

Background: In-frame deletion mutation (Del-L955) in NaV1.7 sodium channel from a kindred with erythromelalgia hyperpolarizes activation.
Results: Del-L955 twists the S6 helix, displacing the Phe960 activation gate. Replacement of Phe960 at the correct helical position depolarizes activation.
Conclusion: Radial tuning of the activation gate is critical to the activation of NaV1.7 channel.
Significance: Structural modeling guided electrophysiology reveals the functional importance of radial tuning of the S6 segment.
Keywords: Biophysics, Electrophysiology, Ion Channels, Molecular Modeling, Mutagenesis, Pain, Sodium Channels
Abstract
Voltage-gated sodium (NaV) channels are membrane proteins that consist of 24 transmembrane segments organized into four homologous domains and are essential for action potential generation and propagation. Although the S6 helices of NaV channels line the ion-conducting pore and participate in channel activation, their functional architecture is incompletely understood. Our recent studies show that a naturally occurring in-frame deletion mutation (Del-L955) of NaV1.7 channel, identified in individuals with a severe inherited pain syndrome (inherited erythromelalgia) causes a substantial hyperpolarizing shift of channel activation. Here we took advantage of this deletion mutation to understand the role of the S6 helix in the channel activation. Based on the recently published structure of a bacterial NaV channel (NaVAb), we modeled the WT and Del-L955 channel. Our structural model showed that Del-L955 twists the DII/S6 helix, shifting location and radial orientation of the activation gate residue (Phe960). Hypothesizing that these structural changes produce the shift of channel activation of Del-L955 channels, we restored a phenylalanine in wild-type orientation by mutating Ser961 (Del-L955/S961F), correcting activation by ∼10 mV. Correction of the displaced Phe960 (F960S) together with introduction of the rescuing activation gate residue (S961F) produced an additional ∼6-mV restoration of activation of the mutant channel. A simple point mutation in the absence of a twist (L955A) did not produce a radial shift and did not hyperpolarize activation. Our results demonstrate the functional importance of radial tuning of the sodium channel S6 helix for the channel activation.
Introduction
The NaV1.7 channel,2 a member of the voltage-gated sodium channel family, preferentially expressed in dorsal root ganglia and sympathetic ganglia neurons, plays a critical role in pain signaling (1, 2). The NaV1.7 channel amplifies subthreshold membrane depolarizations, contributes to the generation of action potentials (3, 4), and may facilitate neurotransmitter release at the central terminals of dorsal root ganglia neurons within the spinal cord (5). Loss-of-function NaV1.7 channel mutations cause congenital indifference to pain (6) whereas gain-of-function missense NaV1.7 mutations cause several painful disorders including inherited erythromelalgia (7–9), paroxysmal extreme pain disorder (10, 11), and small fiber neuropathy (12–14). These gain-of-function missense mutations represent experiments of nature that may shed light on the structural basis of sodium channel function.
The large pore-forming α-subunit of the mammalian NaV1.7 channel, similar to all members of this channel family, consists of four homologous domains (I–IV) linked by three intracellular loops (L1–L3). Each domain of the α-subunit has six transmembrane helices (S1–S6). The S1–S4 helices form the voltage sensor domain whereas the S5 and S6 helices, together with a membrane-reentrant pore loop (P-loop) between S5 and S6, form the ion-conducting pathway. The S6 helices are the backbone of the ion-conducting pathway, and multiple residues in the S6 helices have been implicated in channel activation and inactivation. However, there is still not a full understanding of the contribution of the S6 helices to channel activation. We recently identified a gain-of-function in-frame deletion (Del-L955) within S6 of domain II of NaV1.7 channel from an inherited erythromelalgia family, which causes a robust hyperpolarizing shift of activation and slow inactivation (15). Here, we took advantage of the structural change within the S6 helix produced by Del-L955 to investigate the contribution of the S6 helix to the channel activation. Using structural modeling-guided mutagenesis and patch clamp electrophysiology, we demonstrate that the radial orientation of the DII activation gate residue (Phe960) within S6 is essential for channel activation. The displaced Phe960 in the Del-L955 mutant channel disrupts the activation gate, leading to a disease-causing hyperpolarizing shift of channel activation. Our results demonstrate the importance of radial tuning of the S6 helix for the NaV1.7 channel activation and suggest that structural modeling-guided mutagenesis can contribute to our understanding of the functional architecture of voltage-gated sodium channels.
