

Background: FGFR1 gene expression regulates myoblast proliferation and differentiation, and its expression is controlled by Krüppel-like transcription factors.
Results: KLF10 interacts with the FGFR1 promoter, repressing its activity and cell proliferation.
Conclusion: KLF10 represses FGFR1 promoter activity and thereby myoblast proliferation.
Significance: A model of transcriptional control of chicken FGFR1 gene regulation during myogenesis is presented.
Keywords: Cell Proliferation, DNA Transcription, Fibroblast Growth Factor Receptor (FGFR), Gene Regulation, Myogenesis
Abstract
Skeletal muscle development is controlled by regulation of myoblast proliferation and differentiation into muscle fibers. Growth factors such as fibroblast growth factors (FGFs) and their receptors (FGFRs) regulate cell proliferation and differentiation in numerous tissues, including skeletal muscle. Transcriptional regulation of FGFR1 gene expression is developmentally regulated by the Sp1 transcription factor, a member of the Krüppel-like factor (KLF) family of transcriptional regulators. Here, we show that another KLF transcription factor, KLF10, also regulates myoblast proliferation and FGFR1 promoter activity. Expression of KLF10 reduced myoblast proliferation by 86%. KLF10 expression also significantly reduced FGFR1 promoter activity in myoblasts and Sp1-mediated FGFR1 promoter activity in Drosophila SL2 cells. Southwestern blot, electromobility shift, and chromatin immunoprecipitation assays demonstrated that KLF10 bound to the proximal Sp factor binding site of the FGFR1 promoter and reduced Sp1 complex formation with the FGFR1 promoter at that site. These results indicate that KLF10 is an effective repressor of myoblast proliferation and represses FGFR1 promoter activity in these cells via an Sp1 binding site.
Introduction
Vertebrate skeletal muscle development is partly characterized by expansive proliferation of muscle precursor cells or myoblasts followed by coordinated differentiation of these cells at specific times in development. During differentiation, mononucleated myoblasts form multinucleated muscle fibers, and the muscle nuclei become irreversibly withdrawn from the cell cycle (1). Therefore, vertebrate myogenesis provides an appropriate and amenable system to investigate mechanisms that control cell proliferation versus differentiation.
Members of the family of fibroblast growth factors (FGFs) regulate myoblast proliferation and differentiation by interaction with specific cell surface receptors. FGF1 and FGF2 possess mitogenic activity, stimulate myoblast proliferation, and delay myogenic differentiation (2, 3). These effects on cell proliferation and differentiation are mediated by a high affinity FGF receptor, FGFR1. The members of the family of FGFRs2 (FGFR1–4) are receptor tyrosine kinases that typically activate the mitogen-activated protein kinase (MAPK) signaling pathway in a variety of cell types throughout development. FGFR1 is expressed in developing bone, skin, brain, cardiac muscle, and skeletal muscle (4).
A number of studies have reported that FGFR1 gene expression is developmentally regulated in skeletal muscle cells. Proliferating and migratory myoblasts in vitro and in vivo express the FGFR1 gene, and FGFR1 gene expression at the protein and mRNA levels declines during myogenic differentiation into postmitotic muscle fibers (5–9). FGFR1 gene expression levels are reduced but still detectable after cardiac muscle development, and some data suggest that a minimal level of FGFR1 gene expression persists in skeletal muscle after differentiation in vivo (3, 8). The functional significance of the developmental regulation of FGFR1 gene expression is apparent by disruption of normal myogenesis in embryos with altered FGFR1 gene expression. Myoblasts that constitutively expressed wild type FGFR1 were repressed or delayed in differentiation both in vitro and in vivo (10, 11). Conversely, myoblasts that expressed a dominant negative FGFR1 mutant displayed decreased proliferation and accelerated differentiation. Insufficient FGFR1-mediated cell signaling, reduced myoblast proliferation, and concomitant precocious differentiation may be responsible for the observed reduction in skeletal muscle mass in chick embryos expressing the dominant negative FGFR1 variant (10, 12).
Many growth factor receptor genes possess similar structural motifs in their transcriptional regulatory regions. Promoter regions of growth factor receptor genes are typically GC-rich and often lack consensus TATA boxes. For example, the promoters for the rat transforming growth factor β (TGFβ) receptor type III and the human FGFR3 genes are 69 and 82% GC-rich, respectively (13, 14). Rather than TATA boxes, these promoters often contain multiple potential Sp factor binding sites. These GC boxes (GGGCGG) and CT elements ((CCT)4CGG(CCT)2) are usually clustered near the start of transcription and are thought to functionally substitute for the lack of basal (e.g. TATA and CCAAT elements) cis-regulatory components (15).
The small family of Sp transcription factors (Sp1–4) belongs to a larger extended family of transcriptional regulators known as Krüppel-like factors (KLFs) (16). These proteins contain highly conserved C2H2 zinc finger motifs in their carboxyl-terminal halves and bind to GC-rich sites via these motifs. Although KLFs have significant sequence similarity, the extensive KLF family membership does display divergence in the amino-terminal sequences, providing heterogeneity in structure and function. Many KLF and Sp-like proteins activate transcription, and perhaps the best characterized among these activators is Sp1 (17). Sp1 is broadly expressed and activates a wide variety of constitutively expressed and differentially regulated genes. For example, Sp1 activates the avian FGFR1 promoter in proliferating myoblasts (18). However, other Sp and KLF proteins (e.g. Sp3, KLF9, KLF10, KLF13, and KLF16) repress transcription via specific Sin3 domains within the amino-terminal region that recruit histone deacetylase transcriptional repressor complexes (reviewed in Ref. 19).
The TGFβ-inducible early gene 1 (TIEG1) was first identified in human osteoblast cells (20). Sequence analysis revealed that it also contains three C2H2 zinc finger domains, and it is designated KLF10. Immunohistochemical studies have localized KLF10 protein in a variety of tissues including placenta, breast, pancreas, bone, cardiac muscle, and skeletal muscle (21). Overexpression of KLF10 in human osteosarcoma and pancreatic carcinoma cells mimicked the antiproliferative and apoptotic effects of TGFβ on the cells (22, 23). KLF10 also exhibits antiproliferative effects on breast cancer cells (24). KLF10 null mice display an osteopenic phenotype with weakened bones and decreased expression of RUNX2 and SMAD2, indicating reduced osteoblast differentiation (25). These mice also develop hypertrophic cardiomyopathies with increased fibrosis (26).
