

Background: Sunflower trypsin inhibitor-1 (SFTI-1) and Momordica cochinchinensis trypsin inhibitor-II (MCoTI-II) are potent protease inhibitors comprising a cyclic backbone.
Results: Elucidation of structure-activity relationships for SFTI-1 and MCoTI-II was used to design inhibitors with enhanced inhibitory activity.
Conclusion: An analog of MCoTI-II is one of the most potent inhibitors of matriptase.
Significance: These results provide a solid basis for the design of selective peptide inhibitors of matriptase with therapeutic potential.
Keywords: Molecular Modeling, Nuclear Magnetic Resonance, Peptide Conformation, Peptides, Protease Inhibitor
Abstract
The type II transmembrane serine protease matriptase is a key activator of multiple signaling pathways associated with cell proliferation and modification of the extracellular matrix. Deregulated matriptase activity correlates with a number of diseases, including cancer and hence highly selective matriptase inhibitors may have therapeutic potential. The plant-derived cyclic peptide, sunflower trypsin inhibitor-1 (SFTI-1), is a promising drug scaffold with potent matriptase inhibitory activity. In the current study we have analyzed the structure-activity relationships of SFTI-1 and Momordica cochinchinensis trypsin inhibitor-II (MCoTI-II), a structurally divergent trypsin inhibitor from Momordica cochinchinensis that also contains a cyclic backbone. We show that MCoTI-II is a significantly more potent matriptase inhibitor than SFTI-1 and that all alanine mutants of both peptides, generated using positional scanning mutagenesis, have decreased trypsin affinity, whereas several mutations either maintain or result in enhanced matriptase inhibitory activity. These intriguing results were used to design one of the most potent matriptase inhibitors known to date with a 290 pm equilibrium dissociation constant, and provide the first indication on how to modulate affinity for matriptase over trypsin in cyclic peptides. This information might be useful for the design of more selective and therapeutically relevant inhibitors of matriptase.
Introduction
Serine proteases are one of the largest known family of proteases (1), and are involved in a range of cellular processes such as apoptosis, inflammation, blood coagulation, and extracellular matrix remodeling (2). Several mechanisms are involved in controlling the activity of serine proteases, including synthesis as inactive zymogens and production of specific protease inhibitors. The deregulation of these endogenous controls has dramatic consequences, and can lead to autoimmune and metabolic diseases and to an increased susceptibility to infections and cancer (3).
Matriptase is a type II transmembrane serine protease that is expressed strongly in the human epithelia (4). The expression of this protease, both at the RNA and protein level increases significantly during the progression of prostate cancer (5). The oncogenic potential of matriptase has been demonstrated in transgenic mice where overexpression of matriptase caused spontaneous squamous cell carcinoma. However, overexpression together with a matriptase inhibitor counteracted the oncogenic effects (6), indicating that inhibition of matriptase has significant potential as a therapeutic strategy. Administration of inhibitors in a pancreatic tumor model in mice led to the inhibition of matriptase in vivo for at least 24 h (7), and in a prostate tumor mouse model inhibitors reduced primary tumor growth by 40% as well as reducing the prevalence of metastases (8). Genetic reduction of matriptase in mice resulted in reduced tumor growth, invasiveness, and migration in vitro (9) and correlated with in vivo studies where matriptase-deficient PC3 and DU-145 cells exhibited reduced growth and development compared with control cells when explanted into nude mice (10). Overall, these studies highlight the potential of using matriptase inhibitors as a treatment approach to halt growth and spread of cancer cells.
A major challenge in the design of protease inhibitors is selectivity (11). Broad range inhibitors have caused detrimental effects in clinical trials as a result of interference with signaling pathways and processes that were not foreseen (12). Therefore, when designing inhibitors for pharmaceutical purposes, it is essential that they are selective for the desired target, whereas also having high affinity. Several studies have focused on small molecules for the design of matriptase inhibitors, and have resulted in potent inhibitors with selectivity over other serine proteases such as thrombin (8). However, limited information is available regarding selectivity relative to other closely related serine proteases, particularly the prototypic trypsin.
The active sites of matriptase and trypsin are very similar in structure and both enzymes belong to the S1 family and subfamily A according to the MEROPS database (13). However, there are some important differences, as shown in Fig. 1, which could potentially be exploited for the design of novel inhibitors. The active site of all serine proteases is surrounded by eight loops (1) that will be referred to as I to VIII in this article. Loop II of matriptase has 10 more residues than that of trypsin, resulting in an additional bulge that constitutes the most obvious topological difference between the two protease active sites (Fig. 1). Loops I, III, and V also display different sequences between the two proteases, resulting in slightly different conformations. In contrast, loops IV, VI, and VIII, which cluster at one end of the active site, have similar conformations in matriptase and trypsin. The active site of matriptase is much more negatively charged than that of trypsin, mostly because of two additional aspartate residues in loop IV and three aspartate residues in the extended loop II. The negative charges borne by matriptase loop II are partly compensated by two positively charged arginine residues located at the tip of the loop.
FIGURE 1.

Comparison of trypsin and matriptase active sites. The crystallographic structures of SFTI-1 in complex with trypsin (PDB code 1sfi) and in complex with matriptase (PDB code 3p8f) are represented. The protein backbone of SFTI-1 is in cyan and the functional Lys is displayed in stick representation. The eight loops that surround the active sites are named loops I to VIII. The solvent accessible surfaces of the proteases are colored according to the Poisson-Boltzmann electrostatic potential they generate, as computed by the APBS software (49), with a scale ranging from −5 kT/e (red) to +5 kT/e (blue). The charged positions in each loop are highlighted. The numberings of trypsin and matriptase positions are according to the sequence of bovine trypsin (UniProt P00760) and human matriptase SP1 (Q9Y5Y6), respectively. The figure was generated using PyMol.
Naturally occurring peptidic inhibitors are a promising starting point for the design of novel matriptase inhibitors as they can provide a relatively large surface area to enhance the potential of designing selective analogs. The use of naturally occurring amino acids also leaves open the possibility of using plants as “factories” for producing the modified inhibitors via a low cost route (14). Sunflower trypsin inhibitor-1 (SFTI-1)4 is a 14-residue sunflower peptide that is a potent inhibitor of both trypsin and matriptase, with an inhibition constant against matriptase of 0.92 nm (15). However, SFTI-1 is also active against other serine proteases within the trypsin clan, which limits its therapeutic application. Despite this limitation, the size, versatility, and stability of the SFTI-I backbone makes it an appealing scaffold for the design of drugs (16). Swedberg et al. (17), for example, enhanced both the affinity and specificity of the SFTI-1 backbone against kallikrein 4 (KLK4) through modifications of the inhibitor. They used positional scanning of a synthetic combinatorial library to study protease specificity and generated a mutant of SFTI-1 with an inhibition constant of 3.59 nm against KLK4 compared with no inhibition by native SFTI-1 (17). Although this mutant was not tested in vivo it retained high inhibitory potential against KLK4 after days of incubation in serum, highlighting the advantages of the SFTI-1 scaffold in the design of drugs.