EXPERIMENTAL PROCEDURES
Structural Modeling
Structural modeling was performed as described previously (16, 17). Briefly, a two-step method was used for construction of hNaV1.7 channel. First, four transmembrane domain structural models were generated by a membrane-bound protein predication algorithm GPCR-ITASSER (18, 19). Then, each single domain model was aligned to the recently solved bacterial sodium channel tetramer (Protein Data Bank ID code 3RVY) (20) by TM-align (21). The four transmembrane domains models were assembled in a clockwise order viewed from extracellular side as suggested previously (22, 23). The resulting four domain complex structural model was finally refined by Fragment-Guided Molecular Dynamics simulation (FG-MD) to remove interdomain clashes and improve model quality (19, 24, 25).
Plasmid Preparation and HEK293 Cell Transfection
Tetrodotoxin-resistant human NaV1.7 wild-type (WT) channel (hNaV1.7r) was constructed based on the hNaV1.7 (mRNA, NM_002977.3; protein, NP_002968.1 of NCBI database) (26). Del-L955 and other mutant channels were constructed on the hNaV1.7r background. These channels were transfected into HEK293 cells together with human β-1 and β-2 subunits (27) using Lipofectamine (Invitrogen), as described previously (16). HEK293 cells were maintained in 1:1 Dulbecco's modified Eagle's medium (DMEM)/F-12 supplemented with 10% fetal bovine serum (FBS; Hyclone) in a humidified 5% CO2 incubator at 37 °C. HEK293 cells were seeded onto poly-l-lysine-coated glass coverslips (BD Biosciences) in a 24-well plate 1 day before recording. The functions of all the mutant constructs of the NaV1.7 channel were screened using the PatchXpress automated parallel patch clamp system (Molecular Devices) (28).
Voltage Clamp Recording
Whole cell voltage clamp recordings were obtained after 1 day of transfection as described previously (16, 29). The extracellular solution contained the following: 140 mm NaCl, 3 mm KCl, 1 mm MgCl2, 1 mm CaCl2, 20 mm dextrose, and 10 mm HEPES, pH 7.3, with NaOH (320 mosm adjusted with dextrose). The pipette solution contained the following: 140 mm cesium fluoride, 10 mm NaCl, 1.1 mm EGTA, 10 mm HEPES, 20 mm dextrose, pH 7.3, with CsOH (310 mosm adjusted with dextrose). Patch pipettes had a resistance of 1–2 megaohms when filled with pipette solution. After achieving whole cell recording configuration, the pipette and cell capacitance were manually minimized using the Axopatch 200B (Molecular Devices) compensation circuitry. Series resistance and prediction compensation (80–90%) were applied to reduce voltage errors. Recorded currents were digitized using pClamp software and a digidata 1440A interface (Molecular Devices) at a rate of 50 kHz after passing through a low pass Bessel filter setting of 10 kHz. The recording was initiated after a 5-min equilibration period after establishing whole cell configuration. To generate activation curves, cells were held at −140 mV and stepped to potentials of −80 to +40 mV in 5-mV increments for 100 ms. Peak inward currents were automatically extracted by Origin and fitted with Boltzmann function to determine the half-activation (V1/2), activation curve slope at half-activation (Z) and reversal potential (ENa) for each recording. Conductance was calculated as G = I/(Vm − ENa) and were normalized by the maximum conductance value and fit with a Boltzmann equation.
Data Analysis
Data were analyzed with Clampfit 9.2 (Molecular Devices) and OriginPro 8.5 (Microcal Software). Student's t test was used, and statistical significance was accepted when p < 0.05. Data are presented as means ± S.E.
RESULTS
The Del-L955 Deletion Mutation Hyperpolarizes Activation of NaV1.7 Channel
Our previous work has demonstrated that an in-frame deletion (NaV1.7 Del-L955) within the DII/S6 of the NaV1.7 channel (Fig. 1A) produces inherited erythromelalgia (15). This deletion occurs in the highly conserved S6 region (Fig. 1A) and results in a dramatic (∼25 mV) hyperpolarizing shift in the voltage dependence of activation (15). We evaluated channel activation from cells expressing WT (Fig. 1B) and Del-L955 mutant channels (Fig. 1C) using a recording protocol for voltage dependence of activation. We confirmed a robust shift in the activation V1/2 (voltage at half-activation) for NaV1.7 Del-L955 channel (Del-L955: −50 ± 1.6 mV, n = 11) compared with NaV1.7 WT channel (−23.8 ± 1.4 mV, n = 7, p < 0.001, Fig. 1D). Inactivation kinetics was also analyzed. Within the range more positive than −45 mV, where WT channels begin to activate, inactivation kinetics was more rapid for Del-L955 mutant channel (e.g. fast inactivation time constant τ = 1.53 ± 0.17 at −35 mV, n = 7) compared with WT (τ = 2.79 ± 0.32 at −35 mV, n = 6, p < 0.01). Between −35 mV and 0 mV, where Del-L955 and WT channel are both activated, no significant differences were found. This result is consistent with our previous report (15).