This study addresses the function of KLF10 in skeletal myogenesis. We report that the KLF10 gene is expressed during myogenic differentiation and that KLF10 gene expression suppresses myoblast proliferation in vitro. Because FGFR1 gene expression regulates myoblast proliferation and differentiation, we hypothesized that KLF10 suppresses myoblast proliferation by transcriptional repression of the FGFR1 gene. We report that FGFR1 gene promoter activity is repressed by KLF10 binding to a specific Sp binding site in the FGFR1 proximal promoter. A model of FGFR1 gene transcriptional regulation invoking multiple, functionally redundant levels of transcriptional activation and repression is presented.
EXPERIMENTAL PROCEDURES
Cell Culture
Chick myoblasts were isolated from embryonic day (ED) 13 hindlimbs and placed in collagen-coated dishes as described previously (27, 28). Cells were incubated in F-10 base medium (Invitrogen) supplemented with 10% horse serum (HyClone Laboratories), 5% chick embryo extract, 1.32 mm CaCl2, 2.9 mm glutamine, and penicillin/streptomycin/Fungizone (Invitrogen) at 37 °C in a 5% CO2 humidified incubator. Medium was replaced every other day. Myotube cultures were maintained in medium containing β-arabinofuranoside hydrochloride (AraC) as described previously (9). Drosophila melanogaster SL2 cells were grown in serum-free medium (Invitrogen) supplemented with 2 mm glutamine and penicillin/streptomycin/Fungizone at 25 °C in ambient atmosphere (18).
RT-PCR
Total RNA was isolated using RNA STAT 60 (Tel-Test) from ED13 myogenic cultures at 0, 24, 48, and 72 h as well as 10 days in cell culture. KLF10 transcripts were reverse-transcribed and amplified using the Access RT-PCR kit (Promega). Reverse transcription was carried out at 48 °C for 45 min with the reverse primer (CTGCGCAGCCGCCGACGGAGGG). Amplification was carried out for 35 cycles with the above reverse primer and forward primer (ATGTCTTACAAGCACTGGAAGCAGGGCC) and annealing temperature of 55 °C. PCR products were resolved in a 1% agarose gel.
Immunostaining
ED13 chicken myoblasts were fixed in 100% methanol for 10 min, washed with phosphate-buffered saline (PBS), and then incubated in blocking solution (5% horse serum, 2% bovine serum albumin in PBS) for 1 h at room temperature. Cells were then incubated with primary antibodies directed against proliferating cell nuclear antigen (PCNA; Santa Cruz Biotechnology) and the FLAG epitope (Sigma), diluted 1:100 and 1:2000, respectively, in blocking solution for 1 h at room temperature. The PCNA antibody was conjugated to FITC. Cells were washed with PBS and then incubated in Texas Red-conjugated anti-mouse IgG (Vector Laboratories) diluted 1:100 in blocking solution for 1 h at room temperature. DAPI (2 μm) in PBS was applied to the cells for 10 min, and the cells were then washed in PBS.
DNA Constructs, Transfections, and Promoter Activity Assays
pCMVKLF10 contains the full-length human KLF10 cDNA linked to the FLAG epitope tag (kindly provided by R. Urrutia). The 3284FGFR1CAT expression construct contains the full-length wild type FGFR1 promoter and has been described previously (9). The 3284FGFR1Luc construct contains the same wild type FGFR1 promoter linked to the firefly luciferase gene. m23FGFR1Luc contains the full-length FGFR1 promoter with a mutation of the most proximal (−23-bp) Sp1 binding site described previously (18). For immunostaining of KLF10, ED13 chicken myoblasts were transfected with pCMVKLF10 (4 μg/35-mm culture dish) for 3 h at 37 °C in growth medium without antibiotic supplements using Lipofectamine 2000 (Invitrogen). For analyses of FGFR1 promoter activities, ED13 chicken myoblasts were transfected with 3284FGFR1CAT (1.5 μg/35-mm culture dish) and increasing amounts (0–6 μg/35-mm culture dish) of pCMVKLF10. For transfection of Drosophila SL2 cells, 3284FGFR1CAT (2.5 μg/35-mm culture dish) and pPacSp1 (750 ng) along with increasing amounts of pCMVKLF10 (0–6 μg) were co-transfected using Cellfectin (Invitrogen) as described previously (29). For all transfections related to promoter activities, pRSVβGAL (1 μg) was co-transfected to normalize for variations in transfection efficiencies. Also, pBSKS plasmid DNA (Stratagene) was used to maintain constant DNA amounts in transfections. Promoter activity assays were performed as described previously (29).
Western Blots
Nuclear extracts (100 μg) from control, nontransfected ED13 myoblasts and myotubes and from myoblasts transfected with pCMVKLF10 as above were electrophoresed in a 10% SDS-polyacrylamide gel and transferred to nitrocellulose membrane. The blots were incubated in blocking solution (2% nonfat dry milk, 0.5% Tween 20 in PBS) overnight at 4 °C. Primary antibodies included Sp1 (Santa Cruz Biotechnology; diluted 1:1000), FLAG epitope (Sigma; diluted 1:2000), KLF10 (Abcam; diluted 1:1000), and E47 (Santa Cruz Biotechnology; diluted 1:100). Blots were incubated in primary antibody diluted in blocking solution for 1 h at room temperature. Blots were washed with PBS-Tween 20 and then incubated with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) diluted 1:2000 in blocking solution for 1 h at room temperature. Blots were washed as before and developed by chemiluminescence (Pierce).
Southwestern Analysis and Electromobility Shift Assays
Southwestern blot analysis was performed as described previously (29). Nuclear extracts (75 μg) from control, nontransfected myoblasts and from myoblasts transfected with pCMVKLF10 as above were electrophoresed in a 7.5% SDS-polyacrylamide gel and electroblotted to nitrocellulose membranes. These membranes were then incubated with end-labeled wild type or mutated FGFR1 promoter double-stranded oligonucleotide sequence. The mutated sequence contained a dinucleotide substitution within the most proximal (−23-bp) Sp binding site (5′-CTGCCCTGACTCTCTTTCTCCTATCCACAGCTCACAGCGCC-3′). Bold, italicized nucleotides replaced wild type sequence (5′-CTGCCCTGACTCTCTTTCTCCCCTCCACAGCTCACAGCGCC-3′).
