfection experiments with at least 100 cells per coverslip analyzed. Bars represent mean ± S.E. Asterisk denotes significance from AHI-WT (*, p < 0.0005; **, p < 0.02).
Human AHI1 Directly Interacts with Human HAP1
Recent studies have shown that one of the main interactors of murine Ahi1 is Hap1, and have identified a region located at the N terminus of mouse Ahi1 (1–284 aa) that binds Hap1 (14). Previously, we have shown that Ahi1 is localized at stigmoid bodies in neurons (10); an organelle that is also highly enriched for Hap1 (29). Given the interaction of mouse Ahi1 and Hap1, and the JBTS-like cerebellar abnormalities that have been reported in Hap1−/− mice (14), this suggested a shared pathway for human HAP1 and AHI1 that is involved in brain development. How these two proteins are involved in mediating their effects is unknown, but disruption of AHI1 and HAP1 binding might possibly lead to the neuropathological phenotypes observed in JBTS. To test this hypothesis, we confirmed the binding of mouse Ahi1 and Hap1 in brain (Fig. 6) and extended this to human AHI1 and HAP1 proteins. This was an important step because human AHI1 possesses an N-terminal coiled-coil domain (∼140 aa) that does not exist in the mouse Ahi1 protein (7, 8), and therefore was not considered in the previous publication. To test this, we performed directed yeast two-hybrid analyses and co-immunoprecipitation studies and observed binding of human AHI1 and HAP1 (Fig. 7, A, C, and E). To define the region of AHI1 that binds HAP1, four individual fragments of AHI1 (AHI1-F1, 1–140 aa; AHI1-F2, 121–580 aa; AHI1-F4, 551–940 aa; AHI1-F5, 911–1196 aa) were examined for direct binding to HAP1 via directed yeast two-hybrid assays (Fig. 7, A and C). Only one AHI1 fragment, AHI1-F2, exhibited a positive interaction with HAP1 (Fig. 7C), which was also supported by co-immunoprecipitation studies indicating that AHI1-F2 formed a complex with full-length HAP1 (Fig. 7F). We also determined which region of HAP1 binds AHI1 by yeast two-hybrid analysis and found that the N-terminal region of HAP1 (1–320 aa) (Fig. 7, B and D), which contains a TATA-binding protein interacting region, but not the huntingtin protein binding region (30), binds AHI1 (Fig. 7, B and D).
FIGURE 6.
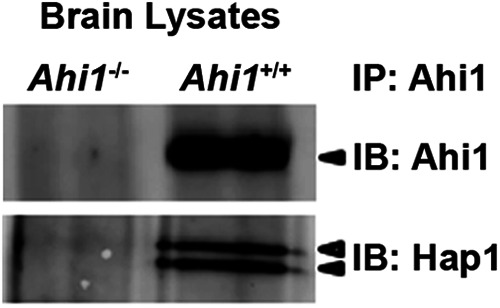
Ahi1 and Hap1 interact in mouse brain. Ahi1 antibodies were used to co-immunoprecipitate Hap1 in mouse brain lysate. Upon probing the immunoprecipitate, we resolved the expected Ahi1 band, in addition to the expected two known isoforms of Hap1. Importantly, we could not pulldown Hap1 in Ahi1 knock-out brain lysate, demonstrating the specificity of our co-immunoprecipitation. IB, immunoblot.
FIGURE 7.

Human AHI1 and HAP1 directly interact. A, diagrams representing full-length AHI1 and five fragments of AHI1 for assaying its binding to HAP1. B, diagram representing full-length HAP1 and two HAP1 fragments that were used to determine the AHI1 binding region. C, full-length AHI1 and full-length HAP1 directly interact as assessed by directed yeast two-hybrid analyses. The coiled-coil domain (AHI1-F1), the WD40 repeat domain (AHI1-F4), and the SH3 domain (AHI1-F5) of AHI1 did not exhibit any direct interactions with HAP1. However, a region of AHI1 between the coiled-coil domain and the WD40 repeats (AHI1-F2) that appeared to not have any definitive protein motif does demonstrate a yeast two-hybrid interaction with HAP1. D, directed yeast two-hybrid assays showed that only HAP1-F1, which contains the N-terminal region of HAP1, exhibited a positive binding with AHI1-F2 as demonstrated by a positive result in an X-Gal filter lift-off assay. E, positive interaction of full-length AHI1 and HAP1 was demonstrated in co-immunoprecipitation assays from lysates of HEK293T cells transiently co-expressing myc-HAP1 and HA-tagged full-length AHI1. Myc antibodies were used for the immunoprecipitation. F, AHI1-F2 binds HAP1 by co-immunoprecipitation with myc. G, a smaller fragment of AHI1-F2, AHI1-F3, still binds HAP1 by co-immunoprecipitation with myc. CC, coiled-coil domain; SH3, SH3 binding motif; TBP-IR, TATA-binding protein interacting region; IB, immunoblot.
Last, we identified the minimal region of AHI1 that binds HAP1 by co-immunoprecipitation. We determined that a smaller region of AHI1-F2, denoted as AHI1-F3 (141–434 aa), is still able to bind HAP1 (Fig. 7, A and G). This sequence is homologous to the previously identified region of mouse Ahi1 that binds Hap1 (14).
Hap1 Knock-out Cells Display Normal Cilium Formation and Function
Considering that JBTS is now considered a ciliopathy and that the Hap1 knock-out mouse appears to display JBTS-like neuroanatomical abnormalities (14), we hypothesized that Hap1 might also function at the primary cilium, possibly through Ahi1. To test this, we isolated both fibroblasts and neurons (hypothalamus, hippocampus, cortex, and cerebellum) from Hap1−/− embryos to evaluate cilia formation. No differences in ciliogenesis were found between Hap1−/− and Hap1+/+ neuronal cultures from these four brain regions (Fig. 8, A and B) or fibroblast cultures (Fig. 9A (0 time point)). Moreover, cilia lengths in neurons were also similar between the genotypes (Fig. 9B).