FIGURE 1.

The Del-L955 deletion mutation hyperpolarizes activation of NaV1.7 channel. A, schematic of the human NaV1.7 channel topology showing residue Leu955. All of the rescue mutations introduced in this study are listed in below. B and C, representative traces of current families recorded from HEK293 cells expressing WT (B) and Del-L955 (C) channel. D, Del-L955 mutant channel studied in parallel with WT channel in HEK293 cells using a voltage dependence of activation protocol. Del-L955: −50 ± 1.6 mV, n = 11; NaV1.7 WT: −23.8 ± 1.4 mV, n = 7, p < 0.001. Stimulus protocol for activation is also shown as an inset.
Structural Modeling Reveals Radial Displacement of the Activation Gate (Phe960) in the NaV1.7 Del-L955 Mutant Channel
To understand the structural basis of this change of channel activation, we constructed an atomic-level structural model of the WT and Del-L955 NaV1.7 channel based on the bacterial voltage-gated sodium channel NaVAb (PDB ID code 3RVY) (20) using our previously published methods (16). As can be seen from the structural model (Fig. 2A), the activation gate in WT NaV1.7 channel consists of four aromatic residues, one from each S6 helix (DI, Tyr405; DII, Phe960; DIII, Phe1449; and DIV, Phe1752) (30). Key residues investigated in this study are colored and circled for highlighting: Phe960 is red, and Ser961 is yellow. Leu955 is located 5 residues upstream of the activation gate residue (Phe960). When the Leu955 residue is deleted, the orientation of Phe960 (red) shifts radially toward the S6 of domain III (Fig. 2B). Del-L955 also rotates Ser961 (yellow) to the previous location of Phe960 so that the side chain of Ser961 now points to the pore, facing the other three activation gate residues (Fig. 2B). Side (Fig. 2C) and top (Fig. 2D) views of the four S6 helices of Del-L955 channel show that the hydrophobic ring which forms the intact activation gate in WT channels is disrupted in Del-L955 channels because of the displaced Phe960 (Fig. 2, C and D). According to our model, the DII/S6 helix extends a few residues below the activation gate residue and becomes a flexible loop as it transitions to L3 (the third intracellular loop). As we previously demonstrated that the intact activation gate is essential for wild-type channel gating (30), we hypothesized that the radial displacement of Phe960 is a structural substrate that contributes strongly to the altered activation of the Del-L955 channel.
FIGURE 2.

Radial displacement of the activation gate (Phe960) in Del-L955 channel. A, structural model of WT NaV1.7 channel shows four residues, one from each domain (Tyr405 of DI, Phe960 of DII, Phe1449 of DIII, and Phe1752 of DIV), forms the activation gate. Phe960 is colored red, and Ser961 is colored yellow; both of them are circled for highlighting. B, in the Del-L955 structural model, when Leu955 is deleted, the orientation of Phe960 rotates radially toward the S6 of domain III. The Leu955 deletion also rotates Ser961 to the previous location of Phe960 so that the side chain of Ser961 now points to the pore, facing the other three activation gate residues. Phe960 of Del-L955 channel is shown in red, and Ser961 is shown in yellow. Both are circled for highlighting. C, intramembrane view shows four S6 helices of the Del-L955 mutant channel structural model. All other structural elements are omitted for clarity. D, extracellular view shows the four S6 helices of the Del-L955 mutant channel structural model.
Restoring Phe in the Activation Gate Location Depolarizes Activation
Our structural model reveals that in the Del-L955 channel, the side chain of Ser961 at DII/S6 becomes aligned with the other activation gate residues from DI, DIII, and DIV (Fig. 2B). The side chain of Ser961, however, is not likely to contribute to maintaining the hydrophobic ring needed for an effective activation gate (Fig. 2B) (30). We hypothesized that the disrupted activation gate contributes to the hyperpolarizing shift of activation in Del-L955 channel. We tested this hypothesis by asking whether restoration of the hydrophobic ring, by mutating Ser961 back to Phe, would depolarize activation of the Del-L955 channel. Structural modeling (Fig. 3A) shows that this newly introduced Phe (yellow) would be situated in a location similar to the original Phe, so that it would be expected to interact with the other three hydrophobic residues to restore the activation gate (Fig. 3A). Note that the displaced Phe960 is in red. For this experiment, we created Del-L955/S961F double mutation and compared it with the Del-L955 channel. NaV1.7 WT channel activation is shown for comparison (−23.8 ± 1.4 mV, n = 7). As shown in Fig. 3B, the Del-L955/S961F mutant channel displayed a robust ∼+10 mV depolarizing shift in activation compared with that of Del-L955 (Del-L955: −50 ± 1.6 mV, n = 11, versus Del-L955/S961F: −40.2 ± 1.5 mV, n = 11, p < 0.001), suggesting that introduction of a Phe to restore the activation gate depolarize channel activation.