Nuclear extracts from myoblasts and myotubes were prepared as described previously (18), and protein content was determined by BCA protein assay (Pierce). Electromobility shift assays were carried out as described previously (18).
Chromatin Immunoprecipitation
Chromatin from ED13 chick myoblast cultures at 0, 24, 48, and 72 h was prepared. In addition, myoblasts were allowed to differentiate in vitro for 10 days, and the resulting myotubes were maintained in cell culture medium supplemented with AraC as described previously (9). Chromatin immunoprecipitation was then conducted as described previously (29). Chromatin was immunoprecipitated with 4 μg of Sp1 or KLF10 antibody (Abcam). As a control, 4 μg of Gaq antibody (Santa Cruz Biotechnology) was used in parallel immunoprecipitation procedures. Precipitated DNA was amplified for 25 cycles using FGFR1 promoter-specific forward primer (CTGTTTTCAGTGCCAACT) and reverse primer (CATGGGGCCCCGTCGGCCGCTG). For transient chromatin immunoprecipitation, ED13 chick myoblasts at a density of 3 × 106 cells/10-cm dish were transfected with 8 μg of pCMVKLF10 using Lipofectamine 2000 (Invitrogen). Myotube cultures were maintained in cell culture medium supplemented with AraC as described previously for a total of 10 days (9). Chromatin immunoprecipitation was then conducted as described previously (29). For chromatin immunoprecipitation experiments using wild type and mutated FGFR1 promoter DNA constructs, 8 μg of pCMVKLF10 and 4 μg of wild type 3284FGFR1Luc or m23FGFR1Luc were transfected into myoblasts. Cell culture and chromatin immunoprecipitation from myotubes was carried out as described above. Precipitated DNA was amplified for 30 cycles using the FGFR1 promoter-specific forward primer (CTGTTTTCAGTGCCAACT) and a luciferase gene-specific reverse primer (CAGCGGTTCCATCATCCAGCGGATAG). DNA was resolved in a 1% agarose gel.
RESULTS
KLF10 Gene Expression Reduces Myoblast Cell Proliferation
The expression of KLF10 affects the proliferative capacity and differentiation of a number of cell types (23, 24). To determine whether KLF10 gene expression affects the proliferative capacity of skeletal myoblasts from developing fetal muscle, KLF10 was overexpressed. ED13 chick myoblasts were isolated and transfected with pCMVKLF10, a DNA construct that constitutively drives full-length human KLF10 gene transcription. Thirty-six hours after transfection, cells were fixed and immunostained. Proliferating cells were identified by immunodetection of PCNA, and transfected cells overexpressing KLF10 were identified by immunodetection of the FLAG epitope tag linked to the KLF10 gene product. Cells were also stained with DAPI to visualize all nuclei.
Cells that expressed KLF10 did not immunostain for PCNA (Fig. 1A), although neighboring cell nuclei were immunostained with the PCNA antibody. To assess the effects of KLF10 gene overexpression on overall proliferation, the percentage of pCMVKLF10 transfected and nontransfected cells that immunostained with the PCNA antibody were counted (Fig. 1B). Approximately 22% of nontransfected cells were PCNA-positive. However, only ∼3% of KLF10-overexpressing cells were PCNA-positive (an 86% reduction from nontransfected cells), indicating that KLF10 overexpression reduced the number of proliferative cells.
FIGURE 1.

Effect of KLF10 gene expression on myoblast proliferation. A, immunodetection of proliferating myoblasts and cells transfected with pCMVKLF10. Myoblasts were transfected with pCMVKLF10, and transfected cells were identified using a FLAG epitope tag antibody followed by Texas Red-conjugated secondary antibody. Proliferating cells were identified by immunodetection of PCNA using a fluorescein-conjugated anti-PCNA antibody. Nuclei were visualized by DAPI staining. Arrows indicate the nucleus of a transfected cell expressing the KLF10 gene but not PCNA. B, the percentage of PCNA-expressing cells with and without transfection of pCMVKLF10. Nontransfected and transfected cells were scored for expression of PCNA based on immunostaining with the PCNA antibody. Transfection of pCMVKLF10 significantly reduced the percentage of PCNA-positive cells (p < 0.01). Error bars indicate S.D.
KLF10 Is Expressed in Both Myoblasts and Myotubes
The expression of the KLF10 gene in proliferating myoblasts and differentiated myotubes was assessed by RT-PCR. Myoblasts were isolated and cultured for 0, 24, 48, and 72 h as well as 10 days. After 24 h in cell culture, myoblasts are still proliferative (Fig. 1). By day 10, myoblasts exit the cell cycle and differentiate into multinucleated myotubes. Total RNA was isolated from myoblasts and myotubes and amplified using KLF10 gene-specific primers (Fig. 2A). PCR product was obtained from both myoblast and myotube RNA extracts at all time points tested.
FIGURE 2.
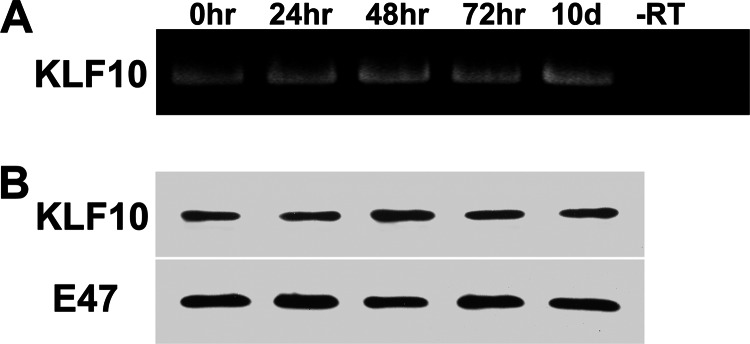
Expression of KLF10 gene in myoblasts and myotubes. A, RNA was isolated from myogenic cultures at 0, 24, 48, and 72 h as well as 10 days of incubation. KLF10 mRNA was reverse-transcribed, and the DNA was amplified using KLF10-specific primers (see “Experimental Procedures”). The control reaction did not include reverse transcriptase (−RT). KLF10 RNA was detected in extracts from both myoblasts and myotubes. B, KLF10 protein was detected by Western blot analysis of nuclear extracts from myogenic cultures at 0, 24, 48, and 72 h as well as 10 days of incubation. Immunodetection of E47 protein was used as a control for amount of nuclear extract in each analyzed sample. KLF10 protein was detected at the indicated time points.
