

Background: Base excision repair is hindered by nucleosomes.
Results: Outwardly oriented uracils near the nucleosome center are efficiently cleaved; however, polymerase β is strongly inhibited at these sites.
Conclusion: The histone octamer presents different levels of constraints on BER, dependent on the structural requirements for enzyme activity.
Significance: Chromatin remodeling is necessary to prevent accumulation of aborted intermediates in nucleosomes.
Keywords: Base Excision Repair, DNA Enzymes, DNA Polymerase, Histones, Nucleosome
Abstract
Packaging of DNA into chromatin affects accessibility of DNA regulatory factors involved in transcription, replication, and repair. Evidence suggests that even in the nucleosome core particle (NCP), accessibility to damaged DNA is hindered by the presence of the histone octamer. Base excision repair is the major pathway in mammalian cells responsible for correcting a large number of chemically modified bases. We have measured the repair of site-specific uracil and single nucleotide gaps along the surface of the NCP. Our results indicate that removal of DNA lesions is greatly dependent on their rotational and translational positioning in NCPs. Significantly, the rate of uracil removal with outwardly oriented DNA backbones is 2–10-fold higher than those with inwardly oriented backbones. In general, uracils with inwardly oriented backbones farther away from the dyad center of the NCP are more accessible than those near the dyad. The translational positioning of outwardly oriented gaps is the key factor driving gap filling activity. An outwardly oriented gap near the DNA ends exhibits a 3-fold increase in gap filling activity as compared with one near the dyad with the same rotational orientation. Near the dyad, uracil DNA glycosylase/APE1 removes an outwardly oriented uracil efficiently; however, polymerase β activity is significantly inhibited at this site. These data suggest that the hindrance presented by the location of a DNA lesion is dependent on the structural requirements for enzyme catalysis. Therefore, remodeling at DNA damage sites in NCPs is critical for preventing accumulation of aborted intermediates and ensuring completion of base excision repair.
Introduction
Maintaining genomic integrity is crucial for the survival of all living organisms; however, this is not a trivial task given that as many as 100,000 spontaneous lesions/cell/day can be formed, 10,000 of which are due to depurination (1, 2). Base excision repair (BER)2 is a critical repair pathway responsible for identifying and removing the vast majority of endogenous damage (2–5). The importance of this pathway is highlighted by the findings that knock-out mice of several BER genes are embryonically lethal (6, 7). Notably, in human cells, G:C→A:T transitions (at CpG regions) in the tumor suppressor gene p53 are associated with bladder cancer (8). Because DNA polymerases efficiently incorporate A opposite to U in the template, mispairs of G:U, due to the deamination of cytosine, can also lead to the mutagenic G:C→A:T transition if uracil is not removed before the next round of replication (9).
Base excision repair is a coordinated stepwise process that efficiently removes uracil and other chemically modified bases (10). In the first step, a DNA glycosylase, specific for a particular base modification, identifies the damaged base and removes it (1, 13). Monofunctional glycosylases such as the Escherichia coli and human uracil DNA glycosylases (UDG) undergo a conformational change that directs a “flipping out” of uridine from the base stack and removal of the uracil by cleavage of the N–C1′ glycosydic bond between the target base and deoxyribose sugar (11). This generates an abasic (apurinic/apyrimidinic) site, recognized by apurinic/apyrimidinic endonuclease I (APE1), which cleaves the DNA backbone 5′ to the abasic site, generating a 3′-hydroxyl and a 5′-2′-deoxyribose-5′-phosphate (5′-dRP) (12). Polymerase β (pol β) utilizes the 3′-hydroxyl to fill the gap through template-directed synthesis (13). Depending on the number of nucleotides added, BER proceeds in one of two directions. Single nucleotide addition is called short patch repair. In short patch BER, pol β's intrinsic dRP-lyase activity removes the 5′-dRP (13, 14). Addition of more than one nucleotide (up to 13) constitutes long patch BER and requires assistance of flap endonuclease I (FEN I) to remove the displaced 5′-flap structure (2, 12, 13). Restoration of the DNA backbone is subsequently completed by DNA ligase I or the DNA ligase III-XRCC1 complex. Notably, failure to complete BER can be more detrimental to the cell than the initial base modifications themselves as the BER intermediates impede replication and can signal apoptosis (10). Consequently, once BER is initiated, it is critical that the downstream enzymes process the cytotoxic intermediates with similar efficiency to decrease the accumulation of aborted intermediates. To ensure this is the case, UDG, APE1, and pol β bind with high affinity to their respective products (11, 15–17), thereby channeling the substrate and increasing recognition efficiency to downstream enzymes (10). The activity of how these enzymes coordinate with one another to complete repair has been well characterized using naked DNA substrates (18, 19), but less is known about their function in the context of chromatin.
At the primary level of chromatin organization, 147 bp of DNA are wrapped in 1.65 superhelical turns around a histone octamer forming a nucleosome core particle (NCP) (20). This octameric complex is composed of an H3-H4 tetramer that is flanked by two dimers of H2A and H2B, where the tetramer interacts with the central 60 bp more strongly as compared with those interactions mediated by the two dimers near the ends of the DNA (20, 21). Inherently, the DNA near the ends is more flexible and able to rotate more freely along its longitudinal axis. Biased intrinsic accessibility of DNA near the ends is also attributed to spontaneous unwrapping events, where the DNA ends transiently lift off of the histone octamer (22, 23). Accessibility to buried DNA by these events varies by orders of magnitude with greater accessibility near the ends and is dependent on both DNA sequence and the post-translational modification status of the histones (23–25).
Despite these apparent intrinsic differences in accessibility, less is known about the effects of translational positioning of DNA lesions on BER as most studies have focused on addressing the effects of rotational setting on BER efficiency (26–31). More recently, Cole et al. (32) investigated the effects of rotational and translational position, relative to the nucleosome dyad center, on the removal of uracil. In agreement with previous studies, these authors found that UDG efficiently removes uracil from within the nucleosome core DNA where the phosphate backbone is oriented away from the histone octamer surface and exhibited significantly reduced activity at uracils oriented toward the histone surface (32). Additionally, they found that cleavage was enhanced at uracils farther away from the dyad center (32). Using a similar nucleosome-positioning sequence, this group had earlier found that ligation of outwardly oriented nicks was decreased ∼10-fold at the dyad center and only 4–6-fold at the ends of nucleosome core DNA (26). Both of these studies addressing the effects of translational position of DNA lesions on BER used a DNA fragment containing the Xenopus borealis 5 S nucleosome-positioning element. Because the DNA sequence is a strong determinant of nucleosome stability (33) and consequently can influence rotational freedom of DNA as well as unwrapping events (34), it is important to address the effects of the rotational and translational position using different DNA sequences (35). Indeed, Ye et al. (36) recently reported that local structural features, in addition to rotational setting and translational position, were critical parameters affecting the overall efficiency of uracil removal.
Not all enzymes in the BER pathway have equal access to the same site, and in rare cases some enzymes are not inhibited by the presence of the histone octamer. For example, Hayes and co-workers (30) observed higher endonuclease activity by FEN1 (under low enzyme concentration) on DNA flaps oriented outwardly when compared with free DNA. A similar finding was also reported by Ye et al. (36), who observed some sites in the 601-NCP to have equal or greater reactivity than free DNA in the removal of uracil. Given the numerous parameters that can influence accessibility to DNA damage, completion of the BER pathway to address how these parameters influence repair efficiency is essential in identifying the rate-limiting steps of BER in vivo. This may be particularly important for the pol β step because the requirement to bend the DNA template opposite the gap by ∼90° (37) might not be easily achieved in the presence of the histones. Indeed, Odell et al. (38) recently reported that pol β can act on single nucleotide gaps with “in” and “out” rotational orientations, having greater extension activity on outwardly oriented gaps in NCPs containing Lytechinus variegatus 5 S DNA. Importantly, the lesions examined in this latter study were near the DNA ends in both the 5 S and 601 DNA positioning sequences, where there is greater DNA accessibility, and the constraints for DNA bending are not expected to be as important (because of weaker histone-DNA interactions) (39, 40).
To gain a better understanding of the aforementioned parameters that play critical roles in BER, we have determined the effects of rotationally and translationally positioning uracil and single nucleotide gaps on BER efficiency using the strong “Widom 601” positioning sequence and purified recombinant enzymes. Substrate handoff from APE1 to pol β on pre-incised nucleosomal DNA was also studied. Our results indicate that both rotational and translational positioning of DNA lesions strongly influence BER. Formaldehyde cross-linking experiments suggest that the rotational flexibility of DNA is also important for the repair of occluded lesions near the dyad. Local structural features contribute to the overall efficiency of uracil removal and pol β extension, albeit to a lesser extent than rotational setting and translational positioning. Surprisingly, the presence of APE1 did not affect pol β extension independent of translational and rotational positioning of the DNA gap.