FIGURE 3.

Restoring the activation gate and removing the displaced Phe depolarize activation. A, structural modeling suggests that mutating Ser961 to Phe may restore the activation gate composition. Phe960 is shown in red, and S961F is shown in yellow; both of them are circled for highlighting. Note that Phe960 (red) is in a displaced position. B, Del-L955/S961F mutant channel was created and studied in parallel with Del-L955 channel in HEK293 cells. Del-L955/S961F displayed a robust ∼+10 mV depolarizing shift in activation V1/2 compared with that of Del-L955 (Del-L955: −50 ± 1.6 mV, n = 11, versus Del-L955/S961F: −40.2 ± 1.5 mV, n = 11, p < 0.001). Both Del-L955 and WT curves (here and in the following figures) are a replot of Fig. 1. C, structural model shows the correction of a displaced Phe960 into Ser (circled and red) together with an introduced Phe (circled and yellow) in the activation gate helical position. D, the displaced Phe960 was mutated to Ser, which was the original residue at this location. This additional mutation further shifted activation V1/2 by ∼5 mV (Del-L955/S961F: −40.2 ± 1.5 mV, n = 11; Del-L955/S961F/F960S: −35.1 ± 1.5 mV, n = 9; p < 0.05). Phe960 was also mutated to Ala (Del-L955/S961F/F960A), and the activation V1/2 of this mutation (−34.4 ± 1.1 mV, n = 9) was comparable with that of Del-L955/S961F/F960S mutation but significantly different from Del-L955/S961F mutation (p < 0.01).
Correcting the Displaced Phe Further Depolarizes Activation
Structural modeling also suggested that the displaced Phe960 (red) may influence the conformation of S6 helix of DIII that contributes to altered activation of the Del-L955 channels (Fig. 3A). To test this hypothesis, we mutated this displaced Phe960 into Ser (red, Fig. 3C), which was the original residue at this location. Voltage clamp analysis showed that this additional mutation further shifted the activation V1/2 by ∼+5 mV (Del-L955/S961F: −40.2 ± 1.5 mV, n = 11; Del-L955/S961F/F960S: −35.1 ± 1.5 mV, n = 9; p < 0.05) (Fig. 3D). To further test the effect of residue size at this site, we mutated the Phe960 to Ala, a small residue predicted to have a minimal structural effect, and created a Del-L955/S961F/F960A mutation. The activation V1/2 of this mutation was comparable with that of Del-L955/S961F/F960S mutation, shifting the V1/2 by ∼6 mV (−34.4 ± 1.1 mV, n = 9, p < 0.01 compared with Del-L955/S961F) (Fig. 3D). These results suggest that a displaced bulky Phe may influence the adjacent DIII/S6 helix lining the pore, thus affecting channel activation. Taken together, introducing a new Phe into the appropriate location within the activation gate and replacement of the displaced Phe with a smaller residue corrected the activation V1/2 of Del-L955 channel by >60%.
A Point Mutation of Residue 955, in the Absence of a Twist, Does Not Cause a Hyperpolarizing Shift of Activation
As we hypothesized that the strong hyperpolarizing shift of the Del-L955 channel is due to an in-frame deletion causing the twist of S6 helix, especially the displacement of the activation gate residue Phe960 at the more distal portion of the S6 helix, we further asked whether a point mutation of Leu955, in the absence of a twist, could hyperpolarize channel activation. If a point mutation of Leu955 were not able to hyperpolarize channel activation, it would further suggest that the twist of the S6 helix by Leu955 deletion, rather than a residue change of Leu955, is likely to be the underlying mechanism of the activation shift. Alanine was chosen due to its small effect on the protein structure and because it has been found to be frequently located in transmembrane helices (31). Structural modeling showed that Leu955 in WT channel pointed toward a space formed by S4–5 linker and S6 helix (Fig. 4A). When Leu955 was mutated to Ala, the side chain was smaller and the structural effect was subtle (Fig. 4, A and B). Interestingly, the activation of L955A channel (L955A: −19.1 ± 2.2 mV, n = 7) was close to that of WT channel (WT: −23.8 ± 1.4 mV, n = 7, p > 0.05, Fig. 4C), which is markedly different from Del-L955/S961F/F960A channel or all the other mutant channels introduced in this study (p < 0.01, Fig. 4C). Therefore, our data strongly suggest that the twist of the S6 helix, rather than the local change of the Leu955 residue, underlies the strong shift of activation seen in Del-L955 mutant channels.