Western blot analysis was also conducted to detect KLF10 gene expression (Fig. 2B). KLF10 protein was detected in nuclear extracts from myoblasts and myotubes. Therefore, both RT-PCR and Western blot analyses confirm that the KLF10 gene is expressed in both proliferating myoblasts and differentiated myotubes.
KLF10 Represses Sp1-mediated Transcription of the FGFR1 Gene
Myoblast proliferation and differentiation are tightly linked to the developmentally regulated expression of the FGFR1 gene (5, 9–11). We have previously shown that FGFR1 gene expression in myoblasts is transcriptionally regulated by multiple Sp factor binding sites in the FGFR1 promoter and that these sites bind the Sp1 transcription factor (18). Sp1 interaction with these sites, located at −23, −48, and −59 bp in the FGFR1 promoter, activates transcription in myoblasts. However, FGFR1 gene expression declines during myogenic differentiation (5, 9), and this may be mediated by transcriptional regulators such as KLF10.
To determine whether expression of KLF10 affects FGFR1 promoter activity, myoblasts were transfected with the transcription reporter construct, 3284FGFR1CAT, which contains the full-length wild type chicken FGFR1 promoter. This promoter has previously been shown to exhibit developmentally regulated transcriptional control similar to the endogenous FGFR1 gene (18). Myoblasts were also transfected with increasing amounts of pCMVKLF10 expression plasmid. FGFR1 promoter activities in myoblasts significantly declined with increasing amounts (0–6 μg/plate) of transfected pCMVKLF10 (Fig. 3A). When compared with FGFR1 promoter activity in the absence of pCMVKLF10, relative promoter activity declined to ∼40% in the presence of pCMVKLF10. The reduced promoter activity was not due to decreased Sp1 protein levels as determined by Western blot analysis of nuclear extracts (not shown).
FIGURE 3.

Repression of FGFR1 promoter activity by KLF10. A, ED13 myoblasts were transfected with 3284FGFR1CAT and increasing amounts of pCMVKLF10. FGFR1 promoter activity was significantly reduced by KLF10 expression (p < 0.02). Error bars indicate S.D. B, Drosophila SL2 cells were transfected with 3284FGFR1CAT, a constant amount of pPacSp1 to activate FGFR1 promoter activity, and increasing amounts of pCMVKLF10. Expression of KLF10 decreased FGFR1 promoter activity (p < 0.02). Error bars indicate S.D.
Myoblasts express Sp1, which is required for FGFR1 promoter activation. To determine whether KLF10 gene expression repressed FGFR1 promoter activity by inhibition of Sp1-mediated transcriptional activation, Drosophila SL2 cells were employed. SL2 cells lack endogenous expression of genes encoding Sp1, Sp2, Sp3, and Sp4 and therefore serve as a useful model system for the study of Sp factor interactions and transcriptional regulation (30). SL2 cells were transfected with 3284FGFR1CAT and a constant amount of pPacSp1 for exogenous expression of Sp1. As shown in Fig. 3B, co-transfection of pPacSp1 activated the FGFR1 promoter in SL2 cells, in agreement with previous findings (18). However, co-transfection of increasing amounts of pCMVKLF10 reduced Sp1-mediated transcriptional activation of the FGFR1 promoter. Maximal amounts of KLF10 expression resulted in ∼40% of Sp1-mediated promoter activity in SL2 cells relative to promoter activity without KLF10 gene expression. These results indicate that Sp1-mediated transcriptional activation of the FGFR1 promoter in SL2 cells is repressed by KLF10.
Overexpressed KLF10 Interacts with the Proximal Sp Site of the FGFR1 Promoter, Displacing a Protein-DNA Complex in Myoblasts
Previous studies have demonstrated that Sp1 binds to the proximal Sp factor binding site of the FGFR1 promoter in proliferating myoblasts (18). Sp1 did not occupy this site in differentiated myotubes. Therefore, we sought to determine whether KLF10 could bind to the proximal Sp factor binding site located at −23 bp within the FGFR1 promoter. Binding of KLF10 to the Sp factor binding site was initially assessed by Southwestern blot analysis. Myotube nuclear extracts were electrophoresed in a 7.5% polyacrylamide gel, transferred to nitrocellulose membrane, and probed with oligonucleotides containing either the wild type or the mutated sequences of the −23-bp Sp factor binding site (Fig. 4). A single band was detected when probed with the wild type Sp factor binding site. This protein had the same relative mobility as KLF10, detected in Western blot. This band was not detected with the oligonucleotide containing the mutated Sp factor binding site.
FIGURE 4.
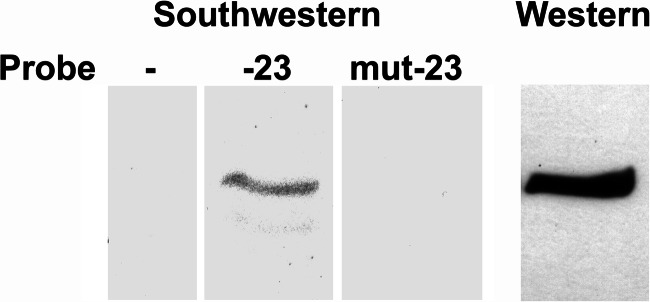
Southwestern blot analysis of KLF10 binding to the −23-bp Sp1 binding site. Proteins within nuclear protein extract from differentiated myotubes were resolved in a polyacrylamide gel and probed with double-stranded oligonucleotides containing the wild type (−23) or mutated (mut-23) sequence of the proximal (−23-bp) Sp1 binding site. A control lane that was not hybridized to an oligonucleotide probe is included. Also included is a Western blot of KLF10, detected with the FLAG epitope tag antibody. The protein band detected with the wild type oligonucleotide has the same relative mobility as KLF10.