EXPERIMENTAL PROCEDURES
Construction of DNA Substrates Containing Uracil and Single Nucleotide Gaps
The 147-bp 601 DNA sequence (41) was slightly modified to introduce a single uracil at distinct positions as shown in supplemental Table S1. Synthetic oligomers were purchased from Midland Certified Reagent Co. or Integrated DNA Technologies. A total of seven oligonucleotides were purchased, six of which contained a single uracil in the strand corresponding to the I chain in the NCP 601 crystal structure reported by Vasudevan et al. (42). Nomenclature of the substrates follows the same convention as reported by Fernandez and Anderson (41), where “+” and “−” indicate the number of nucleotides away from the dyad toward the 5′ and 3′ ends, respectively. Each of these oligonucleotides was radiolabeled with [γ-32P]ATP (PerkinElmer Life Sciences) at the 5′ end using T4 polynucleotide kinase (Invitrogen) and was annealed (1:1) with its complementary or partner strand by heating to 95 °C for 10 min and slow cooling in a buffer containing 30 mm Tris (pH 7.5) and 100 mm potassium acetate. The generated dsDNA was then purified using the QIAquick nucleotide removal kit or the PCR purification kit (Qiagen) to remove excess unincorporated radioactive nucleotides. The single nucleotide gap DNA was generated in a similar manner, except that after the DNA purification, the DNA was treated with UDG and APE1 (New England BioLabs) at 30 and 10 nm, respectively, for 90 min at 37 °C to ensure the cleavage of all uracils. The DNA was then treated 1:2 (v/v) with phenol/chloroform/isoamyl alcohol (PCI; 25:24:1) followed by a chloroform/isoamyl alcohol extraction (25:24) and standard ethanol precipitation.
Nucleosome Core Particle Reconstitutions
Nucleosome core particles were reconstituted by salt gradient dialysis using chicken erythrocyte core particles, which were purified as described previously (28). First, the radiolabeled DNA (∼3.2 pmol) was mixed in a solution containing 10 mm Tris-HCl (pH 7.5), 1 mm EDTA, and 1 m NaCl. Then the chicken nucleosome core particles were added to the mixture (on ice) to a final molar ratio 1:100 of DNA/chicken erythrocyte core particles. The mixture was then placed in a dialysis button as described by Thastrom et al. (43), except that the dialysis membrane with molecular mass cutoff of 6–8 kDa was used (Spectrum Labs). The mixture was dialyzed against three buffers containing 1 mm EDTA, 10 mm Tris (pH 7.5), 0.05% Nonidet P-40 (v/v), 5 mm β-mercaptoethanol, and decreasing amounts of NaCl (1 m, 600 mm, and 50 mm NaCl). Each dialysis step was performed for 1 h at 4 °C. The reconstitution products were then evaluated by electrophoresis in a 6% native polyacrylamide gel in 0.5× TBE buffer, exposed to phosphorimager screens (GE Healthcare), and visualized on a STORM 840 PhosphorImager (Amersham Biosciences). Images were analyzed using ImageQuant 5.2 software (GE Healthcare).
Hydroxyl Radical Footprinting
Hydroxyl radical footprinting of the NCPs was performed as described previously with minor modifications (31, 44). The reaction was quenched with glycerol to a final concentration of 6%. A PCI extraction was performed immediately to remove the histones. The DNA was then precipitated in ethanol, washed twice with 70% ethanol, air-dried, and resuspended in a 1:1 ratio of 1× TE and Hi-DiTM formamide. Samples were boiled for 10 min to denature the DNA, chilled on ice, and separated by electrophoresis in an 8% denaturing (7 m urea) polyacrylamide gel (19:1). The gel was run at 60 watts for 2.5 h, dried, exposed on a phosphor screen, and visualized on a STORM 840 PhosphorImager (Amersham Biosciences). Analysis of the image was performed using ImageQuant 5.2 software (Amersham Biosciences). DNAs containing uracil at different locations were each treated with 40 units of E. coli UDG and 20 units of APE1 and incubated at 37 °C for 90 min for cleavage to take place. Cleavage reaction was stopped by performing a PCI extraction, and the DNA was precipitated in ethanol, washed with 70% ethanol, dried, and resuspended in 1× TE. A 1:1 ratio of sample and Hi-DiTM formamide was mixed, boiled for 10 min to denature DNA, chilled on ice, and loaded onto the sequencing gel.
UDG and APE1 Digestion
Free DNA or NCPs (17.4 ng) were treated with E. coli UDG and human APE1 (both from New England Biolabs) at either low equimolar enzyme concentrations of 0.2 nm or high enzyme concentrations of 30 nm UDG and 10 nm APE1 as shown in the figures or figure legends. These cleavage reactions were performed in the presence of the repair reaction buffer containing 25 mm HEPES (pH 7.5), 2 mm DTT, 100 μg/ml BSA, 10% glycerol, 5 mm MgCl2, 200 μm EDTA (pH 8), and 4 mm ATP. Reaction mixtures were incubated at 37 °C for the specified times from 0 to 60 min and stopped by adding PCI (1:2, sample/PCI, v/v). DNA was ethanol-precipitated, resuspended in 1× TE, and mixed with an equal volume of formamide-containing loading buffer. Digested products were separated by electrophoresis on 10% polyacrylamide (19:1), 7 m urea denaturing gels in 1× TBE buffer and analyzed as described above. To account for minor sequence preferences by UDG, cleavage of uracil in the NCPs was normalized to the maximum percent cleavage of their respective free DNA substrates. Normalized data points were fitted to a single phase exponential curve using GraphPad Prism version 6 and the equation Y = Ymax (1 − e−kobst), as performed previously by others (32). The initial rate of product formation was calculated by multiplying the calculated rate constant (kobs) and the corresponding Ymax. Initial rates of product formation are shown in Tables 1 and 2.
TABLE 1.
Comparison of initial rates of cleavage of uracil by UDG and APE1 in Free DNA and NCPs
Free DNA (data not shown) and NCPs (Fig. 2B) were treated with UDG and APE1 at equimolar concentrations (0.2 nm). The initial rate of cleavage was calculated by fitting the cleavage reaction data to a single phase exponential equation (solid lines in Fig. 2B) and multiplying each of the rate constants by the corresponding Ymax as described under “Experimental Procedures.”
| Substrate | kDNA | kNCP | kNCP/kDNA |
|---|---|---|---|
| % cleaved/min | % cleaved/min | ||
| UO (+10) | 16.71 ± 4.30 | 6.06 ± 0.68 | 0.36 ± 0.10 |
| UO (+29) | 10.90 ± 2.13 | 0.56 ± 0.12 | 0.05 ± 0.01 |
| UO (−35) | 21.59 ± 8.91 | 5.61 ± 2.36 | 0.26 ± 0.15 |
TABLE 2.
Comparison of initial rates of cleavage of uracil by UDG and APE1 in NCPs
As in Table 1, the initial rates were calculated from the kinetic parameters obtained by fitting the data in Fig. 2A to a single-phase exponential equation (solid lines). Minor groove widths were estimated from the minor groove width plot reported by Ye et al. (36) for the DNA strand they designated as strand 2, which corresponds to our uracil-containing strand (supplemental Table S1).
| Substrate | kNCP | Minor groove width |
|---|---|---|
| % cleaved per min | Å | |
| NCP-UI (+4) | 4.48 ± 1.50 | 10 |
| NCP-UI (+25) | 2.37 ± 0.47 | 8 |
| NCP-UI (−29) | 5.11 ± 2.18 | 12 |
| NCP-UI (−49) | 10.78 ± 5.34 | 13 |
| NCP-UO (+10) | 40.36 ± 4.25 | 7 |
| NCP-UO (+29) | 24.56 ± 1.01 | 12 |
| NCP-UO (−35) | 23.64 ± 6.28 | 10 |
pol β Gap Filling Assays
Extension activity by pol β was assessed using two methods. In the two-label assay, the complementary strand (partner strand) opposite the DNA gap was first radiolabeled with [γ-32P]ATP (PerkinElmer Life Sciences) at the 5′ end using T4 polynucleotide kinase (Invitrogen) and annealed (1:1) with its complementary uracil-containing strand. The DNA was then treated with UDG (30 nm) and APE1 (10 nm) for 90 min at 37 °C to ensure cleavage of all uracils. The cleavage reaction was stopped using PCI as before, and the gapped DNA was ethanol-precipitated, dried, and resuspended in 1× TE. Reconstitution as described above was then performed. After the reconstitution efficiency was confirmed, the pol β repair reaction was immediately performed. The repair reaction mixture contained either gapped DNA (17.4 ng) or gapped NCP (17.4 ng), repair reaction buffer (25 mm HEPES (pH 7.5), 2 mm DTT, 100 μg/ml BSA, 10% glycerol, 5 mm MgCl2, 200 μm EDTA (pH 8), and 4 mm ATP), [α-32P]dCTP at 40 nCi/μl (PerkinElmer Life Sciences), and freshly diluted pol β to a final concentration of 5 nm. Reaction mixtures were incubated at 37 °C for the specified times and quenched using PCI. Extension products were separated on 10% polyacrylamide (19:1), 7 m urea denaturing gels in 1× TBE buffer and analyzed as described above.