FIGURE 8.

Hap1-deficient neurons display normal cilium formation. A, normal cilium formation and morphology was observed in Hap1−/− neurons. Formation of primary cilia (green, labeled by Arl13b) from primary hypothalamic, hippocampal, cortical, and cerebellar cultures from Hap1+/+ and Hap1−/− embryos was evaluated by immunolabeling. DNA was visualized with Hoechst 33258 (blue). Scale bar = 5 μm. B, quantification of the number of Arl13b-positive primary cilia from at least 100 neuronal cells (determined by immunolabeling with the neuronal specific protein, NeuN (red)) per embryo (n = 3–8/genotype) from Hap1+/+ and Hap1−/− hypothalamus, hippocampus, cerebral cortex, and cerebellum. The bar chart on the right is the quantification of the number of Arl13b-positive primary cilia on all cerebellar neurons (both NeuN- and non-NeuN-positive cells) from Hap1+/+ and Hap1−/− cerebellum. Error bars represent the S.E.
FIGURE 9.

Primary cilia on Hap1-deficient cells have normal lengths and can properly undergo cilia disassembly under growth factor stimulation conditions. A, the percentage of ciliated MEFs from Hap1+/+ and Hap1−/− mice show similar declining patterns during serum stimulation. MEFs isolated from Hap1+/+ (n = 4) and Hap1−/− (n = 6) embryos were cultured in serum starved conditions for 24 h prior to serum stimulation. After various incubation times with serum, MEFs were fixed for Arl13b cilium immunolabeling (>100 cells were counted from each coverslip). No significant differences were found between Hap1+/+ and Hap1−/− MEFs. Error bars represent the S.E. B, in Hap1−/− neurons, primary cilia lengths were similar to wild type cells. Hypothalamic cells from Hap1+/+ (n = 3) and Hap1−/− (n = 3) embryos were immunolabeled with the ciliary marker, AC3, and cilia lengths were measured using AxioVision software. Range of cilia lengths: Hap1+/+, 1.29–8.22 μm and Hap1−/−, 1.10–8.34 μm. Error bars represent the S.E.
We also examined primary cilia formation by immunolabeling with the neuronal primary cilia marker, adenylyl cyclase III (AC3). Hypothalamic and cortical neurons from Hap1−/− and Hap1+/+ cultures both showed similar percentages of ciliated cells (Figs. 10, A and B, and 11). These results further indicate that deletion of Hap1 in neurons does not disrupt the formation and morphology of primary cilia.
FIGURE 10.

Proteins and receptors are normally localized to primary cilia in Hap1-deficient neurons. A and B, protein and receptor distribution at primary cilia is not significantly different in Hap1+/+ and Hap1−/− hypothalamic neurons. A, AC3 and MchR1 localization to primary cilia was evaluated by immunolabeling (scale bars = 5 μm; arrowheads point to primary cilia), and B, the percentage of cells having primary cilia localization of these markers was determined from at least 100 cells per coverslip (n = 3–8). Error bars represent the S.E. C and D, primary cilia formation is not affected in subsets of hypothalamic neurons in Hap1 knock-out cultures. C, specific subpopulations of hypothalamic neurons, positive for neuropeptide-Y (NPY) and positive for POMC, were examined by immunolabeling (scale bars = 5 μm; arrowheads point to primary cilia), and D, the percentage of NPY- and POMC-positive hypothalamic neurons cells having Arl13b-positive primary cilia was determined from at least 100 cells per coverslip (n = 3). Error bars represent the S.E.
FIGURE 11.
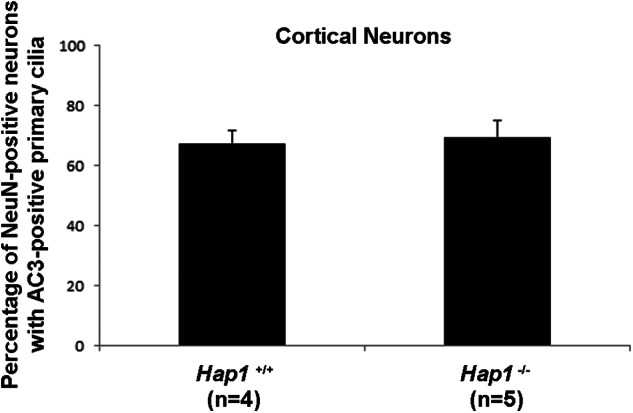
Hap1-deficient cortical neurons display normal cilium formation as assessed by AC3 immunolabeling. Quantification of the number of NeuN-positive cortical neurons that had AC3-positive primary cilia. A minimum of least 100 neuronal cells per coverslip per genotype were assayed (comparing Hap1+/+ and Hap1−/− cerebral cortical cultures). Error bars represent the S.E.
Additionally, we also examined ciliary G-protein coupled receptor distribution in Hap1−/− hypothalamic neurons, because we have observed a loss of melanin-concentrating hormone receptor-1 (MchR1) in primary cilia of Ahi1−/− neurons.4 We did not find a change in ciliary MchR1 localization in Hap1−/− neurons (Fig. 10, A and B), suggesting that Hap1, unlike Ahi1, is not involved in the ciliary targeting of MchR1 and possibly other G-protein coupled receptors.
Hap1 has been implicated in murine feeding behavior (31), and because particular subpopulations of hypothalamic neurons are involved in energy homeostasis (32), we examined primary cilia formation in POMC and neuropeptide Y neurons by immunolabeling. The percentage of POMC-positive and neuropeptide Y-positive Hap1−/− neurons with cilia were not significantly different from Hap1+/+ neurons (Fig. 10, C and D). These data further support our results demonstrating that Hap1 is not involved in the formation of primary cilia.