Next we sought to determine whether KLF10 could not only bind to the −23-bp Sp factor binding site, but also affect Sp1 complex formation with this site. Myoblasts were transfected with pCMVKLF10 as before, and nuclear extracts from transfected and nontransfected myoblasts were prepared. These extracts were then incubated with oligonucleotides containing either the −23-bp wild type Sp factor binding site of the FGFR1 promoter or the consensus Sp1 binding site (GGCGGG). Protein-DNA complexes were resolved in polyacrylamide gels (Fig. 5A). Similar to our previous findings (18), the nontransfected control nuclear extracts formed a single prominent protein-DNA complex with the consensus Sp1 binding site oligonucleotide. This complex was diminished by incubation of the consensus Sp1 binding site oligonucleotide in nuclear extract derived from myoblasts transfected with pCMVKLF10. Similar results were obtained when nuclear extracts were incubated with the −23-bp Sp factor binding site of the FGFR1 promoter. Nuclear extracts from nontransfected control myoblasts formed protein-DNA complexes indicative of Sp1 binding (18). These Sp1-DNA complexes were diminished when nuclear extracts from myoblasts transfected with pCMVKLF10 were included. Rather, formation of a different protein-DNA complex was enhanced. It is unlikely that these results are due to reduced overall Sp1 protein levels because Western blot analysis did not detect noticeable differences in Sp1 protein content between myoblasts transfected with pCMVKLF10 versus nontransfected cells (Fig. 3). In total, these results indicate that KLF10 is able to bind to consensus Sp1 binding sites and to the −23-bp Sp factor binding site of the FGFR1 promoter. Furthermore, KLF10 binding to these sites reduced Sp1 complex formation at these sites.
FIGURE 5.

Electromobility shift analysis of protein complex formation with the proximal Sp1 binding site. A, nuclear protein extracts were prepared from nontransfected, control myoblasts (Con Extract) and from myoblasts transfected with the pCMVKLF10 expression construct (KLF10 Extract). These extracts were incubated with double-stranded oligonucleotides containing the wild type −23-bp Sp1 binding site sequence (−23 Sp Site) and the consensus Sp1 binding site (Sp1 Con Site). Arrows indicate protein-DNA complexes formed by incubation of control extract with either the −23-bp Sp Site or the Sp1 consensus site. These complexes were reduced when control extract was replaced by KLF10 extract. The arrowhead indicates a protein-DNA complex that was enhanced in the presence of KLF10 extract, particularly with the −23-bp Sp Site. B, nuclear extract from differentiated myotubes was incubated with oligonucleotides containing either the wild type (−23 Sp Site) or the mutated binding site (m23 Sp Site). Extract was also incubated with the KLF10 antibody (KLF10 Ab) prior to incubation with the wild type Sp binding site oligonucleotide. Arrows indicate protein-DNA complexes with lower relative mobility due to the addition of the KLF10 antibody.
Endogenous KLF10 Binds the Proximal Sp Factor Bind Site of the FGFR1 Promoter in Differentiated Myotubes
The interaction of endogenous KLF10 with the FGFR1 promoter in differentiated myotubes was assessed by electromobility shift assays (Fig. 5B). Nuclear extracts from differentiated myotube cultures were incubated with either the wild type −23-bp Sp factor binding site oligonucleotide or the oligonucleotide containing the mutated Sp binding site. PAGE resolution of the protein-DNA complexes revealed two prominent complexes. Neither protein-DNA complex formed with the mutated Sp binding site oligonucleotide. The protein-DNA complexes were incubated with the KLF10 antibody to determine whether KLF10 was a component of either protein-DNA complex. The addition of the KLF10 antibody resulted in supershifts of both protein-DNA complexes. The results of these assays indicate that endogenous KLF10 potentially binds to the proximal Sp factor binding site of the FGFR1 promoter in differentiated myotubes.
To further investigate the interaction of KLF10 with the FGFR1 promoter, chromatin immunoprecipitation studies were performed. Chromatin from myogenic cells cultured from 0 to 72 h was obtained and immunoprecipitated with either Sp1 or KLF10 antibodies. DNA was amplified using specific primers that flank the Sp binding sites within the proximal FGFR1 promoter (Fig. 6A). PCR product size was verified by amplification of DNA contained with chromatin prior to immunoprecipitation (input chromatin) as well as plasmid DNA containing the full-length FGFR1 promoter. During the 72-h culture period, proliferative myoblasts fused into differentiated myotubes. FGFR1 chromatin from newly isolated myoblasts (0 h of culture) was immunoprecipitated with the Sp1 antibody, and no detectable chromatin was immunoprecipitated with the KLF10 antibody. FGFR1 chromatin was immunoprecipitated at subsequent time points in cell culture (24–72 h) with the KLF10 antibody. Conversely, Sp1 antibody did not immunoprecipitate FGFR1 chromatin by 72 h of cell culture when myoblasts had differentiated into myotubes. These results indicate that KLF10 replaces Sp1 in the FGFR1 proximal promoter region as proliferative myoblasts transition to differentiated myotubes.
FIGURE 6.