In the second method used, one-label assay, the uracil-containing strand was radiolabeled at its 5′ end, and the gapped substrate was generated as described above. The repair reaction was then performed as described previously except that 44 ng of DNA or NCP substrate was used as well as nonradioactive dCTP (100 μm). The reaction was also performed in the presence or absence of APE1 to a final concentration of 10 nm. Final pol β enzyme concentration ranged from 50 to 200 nm, and all extension products were separated in a denaturing sequencing gel containing 8% polyacrylamide (19:1) and 7 m urea in 1× TBE buffer and analyzed as described previously. The percent extended was calculated as shown in Equation 1,
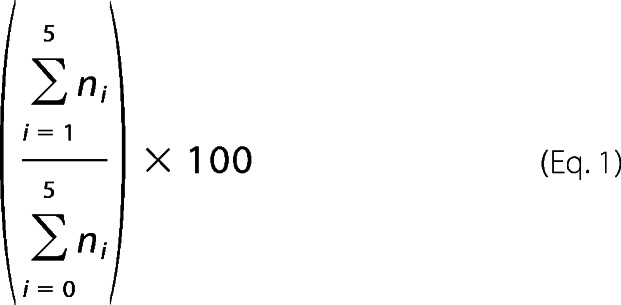 |
where n0 corresponds to the cleavage band, and the n1→5 bands correspond to the gap filling activity of pol β of up to five nucleotides. The pol β extension activity in NCPs was normalized to the maximum extension activity by pol β on the corresponding free DNA to account for sequence preferences. Normalized data from either assay were fitted to a single phase exponential curve using GraphPad Prism version 6 and the equation Y = Ymax (1 − e−kobst), as performed previously by others (32, 45). The initial rate of extension by pol β was calculated by multiplying kobs and the corresponding Ymax values.
Formaldehyde Cross-linking of NCPs
To covalently cross-link DNA and histones, NCPs containing intact or gapped radiolabeled DNA were treated with cross-linking reaction buffer containing 5 mm HEPES, 10 mm NaCl, 100 μm EDTA, 50 μm EGTA, and 1.1% formaldehyde (Mallinckrodt Baker) at room temperature for 2 min. The cross-linking reaction was stopped by adding glycine (500 mm final concentration). Cross-linked and mock-treated (minus formaldehyde) samples were then dialyzed against the last reconstitution buffer containing 10 mm Tris (pH 7.5), 1 mm EDTA, and 50 mm NaCl for 1 h at 4 °C to remove excess formaldehyde and glycine. Digestions with UDG and APE1 or pol β gap filling assays were performed as described above except that reactions were terminated by adding EDTA to a final concentration of 25 mm and boiled for 4 min to kill the enzymes. To reverse the cross-links, samples were then treated 1:1 (v/v) with elution buffer (50 mm Tris (pH 7.5), 10 mm EDTA, 1% SDS) and incubated overnight at 65 °C. DNA was subsequently isolated by PCI, separated by electrophoresis on 10% polyacrylamide (19:1), 7 m urea denaturing gels in 1× TBE buffer, and analyzed as described previously.
Restriction Enzyme Accessibility Assay
Restriction enzymes HaeIII- and HhaI-containing restriction sites corresponding to +51 and +2, respectively, were used to determine the accessibility of these sites in intact and gap-containing NCPs at three sites: gI (+4), gO (+10), and gI (−49), where “I” and “O” correspond to rotational settings “in” and “out,” respectively. Intact and gapped DNAs and NCPs were incubated with 10 units of each enzyme at 37 °C for 2 h followed by standard PCI and chloroform/isoamyl alcohol isolation of DNA. Cleavage products were separated on a 16% native PAGE. Visualization and quantification of results were performed as described previously.
RESULTS
Assembly of Nucleosome Core Particles and Verification of Uracil Rotational Orientation
The design of the substrates is critical in correctly addressing the effects of rotational and translational positioning of DNA lesions on BER efficiency. By selecting a DNA sequence with strong nucleosome positioning power, such as the 601, the effects due to translational positioning can be discerned with greater confidence because it generates more homogeneously positioned NCPs. Additionally, the crystal structures previously generated with this sequence enhance understanding of how unique structural features in NCPs may contribute to accessibility and repair (42, 46). The presence of uracil in several different nucleosome-positioning sequences does not affect NCP formation (28, 32). In fact, even the presence of a single nucleotide gap does not affect the reconstitution efficiency, independent of its rotational orientation (data not shown). The rotational positioning of lesions was determined by comparing the helical orientation of the DNA strand of interest, using hydroxyl radical footprinting, to the location of the UDG/APE1 cleavage site of the uracil-containing strand. The sites were chosen for definitive rotational settings with DNA backbones oriented out or in from the histone surface (Fig. 1A), spanning three DNA flexibility domains that we have arbitrarily assigned based on histone-DNA interactions. The 147-bp 601 DNA positioning sequence reported in Fernandez and Anderson (41) was designed to accommodate the placement of uracil at seven different sites (supplemental Table S1). The 5′ end-labeled uracil-containing oligomers were annealed with the undamaged complementary strand and reconstituted using purified chicken erythrocyte NCPs as described previously (28, 47). The rotational setting of each uracil was verified by treatment of each construct with UDG and APE1 to generate a single strand cleavage at the uracil site of interest, followed by comparison with the •OH footprint of NCPs on DNA sequencing gels (Fig. 1B). Intensity scans of these gels confirmed the rotational setting of the designed substrates (Fig. 1B, right).
FIGURE 1.

Uracil rotational orientation and translational position in NCP substrates. A, using MacPyMOL2, the crystal structure of the 601 NCP Protein Data Bank code 3LZ0 (42) was modified to highlight the positioning of uracil (shown in black). The uracils whose DNA backbone faces the histone octamer have an in rotational orientation, and those with their DNA backbone facing the solvent are outwardly oriented (out). The translational position of the uracils was designated relative to the dyad, translational position 0, where the number in parentheses indicates the number of nucleotides toward the 5′ end (+) or toward the 3′ end (−). The flexibility of DNA increases with distance from the dyad, and thus, in terms of flexibility, domain I < II < III. B, DNA sequencing gel of hydroxyl radical footprints of the equivalent uracil-containing strand associated with the histone octamer. Lane M corresponds to the radiolabeled ([γ-32P]ATP) 10-bp DNA ladder. Lanes 1 and 6, undamaged 601-NCP treated with hydroxyl radicals. In order, lanes 2–5 show uracil-containing DNA: UO (+29), UO (+10), UO (−35), UI (−49), all treated with UDG and APE1, which cleaves 5′ to the AP site. Therefore, the location of the uracils with respect to the NCP footprint will be one nucleotide longer or shifted to the left in the intensity scan shown on the right.
Rotational and Translational Positioning of Uracil Plays an Important Role in UDG/APE1 Excision
Structural and biochemical studies on the excision of uracil by eukaryotic and E. coli UDG have revealed a DNA sequence preference that can range by as much as 10–15-fold in excision repair efficiency (48–50). Therefore, we first cleaved 601 DNA at low equimolar concentrations of UDG and APE1 (0.2 nm) to determine the sequence bias of UDG. The results show a clear difference in cleavage efficiency (data not shown), in agreement with the predicted sequence bias described by others (48–50). As this could not only reflect sequence preferences but also minor differences in uracil incorporation during DNA synthesis, each of the substrates was incubated at high enzyme concentrations to determine maximal uracil cleavage at each of the sites. Cleavage data on free DNA was normalized to this maximum value, which ranged from 82 to 95% (data not shown). Given that these normalized data exhibit a difference in cleavage efficiency, our results agree with previous reports indicating sequence-biased cleavage by UDG. (We note that APE1 activity shows little (or no) sequence bias (51).) Therefore, cleavage of uracil in all NCP experiments was normalized to the maximum percent cleaved of their respective free DNAs.