Several other researchers have also explored the structure-activity relationships of SFTI-1 and shown the importance of particular residues for a range of proteases. The introduction of naturally and non-naturally occurring residues has been explored, as have the roles of the disulfide bonds in activity (18–22). Enhancements in activity and selectivity have been obtained, highlighting the potential of this cyclic scaffold. However, further study is required to determine whether significant selectivity between trypsin and matriptase is achievable.
The stability and versatility of cyclic peptides is not limited to SFTI-1. Other cyclic peptides, such as members of the cyclotide family, have functions as diverse as insecticidal activity (23), HIV inhibition (24), and hemolytic activity (25). Their extraordinary stability to pH, heat, and biological fluids also makes them ideal scaffolds for pharmaceutical design (14, 17). In the current study we have discovered that a member of the trypsin inhibitory subfamily of cyclotides, Momordica cochinchinensis trypsin inhibitor-II (MCoTI-II) (26), a 34-residue cyclic peptide, is also a potent inhibitor of matriptase. Furthermore, we have used alanine scanning and point mutations to design inhibitors, based on SFTI-1 and MCoTI-II, which have enhanced potency and modified selectivity against matriptase. The sequences and structures of SFTI-1 and MCoTI-II are given in Fig. 2 and highlight the sequence and structural diversity of these cyclic peptidic trypsin inhibitors.
FIGURE 2.
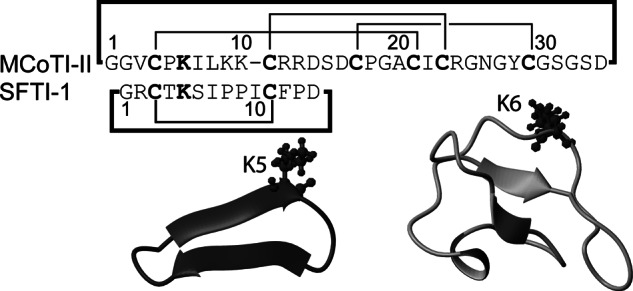
Comparison of the sequences and structures of SFTI-1 and MCoTI-II. The structures were determined based on NMR spectroscopy (PDB codes 1jbl and 1ib9, respectively) and drawn using MolMol.
EXPERIMENTAL PROCEDURES
Peptide Synthesis and Purification
Peptides were synthesized on a 0.5-mmol scale using manual solid-phase peptide synthesis with Boc chemistry. A phenylacetamidomethyl-Gly-Boc resin was used with an S-tritylmercaptopropionic acid linker to facilitate cyclization, as described previously (27, 28). Crude peptides were purified using a series of Phenomenex C18 columns on RP-HPLC. Gradients of 1% min−1 of 0–80% solvent B (90% acetonitrile in 0.045% TFA in H2O) and solvent A (aqueous 0.05% TFA in H2O) were employed and the eluant was monitored at 215 and 280 nm. The purity of the peptides was examined by analytical RP-HPLC on a Phenomenex Jupiter 5μ C18 300 Å 150 × 2.0-mm column and masses were determined by electrospray mass spectrometry. Grafted and native SFTI-1 peptides were folded in solution at 0.1 mg/ml using a range of buffer conditions. Synthetic native SFTI-1 peptide was cyclized and oxidized in a two-step process. Cyclization was achieved in the presence of 0.1 m tris(2-carboxyethyl)phosphine incubated for 24 h and the cyclic product then purified by RP-HPLC. The cyclic reduced peptide was oxidized in 0.1 m ammonium bicarbonate (pH 8.0). By contrast, cyclization and oxidation of MCoTI-II grafted peptides was done in a single step reaction with 0.1 m ammonium bicarbonate (pH 8.5) for 12 h. SFTI-1 grafted peptides were oxidized in 0.1 m ammonium bicarbonate (pH 8.5).
NMR Analysis
Peptides were dissolved in 90% H2O, 10% D2O (v/v) (1 mm). D2O (99.9%) was obtained from Cambridge Isotope Laboratories, Woburn, MA, for 1H NMR measurements. Spectra were recorded at 290–298 K on Bruker Avance 500 and 600 MHz spectrometers. Two-dimensional spectra included TOCSY, NOESY, and COSY. NOESY mixing times of 200–300 ms were used. Spectra were analyzed using SPARKY (29). The sequential assignment procedure pioneered by Wüthrich (30) was used to sequence specifically assign the amino acids, using TOCSY and NOESY spectra.
Enzyme Kinetics
Titration
The concentrations of active inhibitors were determined by titration against burst-titrated bovine trypsin assuming a one to one interaction between the enzyme and the inhibitor. Reactions were conducted in 96-well plates using buffer (50 mm Tris-HCl, 150 mm NaCl, 0.01% Triton X-100, 0.01% sodium azide, pH 7.6) and serial dilutions of each inhibitor with trypsin (25 nm). After preincubation at 37 °C the fluorogenic substrate Benzoyl-l-arginine-4-methylcoumaryl-7-amide (75 μm) was added. Hydrolysis of the substrate was then measured over 10 min with λem and λex of 360 and 465 nm, respectively, on a HTS 7000 Bio assay reader (PerkinElmer Life Sciences). The concentrations of inhibitors with a large Ki,app could not be determined by titration.
Ki Determination
Inhibition constants of the inhibitors against matriptase and trypsin were calculated by preincubating the recombinant human matriptase catalytic domain (2 pm, R&D systems, Inc.) and bovine trypsin (10 pm), respectively, with serial dilutions of the inhibitors in buffer (50 mm Tris-HCl, 150 mm NaCl, 0.01% Triton X-100, 0.01% sodium azide, pH 8.0). After preincubation of 60 min the substrate Boc-QAR-AMC (5 μm) or tos-GPR-AMC (5 μm), respectively, was added to the reaction mixture and hydrolysis was quantified as described above. Additional experiments were performed with preincubation times between 60 and 180 min to verify that each inhibitor was in equilibrium with the protease. The inhibition curves generated were fitted to Morrison's equation (31), to obtain the inhibition constant for each of the tested peptides. The results presented are mean ± S.E. of ≥4 independent experiments.
Three-dimensional Structure Calculations
Preliminary three-dimensional structures were calculated using automated nuclear Overhauser effect assignment within CYANA (32). A final set of 100 structures was calculated and the 20 lowest energy structures were selected for further analysis. Structures were analyzed using MolProbity, and MolMol (33) and PyMol (The PyMOL Molecular Graphics System, version 1.5.0.4, Schrödinger, LLC) were used to display the structural ensembles and surfaces of the peptides, respectively.