Chromatin immunoprecipitation assays of KLF10 interaction with the FGFR1 promoter. A, chromatin was isolated from myogenic cells at 0, 24, 48, and 72 h of cell culture and immunoprecipitated with Sp1 (Sp1 Ab) or KLF10 antibodies (KLF10 Ab). FGFR1 DNA was amplified using FGFR1 promoter-specific forward and reverse primers that flanked all three proximal Sp1 binding sites. Lane 1, input chromatin prior to immunoprecipitation. Lane 2, immunoprecipitation without antibody (No Ab). Lane 3, immunoprecipitation with a nonspecific antibody (N.S. Ab). Lane 4, immunoprecipitation with the Sp1 antibody. Lane 5, immunoprecipitation with the KLF10 antibody. Lane 6, amplification of plasmid DNA containing the FGFR1 promoter sequence (Control). PCR products (input, light gray bars; Sp1 antibody precipitated, gray bars; KLF10 antibody precipitated, black bars) from 0 to 72 h were quantitated. Error bars indicate S.D. B, endogenous FGFR1 chromatin was prepared from nontransfected myotubes and immunoprecipitated using the KLF10 antibody. Chromatin was also prepared from myotubes transfected with pCMVKLF10, which expresses KLF10 coupled to the FLAG epitope tag. Myotubes were also transfected with the wild type FGFR1 promoter coupled to the luciferase reporter gene (3284FGFR1Luc) or the FGFR1 promoter-luciferase construct containing the mutation of the −23-bp Sp binding site (m23FGFR1Luc). Chromatin from transfected cells was immunoprecipitated with the FLAG epitope tag antibody. Endogenous FGFR1 DNA was amplified using FGFR1 promoter-specific forward and reverse primers that flanked all three proximal Sp1 binding sites. DNA containing either the wild type or the mutated FGFR1-luciferase sequence was amplified using the same FGFR1 promoter-specific forward primer and a luciferase gene-specific reverse primer. Lane 1, input chromatin prior to immunoprecipitation. Lane 2, immunoprecipitation without antibody. Lane 3, immunoprecipitation with a nonspecific antibody. Lane 4, immunoprecipitation with the KLF10 or FLAG antibody. Lane 5, amplification of plasmid DNA containing either the wild type or the mutated FGFR1 promoter sequence. PCR products (input, light gray bars; KLF10 antibody precipitated, black bars) from endogenous (Endo.), 3284FGFR1Luc (3284), and m23FGFR1Luc (m23) DNAs were quantitated. Error bars indicate S.D.
To further determine whether KLF10 interacted specifically with the most proximal −23-bp Sp binding site as suggested by the electromobility shift assays (Fig. 5) and endogenous chromatin immunoprecipitation, ED13 myoblasts were transfected with plasmid constructs containing the full-length wild type FGFR1 promoter (3284FGFR1Luc) and the mutation of the −23-bp Sp binding site within the FGFR1 promoter (m23FGFR1Luc). The resulting myotube cultures were maintained for a total of 10 days. Chromatin immunoprecipitations were repeated as above, except that immunoprecipitated DNA was amplified using an FGFR1 promoter-specific primer and a firefly luciferase-specific primer (see “Experimental Procedures”) to distinguish amplification of transfected DNA versus endogenous FGFR1 promoter sequence (Fig. 6B). Similar to the result of endogenous FGFR1 chromatin immunoprecipitation, exogenous wild type FGFR1 chromatin was immunoprecipitated by the KLF10 antibody and amplified using FGFR1 and luciferase gene-specific primers. However, PCR product containing the mutated −23-bp Sp binding site was not detected. These results indicate that KLF10 interacts with the most proximal Sp binding site located at −23 bp within the FGFR1 promoter. These results also indicate that the KLF10 interaction with the FGFR1 proximal promoter is specific to this site relative to the two other Sp binding sites located at −48 and −59 bp because the primers used for amplification flank all three Sp binding sites.
DISCUSSION
Skeletal myoblast proliferation and differentiation are tightly regulated by expression of the FGFR1 gene. The presence of FGFR1 on the cell surface of proliferating myoblasts allows for cellular response to the mitogenic signals initiated by FGF ligand binding to the receptor. Therefore, the molecular mechanisms that control FGFR1 gene expression are also likely to control myoblast proliferation and differentiation. Both transcriptional activators and repressors regulate the FGFR1 promoter. We have previously found that Sp1 activates the FGFR1 promoter (9). In total, five Sp1 binding sites have been identified in the chicken FGFR1 promoter. Three of these sites are located within the proximal (<100-bp) promoter, and two sites are located more than 1 kb upstream from the start of transcription (9, 18, 31). The proximal Sp1 binding site located at −23 bp was the most potent Sp1 activator site (18).
We hypothesized that other KLF family members may function as transcriptional repressors of FGFR1 promoter activity to direct cessation of myoblast proliferation. KLF10 is a C2H2 zinc finger KLF transcription factor that exhibits cell-specific antiproliferative effects (22–24). Indeed, increased KLF10 gene expression in myoblasts significantly reduced proliferation. Given the mechanistic link between myoblast proliferation and FGFR1 gene expression, the effect of KLF10 gene expression on FGFR1 promoter activity was then assessed. Increased KLF10 expression reduced FGFR1 promoter activity in myoblasts. Furthermore, Sp1-mediated activation of the FGFR1 promoter in Drosophila SL2 cells was repressed by KLF10. These results suggested that KLF10 interacts with one or more of the Sp factor binding sites, known to potentially bind other KLF-related transcription factors.
KLF10 was shown to interact with the most proximal (−23-bp) Sp binding site by Southwestern blot, electromobility shift, and chromatin immunoprecipitation assays. There are two aspects of this interaction that are particularly interesting relative to promoter function and the specificity of interaction. First, as mentioned above, the −23-bp proximal Sp binding site displays the strongest transcription-activating potential relative to the other two proximal Sp binding sites. Therefore, it can be considered that modification of the transcriptional regulators at this site will have relatively profound effects on promoter activity. This would be particularly true if a transcriptional repressor such as KLF10 replaced the transcriptional activator, Sp1. Our studies demonstrate that KLF10 does occupy this site and effectively represses FGFR1 promoter activity. Secondly, the chromatin immunoprecipitation results indicate the specificity of KLF10 for interaction with the −23-bp Sp site. We were not able to detect KLF10 interaction with the −48- or −59-bp Sp binding sites. The basis for this specificity remains unclear. However, some insight may be obtained from the binding site sequences. The Sp binding sites at −48 and −59 bp contain the sequence CTGCCC, whereas the −23-bp Sp site contains the sequence CCCCTC. KLF10 may display preference for this −23-bp Sp binding site sequence. In addition, flanking sequence around each binding site may contribute to transcription factor binding site specificity.