Although primary cilia can be differentiated in Hap1−/− neurons, function of primary cilia could be impaired. Several studies have demonstrated that disruption of some ciliary proteins does not necessarily block cilium formation, but compromises ciliary function. For instance, MEFs isolated from mice with deletions in Inpp5e, mutations in its human orthologue are associated with JBTS, are still able to form primary cilium, but show an increased ciliary instability upon stimulation by growth factors (33, 34). Therefore, we examined whether the primary cilia in Hap1−/− cells have similar defects in responding to growth factors. To test this, MEFs isolated from Hap1−/− mice were cultured in serum-starved conditions that favor cilium formation. Ciliated MEFs were then stimulated by adding serum back to the culture medium at various times prior to cilia immunolabeling (4, 8, and 24 h). Once cells were treated with serum, the percentages of ciliated cells in wild type MEFs gradually decreased with longer incubation times (Fig. 9A). This is due to re-absorption of primary cilia before entry back into the cell cycle in response to growth factor stimulation. In serum-treated Hap1−/− MEFs, a similar declining pattern of ciliated populations was observed (Fig. 9A). This indicates that MEFs from Hap1−/− mice and their wild type littermates display similar abilities in cilium formation. This result indicates that Hap1-deficient cells can regulate cilium disassembly similar to wild type cells during growth factor stimulation. Taken together, formation and function of primary cilia do not appear to be affected in cells with Hap1 deficiency, and these results further suggest that the JBTS-like cerebellum abnormality reported in Hap1−/− mice (14) is unlikely due to defective primary cilium formation.
The Ahi1 and Hap1 Interaction Stabilizes Their Protein Levels
Given that there were no observed effects on ciliogenesis in Hap1 knock-out mice, this suggested an alternative mechanism to explain the effects of the Ahi1 and Hap1 interaction. Previously, it was reported that Ahi1 levels are decreased in Hap1−/− mice, and that Hap1 levels are decreased in Ahi1−/− mice, suggesting that Ahi1 and Hap1 stabilize each other (14).
Using our Ahi1 knock-out mouse, we have confirmed an Ahi1-Hap1 stabilization effect. Loss of Ahi1 resulted in a reduction of Hap1 immunostaining in Ahi1−/− brain sections and a lack of stigmoid body staining by Hap1 in the hypothalamus (Fig. 12A). We also quantified the Hap1 levels in brain tissue from Ahi1+/+ and Ahi1−/− mice by Western blotting, and observed a significant decrease in Hap1 levels in Ahi1−/− mice (Fig. 12B). Because murine Ahi1 lacks the coiled-coil domain present in human AHI1, we further analyzed the AHI1-HAP1 stabilization by transfecting increasing amounts of myc-AHI1-WT into PC12 cells and evaluated the expression of Hap1. We found that Hap1 levels are increased when more AHI1 is present (Fig. 12C), confirming the hypothesis that AHI1 and Hap1 stabilize each other.
FIGURE 12.

AHI1 stabilizes HAP1. A, brain midsagittal sections from Ahi1+/+ and Ahi1−/− mice immunostained with Hap1 showing a reduction in Hap1 levels in the absence of Ahi1. Scale bar = 500 μm. Higher magnification images of the hypothalamus showed the absence of stigmoid bodies in Ahi1−/− tissue. Arrowheads point to some of the stigmoid bodies. Scale bar = 10 μm. B, Hap1 levels in brains from Ahi1−/− and Ahi1+/+ mice were analyzed by Western blot and graphically displayed. β-Tubulin III was used as a loading control. Hap1 levels were decreased in Ahi1−/− brains compared with Ahi1+/+ (p < 0.05). Results are expressed as mean ± S.E. (n = 4). C, PC12 cells were transfected with increasing amounts of myc-AHI1-WT (human) and lysed after 24 h. Myc-AHI1-WT and Hap1 levels were analyzed by Western blot using β-tubulin as a loading control. Results are expressed as mean ± S.E. (n = 3). Asterisk denotes significance from control (non-transfected) (p < 0.05).
The JBTS-causing Mutation, V443D, in AHI1 Alters Binding to HAP1
To further explore the binding of AHI1 and HAP1, which occurs in a region of AHI1 that appears not to contain any known protein motif, we examined the known JBTS-causing mutation in AHI1, V443D, that is located in this region. Given that Hap1−/− mice exhibit a JBTS-like phenotype in the cerebellum (14), we hypothesized that the V443D mutation in AHI1 may interrupt its binding to HAP1. Co-immunoprecipitation studies demonstrated that there was an interaction between HAP1 and full-length AHI1-V443D (Fig. 13C) and AHI1-F2-V443D (Fig. 13D). Similar HAP1 and AHI1 interactions were observed when the myc and HA tags were switched. We further examined this interaction by using a directed yeast two-hybrid assay and found a positive interaction between AHI1-F2-V443D and HAP1-F1; however, there appeared to be a reproducible decrease in the β-galactosidase activity as compared with the interaction with HAP1-F1 to AHI1-F2-wild type, AHI1-F2-D330G (mutation producing mild neurological lesions in patients with nephronophthisis)(24), or AHI1-F2-D358N (control mutation) (Fig. 13B). This suggests that the V443D mutation possibly decreases the binding of AHI1 and HAP1.
FIGURE 13.