To address the effect of rotational and translational positioning of uracil on repair, NCPs were incubated with UDG (30 nm) and APE1 (10 nm) or minimal enzyme concentrations required to detect repair of uracils facing inward on the histone surface. Low equimolar enzyme concentrations (0.2 nm) previously used to repair free DNA yielded no measurable repair for inwardly oriented uracils (data not shown). Importantly, at this low enzyme concentration uracils with outwardly oriented backbones cleave at ∼3–19-fold lower rates as compared with free DNA (Table 1). The Ymax for these outwardly oriented uracils also varies significantly (Fig. 2). Consequently, minor rotational variants may affect accessibility due to enhanced DNA flexibility at different sites. Thus, the ability to locate uracil in suboptimal rotational orientations by UDG appears to be rate-limiting. Moreover, with single-turnover kinetics (e.g. at 150× higher UDG concentration), Ymax increases to nearly 100% (Fig. 2A). However, under these same conditions, sterically occluded uracils are significantly resistant to cleavage (Fig. 2A and Table 2), exhibiting a Ymax of 3–5-fold lower as compared with NCP-UOs. Given that the difference in kobs between NCP-UIs and NCP-UOs is <2-fold, the differences in initial rates (Table 2) of NCP-UIs and NCP-UOs can be attributed to a reduction in substrate availability. Alternatively, this could also suggest UDG and APE1 digest a significant fraction of the substrate at a much slower rate. These results are in agreement with previous studies, indicating the importance of helical orientation for glycosylase activity (28, 31, 32, 38, 52).
FIGURE 2.

Assessment of the removal of rotationally and translationally positioned uracils by UDG and APE1. A, NCPs containing a single uracil at different sites were incubated with UDG and APE1. Open symbols represent in uracils as follows: red square, NCP-UI (+4) and open blue circle, NCP-UI (+25). Filled symbols correspond to out uracils as follows: solid blue circle, NCP-UO (+29) and purple triangle, NCP-UO (−35). B, NCPs containing uracil with DNA backbones outwardly oriented were treated with low equimolar enzyme concentrations of 0.2 nm. Data points represent the mean ± 1 S.D. of at least three independent experiments and were fitted to a single phase exponential curve as described under “Experimental Procedures.” For data points where the error bars are not visible, the standard deviations were smaller or similar in magnitude to the size of the symbols.
We also observed an effect due to translational positioning, where the rate of uracil removal at NCP-UI (−49) is ∼5-fold greater than at NCP-UI (+25) and ∼2-fold greater than at NCP-UI (+4) (Fig. 2A and Table 2). Uracils with outwardly oriented DNA backbones exhibit a similar trend, where the cleavage rate at NCP-UO (−35) is ∼10-fold greater than at NCP-UO (+29) (Fig. 2B). However, NCP-UO (+10) was the most reactive of the three outwardly oriented uracils (Fig. 2B and Table 1), indicating there are other local structural features contributing to repair in addition to site exposure.
Formaldehyde Cross-linking Decreases Accessibility to Uracil Independent of Rotational Orientation
To address the impact of rotational flexibility defined as the ability of DNA to rotate along its longitudinal axis (Fig. 3A) on the removal of uracil, the dynamics of histone-DNA interactions were reduced through formaldehyde cross-linking (53–55). The advantages of using formaldehyde to freeze histone-DNA interactions include little (or no) reactivity with DNA (56) and minimal distortion of nucleosome structure (57). This is also revealed in hydroxyl radical footprinting of formaldehyde cross-linked NCPs, where the cleavage pattern was found to be identical to untreated NCPs (data not shown).
FIGURE 3.

Effect of formaldehyde cross-linking on the removal of uracil. A, cross-linking by formaldehyde reduces the rotational flexibility of DNA or the freedom to rotate along its longitudinal axis (black dot). The crystal structure Protein Data Bank code 3LZ0 was modified with MacPyMOL2 to illustrate some potential cross-linking regions (black X) near UO (+10) and UI (+4), where the red spheres represent cross-linkable amino acids (Lys or Arg) in proximity to A, G, or C (shown in green). B and D, cross-linked and noncross-linked NCPs containing outwardly oriented uracils near the dyad axis, domain I (B) or near the ends (D), were incubated at low UDG and APE1 equimolar concentrations (0.2 nm). Inwardly oriented uracils were incubated at a high enzyme concentration of 30 nm UDG and 10 nm APE1. C, all NCPs were incubated at high enzyme concentration (30 nm UDG and 10 nm APE1). Data points represent the mean ± 1 S.D. of at least three independent experiments and were fitted to a single phase exponential curve as described under “Experimental Procedures.”
After confirmation of formaldehyde cross-linking efficiency by electrophoretic mobility shift assays (EMSA) (data not shown), cross-linked and noncross-linked NCPs were incubated with UDG and APE1 for different times (0–40 min). The results indicate that rotational flexibility of DNA on the NCP surface plays an important role in the removal of uracil (independent of rotational setting; Fig. 3, B–D). Interestingly, our results show that near the dyad this rotational flexibility is critical for repair of the occluded uracil NCP UI (+4), with much less contribution to repair of NCP-UO (+10) (Fig. 3B). The rotational flexibility is nevertheless also important for repair of outwardly oriented uracils at sites where the histone-DNA interactions are weaker and where there is little effect due to unwrapping (domain II), as for NCP-UO (+29) (Fig. 3C). Thus, even an outwardly oriented uracil at this site may spend more time in less optimal orientations due to increased rotational flexibility. This also implies that the effect of formaldehyde cross-linking at that region has a less pronounced effect for an inwardly oriented uracil as compared with one near the dyad, which is in agreement with our results (Fig. 3, B and C). It is possible that the effects of formaldehyde cross-linking near the DNA ends (Fig. 3D) are not only due to decreased rotational flexibility but also to reduced DNA breathing. Taken together, our results indicate the dynamic histone-DNA interactions are important for accessibility and repair; however, the rotational setting is the predominant factor in the removal of uracil by UDG and APE1.
pol β Extension Activity Is Markedly Affected by Different Rotational and Translational Settings in Nucleosome DNA
To determine whether the effects of rotational and translational position persisted after lesion recognition and removal, we designed four DNA substrates containing a single nucleotide gap at UO (+10), UI (+4), UO (−35), and UI (−49), designated gO (+10), gI (+4), gO (−35), and gI (−49), respectively. We first examined the effect of these single strand gaps on the rotational orientation of DNA in NCPs. As mentioned earlier, the reconstitution efficiency of 601 DNA containing these DNA gaps does not change (data not shown); however, the question remains whether they maintain the same helical orientation as the uracil-containing substrates. Therefore, we performed •OH footprinting of the undamaged complementary strand opposite to a single nucleotide gap or uracil. Our results show that the same rotational orientation is obtained whether the DNA contained a uracil or single nucleotide gap, and importantly, this result is the same for a single nucleotide gap inwardly oriented near the dyad, NCP-gI (+4), or near the ends, NCP-gI (−49) (data not shown). These results are in agreement with Hayes and co-workers (26, 30), who found that NCPs containing nicked templates and outwardly oriented 5-nucleotide gaps adopt the same translational and rotational positions as those reconstituted with intact 5 S rDNA fragments.
To test DNA pol β activity on different gap orientations in NCPs, 5′ end-labeled DNAs and NCPs containing single nucleotide gaps were incubated with pol β (200 nm, for 30 min), and extension activity was measured by the appearance of slower migrating bands relative to the cleaved band (Fig. 4A, n0). Gap filling activity by pol β in these NCPs is significantly inhibited by 2.5- and 3-fold at NCP-gO (+10) and NCP-gI (+4), respectively (Fig. 4A). Interestingly, the rotational setting does not have the same effect on the rate of gap filling activity by pol β as it does on the removal of uracil at these two sites (Fig. 2A). In this case, the rate of pol β extension activity is marginally (≤2-fold) greater at NCP-gI (+4) as compared with NCP-gO (+10) (Fig. 4A, lanes 8 and 10). These results are independent of enzyme concentration when pol β is in excess relative to the nucleosomal substrate (Fig. 4B), and by using this type of assay, we were unable to detect extension when pol β is limiting.
FIGURE 4.
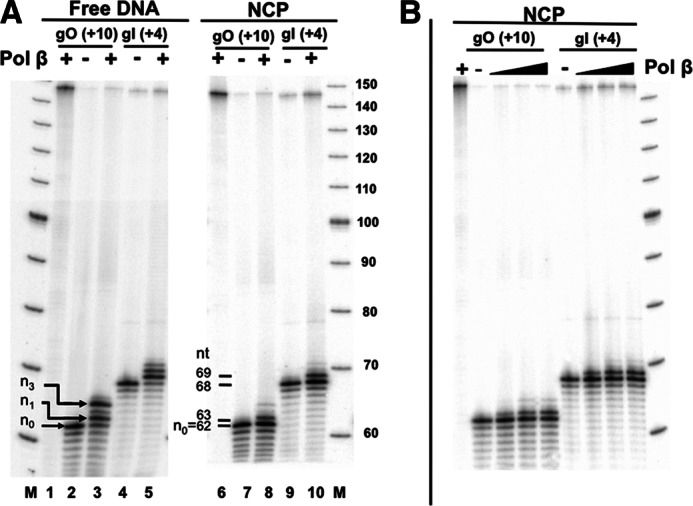
Polymerase β extension activity in free and nucleosomal DNA. A, intact or gap-containing free DNA and NCPs were incubated with pol β (200 nm), indicated by +, for 30 min at 37 °C. Lane M corresponds to the radiolabeled 10-bp DNA ladder. Lanes 1 and 6 correspond to intact DNA-UO (+10) and NCP-UO (+10) respectively. pol β extension was quantified as the appearance of slower migrating bands relative to the untreated (− pol β) lane. The percent extended was calculated as described under “Experimental Procedures.” B, NCPs incubated with increasing amounts of pol β (50, 200, and 500 nm) at 37 °C for 30 min.