Molecular Modeling
The three-dimensional structures of the complexes between SFTI-1 and trypsin (PDB identifier 1sfi), between SFTI and the protease domain of matriptase (PDB identifier 3p8f), and between MCoTI-II and trypsin5 were determined experimentally using x-ray crystallography independently by different research groups (16, 34). The three-dimensional structure of MCoTI-II in complex with the protease domain of matriptase was built in this study by homology using Modeler9v11 (35). The three previously mentioned x-ray structures were used as structural templates to generate 100 models of the matriptase·MCoTI-II complex. The model with the lowest DOPE score (36) was selected and was then refined using molecular dynamic (MD) simulations. MD simulations were performed using the program PMEMD from the AMBER 12 package with the ff12SB force field (37). The protease domain of the matriptase·MCoTI-II complex (274 residues) was solvated in a truncated octahedral periodic box with ∼4900 TIP3P water molecules. Three sodium ions were added to neutralize the system. The system was then equilibrated using a similar strategy to Cerutti et al. (38). A steepest descent minimization of 2000 steps was first carried out with the solute restrained to its positions and then a further 2000 steps of steepest descent minimization was performed with the solvent restrained to its position. MD simulations were then performed with the solute atoms restrained to their position by a harmonic potential whose spring constant was progressively decreased from 16 to 1 kcal/mol Å2 over 150 ps in the NVT ensemble, before it was progressively decreased from 1 to 0 over 300 ps of standard NPT simulations (300 K, 1 atm). An MD simulation of 300 ps with a 1-fs time step was performed followed by a production run of 20 ns with a 2-fs time step. All bonds involving hydrogen atoms were constrained with the SHAKE algorithm (39). Long-range electrostatic interactions were simulated with the particle-mesh Ewald method (40). Similarly prepared 20-ns MD simulations starting from the crystallographic structures of SFTI-1/matriptase (3p8f), SFTI-1/trypsin (1sfi), and MCoTI-II/trypsin were also performed. These simulations gave information on the dynamics of the interactions at the interface. All simulations were performed in triplicates using different random seeds.
The three-dimensional structures of complexes involving mutants of SFTI-1 and MCoTI-II were modeled using the mutation procedure implemented in Modeler9v11. Each homology model was subjected to 4000 steps of minimization using PMEMD. The dynamics of complexes involving a selection of mutants, including [I10R]SFTI-1 [I10D]SFTI-1, [I10K]SFTI-1, [I10R]SFTI-1, [I7A+I10R]SFTI-1, [V3R]MCoTI-II, [I7A]MCoTI-II, and [V3R + I7A]MCoTI-II, were studied using 5-ns MD.
RESULTS
Synthesis and Characterization of Peptides
Variants of SFTI-1 and MCoTI-II were synthesized to identify residues that are essential for inhibition of matriptase or trypsin. The mutants were synthesized using Boc chemistry and thioester-mediated backbone cyclization and included alanine substitutions as well as several other single or double amino acid changes. In general, the SFTI-1 mutants were cyclized and oxidized in separate steps as cyclization occurs more efficiently in the presence of a reducing agent. By contrast, the MCoTI-II analogs could be cyclized and oxidized in a single step. It is possible that the presence of three disulfide bonds in MCoTI-II compared with one in SFTI-1 facilitates the cyclization process via a thia-zip mechanism (41). All peptides were purified by RP-HPLC and analyzed by mass spectrometry. The structures of the peptides were analyzed using NMR chemical shift analysis (Fig. 3A) to confirm the native fold was present, as shown for [R2A]SFTI-1 and [I10R]-SFTI-1 in Fig. 3B. The similarity in αH chemical shifts with the native peptides indicates that the overall fold was maintained despite the mutations.
FIGURE 3.

A, comparison of the secondary shifts of selected SFTI-1 and MCoTI-II mutants. The shifts were generated by subtracting the αH shifts from random coil shifts (50). B, alignment of NMR structures of SFTI-1 backbone with the SFTI-1 variants I10R (left) and R2A (right). The parent peptide SFTI-1 is displayed in pale cyan. The variants I10R and R02A are displayed in green and purple respectively.
Enzyme Kinetics
Inhibition constants for the SFTI-1 and MCoTI-II mutants against matriptase and trypsin are given in Table 1, and graphically presented in Fig. 4. Several mutations had selective influences on the inhibitory activity as outlined below for the SFTI-1 and MCoTI-II mutants.
TABLE 1.
Equilibrium dissociation constant Ki for the inhibition of trypsin and matriptase by SFTI-1, MCoTI-II, and mutants (mean ± S.E., n ≥4)
| Inhibitor name |
Ki |
|
|---|---|---|
| Trypsin | Matriptase | |
| nm | ||
| SFTI-1 native | 0.0017 ± 0.00026 | 200 ± 22 |
| SFTI-1 alanine mutants | ||
| G1A | 0.0048 ± 0.00032 | 190 ± 26 |
| R2A | 0.16 ± 0.016 | ∼10,000 |
| T4A | 0.16 ± 0.016 | 1,500 ± 170 |
| K5A | >1000 | >10,000 |
| S6A | 0.15 ± 0.018 | 1600 ± 390 |
| I7A | 0.89 ± 0.21 | 84 ± 15 |
| P8A | 0.035 ± 0.0044 | 27 ± 1.4 |
| P9A | 0.0017 ± 0.0001 | 370 ± 58 |
| I10A | 0.086 ± 0.016 | 73 ± 11 |
| F12A | 0.21 ± 0.031 | 320 ± 35 |
| P13A | 0.0037 ± 0.0003 | 240 ± 37 |
| D14A | 0.01 ± 0.0014 | 1,500 ± 150 |
| SFTI-1 additional mutants | ||
| R2K | 0.002 ± 0.00003 | 1,200 ± 160 |
| K5R | 0.0027 ± 0.00078 | 310 ± 52 |
| I7R | 0.01 ± 0.00095 | 4,500 ± 500 |
| I10D | 0.051 ± 0.006 | ∼10,000 |
| I10G | 0.081 ± 0.011 | 3,700 ± 490 |
| I10R | 0.0038 ± 0.00062 | 6.4 ± 1.3 |
| I10K | 0.0057 ± 0.0015 | 40 ± 5.8 |
| I7A + I10R | 1.2 ± 0.25 | 51 ± 4.9 |
| MCoTI-II native | 0.0023 ± 0.0007 | 2.8 ± 0.51 |
| MCoTI-II alanine mutants | ||
| G02A | 0.38 ± 0.043 | 180 ± 17 |
| V03A | 0.15 ± 0.028 | 2.3 ± 0.12 |
| P05A | 0.056 ± 0.0076 | 39 ± 5.4 |
| K06A | >1,000 | >10,000 |
| I07A | 1.2 ± 0.22 | 9.8 ± 1.9 |
| L08A | 0.13 ± 0.011 | 12 ± 1.9 |
| K09A | 0.0034 ± 0.0048 | 76 ± 14 |
| K10A | 0.051 ± 0.0095 | 4.1 ± 1 |
| N26A | 0.008 ± 0.0027 | 12 ± 0.41 |
| Y28A | 0.15 ± 0.021 | 11 ± 1.3 |
| G30A | 0.0032 ± 0.00036 | 3.7 ± 0.033 |
| S31A | 0.069 ± 0.0079 | 98 ± 10 |
| MCoTI-II additional mutants | ||
| V03R | 0.01 ± 0.0025 | 0.29 ± 0.054 |
| K6R | 0.13 ± 0.0055 | 39 ± 4.4 |
| I7R | 5.2 ± 0.62 | 110 ± 8.5 |
| V3R + I7A | 2.8 ± 0.23 | 3.5 ± 0.32 |
FIGURE 4.