The V443D mutation in AHI1, which causes Joubert syndrome, alters its binding to HAP1. A, alignment of amino acids from various vertebrate species that surrounds the conserved valine (red) residue that is mutated in AHI1 causing JBTS (V443D) using ClustalW. Sequences were from NP_001128303 (Homo sapiens), XP_001170580 (Pan troglodytes), XP_001099136 (Macaca mulatta), XP_001788246 (Bos taurus), XP_533417 (Canis familiaris), XP_001503573 (Equus caballus), NP_001002277 (Rattus norvegicus), NP_080479 (Mus musculus), XP_001521581 (Ornithorhynchus anatinus), CAG13180 (Tetraodon nigroviridis), XP_002189154 (Taeniopygia guttata), and NP_001071029 (Danio rerio). The asterisk indicates identical amino acids. B, directed yeast two-hybrid assays showed that in HAP1-F1, whereas exhibiting positive binding with AHI1-F2 (D330G), AHI1-F2 (D358N), and AHI1-F2 (V443D), there was a reproducible decrease in binding of AHI1-F2 (V443D) with HAP1-F1. C, positive interaction of full-length AHI1-V443D and HAP1 was demonstrated in co-immunoprecipitation assays from lysates of HEK293 cells transiently co-expressing myc-HAP1 and HA-tagged full-length AHI1-WT or -V443D. The wild type blots from Fig. 6 are reproduced here for comparison. D, AHI1-F2 wild type or -V443D binds HAP1 by co-immunoprecipitation. The wild type blots from Fig. 6 are reproduced here for comparison. E, Western blots and plots showing the band intensities against the fraction number of AHI1-WT/HAP1 soluble proteins derived from HEK293 cells transiently expressing full-length HA-HAP1 and myc-AHI1-WT and subjected to gel filtration chromatography. Fractions 16–29 corresponding to 750–150 kDa were analyzed by Western blotting. The data were fitted to a multi-Gaussian function. The intensity distribution curve denotes that AHI1-WT and HAP1 co-migrate in a single complex of 470 kDa, corresponding to a heterotetramer. The X refers to the binding of an unknown protein (50 kDa) to AHI1. Western blot images are representative of three independent experiments. F, Western blots and plots showing the quantified band intensities against the fraction number of AHI1-V443D/HAP1 soluble proteins derived from HEK293 cells transiently expressing full-length HA-HAP1 and myc-AHI1-V443D and subjected to gel filtration chromatography. Fractions 16–29 corresponding to 750–150 kDa were analyzed by Western blotting. The data were fitted to a multi-Gaussian function. The intensity distribution curve denotes that the V443D mutation in AHI1 alters the association of AHI1 and HAP1 with the emergence of a new peak corresponding to the homohexamer of HAP1. Fraction 24 contains no AHI1 or HAP1. The X refers to the binding of an unknown protein (80 kDa) to AHI1. Asterisks denote that this is the mutant AHI1-V443D in this subfigure. Western blot images are representative of three independent experiments. IB, immunoblot.
To examine the possible decrease in binding of AHI1-V443D with HAP1, we co-transfected HEK293 cells with HA-HAP1 and myc-AHI1-WT or -V443D. Cells were lysed and the soluble proteins were subjected to gel filtration chromatography to separate the different protein complexes by size. Fig. 13, E and F, show the distribution of AHI1 and HAP1 in the different fractions of soluble proteins from the co-transfected cell with HAP1 and either AHI1 wild type (Fig. 13E) or mutant AHI1-V443D (Fig. 13F). For wild type AHI1, AHI1 is mostly found in fractions 20–22 and 27, whereas HAP1 was found in fractions 19–23 (Fig. 13E, bottom). The graphical display in Fig. 13E shows that AHI1 and HAP1 co-migrate in a complex of 470 kDa, which suggests that they are able to form a heterotetramer (AHI12-HAP12). A single peak of AHI1 is also observed at 180 kDa, which could correspond to an AHI1 homodimer. However, the molecular weight of the peak is smaller than the expected weight of the dimer (Fig. 13E, top). Conversely, for AHI1-V443D, AHI1 is mainly present in fractions 20–23 with HAP1 in fractions 19–23 (Fig. 13F, bottom). The intensity distribution of AHI1-V443D and HAP1 (Fig. 13F) shows that the mutation alters the association of these two proteins, even when the heterotetramer of AHI12-HAP12 is still present. There is a gain of a HAP1 peak at 550 kDa, corresponding to a hexamer indicating that a fraction of HAP1 does not bind AHI1-V443D (Fig. 13F, top). Taken together, these data demonstrate that human AHI1 with a mutation at V443D interacts with a decreased binding efficiency to HAP1, and may associate with new protein interactors.
The ability of AHI1 to self-associate, as suggested by previous data with NPHP1 (Fig. 3) and HAP1 (Fig. 13), has not been previously shown, so we examined this self-association directly by co-immunoprecipitation analysis (Fig. 14). HEK293 cells were co-transfected with AHI1 fused to a C-terminal GFP (CGFP-AHI1) and myc-AHI1. Cell lysates were prepared and an anti-myc antibody was used to pull-down protein complexes, which were analyzed by Western blotting. Anti-GFP and anti-myc antibodies revealed the presence of both AHI1-tagged versions when immunoprecipitating the myc tag. These results demonstrate that AHI1 is capable of self-association; however, the V443D mutation in AHI1 did not disrupt the self-association of AHI1 (Fig. 14).
FIGURE 14.
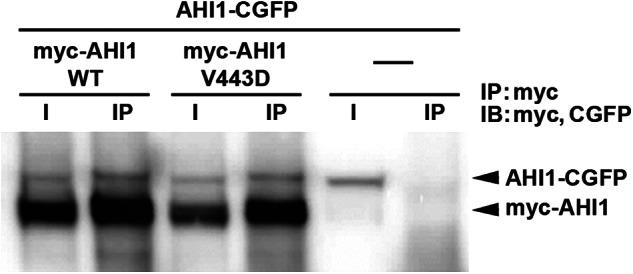
AHI1 self-associates. AHI1 with a C-terminal-tagged GFP (AHI1-CGFP) was co-transfected with either myc-tagged AHI1-WT or AHI1-V443D and co-immunoprecipitated (IP) with antibodies to myc. Blots were subsequently probed with antibodies to both GFP and myc. A representative blot demonstrating that AHI1 was able to form a complex with itself, and that the V443D in AHI1 did not affect its binding to wild type AHI1 is shown.