FIGURE 4.
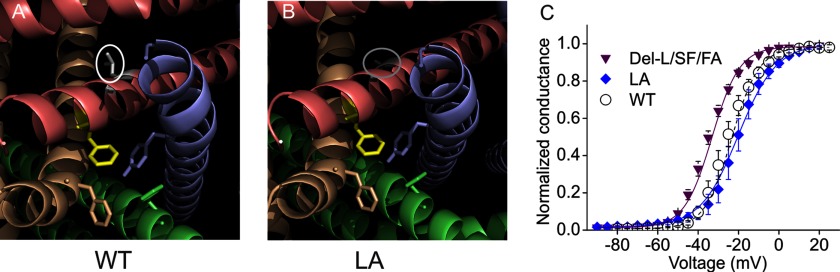
A point mutation of residue 955 that does not introduce a twist does not hyperpolarize activation. A and B, structural model of WT and L955A mutant channel. Leu955 is highlighted in white and circled (A). Ala955 is highlighted in gray and circled (B). No obvious structural difference was observed. C, the L955A point mutation was created and studied. The activation V1/2 of L955A mutant channel (−19.1 ± 2.2 mV, n = 7) was comparable with that of WT channel (−23.8 ± 1.4 mV, n = 7, p > 0.05) but very much different from Del-L955/S961F/F960A or all the other mutant channels introduced in this study (p < 0.01).
DISCUSSION
In this study, we took advantage of a naturally occurring deletion (Del-L955) mutation of the NaV1.7 sodium channel that produces a large hyperpolarizing shift in channel activation and employed a combined approach of structural modeling, mutagenesis, and voltage clamp electrophysiology to understand the contribution of the functional architecture of the S6 transmembrane helix to channel activation. Using structural modeling, we found that this in-frame deletion results in a twist of the DII S6 helix beginning several residues above the activation gate residue (Phe960), causing a radial shift of this residue toward the S6 helix of DIII instead of the ion-conducing pore. Virtual mutagenesis predicted that the substitution Ser961 to Phe (Del-L955/S961F) might restore the WT composition of the activation gate and thus partially rescue activation. This manipulation indeed depolarized activation V1/2 by ∼10 mV, supporting our hypothesis. As modeling further suggested that the originally displaced Phe960 might have a functional effect on the S6 of DIII, we additionally mutated the displaced Phe960 into a Ser (Del-L955/S961F/F960S) to mimic the original residue composition of this part of the DII/S6. This substitution resulted in an additional ∼+5 mV shift of activation V1/2 toward the WT value. Consistent with a steric influence, we observed a depolarization of the activation V1/2 with Del-L955/S961F/F960A. To understand whether the local change of the Leu955 residue contributes to this hyperpolarizing shift of activation, we created and assessed a point mutation (L955A) that did not introduce a twist. This manipulation placed the activation V1/2 within the range of that of the WT NaV1.7 channel, with a slight depolarizing tendency that did not reach statistical significance. Taken together, these results indicate that the structural integrity of the helix rather than a single amino acid change in this region is responsible for the large hyperpolarizing shift of activation. Our results support a model in which radial orientation of critical residues within S6 plays a key role in shaping channel activation.
The S6 helix directly lines the ion-conducing pathway and contributes to several aspects of channel gating. Point mutations of the NaV1.7 channel in patients with inherited erythromelalgia reported so far all hyperpolarize activation (1, 2). These point mutations, although informative for understanding channel gating, do not provide insight into the functional architecture of the S6 helix. An in-frame deletion such as Del-L955, on the other hand, causes a more significant change to the overall architecture. We therefore used this deletion as a tool to identify key features of the S6 helix. We demonstrated that a radial displacement of Phe960 in the Del-L955 mutant channel disrupted the activation gate and that the displaced bulky Phe appears to influence a neighboring helix. Interestingly, in our previous study, we found that direct mutation of activation gate Phe960 (F960V) leads to a ∼6-mV hyperpolarizing shift of activation (30), emphasizing the importance of intact activation gates. The current study reinforces this finding and additionally shows that for the DII activation gate, radial orientation is essential. Interestingly, our earlier study suggests that replacing Phe with a larger Trp would not produce additional effects regarding the channel activation (30). Although the mutations we introduced in this study depolarize activation significantly toward WT channel, the effect is incomplete. This may be because the in-frame deletion may cause more profound changes to channel structure in additional to the twist of the activation gate (e.g. the orientation of all the residues of the DII/S6 helix downstream of Phe960 was shifted as well). Addition of another residue after the Phe may not resolve this issue, because neither crystal structure nor molecular biophysical analysis has definitively identified the end of the S6 helix of the hNaV1.7 channel yet.