Comparison of the differences in inhibitory activity of the SFTI-1 (A) and MCoTI-II (B) mutants relative to the native peptides. The side chains are highlighted on the structures shown on the right of the diagram. The disulfide bonds are shown as yellow sticks. The surface diagrams were generated using PyMol.
SFTI-1 Mutants
The trypsin inhibitory activity for the SFTI-1 alanine mutants was consistent with our previous results (27). Several of the alanine mutants displayed decreased potency compared with the native peptide for both trypsin and matriptase. For example, the alanine substitutions at positions 2, 4, 5, 6, and 14 led to a 7.5–50-fold increase in the Ki against matriptase. Although the R2A mutant had significant loss of inhibition against matriptase, it was still a potent inhibitor of trypsin (Ki 160 pm compared with 1.7 pm for the native molecule). Substitution of the active site residue, Lys-5, abolished inhibitory activity for both enzymes. Additional substitutions were tested to assess the charge requirements at positions 2 and 5. Substitution of Arg-2 with Lys had no effect on trypsin inhibitory activity although the Ki for matriptase increased 6-fold. Substitution of Lys-5 with Arg had little effect on trypsin or matriptase inhibition.
[I7A]SFTI-1 exhibited a 700-fold decrease in potency relative to the native peptide against trypsin but was 2-fold more potent against matriptase than the native peptide. Similarly, [P8A]SFTI-1 had a 20-fold decreased trypsin activity but a 7-fold increase in matriptase activity, and [I10A]SFTI-1 a ∼100-fold decrease in potency against trypsin but a 2-fold increase in activity against matriptase. The contrasting effect of these mutations against trypsin and matriptase indicated that these positions are potential sites for exploring selectivity. Additional point mutations were made and the selectivity for matriptase was modulated in [I10R]SFTI-1, which had a 30-fold increase in activity against matriptase and a decrease in trypsin inhibitory activity of 2-fold. Mutating position 10 to an aspartic acid or glycine significantly decreased activity against both matriptase and trypsin. A combination of the I7A and I10R mutations resulted in a peptide with a 700-fold decrease in trypsin inhibitory activity and a 4-fold increase in matriptase activity.
MCoTI-II Mutants
Analysis of the matriptase inhibitory activity of MCoTI-II revealed it is a more potent inhibitor (Ki = 2.8 nm) than SFTI-1 (Ki = 200 nm) (Table 1). In contrast, both inhibitors had similar Ki values against trypsin, consistent with previous studies (27, 42). Alanine mutants in the active site loop of MCoTI-II (loop 1) were synthesized to determine the role each residue plays in enzyme inhibitory activity. Additional alanine substitutions were generated for residues in loops 5 and 6 that were predicted to be in close proximity to the enzyme based on modeling studies using the crystal structure of MCoTI-II in complex with trypsin. Sites from loops 5 and 6 that were mutated included residues 2, 3, 26, 28, 30, and 31 (Table 1).
In general, most of the alanine substitutions led to decreased inhibitory activity, but the influence of the substitutions varied for trypsin and matriptase. As expected, substitution of the trypsin active site residue (Lys-6) with an alanine abolished inhibitory activity for both trypsin and matriptase. Preference for lysine in this position was confirmed with the K6R substitution, which decreased the inhibitory activity against both trypsin and matriptase relative to the native peptide. Interestingly, the mutation of Val-3 in MCoTI-II to an alanine maintained potency against matriptase and decreased potency against trypsin. Based on the selective influence of this mutation at the P4 position of MCoTI-II and the importance of Arg-2 in the P4 position in SFTI-1 for potency against matriptase, a MCoTI-II-V3R mutant was synthesized. Analysis of the kinetics of this mutant indicated that it did improve inhibitory activity against matriptase and resulted in one of the most potent matriptase inhibitors known, with a Ki of 290 pm.
The mutation I7A of MCoTI-II significantly decreased the trypsin inhibitory potency but had a lesser impact on matriptase potency. A combination of the I7A mutation with the V3R mutation resulted in a peptide with a 2-fold decrease in matriptase inhibitory activity relative to the native peptide but a >1000-fold decrease in trypsin inhibitory activity.
Hybrids between SFTI-1 and MCoTI-II
Although MCo-TI-II is a more potent matriptase inhibitor than SFTI-1, SFTI-1 is smaller, which is potentially advantageous for pharmaceutical purposes as it is cheaper to synthesize and easier to modify. A series of grafted peptides was thus generated to investigate the relative importance of the SFTI-1 and MCoTI-II scaffolds and the respective binding loops for the inhibitory activity. Two grafted peptides were generated for the SFTI-1 loop in the MCoTI-II scaffold to investigate the role of the binding loop and loop 5 in interacting with the enzymes. The latter peptide also included the substitution of Lys-6 into alanine to prevent interference of MCoTI-II native loop in our studies. These peptides, MCoTI-II-S1 and MCoTI-II-S5, displayed poor inhibition of both enzymes. The peptide SFMC, which had the MCoTI-II binding loop grafted onto the SFTI-1 scaffold, was as efficient as native SFTI-1 in inhibition of both trypsin and matriptase, as shown in Table 2. However, the SFMC mutant was not as active as native MCoTI-II against matriptase and the additional peptide/enzyme interactions that occur with the larger MCoTI-II scaffold may be important for enhanced enzyme inhibitory activity.
TABLE 2.