DISCUSSION
In our study, we show that the function of AHI1 is affected by one of the known JBTS-associated missense mutations in AHI1 with the resulting amino acid substitution, V443D, causing an inability for AHI1-V443D to localize to the basal body and cell-cell junctions. Moreover, this AHI1 mutation alters its binding to NPHP1 and HAP1. Alterations in AHI1 protein distribution and AHI1 protein-protein interactions, due to this specific amino acid change, also significantly decreases primary cilia formation. Furthermore, the R351L point mutation in AHI1 also results in the inability of AHI1 to localize to the basal body and cell junctions. Overall, these data implicate basal body, primary cilia, and cell junctions in the pathogenesis observed in JBTS.
Several mutations in AHI1 have been identified in individuals with JBTS with most being nonsense mutations, causing an early termination and production of non-functional protein (1). Six of the nine documented missense mutations occur within the WD40 repeat domain present in AHI1, and the effects of these mutations on protein structure and function is predictable. However, only three missense mutations (R351L, V443D, and E1086G) are located in regions that contain no known protein motifs, but still result in the neurological pathologies observed in JBTS. This suggests that these AHI1 mutations cause some alteration in the structure of the protein that affects binding partner interactions, resulting in an abnormally functioning protein. Indeed, our study demonstrated that the V443D and R351L mutations in AHI1, occurring in a region between the coiled-coil domain and WD40 repeats, significantly alter AHI1 subcellular distribution and protein interactions. From a protein structure perspective, the V443D mutation is a substitution from a hydrophobic amino acid to a charged, hydrophilic amino acid, and it is located in the region between two protein domains. In general, a hydrophobic amino acid residue tends to be embedded inside a protein, whereas most of hydrophilic amino acid residues remain on the protein surface. Therefore, the V443D mutation is very likely to perturb this region of the protein structure and make the mutant protein unstable. Indeed, a reduction in protein levels resulting from the V443D mutation was demonstrated in this study, indicating that the mutant AHI1-V443D protein is unstable and may further augment the JBTS-phenotype beyond its effects on cilia formation and localization at cell-cell junctions.
Given the location of the V443D mutation between the coiled-coil domain and the WD40 repeats, it is possible that this substituted charged amino acid residue produces a repulsive force in the interface between these two domains resulting in a subsequent change in protein conformation, thereby affecting the function of this protein. This interpretation can be partially supported by our result demonstrating that the AHI1-V443D mutant protein displays abnormal subcellular localization and protein interactions, and by the finding that AHI1-F3 (141–434 aa) is necessary and sufficient to bind HAP1, because this fragment does not contain the highly conserved valine at the 443 position. Therefore, this residue is not required for the AHI1 and HAP1 interaction, but when it is replaced by an aspartic acid residue, it destabilizes the AHI1-HAP1 interaction.
Coiled-coil domains and WD40 repeats are protein motifs that are commonly involved in protein-protein interactions (35, 36). For example, AHI1, through its WD40 domain, binds NPHP1 (15). Given the proposed function for AHI1 as a scaffold protein, the V443D mutation in AHI1 could result in a repulsive force between these two domains that could distance the coiled-coil domain and WD40 repeats. This would have the consequence of possibly affecting the recruitment and assembly of protein complexes at AHI1 through this mutation altering its structural conformation. Moreover, the V443D mutation is located outside the WD40 domain, which is required for AHI1 binding to NPHP1, but also partially disrupts the interaction of these two proteins. This suggests that mutations in the region between the coiled-coil motif and the WD40 domain in AHI1 can have a profound effect on protein binding, likely through altering the protein structure, even though this region is not required for NPHP1 binding. However, the consequence of the V443D mutation in AHI1 cannot be definitively known until the crystal structure for AHI1 is determined.
With the AHI1-R351L mutation, we have the opposite situation as AHI1-V443D. For AHI1-R351L, the change from a positively charged residue to a branched aliphatic group likely perturbs the structure of the protein in two different ways. It is possible that the positively charged arginine residue stabilizes the structure of the protein by forming a salt bridge and that the change to a leucine completely disrupts this interaction, affecting the internal structure of the protein or the interaction of AHI1 with other proteins. It is also possible that residue Arg-351 is found in a hydrophilic pocket or exposed to the solvent. In this environment, leucine would not be able to establish interactions, which possibly could change the folding pattern, thereby affecting the final structure of the protein. Moreover, leucine is a branched amino acid and its presence could change the interaction with the surrounding amino acids. Taken together, the change from arginine to leucine in the R351L mutation in AHI1 could affect the overall protein structure of AHI1 disrupting its function and interactions with other proteins.
Similarities between AHI1 and HAP1 have been demonstrated in several studies, which have shown co-expression of Ahi1 and Hap1 in identical regions of the brain, in similar neuronal compartments, namely stigmoid bodies, and mouse knock-outs for both genes have a similar postnatal lethality (in a mixed background for Hap1 and only on a C57BL/6J background for Ahi1 (10, 12, 14, 37)). Also, Ahi1 and Hap1 interact and stabilize each other (14), and our results support this and further demonstrate that the AHI1-V443D mutations cause altered interactions with HAP1. Last, the Hap1 knock-out mouse was recently shown to have a JBTS-like phenotype (14). Thus, this suggests that a common pathway involving AHI1 and HAP1 may be important for neuronal development, and may involve the primary cilium. Therefore, the relationship of Hap1 to the primary cilium was also examined here, because Ahi1 is a basal body protein and is involved in cilium formation (11, 15). However, Hap1-deficient neurons display typical cilium morphology and numbers, protein distribution, and have normal ciliary signaling upon growth factor stimulation, indicating that Hap1, unlike Ahi1, is not involved in ciliary formation/function in neurons. Importantly, this further indicates that the JBTS-like cerebellar abnormality observed in Hap1 knock-out mice is not a result of defective cilia formation.