Following the initial report of the structure of the bacterial voltage-gated sodium channel (NaVAb, PDB ID code 3RVY) (20), the crystal structures of other bacterial NaV channels (NaVRh, PDB 4DXW) (32) and (NaVM, 4F4L) (33), as well NaVAb in an inactivated state (PDB 4EKW) (34) have been reported. These new structures suggest an asymmetric organization of the four S6 helices, despite the fact that these are homotetrameric channels. With the availability of new structures, we asked how they may affect our modeling of the activation gate region of the NaV1.7 channel. In Fig. 5, we align both the WT and Del-L955 NaV1.7 structural models with NaVRh (PDB 4DXW). Within this particular region, the activation gate residues (Tyr405 of DI, Phe960 of DII, Phe1449 of DIII, and Phe1752 of DIV) of NaV1.7 align reasonably well with those of NaVRh (Leu219), although it is clear that when the helices extend toward the extracellular direction, they begin to separate. Given that NaVRh and NaVAb are different bacterial sodium channels, it is understandable that notable differences exist between NaVRh and NaVAb (35) and between our NaV1.7 structural model and NaVRh or NaVAb. Nevertheless, within the activation gate region, the structural elements are relatively conserved; therefore, it is not surprising that our modeling provided guidance for our mutagenesis and voltage clamp experiments, which permitted us to dissect the architecture of an S6 helix.
FIGURE 5.

Alignment of NaV1.7 WT and Del-L955 mutant channel structural model with NaVRh structures. A, cytoslic view of the alignment of NaV1.7 structural model with that of NaVRh. NaV1.7 is shown in wheat, and NaVRh is shown in gray. Tyr405 of DI, Phe960 and Ser961 of DII, Phe1449 of DIII, and Phe1752 of DIV are shown in stick configuration. Activation gate of NaVRh (Leu219) is also shown in stick configuration. B, extracellular view of the alignment of NaV1.7 structural model with NaVRh. C, cytoslic view of the alignment of Del-L955 mutant channel structural model with NaVRh. Del-L955 is shown in cyan, and NaVRh is shown in gray. D, extracellular view of the alignment of Del-L955 mutant channel structural model with NaVRh.
Traditional, systematic mutagenesis using alanine (36), cysteine (37), or tryptophan scanning (38, 39) has yielded important information regarding the structure-function relationship of sodium channels. However, this approach is relatively time- and resource-consuming. In the present study, structural modeling provided insights regarding which residues to change via site-directed mutagenesis to test our hypothesis. With the increasing availability of sodium channel crystal structures (20, 32, 34), structural modeling-guided mutagenesis may provide an approach that is direct and time- and resource-efficient.
In summary, our studies indicate that a naturally occurring gain-of-function deletion mutation (Del-L955) causes a radial shift of the S6 helix of NaV1.7 channel, resulting in a disruption of the activation gate and displacement of a bulky residue which affects the neighboring DIII S6 helix. Our results demonstrate an important contribution of radial tuning of S6 to activation and suggest that structural modeling-guided mutagenesis may provide a useful tool for understanding the functional architecture of voltage-gated sodium channels.
Acknowledgments
We thank Dr. Jian Zhang of Dartmouth College and Dr. Yang Zhang of University of Michigan for modeling support; Lynda Tyrrell and Palak Shah for technical assistance; and Dr. Xiaoyang Cheng, Dr. Chongyang Han, Dr. Hyesook Ahn, Dr. Dmytro Vasylyev and Dr. Jianying Huang for valuable comments.
This work was supported by the Medical Research Service and Rehabilitation Research Service, Department of Veterans Affairs and the Erythromelalgia Association (to S. G. W. and S. D. D.-H.). The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America with Yale University.
- NaV
- voltage-gated sodium channel
- PDB
- Protein Data Bank.