Equilibrium dissociation constant Ki for the inhibition of trypsin and matriptase by SFTI-1 and MCoTI-II hybrids
| Peptide | Sequence | Inhibition constant |
|
|---|---|---|---|
| Trypsin | Matriptase | ||
| nm | |||
| SFTI-1 | GRCTKSIPPICFPD | 0.0017 ± 00026 | 200 ± 22 |
| MCoTI-II | GGVCPKILKKCRRDSDCPGACICRGNGY--CGSGSD | 0.0023 ± 0.0007 | 2.8 ± 0.51 |
| SFMC | GRCPKILKKCFPD | 0.0023 ± 0.00018 | 290 ± 34 |
| MCoTI-II-S1 | GGVCTKSIPPCRRDSDCPGACICRGNGY--CGSGSD | 450 ± 27 | 3,500 ± 660 |
| MCoTI-II-S5 | GGVCPAILKKCRRDSDCPGACICTKSIPPICGSGSD | 68 ± 4.5 | >10,000 |
Structure Determination Using Nuclear Magnetic Resonance Spectroscopy
NMR structures were determined for selected SFTI-1 variants to enhance the understanding of the structure-activity relationships and for use in molecular modeling of inhibitor·enzyme complexes. [I10R]SFTI-1 was chosen because it is the most potent SFTI-1 mutant against matriptase, and R2A was chosen because it was one of the least active analogs against matriptase, aside from the K5A mutant. Structures were determined using torsion angle dynamics in the program CYANA and the 20 lowest energy structures chosen to represent the fold. Energetic and geometric statistics are given in Table 3. The structures were analyzed using PROMOTIF and revealed that the major element of the secondary structure in both R2A and I10R is a β-hairpin with the strands involving residues 2–4 and 10–12. Comparison of the structures with the native peptide confirmed the overall fold was maintained and therefore the observed effects of the mutations on the inhibition constants are likely to arise from interactions between the side chains of the substituted amino acids and the proteases (Fig. 3B). However, several hydrogen bonds were missing from the mutant structures compared with the native peptide, suggesting that the overall fold had been destabilized, which might also influence the inhibitory activity.
TABLE 3.
Structural statistics for SFTI-1 mutant structures
| I10R | R2A | |
|---|---|---|
| Experimental restraints | ||
| Interproton distances | 95 | 88 |
| Intraresidue | 28 | 28 |
| Sequential | 33 | 36 |
| Medium range, i -- j <5 | 9 | 3 |
| Long range, i -- j ≥5 | 25 | 21 |
| Disulfide bond restraints | 2 | 2 |
| Dihedral bond (φ, ψ, χ1) restraints | 6 | 8 |
| Root mean square deviation from mean coordinate structure (Å) | ||
| Backbone atoms (residues 1–14) | 0.19 ± 0.11 | 0.20 ± 0.09 |
| All heavy atoms (residues 1–14) | 1.05 ± 0.19 | 0.70 ± 0.09 |
| Stereochemical quality | ||
| Residues in most favored Ramachandran region (%) | 87.5 | 85.8 |
| Ramachandran outliers (%) | 12.5 | 14.2 |
| Overall MolProbity scorea | 1.76 ± 0.32 | 2.15 ± 0.47 |
a Determined using MolProbity.
Molecular Modeling
The three-dimensional structures of complexes involving matriptase or trypsin were used to propose explanations for the activity of SFTI-1 and MCoTI-II variants. The structures of complexes between SFTI-1 and trypsin, SFTI-1 and matriptase, and MCoTI-II and trypsin have been determined experimentally by x-ray crystallography (16, 34) and were used here to model the structure of the matriptase·MCoTI-II complex by homology with refinement using 20-ns MD simulations (Fig. 5A). The three other complexes involving wild-type SFTI-1 or MCoTI-II were also simulated for 20 ns by MD (Fig. 5A), and these simulations were used to compare the dynamics of the molecular interactions at the interface. All simulations quickly converged, as indicated by the stabilization of the Cα atoms root mean square deviations from the initial homology model (matriptase·MCoTI-II complex) or from the crystal structures (supplemental Fig. S1). Structural models of the complexes between the peptide variants and the proteases were modeled by comparison based on the wild-type models and refined by 5-ns MD. These simulations allow local conformational change to occur and also provide information on the structural dynamics of the complexes.
FIGURE 5.
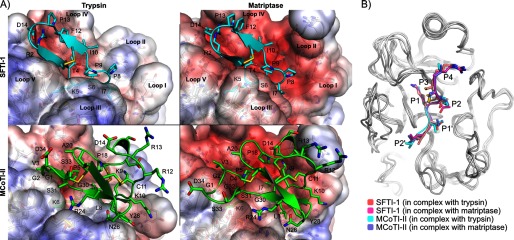
A, binding modes of SFTI-1 (cyan) and MCoTI-II (green) in the active sites of trypsin (left) and matriptase (right). The three major differences between the active sites of trypsin and matriptase are the longest and more charged loop II of matriptase, the different conformation of loop III and the more negatively charged loop IV of matriptase. The structures in this figure are the final conformations of 20-ns molecular dynamics simulations carried out for each complex. SFTI-1 and MCoTI-II adopted similar binding modes in the two protease active sites, but MCoTI-II displayed more conformational variability than SFTI-1. The solvent accessible surfaces of the proteases are colored according to the Poisson-Boltzmann electrostatic potential they generate, as computed by the APBS software (49), with a scale ranging from −5 kT/e (red) to +5 kT/e (blue). B, comparison of the binding modes of the inhibitory loops of SFTI-1 and MCoTI-II when they are in complex with matriptase and trypsin. The backbones of the proteases are shown in white using schematic representations, and the inhibitory loops are in schematic and stick representations. The proteases were superimposed using PyMol. The remaining parts of the SFTI-1 and MCoTI-II beside the inhibitory loops are not shown.
Position 10 of SFTI-1 faces loop II of the proteases (Fig. 5A), which is 10 residues longer in matriptase than trypsin. Several SFTI-1 variants at position 10 were prepared and investigated for their ability to discriminate between the two proteases. The I10A substitution caused small shifts of the positions of SFTI-1 side chains Arg-2, Phe-12, and Asp-14 in both protease complexes, decreasing the distance by ∼2 Å between the positively charged guanidinium group from Arg-2 of SFTI-1 and the negatively charged carboxylic group of matriptase Asp-709 compared with wild-type SFTI-1 (Fig. 6 and supplemental Fig. S2). As a result the electrostatic interactions were improved and at the same time the buried surface area remained globally similar (Table 4). The introduction of a negatively charged residue at position 10 of SFTI-1 (I10D) dramatically decreased the potency for matriptase, and in the corresponding model the substituted aspartate at position 10 had moved away from loops II and IV of matriptase relative to the position of isoleucine 10 in wild-type SFTI-1 (Fig. 6). This probably arose from charge repulsions between Asp-10 of SFTI-1 and Asp-660, Asp-661, and Asp-705. Indeed the distance between the Cα of matriptase Asp-705 and position 10 in SFTI-1 wild-type and the I10D mutant had increased slightly by ∼1 Å (Fig. 6 and supplemental Fig. S2). Furthermore the buried surface area in the mutant had decreased on average by ∼50 Å2 (Table 4). By contrast, the substitution of Ile-10 by the positively charged residues, arginine or lysine, resulted in a more potent inhibitor of matriptase than wild-type SFTI-1, possibly because of positive electrostatic interactions with loop II (Fig. 6). The two variants [I10R]SFTI-1 and [I10K]SFTI-1 exhibited a small decrease in activity for trypsin. The models suggest that [I10R]SFTI-1 is a better inhibitor of matriptase than [I10K]SFTI-1 because an arginine residue at position 10 can establish a salt bridge with matriptase Asp-705, whereas the smaller side chain of lysine does not allow for the formation of this interaction (Fig. 6 and supplemental Fig. S2). During most of the 5-ns simulation of the [I10R]SFTI-1·matriptase complex, the guanidinium group of Arg-10 established at least one hydrogen bond with the carboxylic group of Asp-705 (distance oxygen to nitrogen of ∼3 Å), whereas over the 5-ns simulation of [I10K]SFTI-1/matriptase, Lys-10 did not form a hydrogen bond with Asp-705 (supplemental Fig. S2).