Given that pol β must have access not only to the DNA gap, but also to the templating base opposite the gap, we hypothesized that accessibility to the gap is critical to the rate of enzymatic activity for inwardly oriented gaps. Conversely, for outwardly oriented gaps, accessibility to the templating base and the ability to bend the template strand by ∼90° may be the limiting parameters for catalysis. To further examine extension by pol β at these sites, we performed a similar experiment as above, except incorporation of [α-32P]dCTP was measured with nucleosomal substrate in excess relative to pol β. The results indicate that the rate of incorporation of [α-32P]dCTP for NCP-gI (+4) is still greater (∼5-fold) than the incorporation at NCP-gO (+10) (Fig. 5B).
FIGURE 5.

Effect of formaldehyde cross-linking on polymerase β extension activity in NCPs near the dyad. A, MacPyMOL2 was used to measure the distance between atoms where cross-linking is more likely to occur due to proximity. As shown, there is a predominance of arginines (red) near cross-linkable DNA sites (A, C, and G shown in blue). Because the flanking sequence of gI (+4) faces the histone octamer, this area is in closer proximity for cross-linking than the outwardly oriented DNA regions near gO (+10). B, plot of the mean ± 1 S.D. of three independent experiments of pol β extension activity on cross-linked and noncross-linked NCPs containing a single nucleotide gap. Representative gels show gap filling activity of pol β, which was monitored as the appearance of [α-32P]dCTP. The signal in the extension product (EP) was normalized to the signal of the full length (FL) after background subtraction of each band. The negative control, intact (I), corresponds to NCP-UI (+4), incubated with pol β and [α-32P]dCTP for 60 min at 37 °C. Data points were fitted to a single phase exponential equation as described under “Experimental Procedures.” The initial rates of relative dCTP incorporation/min were calculated from the kinetic fits for cross-linked and noncross-linked NCPs and gave values of 0.47 ± 0.03, 0.08 ± 0.01, 0.09 ± 0.02, and 0.13 ± 0.04 for NCP-gI (+4), F-NCP-gI(+4), NCP-gO (+10), and F-NCP-gO (+10), respectively. For data points where the error bars are not visible, the standard deviations were smaller or similar in magnitude to the size of the symbols.
To determine whether the limiting factors for pol β gap filling activity at NCP-gI (+4) and NCP-gO (+10) are in fact accessibility to the DNA gap and template strand, respectively, we determined gap filling activity by pol β in formaldehyde cross-linked NCPs. Inspection of the 601 NCP for regions of DNA that could be potentially cross-linked suggests that in order for cross-links to occur at the maximal distance of 2–3 Å, the dynamic nature of the histone-DNA interactions must play a significant role because all the calculated distances between potential cross-linking sites exceed the maximal distance (Fig. 5A). (We note that small amounts of polymerized formaldehyde may be present in the reaction and responsible for cross-linking sites of longer distances than 2–3 Å). Importantly, cross-linking by formaldehyde significantly decreases the rate of [α-32P]dCTP incorporation at NCP-gI (+4) by 6-fold, with no effect at NCP-gO (+10) (Fig. 5B), indicating that rotational flexibility near the dyad is critical for repair of inwardly oriented gaps. Because the cross-links are more likely occurring in the DNA strand facing the histone octamer in the case of the NCP-gI (+4), the cross-links restrict accessibility to the gap. However, at NCP-gO (+10), cross-linking restricts accessibility to the templating base and could potentially inhibit bending of the template strand, although no effect at NCP-gO (+10) is observed. These results agree with the effect observed for formaldehyde cross-linking on the removal of uracil near the dyad (Fig. 3B).
Single Nucleotide Gaps Do Not Change Nucleosome Dynamics or Accessibility
Given that DNA gaps do not affect NCP reconstitution or change the helical orientation of DNA on the histone surface (data not shown), it is still possible that they could change nucleosome dynamics and accessibility. Indeed, UV photoproducts destabilize nucleosomes and promote nucleosome unwrapping (58), whereas lesions derived from the polycyclic aromatic hydrocarbon benzo[a]pyrene stabilize NCPs (59). Therefore, we performed restriction enzyme accessibility assays to determine global and local changes in NCP dynamics and accessibility (22). Nucleosome core particles containing either uracil or its corresponding DNA gap were digested with either HaeIII or HhaI, and the digestion products were separated on native polyacrylamide gels. As shown in Fig. 6, A and C, restriction enzyme accessibility near the dyad (HhaI cut site at +2) is unchanged in the presence of a single nucleotide gap at +10, +4, and −49. Similarly, cleavage by HaeIII at +51 (or near the DNA ends) is not significantly different in NCP DNA with these different gaps (Fig. 6, B and D).
FIGURE 6.

Restriction enzyme accessibility assay. A, representative polyacrylamide native gels of restriction enzyme accessibility near the DNA ends for intact (I), which contains a single uracil, or a single nucleotide gap (g) at the corresponding uracil location. Lanes M represent a radiolabeled 25-bp DNA ladder. HaeIII cleaves at +51. B, representative 16% polyacrylamide native gels of restriction enzyme accessibility near the dyad with HhaI, which cleaves at +2. C and D, plot of the mean ± 1 S.D. of three independent experiments.
APE1 Does Not Influence pol β Extension Activity in NCPs
During BER, substrate coordination has been observed with free DNA (17, 19) and recently in NCPs (31, 38). To study substrate coordination at the APE1-pol β step of BER, we incubated single nucleotide gap NCPs with pol β (100 nm) in the presence and absence of APE1 (10 nm), and we measured gap filling activity as described above (see Fig. 4). pol β activity in NCPs was normalized to the maximum extension activity on free DNA for the corresponding substrate to account for minor sequence preferences. The normalized data were fitted to a single exponential equation, as described by others (60), and the initial rates were calculated as described under “Experimental Procedures.” Our results indicate that APE1 does not influence the rate of product formation by pol β (Figs. 7 and 8). However, the different sites exhibit a significant difference in the rate of extension by pol β, where the translational positioning plays an important role for outwardly oriented gaps. The rate of extension by pol β at NCP-gO (−35) is 3-fold greater than at NCP-gO (+10) (Figs. 7 and 8). Nevertheless, rotational positioning of the DNA gap seems to play less of a role in the rate of extension, as pol β exhibits a marginally greater rate (<2-fold) at NCP-gI (+4) as compared with NCP-gO (+10) (Fig. 7C). Similarly, near the DNA ends, the initial rate of extension by pol β is <2-fold different between NCP-gO (−35) and NCP-gI (−49) (Fig. 8C), indicating that translational positioning plays a key role in the repair of single nucleotide gaps by pol β.
FIGURE 7.

Polymerase β extension activity in NCPs near the dyad. A, representative gels for NCP-gO (+10) and NCP-gI (+4) pol β (100 nm) extension in the absence of APE1. B, NCP-gO (+10) and NCP-gI (+4) were incubated with pol β and APE1 at 100 and 10 nm, respectively, for the specified times. C, plot of the mean ± 1 S.D. of three independent experiments normalized to pol β (200 nm) extension on the corresponding free DNA. Normalized data points were fitted to a single phase exponential equation as described under “Experimental Procedures.” The initial rates were calculated for extension in the absence of APE1 with values of 14.1 ± 1.6 and 9.1 ± 1.2% extended per min for NCP-gI (+4) and NCP-gO (+10), respectively. For data points where the error bars are not visible, the standard deviations were smaller or similar in magnitude to the size of the symbols.
FIGURE 8.

Polymerase β extension activity in NCPs near DNA ends. A, representative gels for NCP-gO (−35) and NCP-gI (−49) pol β (100 nm) extension in the absence of APE1. B, NCP-gO (−35) and NCP-gI (−49) were incubated with pol β and APE1 at 100 and 10 nm, respectively, for the specified times. C, plot of the mean ± 1 S.D. of three independent experiments normalized to the maximum pol β extension on their respective free DNAs, which was performed at an enzyme concentration of 200 nm for 60 min at 37 °C. Normalized data points were fitted as described under “Experimental Procedures.” The initial rates were calculated for extension in the absence of APE1 with values of 27.1 ± 2.0 and 16.1 ± 2.1% extended per min for NCP-gO (−35) and NCP-gI (−49), respectively. Data points where the error bars are not visible, the standard deviations were smaller or similar in magnitude to the size of the symbols.