REFERENCES
- 1. Dib-Hajj S. D., Cummins T. R., Black J. A., Waxman S. G. (2010) Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 33, 325–347 [DOI] [PubMed] [Google Scholar]
- 2. Dib-Hajj S. D., Yang Y., Black J. A., Waxman S. G. (2013) The NaV1.7 sodium channel: from molecule to man. Nat. Rev. Neurosci. 14, 49–62 [DOI] [PubMed] [Google Scholar]
- 3. Waxman S. G. (2006) Neurobiology: a channel sets the gain on pain. Nature 444, 831–832 [DOI] [PubMed] [Google Scholar]
- 4. Cummins T. R., Howe J. R., Waxman S. G. (1998) Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J. Neurosci. 18, 9607–9619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Minett M. S., Nassar M. A., Clark A. K., Passmore G., Dickenson A. H., Wang F., Malcangio M., Wood J. N. (2012) Distinct NaV1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat. Commun. 3, 791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cox J. J., Reimann F., Nicholas A. K., Thornton G., Roberts E., Springell K., Karbani G., Jafri H., Mannan J., Raashid Y., Al-Gazali L., Hamamy H., Valente E. M., Gorman S., Williams R., McHale D. P., Wood J. N., Gribble F. M., Woods C. G. (2006) An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang Y., Wang Y., Li S., Xu Z., Li H., Ma L., Fan J., Bu D., Liu B., Fan Z., Wu G., Jin J., Ding B., Zhu X., Shen Y. (2004) Mutations in SCN9A, encoding a sodium channel α subunit, in patients with primary erythermalgia. J. Med. Genet. 41, 171–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cummins T. R., Dib-Hajj S. D., Waxman S. G. (2004) Electrophysiological properties of mutant NaV1.7 sodium channels in a painful inherited neuropathy. J. Neurosci. 24, 8232–8236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dib-Hajj S. D., Rush A. M., Cummins T. R., Hisama F. M., Novella S., Tyrrell L., Marshall L., Waxman S. G. (2005) Gain-of-function mutation in NaV1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 128, 1847–1854 [DOI] [PubMed] [Google Scholar]
- 10. Dib-Hajj S. D., Estacion M., Jarecki B. W., Tyrrell L., Fischer T. Z., Lawden M., Cummins T. R., Waxman S. G. (2008) Paroxysmal extreme pain disorder M1627K mutation in human NaV1.7 renders DRG neurons hyperexcitable. Mol. Pain 4, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fertleman C. R., Baker M. D., Parker K. A., Moffatt S., Elmslie F. V., Abrahamsen B., Ostman J., Klugbauer N., Wood J. N., Gardiner R. M., Rees M. (2006) SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 52, 767–774 [DOI] [PubMed] [Google Scholar]
- 12. Faber C. G., Hoeijmakers J. G., Ahn H. S., Cheng X., Han C., Choi J. S., Estacion M., Lauria G., Vanhoutte E. K., Gerrits M. M., Dib-Hajj S., Drenth J. P., Waxman S. G., Merkies I. S. (2012) Gain of function Nanu1.7 mutations in idiopathic small fiber neuropathy. Ann. Neurol. 71, 26–39 [DOI] [PubMed] [Google Scholar]
- 13. Han C., Hoeijmakers J. G., Liu S., Gerrits M. M., te Morsche R. H., Lauria G., Dib-Hajj S. D., Drenth J. P., Faber C. G., Merkies I. S., Waxman S. G. (2012) Functional profiles of SCN9A variants in dorsal root ganglion neurons and superior cervical ganglion neurons correlate with autonomic symptoms in small fibre neuropathy. Brain 135, 2613–2628 [DOI] [PubMed] [Google Scholar]
- 14. Han C., Hoeijmakers J. G., Ahn H. S., Zhao P., Shah P., Lauria G., Gerrits M. M., te Morsche R. H., Dib-Hajj S. D., Drenth J. P., Faber C. G., Merkies I. S., Waxman S. G. (2012) NaV1.7-related small fiber neuropathy: impaired slow-inactivation and DRG neuron hyperexcitability. Neurology 78, 1635–1643 [DOI] [PubMed] [Google Scholar]
- 15. Cheng X., Dib-Hajj S. D., Tyrrell L., Te Morsche R. H., Drenth J. P., Waxman S. G. (2011) Deletion mutation of sodium channel NaV1.7 in inherited erythromelalgia: enhanced slow inactivation modulates dorsal root ganglion neuron hyperexcitability. Brain 134, 1972–1986 [DOI] [PubMed] [Google Scholar]
- 16. Yang Y., Dib-Hajj S. D., Zhang J., Zhang Y., Tyrrell L., Estacion M., Waxman S. G. (2012) Structural modeling and mutant cycle analysis predict pharmacoresponsiveness of a NaV1.7 mutant channel. Nat. Commun. 3, 1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang Y., Shi W., Chen X., Cui N., Konduru A. S., Shi Y., Trower T. C., Zhang S., Jiang C. (2011) Molecular basis and structural insight of vascular KATP channel gating by S-glutathionylation. J. Biol. Chem. 286, 9298–9307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protocols 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang J., Zhang Y. (2010) GPCRRD: G protein-coupled receptor spatial restraint database for 3D structure modeling and function annotation. Bioinformatics 26, 3004–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Payandeh J., Scheuer T., Zheng N., Catterall W. A. (2011) The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y., Skolnick J. (2005) TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dudley S. C., Jr., Chang N., Hall J., Lipkind G., Fozzard H. A., French R. J. (2000) μ-conotoxin GIIIA interactions with the voltage-gated Na+ channel predict a clockwise arrangement of the domains. J. Gen. Physiol. 