The results presented here allow for further development of the mechanism of transcriptional regulation of the FGFR1 promoter activity in proliferating avian myoblasts and differentiated myotubes (Fig. 7). Sp1 is present in myoblasts, binds to multiple sites within the FGFR1 promoter, and activates transcription. We have previously shown that the transcriptional complex composed of E2F4, p107, and Sp1 occupies the E2F binding site, also within the proximal FGFR1 promoter (29). Although E2F4 functions as a transcriptional repressor of FGFR1 promoter activity, its repressor activity is masked by its association with Sp1 via p107 (29). As myogenic differentiation and the formation of myotubes proceeds, a transition in the transcription factors associated with the FGFR1 promoter occurs. As Sp1 protein levels decline, Sp1 interaction with the FGFR1 promoter is correspondingly reduced. This has several effects. First, the lack of Sp1 interaction with the FGFR1 promoter at multiple Sp1 binding sites reduces Sp1-mediated promoter activation. Secondly, the decline of Sp1 protein is coincident with the exchange of p130 and p107 within the E2F4-based transcriptional repressor complex. It is not known whether the reduced Sp1 protein level directly facilitates this exchange. This latter complex is active in its role as a transcriptional repressor. Lastly, the unoccupied Sp1 binding site at the proximal −23-bp location is now accessible for interaction with the transcriptional repressor, KLF10.
FIGURE 7.

Model of transcriptional regulation of FGFR1 promoter activity in proliferating myoblasts and differentiated myotubes.
Repression of FGFR1 gene expression in differentiated muscle fibers is stable. Stable repression is perhaps achieved through KLF10 occupancy of the proximal promoter element and by KLF10-mediated chromatin modification. KLF10 represses transcription of Smad7 by interaction with JARID1B, leading to histone H3 lysine 4 demethylation (32). Additionally, the FOXP3 promoter is transcriptionally silenced by trimethylation of histone H3K27 in KLF10-deficient mice (33). Therefore, replacement of Sp1 by KLF10 in the proximal promoter of the FGFR1 gene may result in epigenetic modifications that add to the repression of FGFR1 promoter activity.
Left unresolved in these studies is the cause for the apparent lack of FGFR1 promoter repression in myoblasts that nonetheless express the KLF10 gene. A relatively straightforward hypothesis can be put forth in which Sp1 occupancy of the −23-bp Sp binding site precludes KLF10 interaction with the same site. Our results provide suggestive support for this hypothesis. Increasing KLF10 gene expression repressed Sp1-mediated activation of the FGFR1 promoter in SL2 cells (Fig. 3). Also, we have previously demonstrated that Sp1 protein levels decline during myogenic differentiation (18). This reduced Sp1 protein content results in lack of Sp1 occupancy of the −23-bp Sp binding site in myotubes, thereby allowing access to this site by KLF10. Alternatively, protein-protein interactions involving KLF10 with other regulators may inhibit its transcriptional repressor function. Similar to E2F4, KLF10 is present in both proliferating myoblasts and differentiated myotubes. However, both transcriptional regulators do not fully repress FGFR1 promoter activity in proliferating myoblasts. Endogenous KLF10, which is present in both myoblasts and myotubes, only occupies the FGFR1 promoter regulatory region as Sp1 occupancy of the promoter declines. Moreover, E2F4 does occupy the E2F binding site of the FGFR1 promoter in myoblasts yet does not exert its full repressor activity. Therefore, components of FGFR1 promoter repression appear to be readily present in proliferating myoblasts, but are temporarily disallowed their full repressive activity until myogenic differentiation.
Footnotes
- FGFR
- FGF receptor
- KLF
- Krüppel-like factor
- AraC
- β-arabinofuranoside hydrochloride
- ED
- embryonic day
- PCNA
- proliferating cell nuclear antigen.
REFERENCES
- 1. Stockdale F. E., Holtzer H. (1961) DNA synthesis and myogenesis. Exp. Cell Res. 24, 508–520 [DOI] [PubMed] [Google Scholar]
- 2. Gospodarowicz D., Weseman J., Moran J. (1975) Presence in brain of a mitogenic agent promoting proliferation of myoblasts in low density culture. Nature 256, 216–219 [DOI] [PubMed] [Google Scholar]
- 3. Kardami E., Spector D., Strohman R. C. (1985) Select muscle and nerve extracts contain an activity which stimulates myoblast proliferation and which is distinct from transferrin. Dev. Biol. 112, 353–358 [DOI] [PubMed] [Google Scholar]
- 4. Wanaka A., Milbrandt J., Johnson E. M. (1991) Expression of FGF receptor gene in rat development. Development 111, 455–468 [DOI] [PubMed] [Google Scholar]
- 5. Olwin B. B., Hauschka S. D. (1988) Cell surface fibroblast growth factor and epidermal growth factor receptors are permanently lost during skeletal muscle terminal differentiation in culture. J. Cell Biol. 107, 761–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Olwin B. B., Hauschka S. D. (1990) Fibroblast growth factor receptor levels decrease during chick embryogenesis. J. Cell Biol. 110, 503–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moore J. W., Dionne C., Jaye M., Swain J. L. (1991) The mRNAs encoding acidic FGF, basic FGF, and FGF receptor are coordinately downregulated during myogenic differentiation. Development 111, 741–748 [DOI] [PubMed] [Google Scholar]
- 8. Grothe C., Brand-Saberi B., Wilting J., Christ B. (1996) Fibroblast growth factor receptor 1 in skeletal and heart muscle cells: expression during early avian development and regulation after notochord transplantation. Dev. Dyn. 206, 310–317 [DOI] [PubMed] [Google Scholar]
- 9. Patel S. G., Funk P. E., DiMario J. X. (1999) Regulation of avian fibroblast growth factor receptor 1 (FGFR-1) gene expression during skeletal muscle differentiation. Gene 237, 265–276 [DOI] [PubMed] [Google Scholar]