DISCUSSION
In this study, we investigated the effect of rotationally and translationally positioned DNA lesions in NCPs on BER by assessing removal of uracil by UDG and APE1 and the extension activity of pol β at single nucleotide gaps. Our results indicate that both rotational and translational positioning of DNA lesions in NCPs are important parameters for accessibility and repair. The effect of these parameters is, however, dependent on the structural requirements and mode of action of each enzyme, suggesting that different DNA lesions, even when located at the same site, present different levels of constraints. For example, although the rate of removal of uracil at NCP-UO (+10) is ∼9-fold greater as compared with NCP-UI (+4) (Fig. 2A and Table 2), there is only a marginal difference (<2-fold) for extension of a single nucleotide gap at these sites (Fig. 7C). More surprising is that pol β extension activity is greater at NCP-gI (+4) as compared with NCP-gO (+10) (Fig. 7C). This suggests that to prevent the negative consequences of having unprotected intermediates, stable binding of APE1 to its product is critical. Nevertheless, recruitment of ATP-dependent remodeling factors is necessary for completion of BER (28).
The difference in the effects of translational positioning on the removal of uracil and the repair of single nucleotide gaps are also noteworthy given that for the removal of uracil the translational positioning played a less significant role as compared with the rotational setting. Uracils with inwardly oriented DNA backbones generally benefited from increased distance from the dyad (Fig. 2A and Table 2), in agreement with previous findings for uracil and thymine glycol excision by UDG and NTH1, respectively (32, 52). The increased distance from the dyad for inwardly oriented uracils did not supersede the constraints imposed by the rotational setting, indicating the rotational setting is the limiting factor for the removal of uracil. This suggests that DNA breathing may only marginally increase accessibility to occluded uracils by capturing the transient exposed states. This is not surprising given that only 1–10% of the time occluded sites are exposed through this mechanism (61). This is in agreement with the findings reported previously on the effects of rotational and translational positioning on uracil removal efficiency (32). The observed increased efficiency in the removal of occluded uracils is not only correlated with increased distance from the dyad but also to the minor groove width at the uracil site (Table 2), estimated from the results recently published by Ye et al. (36). Thus, in agreement with Ye et al. (36), it is feasible that local structural features also play a role in the overall efficiency of uracil removal. However, we do not believe these local structural features take precedence over rotational setting as all outwardly oriented uracils are more efficiently removed as compared with inwardly oriented uracils (Fig. 2A and Table 2) despite the wide range of minor groove measurements (Table 2). Also, we found no correlation of minor groove width and rate of uracil removal at outwardly oriented uracils.
We observe differences in uracil removal at different translational positions among the outwardly oriented uracils under low enzyme concentrations (Fig. 2B); however, the site nearest the dyad, NCP-UO (+10), exhibits the highest initial rate of uracil removal (Tables 1 and 2). Conversely, this is the site where pol β exhibits the least extension activity (Fig. 7C). Inspection of the NCP crystal structure reveals that the tail of H3 interacts with DNA near the dyad, in proximity to NCP-gO (+10) (20). However, because it was previously shown that removal of the histone tails had little (or no) effect on the activity of UDG, APE1, or pol β (28), obstruction at NCP-gO (+10) by the histone H3 tail is not likely to play a significant role in the observed inhibition of pol β gap filling activity at this site as compared with NCP-gI (+4). This site, NCP-gO (+10), is flanked at +5 and +15 by two TA flexible dinucleotides, unlike any other site that was analyzed in this study. Although TA dinucleotides in NCPs tend to compress the minor groove width (42), thus predicting a disadvantage for enzymes like UDG and pol β that make minor groove contacts with DNA (11, 37), it has nevertheless been observed that the activity of UDG is enhanced by the presence of local DNA flexibility (62). Additionally, proteins that bind to the minor groove have less information available and presumably utilize indirect modes that are dependent on the intrinsic shape and mechanical properties of DNA (41). It is conceivable that this structural feature might be responsible for increased recognition by the glycosylase. Because UDG binds to only one face of the DNA helix with the UDG-DNA interface being relatively small, the flexible TA dinucleotides are in the vicinity of but not within the UDG-DNA interface; consequently, this local deformity may be beneficial for detection with no direct effect in overall catalysis. However, for pol β, which binds both faces of the DNA helix and makes contact six nucleotides upstream and downstream of the DNA gap (63), where the TA-flexible dinucleotides are located, the compression of the minor groove by these TA dinucleotides may prevent a stable interaction, resulting in decreased extension by pol β as compared with other sites. Other structural constraints may play a role in pol β extension activity, including the width of the minor groove, not only at the DNA gap site but also at regions where pol β interacts with DNA.
The difference between the increased activity of UDG at outwardly oriented lesions and decreased pol β activity at these same sites is likely associated with the requirement to bend the template strand by ∼90° during the pol β extension step (10). This gives the template strand of inwardly oriented gaps a more optimal orientation away from the occlusion of the histones compared with the template strands of the outwardly oriented gaps. As expected, these structural constraints on the template strand for outwardly oriented gaps are diminished near the DNA ends, where pol β exhibits a 3-fold increase in extension activity at NCP-gO (−35) as compared with NCP-gO (+10). Consistent with this observation, Nilsen et al. (64) also reported an effect on pol β extension activity due to translational position, where the extension activity was reduced at a site closer to the dyad as compared with one 29 nucleotides away from the DNA ends. If DNA breathing was a strong contributor for pol β extension activity, one would expect to observe a similar effect at NCP-gI (−49) (Table 2); however, translational positioning had very little effect at inwardly oriented gaps (Figs. 7C and 8C). We also observed that near the DNA ends, pol β extension activity was greater at NCP-gO (−35) as compared with NCP-gI (−49). This is in agreement with Odell et al. (38), who found pol β to be more active on outwardly facing than inwardly facing gaps near the DNA ends of the X. borealis 5 S-NCP.
The removal of uracil at NCP-UI (+4) and repair of a single nucleotide gap at the same site was significantly inhibited by formaldehyde cross-linking, indicating that the dynamic nature of the histone-DNA interactions is important for repair of occluded DNA lesions near the dyad where the primary mechanism for accessibility appears to be the rotational freedom. Not surprisingly, at outwardly oriented DNA lesions near the dyad, we did not observe a significant effect due to formaldehyde cross-linking, as these lesions are already well positioned substrates for recognition and cleavage.
It is clear that independent of rotational setting, the histone octamer poses a strong barrier for repair even at optimally positioned DNA lesions (Table 1). To facilitate repair, BER efficiency is enhanced through protein-protein interactions between the BER enzymes (17, 31, 38). Therefore, we assessed the activity of pol β in the presence and absence of APE1. The results indicate that APE1 does not have an effect on pol β extension activity (Figs. 7 and 8). The effect of APE1 on pol β extension activity has been previously characterized on free DNA, and although one group found pol β extension activity to be enhanced (19), another group found that the polymerase activity was decreased due to the binding of APE1 to its incised product (65). The enhancement of pol β gap filling activity was only observed when the ternary complex of APE1-pol β-DNA was formed, under conditions where the APE1 concentration was greater than that of pol β (19). As this was not the case in the experiments used for Figs. 7 and 8, we also assessed pol β activity by incorporation of [α-32P]dCTP. In this case, we achieved a 10-fold excess of APE1 relative to pol β, yet even under these conditions APE1 again did not have a significant effect on pol β extension activity (data not shown). In addition, we were unable to detect the NCP-pol β or NCP-APE1-pol β complexes by EMSA (data not shown), and it was difficult to definitively address substrate “handoff” from APE1 to pol β in the NCP. Moreover, although we did not observe an effect on the gap filling activity of pol β, it is still possible that under the right conditions APE1 would influence pol β activity in the NCP, particularly the removal of 5′-dRP, which was not assessed in this study.