FIGURE 6.

Comparison of the binding modes of six SFTI-1 variants modeled in the matriptase and trypsin active sites. The models of the variants were generated by comparison using the structure of the complexes with wild-type SFTI-1 and were simulated by molecular dynamics for at least 5 ns. The proteases in the mutated complex are shown in green using schematic and stick representations, and the SFTI-1 variants are shown in cyan using schematic and stick representations. The structures of the corresponding complexes but without mutations are shown in transparency with the backbone of SFTI-1 in violet and the proteases in white. The mutated position is circled, and residues and loops discussed in the text are highlighted.
TABLE 4.
Average buried surface area in Å2 of SFTI-1 wild-type and variants in their complex with matriptase and trypsin
The buried surface areas were computed over the last 15 ns of the simulations involving wild-type SFTI-1 and over the last 4 ns of the simulations involving SFTI-1 variants using the LCPO approximation implemented in the ccptraj software from the Amber12 package. Standard deviations are provided for each measure.
| Complex with trypsin | Complex with matriptase | |
|---|---|---|
| SFTI-1 wild-type | 1340 ± 49 | 1483 ± 55 |
| [I10A]SFTI-1 | 1465 ± 52 | 1476 ± 54 |
| [I10D]SFTI-1 | 1413 ± 60 | 1431 ± 62 |
| [I10K]SFTI-1 | 1384 ± 59 | 1417 ± 55 |
| [I10R]SFTI-1 | 1407 ± 64 | 1538 ± 81 |
| [I7A]SFTI-1 | 1295 ± 44 | 1428 ± 52 |
| [I7A + I10R]SFTI-1 | 1316 ± 58 | 1414 ± 66 |
| [R2A]SFTI-1 | 1274 ± 62 | 1275 ± 73 |
| MCoTI-II | 1550 ± 64 | 1971 ± 124 |
| [V3R]MCoTI-II | 1637 ± 97 | 2152 ± 62 |
| [I7A]MCoTI-II | 1396 ± 56 | 2021 ± 73 |
| [V3R + I7A]MCoTI-II | 1632 ± 70 | 2081 ± 57 |
Position 7 of SFTI-1 contacts loops I and V (Fig. 5A), which has little sequence conservation between matriptase and trypsin, and this position is therefore potentially important in determining the selectivity for matriptase. According to the modeling, the mutation I7A did not change the orientation of SFTI-1 in the trypsin binding site (Fig. 6), but resulted in the largest decrease in buried surface area (∼50 Å2) observed among all the SFTI-1 variants in complex with trypsin (Table 4). The variant [I7A]SFTI-1 was also a more potent inhibitor of matriptase than the wild-type. Unexpectedly the double mutant [I7A + I10R]SFTI-1 was not a more potent inhibitor of matriptase than [I10R]SFTI-1. The corresponding model suggested that this doubly mutated peptide had a different binding mode to [I10R]SFTI-1, and, in this new binding mode, Arg-10 cannot form a salt bridge with Asp-705 (Fig. 6). By contrast, the effect of the double mutation for inhibition of trypsin was cumulative, in line with the models of [I7A]SFTI-1, [I10R]SFTI-1, and [I7A + I10R]SFTI-1 in complex with trypsin that displayed similar binding modes (Fig. 6). [I7A + I10R]SFTI-1 was the worst inhibitor for trypsin among all SFTI-1 variants tested in this study, and the most selective for matriptase among SFTI-1 variants.
Substitution of Arg-2 of SFTI-1 with an alanine resulted in a loss of potency for both proteases. Arg-2 is involved in charge interactions with Asp-14 (SFTI-1), and also potentially establishes cation-π interactions with several aromatic side chains, including Phe-12 from SFTI-1 and a tryptophan from the proteases (Trp-216 for trypsin and Trp-826 for matriptase). Matriptase has two phenylalanines, i.e. Phe-706 and Phe-708 with proximity to Arg-2 and these residues might also participate in cation-π interactions with the Arg-2 side chain (Fig. 7).
FIGURE 7.

Comparison of complexes involving matriptase by focusing around position 2 of SFTI-1 (A), position 3 of MCoTI-II (B), and position 3 of MCoTI-II V3R (C). These three positions occupy equivalent coordinates in the matriptase active site. SFTI-1, MCoTI-II, and [V3R]MCoTI-II are shown in cyan, and matriptase is in green. Positions discussed in the text are highlighted. The represented structures are the final conformations from molecular dynamics simulations.
MCoTI-II globally displayed more flexibility than SFTI-1 during the MD simulations, and the tip of loop 6 (positions 32, 33, 34, and 1) was the most flexible region of MCoTI-II in the complexes with matriptase and trypsin (supplemental Fig. S3). By contrast, the inhibitory loop (around Lys-5) was the most stable region of the peptide and the conformation of this inhibitory loop was nearly identical between MCoTI-II and SFTI-1 in complex with the two proteases (Fig. 5B). As a consequence, the Cα at position 3 of MCoTI-II and position 2 of SFTI-1 occupies the same region in the active sites, and because Arg-2 of SFTI-1 was shown to be important for the binding affinity, the mutant [V3R]MCoTI-II was predicted to have improved activity. Indeed, the V3R substitution resulted in the best matriptase inhibitor among MCoTI-II variants. For binding to matriptase, the V3R substitution resulted in a large increase of buried surface area (∼ 180 Å2 on average in Table 4) and, similarly to the comments made for the analysis of the mutant [I7A]SFTI-1, Arg-3 can potentially establish positive electrostatic interactions with Asp-709 in matriptase (Fig. 7).