- 10. Scata K. A., Bernard D. W., Fox J., Swain J. L. (1999) FGF receptor availability regulates skeletal myogenesis. Exp. Cell Res. 250, 10–21 [DOI] [PubMed] [Google Scholar]
- 11. Itoh N., Mima T., Mikawa T. (1996) Loss of fibroblast growth factor receptors is necessary for terminal differentiation of embryonic limb muscle. Development 122, 291–300 [DOI] [PubMed] [Google Scholar]
- 12. Flanagan-Steet H., Hannon K., McAvoy M. J., Hullinger R., Olwin B. B. (2000) Loss of FGF receptor 1 signaling reduces skeletal muscle mass and disrupts myofiber organization in the developing limb. Dev. Biol. 218, 21–37 [DOI] [PubMed] [Google Scholar]
- 13. Ji C., Chen Y., McCarthy T. L., Centrella M. (1999) Cloning the promoter for transforming growth factor-β type III receptor: basal and conditional expression in fetal rat osteoblasts. J. Biol. Chem. 274, 30487–30494 [DOI] [PubMed] [Google Scholar]
- 14. Perez-Castro A. V., Wilson J., Altherr M. R. (1997) Genomic organization of the human fibroblast growth factor receptor 3 (FGFR3) gene and comparative sequence analysis with the mouse Fgfr3 gene. Genomics 41, 10–16 [DOI] [PubMed] [Google Scholar]
- 15. DiMario J. X. (2002) Activation and repression of growth factor receptor gene transcription. Int. J. Mol. Med. 10, 65–71 [PubMed] [Google Scholar]
- 16. Pearson R., Fleetwood J., Eaton S., Crossley M., Bao S. (2008) Krüppel-like transcription factors: a functional family. Int. J. Biochem. Cell Biol. 40, 1996–2001 [DOI] [PubMed] [Google Scholar]
- 17. Dynan W. S., Tjian R. (1983) The promoter-specific transcription factor Sp1 binds to upstream sequences in the SV40 early promoter. Cell 35, 79–87 [DOI] [PubMed] [Google Scholar]
- 18. Parakati R., DiMario J. X. (2002) Sp1- and Sp3-mediated transcriptional regulation of the fibroblast growth factor receptor 1 gene in chicken skeletal muscle cells. J. Biol. Chem. 277, 9278–9285 [DOI] [PubMed] [Google Scholar]
- 19. Lomberk G., Urrutia R. (2005) The family feud: turning off Sp1 by Sp1-like KLF proteins. Biochem. J. 392, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Subramaniam M., Harris S. A., Oursler M. J., Rasmussen K., Riggs B. L., Spelsberg T. C. (1995) Identification of a novel TGF-β-regulated gene encoding a putative zinc finger protein in human osteoblasts. Nucleic Acids Res. 23, 4907–4912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Subramaniam M., Hawse J. R., Johnsen S. A., Spelsberg T. C. (2007) Role of TIEG1 in biological processes and disease states. J. Cell. Biochem. 102, 539–548 [DOI] [PubMed] [Google Scholar]
- 22. Tachibana I., Imoto M., Adjei P. N., Gores G. J., Subramaniam M., Spelsberg T. C., Urrutia R. (1997) Overexpression of the TGFβ-regulated zinc finger encoding gene, TIEG, induces apoptosis in pancreatic epithelial cells. J. Clin. Invest. 99, 2365–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cook T., Urrutia R. (2000) TIEG proteins join the Smads as TGF-β-regulated transcription factors that control pancreatic cell growth. Am. J. Physiol. Gastrointest. Liver Physiol. 278, G513–G521 [DOI] [PubMed] [Google Scholar]
- 24. Reinholz M. M., An M. W., Johnsen S. A., Subramaniam M., Suman V. J., Ingle J. N., Roche P. C., Spelsberg T. C. (2004) Differential gene expression of TGFβ inducible early gene (TIEG), Smad7, Smad2, and Bard1 in normal and malignant breast tissue. Breast Cancer Res. Treat. 86, 75–88 [DOI] [PubMed] [Google Scholar]
- 25. Subramaniam M., Gorny G., Johnsen S. A., Monroe D. G., Evans G. L., Fraser D. G., Rickard D. J., Rasmussen K., van Deursen J. M., Turner R. T., Oursler M. J., Spelsberg T. C. (2005) TEIG1 null mouse-derived osteoblasts are defective in mineralization and in support of osteoclast differentiation in vitro. Mol. Cell Biol. 25, 1191–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rajamannan N. M., Subramaniam M., Abraham T. P., Vasile V. C., Ackerman M. J., Monroe D. G., Chew T. L., Spelsberg T. C. (2007) TGFβ inducible early gene-1 (TIEG1) and cardiac hypertrophy: Discovery and characterization of a novel signaling pathway. J. Cell Biochem. 100, 315–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Neill M. C., Stockdale F. E. (1972) A kinetic analysis of myogenesis in vitro. J. Cell Biol. 52, 52–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DiMario J. X., Stockdale F. E. (1995) Differences in the developmental fate of cultured and noncultured myoblasts when transplanted into embryonic limbs. Exp. Cell Res. 216, 431–442 [DOI] [PubMed] [Google Scholar]
- 29. Parakati R., DiMario J. X. (2005) Dynamic transcriptional regulatory complexes, including E2F4, p107, p130, and Sp1, control fibroblast growth factor receptor 1 gene expression during myogenesis. J. Biol. Chem. 280, 21284–21294 [DOI] [PubMed] [Google Scholar]
- 30. Courey A. J., Tjian R. (1988) Analysis of Sp1 in vivo reveals multiple transcriptional domains, including a novel glutamine-rich activation motif. Cell 55, 887–898 [DOI] [PubMed] [Google Scholar]
- 31. Patel S. G., DiMario J. X. (2001) Two distal Sp1-binding cis-elements regulate fibroblast growth factor receptor 1 (FGFR1) gene expression in myoblasts. Gene 270, 171–180 [DOI] [PubMed] [Google Scholar]
- 32. Kim J., Shin S., Subramaniam M., Bruinsma E., Kim T. D., Hawse J. R., Spelsberg T. C., Janknecht R. (2010) Histone demethylase JARID1B/KDM5B is a corepressor of TIEG1/KLF10. Biochem. Biophys. Res. Comm. 401, 412–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xiong Y., Khanna S., Grzenda A. L., Sarmento O. F., Svingen P. A., Lomberk G. A., Urrutia R. A., Faubion W. A. (2012) Polycomb antagonizes p300/CREB-binding protein-associated factor to silence FOXP3 in a Kruppel-like factor-dependent manner. J. Biol. Chem. 287, 34372–34385 [DOI] [PMC free article] [PubMed] [Google Scholar]