The ability of pol β to remove the 5′-dRP is critical for the overall BER efficiency as this has been suggested to be the rate-limiting step during BER (66). The removal of the 5′-dRP structure is dependent, among other factors, on patch size (2). In mammalian cells, if pol β adds more than one nucleotide, the dRP lyase activity of pol β is inhibited, and excision of the displaced flap must be carried out by FEN1 (67). Studies addressing the activity of FEN1 in NCPs have shown efficient removal of 5-nucleotide outwardly oriented flaps near the dyad (30). Interestingly, pol β exhibits a bias for incorporating longer size patches near the DNA ends as compared with near the dyad center (Figs. 7 and 8). Although this can also be due to the sequence context around the gap (Fig. 4A), it is nevertheless interesting to see that near the dyad we observe predominantly extension of only 1 nucleotide (Fig. 7), although near the DNA ends, there is also extension of up to 5 nucleotides, particularly at NCP-gO (−35) (Fig. 8). Further studies are necessary to determine the biological significance of this observation, given that other factors not present in our reactions are capable of influencing patch size, including XRCC1 and FEN1. It is nonetheless tempting to hypothesize that the structural location of the DNA gap, due to structural constraints, may influence patch size and consequently whether BER will proceed via a short or long patch.
The findings in this study strongly suggest that BER near the dyad is limited by the activity of pol β for outwardly oriented uracils, whereas for uracils with inwardly oriented DNA backbones, the UDG and APE1 steps are much slower, limiting overall BER efficiency at these sites. Because there is a significant difference in the efficiency of repair between the first two steps and the third step of BER at NCP-UO (+10), our results also suggest that this site could be a hot spot for greater mutagenic potential due to the persistence of the DNA gaps. Importantly, these results for pol β can further be expanded to those studies where the helical and translational position of the lesion has a similar effect on the glycosylase activity as is the case for the removal of thymine glycol (52). Taken together, our results suggest that positioning of different DNA lesions at discrete translational and rotational orientations on NCPs presents different levels of inhibition, which are dependent on the structural requirement for the enzymes' catalysis. Therefore, it would be interesting to determine how the activity of different DNA glycosylases for the same site is influenced by helical orientation of the DNA lesion near the dyad to determine the types of DNA damage that have the greatest cytotoxic/mutagenic potential. Given that the observed effect of rotational setting of the DNA gap near the dyad may be further enhanced in the presence of histone H1 or higher orders of chromatin structure (32), studies to address the effect of histone H1 on pol β activity would be essential. Therefore, this study has identified unique structural and dynamic features of NCPs that drive the repair of uracil and single nucleotide gaps, and it has provided grounds for investigation of specific factors that would facilitate the completion of BER.
Acknowledgments
We thank Drs. Samuel H. Wilson and Rajendra Prasad (Laboratory of Structural Biology, NIEHS, National Institutes of Health) for providing human DNA polymerase β and critically evaluating this manuscript. We thank Dr. Bill Beard (Laboratory of Structural Biology, NIEHS, National Institutes of Health) for assistance with the kinetic fits of the pol β data. We also thank Drs. Lisa Gloss and Gregory Poon (Washington State University) for helpful discussions on enzyme kinetics. We are grateful to Dr. John Hinz and Rithy Meas (Washington State University) for their critical evaluations of this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant ES004106 from the NIEHS (to M. J. S.) and Training Award T32GM008336 from NIGMS.

This article contains supplemental and Table S1.
- BER
- base excision repair
- NCP
- nucleosome core particle
- UDG
- uracil DNA glycosylase
- pol
- polymerase
- dRP
- 5′-2′-deoxyribose-5′-phosphate
- UI
- uracil in
- UO
- uracil out.
REFERENCES
- 1. Lindahl T., Barnes D. E. (2000) Repair of endogenous DNA damage. Cold Spring Harbor Symp. Quant. Biol. 65, 127–133 [DOI] [PubMed] [Google Scholar]
- 2. Baute J., Depicker A. (2008) Base excision repair and its role in maintaining genome stability. Crit. Rev. Biochem. Mol. Biol. 43, 239–276 [DOI] [PubMed] [Google Scholar]
- 3. Nilsen H., Krokan H. E. (2001) Base excision repair in a network of defence and tolerance. Carcinogenesis 22, 987–998 [DOI] [PubMed] [Google Scholar]
- 4. Zharkov D. O. (2008) Base excision DNA repair. Cell. Mol. Life Sci. 65, 1544–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huffman J. L., Sundheim O., Tainer J. A. (2005) DNA base damage recognition and removal: new twists and grooves. Mutat. Res. 577, 55–76 [DOI] [PubMed] [Google Scholar]
- 6. Friedberg E. C., Walker G. C., Siede W., Wood R. D., Schultz R. A., Ellenberger T. (2005) DNA Repair and Mutagenesis, 2nd Ed., pp 169–214, American Society for Microbiology, Washington, D. C [Google Scholar]
- 7. Wei Q., Li L., Chen . D. J. (2007) in DNA Repair, Genetic Instability, and Cancer (Wei Q., Li L., Chen D. J., eds) 1st Ed., p. 376, World Scientific Publishing, Singapore [Google Scholar]
- 8. Schroeder J. C., Conway K., Li Y., Mistry K., Bell D. A., Taylor J. A. (2003) p53 mutations in bladder cancer: evidence for exogenous versus endogenous risk factors. Cancer Res. 63, 7530–7538 [PubMed] [Google Scholar]
- 9. Krokan H. E., Drabløs F., Slupphaug G. (2002) Uracil in DNA–occurrence, consequences and repair. Oncogene 21, 8935–8948 [DOI] [PubMed] [Google Scholar]
- 10. Wilson S. H., Kunkel T. A. (2000) Passing the baton in base excision repair. Nat. Struct. Biol. 7, 176–178 [DOI] [PubMed] [Google Scholar]
- 11. Parikh S. S., Mol C. D., Slupphaug G., Bharati S., Krokan H. E., Tainer J. A. (1998) Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 17, 5214–5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hegde M. L., Hazra T. K., Mitra S. (2008) Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 18, 27–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robertson A. B., Klungland A., Rognes T., Leiros I. (2009) DNA repair in mammalian cells: Base excision repair: the long and short of it. Cell. Mol. Life Sci. 66, 981–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fromme J. C., Verdine G. L. (2004) Base excision repair. Adv. Protein Chem. 69, 1–41 [DOI] [PubMed] [Google Scholar]
- 15. Bennett R. A., Wilson D. M., 3rd, Wong D., Demple B. (1997) Interaction of human apurinic endonuclease and DNA polymerase β in the base excision repair pathway. Proc. Natl. Acad. Sci. U.S.A. 94, 7166–7169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mol C. D., Hosfield D. J., Tainer J. A. (2000) Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: the 3′ ends justify the means. Mutat. Res. 460, 211–229 [DOI] [PubMed] [Google Scholar]
- 17. Prasad R., Shock D. D., Beard W. A., Wilson S. H. (2010) Substrate channeling in mammalian base excision repair pathways: passing the baton. J. Biol. Chem. 285, 40479–40488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Waters T. R., Gallinari P., Jiricny J., Swann P. F. (1999) Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J. Biol. Chem. 274, 67–74 [DOI] [PubMed] [Google Scholar]
- 19. Liu Y., Prasad R., Beard W. A., Kedar P. S., Hou E. W., Shock D. D., Wilson S. H. (2007) Coordination of steps in single nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase β. J. Biol. Chem. 282, 13532–13541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 21. Davey C. A., Sargent D. F., Luger K., Maeder A. W., Richmond T. J. (2002) Solvent-mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J. Mol. Biol. 319, 1097–1113 [DOI] [PubMed] [Google Scholar]
- 22. Polach K. J., Widom J. (1995) Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J. Mol. Biol. 254, 130–149 [DOI] [PubMed] [Google Scholar]
- 23. Li G., Levitus M., Bustamante C., Widom J. (2005) Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 12, 46–53 [DOI] [PubMed] [Google Scholar]
- 24. Anderson J. D., Widom J. (2000) Sequence and position dependence of the equilibrium accessibility of nucleosomal DNA target sites. J. Mol. Biol. 296, 979–987 [DOI] [PubMed] [Google Scholar]
- 25. Tims H. S., Gurunathan K., Levitus M., Widom J. (2011) Dynamics of nucleosome invasion by DNA-binding proteins. J. Mol. Biol. 411, 430–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chafin D. R., Vitolo J. M., Henricksen L. A., Bambara R. A., Hayes J. J. (2000) Human DNA ligase I efficiently seals nicks in nucleosomes. EMBO J. 19, 5492–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Beard B. C., Stevenson J. J., Wilson S. H., Smerdon M. J. (2005) Base excision repair in nucleosomes lacking histone tails. DNA Repair 4, 203–209 [DOI] [PubMed] [Google Scholar]