Every modification to the inhibition loops of MCoTI-II, i.e. the alanine substitutions in positions 5–8, resulted in a drop of activity for both proteases, which could be explained by the tight fit of the inhibition loop in both active sites (Fig. 5B). Interestingly, the I7A substitution caused a decrease of activity that was much more dramatic for trypsin than matriptase, and this substitution resulted in an important loss of ∼150 Å2 of buried surface area in the models with trypsin. The double mutant [V3R + I7A]MCoTI-II was as specific for matriptase as for trypsin, with each substitution independently contributing to the loss of activity for trypsin. The models showed that the substitutions at positions 3 and 7 should have an independent impact because these residues are distant from each other and did not cause obvious changes of binding mode. This result contrasts with the change in binding mode predicted for the [I7A + I10R]SFTI-1 double mutant.
DISCUSSION
Naturally occurring peptides with cyclic backbones have significant promise in drug design (14, 17, 43), and in this study we have highlighted the potential of the frameworks of cyclic trypsin inhibitors from seeds. In particular, we have discovered that MCoTI-II is a potent inhibitor of matriptase and also generated substantial structure-activity data regarding MCoTI-II and SFTI-1 that has provided insights on how to modulate affinity toward matriptase over the prototypic trypsin.
Alanine scanning of SFTI-1 against trypsin and matriptase highlighted enzyme-specific requirements for high affinity inhibition (Table 1, Fig. 3). In SFTI-1, Arg-2 is indispensable for inhibition of matriptase, whereas for trypsin there is loss of inhibition yet the R2A mutant remains a potent inhibitor with a nanomolar affinity. The importance of this arginine residue was previously highlighted by Long et al. (15) who suggested that it is involved in a cation-π interaction with Phe-706 and Phe-708 of matriptase (15). In addition, It has been reported that Arg-2 is critical for matriptase inhibitory activity, with mutation to a phenylalanine derivative resulting in a 900-fold decrease in potency (20). We further characterized the role of this residue by substituting it with a lysine to determine whether the charge is the requirement at this position. However, this substitution resulted in a significant loss of inhibitory activity against matriptase suggesting that Arg-2 is an absolute requirement. These results are consistent with the substrate specificity of matriptase, as arginine is preferred at the P4 position (44), i.e. the position Arg-2 occupies when bound to the enzyme (Fig. 5).
Mutations in substrate sites P2, P1, and P1′ of SFTI-1 had a major impact on inhibitory activity. As expected, mutation of Lys-5 (P1) had the most significant effect, consistent with a previous study (20). Substitution of the adjacent residues (Thr-4 and Ser-6) also caused a significant loss of affinity against both enzymes. In general, the mutations had more detrimental effects on inhibitory activity against trypsin compared with matriptase, indicating that SFTI-1 has a highly optimized framework in respect to the trypsin inhibitory activity, reflecting its biological function in plants as a deterrent of insect and animal predators. Although substitution of Ser-6 with an alanine has significant effects on trypsin and matriptase activity it produces an SFTI-1 variant with potent inhibitory activity against chymotrypsin (45).
In contrast to the decreased activity observed for the majority of the SFTI-1 alanine mutants against trypsin, the I7A, P8A, and I10A substitutions resulted in enhanced inhibitory activity against matriptase. In addition, F12A had significantly less activity against trypsin but maintained similar activity to wild-type SFTI-1 against matriptase. Additional mutations at positions 7 and 10 were explored, and one, the variant I10R, was identified to have 30-fold enhanced inhibitory activity against matriptase compared with the wild-type peptide. The models generated for the inhibitor [I10R]SFTI-1 in complex with both enzymes illustrated why this substitution was ineffective in the trypsin complex as the side chain of Arg-10 is exposed to the solvent and does not interact with the enzyme. However, in matriptase, Arg-10 establishes a salt bridge with Asp-705 of matriptase (3.1 Å) when bound to the enzyme. The additional electrostatic interaction between these residues is likely to be responsible for stabilizing the complex, which is reflected in enhanced inhibition.
Combining the I7A and I10R mutants for SFTI-1 resulted in a peptide with significantly decreased trypsin activity (700-fold) and enhanced matriptase activity (4-fold) relative to the wild-type peptide. This selective enhancement of matriptase activity argues well for the design of more selective inhibitors using this framework. In particular, further exploration of mutants at positions 8 and 12 might provide useful information for subsequent design studies.
Initial studies using molecular modeling provided significant insight into the binding of SFTI-1 to matriptase and predicted a role for Arg-2, Lys-5, Ile-10, and Phe-12 in binding (15). The importance of Arg-2 and Lys-5 has been confirmed experimentally (20) and is consistent with the current study. The importance of Ile-10 and Phe-12 has also been explored, and both residues influence activity. For example, replacement of Ile-10 with a glutamine enhanced the selectivity for matriptase versus thrombin but decreased the potency against matriptase (20). Ile-10 was also highlighted in an analysis of the crystal structure of SFTI-1 bound to matriptase, and it was suggested that this residue would be a useful site for mutational analysis to improve binding (34). However, it was suggested that the replacement of Ile-10 with a basic residue, arginine or lysine, would not be beneficial because increasing the flexibility may result in a loss of entropy upon binding. Our results confirm that this site is indeed useful for modulating the inhibitory activity against matriptase and that the increase in potency of the I10R and I10K variants indicates that the introduction of more flexible residues does not impact negatively on activity. Very recently an acyclic SFTI-1 analog has been synthesized with an I10R substitution in addition to truncation and introduction of a His residue. This peptide also has enhanced matriptase affinity as well as improved selectivity against trypsin (46).
Consistent with the SFTI-1 alanine mutants, several of the alanine mutants of MCoTI-II had more significant losses of activity against trypsin relative to matriptase. Lys-9 and Lys-10 displayed enzyme-specific dependences with mutation of Lys-9 influencing affinity toward matriptase more than for trypsin. By contrast, mutation of Lys-10 influenced inhibition of trypsin more than that of matriptase. Val-3 was the only MCoTI-II mutant to maintain activity against matriptase, and mutation of this residue to an arginine resulted in one of the most potent inhibitors of matriptase. The model of [V3R]MCoTI-II shows that the mutated Arg-3 residue sits in the substrate S4 pocket (Fig. 7) supporting the strong preference for an arginine at this position. Combining the V3R mutant with I7A, one of the least active inhibitors against trypsin, reversed the trend of having higher trypsin potency relative to matriptase, as a result of a 1200-fold decrease in potency against trypsin, whereas maintaining potency against matriptase. Based on these results, MCoTI-II also appears to be a valuable framework for the development of more selective inhibitors of matriptase.