116, 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li R. A., Ennis I. L., French R. J., Dudley S. C., Jr., Tomaselli G. F., Marbán E. (2001) Clockwise domain arrangement of the sodium channel revealed by μ-conotoxin (GIIIA) docking orientation. J. Biol. Chem. 276, 11072–11077 [DOI] [PubMed] [Google Scholar]
- 24. Zhang J., Liang Y., Zhang Y. (2011) Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure 19, 1784–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang J., Zhang Y. (2010) A novel side-chain orientation dependent potential derived from random-walk reference state for protein fold selection and structure prediction. PloS One 5, e15386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Herzog R. I., Cummins T. R., Ghassemi F., Dib-Hajj S. D., Waxman S. G. (2003) Distinct repriming and closed-state inactivation kinetics of NaV1.6 and NaV1.7 sodium channels in mouse spinal sensory neurons. J. Physiol. 551, 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lossin C., Wang D. W., Rhodes T. H., Vanoye C. G., George A. L., Jr. (2002) Molecular basis of an inherited epilepsy. Neuron 34, 877–884 [DOI] [PubMed] [Google Scholar]
- 28. Estacion M., Choi J. S., Eastman E. M., Lin Z., Li Y., Tyrrell L., Yang Y., Dib-Hajj S. D., Waxman S. G. (2010) Can robots patch-clamp as well as humans? Characterization of a novel sodium channel mutation. J. Physiol. 588, 1915–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Estacion M., Yang Y., Dib-Hajj S. D., Tyrrell L., Lin Z., Yang Y., Waxman S. G. (2013) A new NaV1.7 mutation in an erythromelalgia patient. Biochem. Biophys. Res. Commun. 432, 99–104 [DOI] [PubMed] [Google Scholar]
- 30. Lampert A., O'Reilly A. O., Dib-Hajj S. D., Tyrrell L., Wallace B. A., Waxman S. G. (2008) A pore-blocking hydrophobic motif at the cytoplasmic aperture of the closed-state NaV1.7 channel is disrupted by the erythromelalgia-associated F1449V mutation. J. Biol. Chem. 283, 24118–24127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McPhee J. C., Ragsdale D. S., Scheuer T., Catterall W. A. (1994) A mutation in segment IVS6 disrupts fast inactivation of sodium channels. Proc. Natl. Acad. Sci. U.S.A. 91, 12346–12350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang X., Ren W., DeCaen P., Yan C., Tao X., Tang L., Wang J., Hasegawa K., Kumasaka T., He J., Wang J., Clapham D. E., Yan N. (2012) Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 486, 130–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McCusker E. C., Bagnéris C., Naylor C. E., Cole A. R., D'Avanzo N., Nichols C. G., Wallace B. A. (2012) Structure of a bacterial voltage-gated sodium channel pore reveals mechanisms of opening and closing. Nat. Commun. 3, 1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Payandeh J., Gamal El-Din T. M., Scheuer T., Zheng N., Catterall W. A. (2012) Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486, 135–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang X., Yan N. (2013) The conformational shifts of the voltage sensing domains between NaVRh and NaVAb. Cell Res. 23, 444–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yarov-Yarovoy V., Brown J., Sharp E. M., Clare J. J., Scheuer T., Catterall W. A. (2001) Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na+ channel α subunit. J. Biol. Chem. 276, 20–27 [DOI] [PubMed] [Google Scholar]
- 37. Zarrabi T., Cervenka R., Sandtner W., Lukacs P., Koenig X., Hilber K., Mille M., Lipkind G. M., Fozzard H. A., Todt H. (2010) A molecular switch between the outer and the inner vestibules of the voltage-gated Na+ channel. J. Biol. Chem. 285, 39458–39470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang S. Y., Bonner K., Russell C., Wang G. K. (2003) Tryptophan scanning of D1S6 and D4S6 C-termini in voltage-gated sodium channels. Biophys. J. 85, 911–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Muroi Y., Arcisio-Miranda M., Chowdhury S., Chanda B. (2010) Molecular determinants of coupling between the domain III voltage sensor and pore of a sodium channel. Nat. Struct. Mol. Biol. 17, 230–237 [DOI] [PMC free article] [PubMed] [Google Scholar]