- 28. Beard B. C., Wilson S. H., Smerdon M. J. (2003) Suppressed catalytic activity of base excision repair enzymes on rotationally positioned uracil in nucleosomes. Proc. Natl. Acad. Sci. U.S.A. 100, 7465–7470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Menoni H., Gasparutto D., Hamiche A., Cadet J., Dimitrov S., Bouvet P., Angelov D. (2007) ATP-dependent chromatin remodeling is required for base excision repair in conventional but not in variant H2A. Bbd nucleosomes. Mol. Cell. Biol. 27, 5949–5956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huggins C. F., Chafin D. R., Aoyagi S., Henricksen L. A., Bambara R. A., Hayes J. J. (2002) Flap endonuclease 1 efficiently cleaves base excision repair and DNA replication intermediates assembled into nucleosomes. Mol. Cell 10, 1201–1211 [DOI] [PubMed] [Google Scholar]
- 31. Hinz J. M., Rodriguez Y., Smerdon M. J. (2010) Rotational dynamics of DNA on the nucleosome surface markedly impact accessibility to a DNA repair enzyme. Proc. Natl. Acad. Sci. U.S.A. 107, 4646–4651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cole H. A., Tabor-Godwin J. M., Hayes J. J. (2010) Uracil DNA glycosylase activity on nucleosomal DNA depends on rotational orientation of targets. J. Biol. Chem. 285, 2876–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gansen A., Tóth K., Schwarz N., Langowski J. (2009) Structural variability of nucleosomes detected by single-pair Forster resonance energy transfer: histone acetylation, sequence variation, and salt effects. J. Phys. Chem. B 113, 2604–2613 [DOI] [PubMed] [Google Scholar]
- 34. North J. A., Shimko J. C., Javaid S., Mooney A. M., Shoffner M. A., Rose S. D., Bundschuh R., Fishel R., Ottesen J. J., Poirier M. G. (2012) Regulation of the nucleosome unwrapping rate controls DNA accessibility. Nucleic Acids Res. 40, 10215–10227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Widom J. (2001) Role of DNA sequence in nucleosome stability and dynamics. Q. Rev. Biophys. 34, 269–324 [DOI] [PubMed] [Google Scholar]
- 36. Ye Y., Stahley M. R., Xu J., Friedman J. I., Sun Y., McKnight J. N., Gray J. J., Bowman G. D., Stivers J. T. (2012) Enzymatic excision of uracil residues in nucleosomes depends on the local DNA structure and dynamics. Biochemistry 51, 6028–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pelletier H., Sawaya M. R., Wolfle W., Wilson S. H., Kraut J. (1996) Crystal structures of human DNA polymerase β complexed with DNA: implications for catalytic mechanism, processivity, and fidelity. Biochemistry 35, 12742–12761 [DOI] [PubMed] [Google Scholar]
- 38. Odell I. D., Barbour J. E., Murphy D. L., Della-Maria J. A., Sweasy J. B., Tomkinson A. E., Wallace S. S., Pederson D. S. (2011) Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Mol. Cell. Biol. 31, 4623–4632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hayes J. J., Clark D. J., Wolffe A. P. (1991) Histone contributions to the structure of DNA in the nucleosome. Proc. Natl. Acad. Sci. U.S.A. 88, 6829–6833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tan S., Davey C. A. (2011) Nucleosome structural studies. Curr. Opin. Struct. Biol. 21, 128–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fernandez A. G., Anderson J. N. (2007) Nucleosome positioning determinants. J. Mol. Biol. 371, 649–668 [DOI] [PubMed] [Google Scholar]
- 42. Vasudevan D., Chua E. Y., Davey C. A. (2010) Crystal structures of nucleosome core particles containing the “601” strong positioning sequence. J. Mol. Biol. 403, 1–10 [DOI] [PubMed] [Google Scholar]
- 43. Thåström A., Lowary P. T., Widom J. (2004) Measurement of histone-DNA interaction free energy in nucleosomes. Methods 33, 33–44 [DOI] [PubMed] [Google Scholar]
- 44. Tullius T. D. (1991) DNA footprinting with the hydroxyl radical. Free Radic. Res. Commun. 12, 521–529 [DOI] [PubMed] [Google Scholar]
- 45. Donigan K. A., Sun K. W., Nemec A. A., Murphy D. L., Cong X., Northrup V., Zelterman D., Sweasy J. B. (2012) Human POLB gene is mutated in high percentage of colorectal tumors. J. Biol. Chem. 287, 23830–23839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Makde R. D., England J. R., Yennawar H. P., Tan S. (2010) Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature 467, 562–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kosmoski J. V., Smerdon M. J. (1999) Synthesis and nucleosome structure of DNA containing a UV photoproduct at a specific site. Biochemistry 38, 9485–9494 [DOI] [PubMed] [Google Scholar]
- 48. Eftedal I., Guddal P. H., Slupphaug G., Volden G., Krokan H. E. (1993) Consensus sequences for good and poor removal of uracil from double-stranded DNA by uracil-DNA glycosylase. Nucleic Acids Res. 21, 2095–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nilsen H., Yazdankhah S. P., Eftedal I., Krokan H. E. (1995) Sequence specificity for removal of uracil from U.A pairs and U.G mismatches by uracil-DNA glycosylase from Escherichia coli, and correlation with mutational hotspots. FEBS Lett. 362, 205–209 [DOI] [PubMed] [Google Scholar]
- 50. Slupphaug G., Mol C. D., Kavli B., Arvai A. S., Krokan H. E., Tainer J. A. (1996) A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature 384, 87–92 [DOI] [PubMed] [Google Scholar]
- 51. Berquist B. R., McNeill D. R., Wilson D. M., 3rd (2008) Characterization of abasic endonuclease activity of human Ape1 on alternative substrates, as well as effects of ATP and sequence context on AP site incision. J. Mol. Biol. 379, 17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Prasad A., Wallace S. S., Pederson D. S. (2007) Initiation of base excision repair of oxidative lesions in nucleosomes by the human, bifunctional DNA glycosylase NTH1. Mol. Cell. Biol. 27, 8442–8453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zeng P. Y., Vakoc C. R., Chen Z. C., Blobel G. A., Berger S. L. (2006) In vivo dual cross-linking for identification of indirect DNA-associated proteins by chromatin immunoprecipitation. BioTechniques 41, 694. [DOI] [PubMed] [Google Scholar]
- 54. Barker S., Weinfeld M., Murray D. (2005) DNA-protein cross-links: their induction, repair, and biological consequences. Mutat. Res. 589, 111–135 [DOI] [PubMed] [Google Scholar]
- 55. Orlando V., Strutt H., Paro R. (1997) Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods 11, 205–214 [DOI] [PubMed] [Google Scholar]
- 56. Solomon M. J., Varshavsky A. (1985) Formaldehyde-mediated DNA-protein cross-linking: a probe for in vivo chromatin structures. Proc. Natl. Acad. Sci. U.S.A. 82, 6470–6474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jackson V. (1978) Studies on histone organization in the nucleosome using formaldehyde as a reversible cross-linking agent. Cell 15, 945–954 [DOI] [PubMed] [Google Scholar]
- 58. Duan M. R., Smerdon M. J. (2010) UV damage in DNA promotes nucleosome unwrapping. J. Biol. Chem. 285, 26295–26303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cai Y., Wang L., Ding S., Schwaid A., Geacintov N. E., Broyde S. (2010) A bulky DNA lesion derived from a highly potent polycyclic aromatic tumorigen stabilizes nucleosome core particle structure. Biochemistry 49, 9943–9945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dalal S., Starcevic D., Jaeger J., Sweasy J. B. (2008) The I260Q variant of DNA polymerase β extends mispaired primer termini due to its increased affinity for deoxynucleotide triphosphate substrates. Biochemistry 47, 12118–12125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tims H. S., Widom J. (2007) Stopped-flow fluorescence resonance energy transfer for analysis of nucleosome dynamics. Methods 41, 296–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Donigan K. A., Sweasy J. B. (2009) Sequence context-specific mutagenesis and base excision repair. Mol. Carcinog. 48, 362–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wilson S. H. (1998) Mammalian base excision repair and DNA polymerase β. Mutat. Res. 407, 203–215 [DOI] [PubMed] [Google Scholar]
- 64. Nilsen H., Lindahl T., Verreault A. (2002) DNA base excision repair of uracil residues in reconstituted nucleosome core particles. EMBO J. 21, 5943–5952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wong D., Demple B. (2004) Modulation of the 5′-deoxyribose-5-phosphate lyase and DNA synthesis activities of mammalian DNA polymerase β by apurinic/apyrimidinic endonuclease 1. J. Biol. Chem. 279, 25268–25275 [DOI] [PubMed] [Google Scholar]
- 66. Prasad R., Beard W. A., Strauss P. R., Wilson S. H. (1998) Human DNA polymerase β deoxyribose phosphate lyase. Substrate specificity and catalytic mechanism. J. Biol. Chem. 273, 15263–15270 [DOI] [PubMed] [Google Scholar]
- 67. Prasad R., Dianov G. L., Bohr V. A., Wilson S. H. (2000) FEN1 stimulation of DNA polymerase β mediates an excision step in mammalian long patch base excision repair. J. Biol. Chem. 275, 4460–4466 [DOI] [PubMed] [Google Scholar]