Although our Hap1 data do not support a role for the primary cilium as the mediator of the JBTS-phenotype in mice, other pathways may be responsible. HAP1 has been proposed as playing a regulatory role in intracellular trafficking and receptor recycling pathways, whereas Ahi1 has been similarly proposed as having a function in mediating receptor endocytosis (11, 38). Therefore, defects in intracellular trafficking may be responsible for the Hap1 knock-out JBTS-phenotype, and such mechanisms will need to be explored in the future.
Our results showing a role for AHI1 in primary cilia formation and function in fibroblasts from patients with AHI1 mutations, as well as the similarly observed effects in cells expressing the AHI1-V443D and AHI1-R351L mutations, help to establish that AHI1 does have a significant role in mediating ciliogenesis and primary cilia function. Our data from JBTS patient cells and Ahi1 knock-out mice showing impairments in primary cilia formation (11, 12) are opposite to another reported Ahi1 knock-out mouse (39, 40). Moreover, a previous report has shown a decrease in the number of AC3-positive ciliated cortical neurons in Hap1-deficient mice (41); however, we found no difference in the number of cilia in Hap1 knock-out mice using a variety of ciliary markers and examining multiple brain regions and tissues. In fact, given this report, we specifically examined AC3-positive ciliated cells in cortical neurons and found no differences between Hap1+/+ and Hap1−/− mice. These differences could be attributed to the genetic background of the animals studied. Because the Keryer study (41) did not list the genetic background of their mice, it is difficult to reach a conclusion; however, the previously reported Ahi1 mouse line was on a different background. Therefore, it is possible that a genetic modifier is present in different mouse strains that could alter these phenotypes. Also, these incongruities regarding the variability of results between studies in comparing knock-out mice emphasize that special consideration should be given to the genetic background of the animals studied.
In addition to aberrant protein localization, expression of AHI1-V443D and AHI1-R351L causes a slight, but statistically significant reduction in cilia formation. Importantly, IMCD3 cells have wild type Ahi1 endogenously expressed. Therefore, the result of not seeing a total blockade of cilium formation in IMCD3 cells transfected with mutant AHI1-V443D and AHI1-R351L suggests that these mutations are not likely complete dominant negatives for wild type AHI1 in cilia formation. This could further explain why no apparent clinical phenotypes are observed in individuals carrying the heterozygous mutation of AHI1-V443D or AHI1-R351L.
A reduced interaction of AHI1-V443D and NPHP1 also implicates an important role for AHI1 during cell polarity establishment. NPHP1 and other nephrocystin proteins have been identified at cell junctions and the basal body, as well as having a physical interaction with PCP protein complex proteins (PALS1/PATJ and Par6) (22). In fact, knockdown of Nphp1 in Madin-Darby canine kidney cells results in several polarity defects, including cilium malformation, delayed tight junction formation, and disorganized lumen formation in three-dimensional matrix cultures, indicative of a critical role of NPHP1 during cell polarization (22). Interestingly, previous results from our lab have shown that knockdown of Ahi1 expression in IMCD3 cells results in cell polarity defects, such as abnormal localization of the Golgi, further supporting a role of AHI1 in cell polarity (11). Although mutations in AHI1 causing JBTS often result in mainly CNS defects, a concurrence of CNS abnormalities and renal pathology can be observed in JBTS patients carrying both mutations in AHI1 and NPHP1, implicating a common pathway involving a genetic interaction of AHI1 and NPHP1 in brain and kidney development.
Taken together, the JBTS-associated missense mutation V443D in AHI1, which occurs in a region containing no known protein binding motifs, 1) abrogates its subcellular localization to the basal body of the primary cilium and at cell junctions, 2) has reduced expression due to protein instability, and 3) alters its protein interactions with HAP1 and NPHP1. These results suggest a critical function for AHI1 in these specific subcellular structures and shed light on a pathway shared by AHI1 and NPHP1 for the pathogenesis seen in JBTS. Understanding the effects of JBTS disease proteins, such as AHI1-V443D and AHI1-R351L, can provide an alternative approach for gaining insight into specific mechanisms underlying the clinical pathology observed in JBTS.
Acknowledgments
We thank the families who have contributed to this study. We also thank Dr. Tamara Caspary (Emory) for generously supplying the Arl13b antibody, Dr. Blanca Barquera (RPI) for generously allowing the use of her gel chromatography equipment, and Dr. Livingston Van De Water (AMC) for supplying cells. Last, we thank Linda Crane Bonin for critically reading our manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM50821 (to M. R. L.), R01NS064077 (to D. D.), K01MH71801 and R01NS064283 (to R. J. F.) and March of Dimes Foundation Grant 5-FY09–29 (to R. J. F.).
K. Tuz, Y-C. Hsiao, and R. J. Ferland, unpublished observations.
- JBTS
- Joubert syndrome
- aa
- amino acid
- AC3
- adenylyl cyclase III
- AHI1
- Abelson-helper integration site 1 gene
- HAP1
- Huntingtin-associated protein-1
- Htt
- huntingtin
- IMCD
- inner medullary collecting duct
- MchR1
- melanin-concentrating hormone receptor-1
- MEF
- mouse embryonic fibroblast
- NPHP
- nephronophthisis
- NPHP1
- nephrocystin-1
- NPY
- neuropeptide Y
- POMC
- pro-opiomelanocortin
- RES
- restriction enzyme site
- SH3
- SH3 binding motif
- WD40
- WD40 repeats domain.
REFERENCES
- 1. Parisi M. A. (2009) Clinical and molecular features of Joubert syndrome and related disorders. Am. J. Med. Genet. C Semin. Med. Genet. 151 C(4), 326–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alvarez Retuerto A. I., Cantor R. M., Gleeson J. G., Ustaszewska A., Schackwitz W. S., Pennacchio L. A., Geschwind D. H. (2008) Association of common variants in the Joubert syndrome gene (AHI1) with autism. Hum. Mol. Genet. 17, 3887–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hildebrandt F., Benzing T., Katsanis N. (2011) Ciliopathies. N. Engl. J. Med. 364, 1533–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goetz S. C., Anderson K. V. (2010) The primary cilium. A signalling centre during vertebrate development. Nat. Rev. Genet. 11, 331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Badano J. L., Mitsuma N., Beales P. L., Katsanis N. (2006) The ciliopathies. An emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 7, 125–148 [DOI] [PubMed] [Google Scholar]
- 6. Dixon-Salazar T., Silhavy J. L., Marsh S. E., Louie C. M., Scott L. C., Gururaj A., Al-Gazali L., Al-Tawari A. A., Kayserili H., Sztriha L., Gleeson J. G. (2004) Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am. J. Hum. Genet. 75, 979–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ferland R. J., Eyaid W., Collura R. V., Tully L. D., Hill R. S., Al-Nouri D., Al-Rumayyan A., Topcu M., Gascon G., Bodell A., Shugart Y. Y., Ruvolo M., Walsh C. A. (2004) Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat. Genet. 36, 1008–1013 [DOI] [PubMed] [Google Scholar]
- 8. Jiang X., Hanna Z., Kaouass M., Girard L., Jolicoeur P. (2002) Ahi-1, a novel gene encoding a modular protein with WD40-repeat and SH3 domains, is targeted by the Ahi-1 and Mis-2 provirus integrations. J. Virol. 76, 9046–9059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kroes H. Y., van Zon P. H., Fransen van de Putte D., Nelen M. R., Nievelstein R. J., Wittebol-Post D., van Nieuwenhuizen O., Mancini G. M., van der Knaap M. S., Kwee M. L., Maas S. M., Cobben J. M., De Nef J. E., Lindhout D., Sinke R. J. (2008) DNA analysis of AHI1, NPHP1, and CYCLIN D1 in Joubert syndrome patients from the Netherlands. Eur. J. Med. Genet. 51, 24–34 [DOI] [PubMed] [Google Scholar]
- 10. Doering J. E., Kane K., Hsiao Y. C., Yao C., Shi B., Slowik A. D., Dhagat B., Scott D. D., Ault J. G., Page-McCaw P. S., Ferland R. J. (2008) Species differences in the expression of Ahi1, a protein implicated in the neurodevelopmental disorder Joubert syndrome, with preferential accumulation to stigmoid bodies. J. Comp. Neurol. 511, 238–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hsiao Y. C., Tong Z. J., Westfall J. E., Ault J. G., Page-McCaw P. S., Ferland R. J. (2009) Ahi1, whose human ortholog is mutated in Joubert syndrome, is required for Rab8a localization, ciliogenesis and vesicle trafficking. Hum. Mol. Genet. 18, 3926–3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Westfall J. E., Hoyt C., Liu Q., Hsiao Y. C., Pierce E. A., Page-McCaw P. S., Ferland R. J. (2010) Retinal degeneration and failure of photoreceptor outer segment formation in mice with targeted deletion of the Joubert syndrome gene, Ahi1. J. Neurosci. 30, 8759–8768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Louie C. M., Caridi G., Lopes V. S., Brancati F., Kispert A., Lancaster M. A., Schlossman A. M., Otto E. A., Leitges M., Gröne H. J., Lopez I., Gudiseva H. V., O'Toole J. F., Vallespin E., Ayyagari R., Ayuso C., Cremers F. P., den Hollander A. I., Koenekoop R. K., Dallapiccola B., Ghiggeri G. M., Hildebrandt F., Valente E. M., Williams D. S., Gleeson J. G. (2010) AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat. Genet. 42, 175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sheng G., Xu X., Lin Y. F., Wang C. E., Rong J., Cheng D., Peng J., Jiang X., Li S. H., Li X. J. (2008) Huntingtin-associated protein 1 interacts with Ahi1 to regulate cerebellar and brainstem development in mice. J. Clin. Invest. 118, 2785–2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eley L., Gabrielides C., Adams M., Johnson C. A., Hildebrandt F., Sayer J. A. (2008) Jouberin localizes to collecting ducts and interacts with nephrocystin-1. Kidney Int. 74, 1139–1149 [DOI] [PubMed] [Google Scholar]
- 16. Li X. J., Li S. H., Sharp A. H., Nucifora F. C., Jr., Schilling G., Lanahan A., Worley P., Snyder S. H., Ross C. A. (1995) A huntingtin-associated protein enriched in brain with implications for pathology. Nature 378, 398–402 [DOI] [PubMed] [Google Scholar]
- 17. Li H., Wyman T., Yu Z. X., Li S. H., Li X. J. (2003) Abnormal association of mutant huntingtin with synaptic vesicles inhibits glutamate release. Hum. Mol. Genet. 12, 2021–2030 [DOI] [PubMed] [Google Scholar]
- 18. Wu L. L., Zhou X. F. (2009) Huntingtin associated protein 1 and its functions. Cell Adh. Migr. 3, 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Metzger S., Rong J., Nguyen H. P., Cape A., Tomiuk J., Soehn A. S., Propping P., Freudenberg-Hua Y., Freudenberg J., Tong L., Li S. H., Li X. J., Riess O. (2008) Huntingtin-associated protein-1 is a modifier of the age-at-onset of Huntington's disease. Hum. Mol. Genet. 17, 1137–1146 [DOI] [PubMed] [Google Scholar]
- 20. Rong J., Li S. H., Li X. J. (2007) Regulation of intracellular HAP1 trafficking. J. Neurosci. Res. 85, 3025–3029 [DOI] [PubMed] [Google Scholar]
- 21. Wolf M. T., Hildebrandt F. (2011) Nephronophthisis. Pediatr. Nephrol. 26, 181–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delous M., Hellman N. E., Gaudé H. M., Silbermann F., Le Bivic A., Salomon R., Antignac C., Saunier S. (2009) Nephrocystin-1 and nephrocystin-4 are required for epithelial morphogenesis and associate with PALS1/PATJ and Par6. Hum. Mol. Genet. 18, 4711–4723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Donaldson J. C., Dise R. S., Ritchie M. D., Hanks S. K. (2002) Nephrocystin-conserved domains involved in targeting to epithelial cell-cell junctions, interaction with filamins, and establishing cell polarity. J. Biol. Chem. 277, 29028–29035 [DOI] [PubMed] [Google Scholar]
- 24. Tory K., Lacoste T., Burglen L., Morinière V., Boddaert N., Macher M. A., Llanas B., Nivet H., Bensman A., Niaudet P., Antignac C., Salomon R., Saunier S. (2007) High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis. Potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J. Am. Soc. Nephrol. 18, 1566–1575 [DOI] [PubMed] [Google Scholar]
- 25. Li S. H., Yu Z. X., Li C. L., Nguyen H. P., Zhou Y. X., Deng C., Li X. J. (2003) Lack of huntingtin-associated protein-1 causes neuronal death resembling hypothalamic degeneration in Huntington's disease. J. Neurosci. 23, 6956–6964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Caspary T., Larkins C. E., Anderson K. V. (2007) The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell 12, 767–778 [DOI] [PubMed] [Google Scholar]
- 27. Eubel H., Jänsch L., Braun H. P. (2003) New insights into the respiratory chain of plant mitochondria. Supercomplexes and a unique composition of complex II. Plant Physiol. 133, 274–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schägger H., Pfeiffer K. (2001) The ratio of oxidative phosphorylation complexes I-V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J. Biol. Chem. 276, 37861–37867 [DOI] [PubMed] [Google Scholar]
- 29. Gutekunst C. A., Li S. H., Yi H., Ferrante R. J., Li X. J., Hersch S. M. (1998) The cellular and subcellular localization of huntingtin-associated protein 1 (HAP1). Comparison with huntingtin in rat and human. J. Neurosci. 18, 7674–7686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li S. H., Hosseini S. H., Gutekunst C. A., Hersch S. M., Ferrante R. J., Li X. J. (1998) A human HAP1 homologue. Cloning, expression, and interaction with huntingtin. J. Biol. Chem. 273, 19220–19227 [DOI] [PubMed] [Google Scholar]
- 31. Sheng G., Chang G. Q., Lin J. Y., Yu Z. X., Fang Z. H., Rong J., Lipton S. A., Li S. H., Tong G., Leibowitz S. F., Li X. J. (2006) Hypothalamic huntingtin-associated protein 1 as a mediator of feeding behavior. Nat. Med. 12, 526–533 [DOI] [PubMed] [Google Scholar]
- 32. Wu Q., Palmiter R. D. (2011) GABAergic signaling by AgRP neurons prevents anorexia via a melanocortin-independent mechanism. Eur. J. Pharmacol. 660, 21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bielas S. L., Silhavy J. L., Brancati F., Kisseleva M. V., Al-Gazali L., Sztriha L., Bayoumi R. A., Zaki M. S., Abdel-Aleem A., Rosti R. O., Kayserili H., Swistun D., Scott L. C., Bertini E., Boltshauser E., Fazzi E., Travaglini L., Field S. J., Gayral S., Jacoby M., Schurmans S., Dallapiccola B., Majerus P. W., Valente E. M., Gleeson J. G. (2009) Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidylinositol signaling to the ciliopathies. Nat. Genet. 41, 1032–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jacoby M., Cox J. J., Gayral S., Hampshire D. J., Ayub M., Blockmans M., Pernot E., Kisseleva M. V., Compère P., Schiffmann S. N., Gergely F., Riley J. H., Pérez-Morga D., Woods C. G., Schurmans S. (2009) INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat. Genet. 41, 1027–1031 [DOI] [PubMed] [Google Scholar]
- 35. Neer E. J., Schmidt C. J., Nambudripad R., Smith T. F. (1994) The ancient regulatory-protein family of WD-repeat proteins. Nature 371, 297–300 [DOI] [PubMed] [Google Scholar]
- 36. Burkhard P., Stetefeld J., Strelkov S. V. (2001) Coiled coils. A highly versatile protein folding motif. Trends Cell Biol. 11, 82–88 [DOI] [PubMed] [Google Scholar]
- 37. Chan E. Y., Nasir J., Gutekunst C. A., Coleman S., Maclean A., Maas A., Metzler M., Gertsenstein M., Ross C. A., Nagy A., Hayden M. R. (2002) Targeted disruption of Huntingtin-associated protein-1 (Hap1) results in postnatal death due to depressed feeding behavior. Hum. Mol. Genet. 11, 945–959 [DOI] [PubMed] [Google Scholar]
- 38. Xu X., Yang H., Lin Y. F., Li X., Cape A., Ressler K. J., Li S., Li X. J. (2010) Neuronal Abelson helper integration site-1 (Ahi1) deficiency in mice alters TrkB signaling with a depressive phenotype. Proc. Natl. Acad. Sci. U.S.A. 107, 19126–19131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lancaster M. A., Schroth J., Gleeson J. G. (2011) Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat. Cell Biol. 13, 700–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lancaster M. A., Louie C. M., Silhavy J. L., Sintasath L., Decambre M., Nigam S. K., Willert K., Gleeson J. G. (2009) Impaired Wnt-β-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nat. Med. 15, 1046–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Keryer G., Pineda J. R., Liot G., Kim J., Dietrich P., Benstaali C., Smith K., Cordelières F. P., Spassky N., Ferrante R. J., Dragatsis I., Saudou F. (2011) Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J. Clin. Invest. 121, 4372–4382 [DOI] [PMC free article] [PubMed] [Google Scholar]