Analysis of the hybrid peptides of SFTI-1 and MCoTI-II indicates that the SFTI-1 framework is more amenable to substantial sequence changes than MCoTI-II. Grafting the SFTI-1 active site loop into the MCoTI-II resulted in significant losses in activity against both trypsin and matriptase. By contrast, the loss of activity for the hybrid with the MCoTI-II active site loop grafted into the SFTI-1 framework (SFMC) was minimal, despite having one residue less than the wild-type peptide. A naturally occurring example of a peptide similar to SFTI-1 has been found in sunflower (SFT-L2) that comprises only 12 residues and has significant sequence differences in the bioactive loop (47). Although only containing 12 residues the structure of SFT-L2, braced by a disulfide bond and cyclic backbone, is extremely well defined and has been suggested as a useful framework in drug design. The minimized framework of SFMC might represent a new lead molecule for the design of inhibitors against serine proteases.
In summary, the structure-activity data elucidated in this study are a solid basis for the design of selective peptide inhibitors of matriptase and highlights the versatility of cyclic peptides for drug design. The use of disulfide-rich, cyclic peptides as scaffolds is emerging as a powerful approach for the design of novel drug leads. Using this approach a cyclic cone snail venom peptide has been engineered from the naturally occurring acyclic peptide, which is orally active in a rat model of pain (48). In addition, a modified cyclotide has oral activity in an in vivo mouse model of visceral pain (43). The latter study exemplifies the potential of the cyclotide scaffold for conferring oral activity and suggests that selective inhibitors of matriptase, based on cyclic peptide scaffolds, might hold significant promise for the treatment of cancer.
Acknowledgments
We thank Phillip Walsh and Philip Sunderland for assistance with peptide synthesis and Sabine Streicher for kinetic measurements.
This work was supported in part by a Queensland Government Smart State Fellowship (to N. L. D.), which was co-funded by Hexima Limited, and Project SO 249/1-1, priority program 1394 “Mast cells-promoters of health and modulator of diseases,” of the Deutsche Forschungsgemeinschaft (to C. P. S.).

This article contains supplemental Figs. S1–S3.
N. L. Daly, L. Thorstholm, K. P. Greenwood, G. K. King, K. J. Rosengren, B. Heras, J. L. Martin, and D. J. Craik, unpublished data.
- SFTI-1
- sunflower trypsin inhibitor-1
- COSY
- correlation spectroscopy
- MCoTI-II
- Momordica cochinchinensis trypsin inhibitor II
- MD
- molecular dynamics
- NOESY
- nuclear Overhauser effect spectroscopy
- RP-HPLC
- reverse phase high performance liquid chromatography
- TOCSY
- total correlation spectroscopy
- PDB
- Protein data bank
- Boc
- t-Butyloxycarbonyl.
REFERENCES
- 1. Page M. J., Di Cera E. (2008) Serine peptidases. Classification, structure and function. Cell Mol. Life Sci. 65, 1220–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Heutinck K. M., ten Berge I. J., Hack C. E., Hamann J., Rowshani A. T. (2010) Serine proteases of the human immune system in health and disease. Mol. Immunol. 47, 1943–1955 [DOI] [PubMed] [Google Scholar]
- 3. Conlan B. F., Anderson M. A. (2011) Circular micro-proteins and mechanisms of cyclization. Curr. Pharm. Des. 17, 4318–4328 [DOI] [PubMed] [Google Scholar]
- 4. Oberst M. D., Singh B., Ozdemirli M., Dickson R. B., Johnson M. D., Lin C. Y. (2003) Characterization of matriptase expression in normal human tissues. J. Histochem. Cytochem. 51, 1017–1025 [DOI] [PubMed] [Google Scholar]
- 5. Bergum C., List K. (2010) Loss of the matriptase inhibitor HAI-2 during prostate cancer progression. Prostate 70, 1422–1428 [DOI] [PubMed] [Google Scholar]
- 6. List K., Szabo R., Molinolo A., Sriuranpong V., Redeye V., Murdock T., Burke B., Nielsen B. S., Gutkind J. S., Bugge T. H. (2005) Deregulated matriptase causes ras-independent multistage carcinogenesis and promotes ras-mediated malignant transformation. Genes Develop. 19, 1934–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Napp J., Dullin C., Müller F., Uhland K., Petri J. B., van de Locht A., Steinmetzer T., Alves F. (2010) Time-domain in vivo near infrared fluorescence imaging for evaluation of matriptase as a potential target for the development of novel, inhibitor-based tumor therapies. Int. J. Cancer 127, 1958–1974 [DOI] [PubMed] [Google Scholar]
- 8. Steinmetzer T., Schweinitz A., Stürzebecher A., Dönnecke D., Uhland K., Schuster O., Steinmetzer P., Müller F., Friedrich R., Than M. E., Bode W., Stürzebecher J. (2006) Secondary amides of sulfonylated 3-amidinophenylalanine. New potent and selective inhibitors of matriptase. J. Med. Chem. 49, 4116–4126 [DOI] [PubMed] [Google Scholar]
- 9. Förbs D., Thiel S., Stella M. C., Stürzebecher A., Schweinitz A., Steinmetzer T., Stürzebecher J., Uhland K. (2005) In vitro inhibition of matriptase prevents invasive growth of cell lines of prostate and colon carcinoma. Int. J. Oncol. 27, 1061–1070 [PubMed] [Google Scholar]
- 10. Sanders A. J., Parr C., Davies G., Martin T. A., Lane J., Mason M. D., Jiang W. G. (2006) Genetic reduction of matriptase-1 expression is associated with a reduction in the aggressive phenotype of prostate cancer cells in vitro and in vivo. J. Exp. Ther. Oncol. 6, 39–48 [PubMed] [Google Scholar]
- 11. Swedberg J. E., Harris J. M. (2012) Natural and engineered plasmin inhibitors. Applications and design strategies. Chembiochem 13, 336–348 [DOI] [PubMed] [Google Scholar]
- 12. Overall C. M., Kleifeld O. (2006) Tumour microenvironment-opinion. Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 6, 227–239 [DOI] [PubMed] [Google Scholar]
- 13. Rawlings N. D., Barrett A. J., Bateman A. (2012) MEROPS. The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 40, D343–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Craik D. J., Swedberg J. E., Mylne J. S., Cemazar M. (2012) Cyclotides as a basis for drug design. Expert Opin. Drug Discov. 7, 179–194 [DOI] [PubMed] [Google Scholar]
- 15. Long Y. Q., Lee S. L., Lin C. Y., Enyedy I. J., Wang S., Li P., Dickson R. B., Roller P. P. (2001) Synthesis and evaluation of the sunflower derived trypsin inhibitor as a potent inhibitor of the type II transmembrane serine protease, matriptase. Bioorg. Med. Chem. Lett. 11, 2515–2519 [DOI] [PubMed] [Google Scholar]
- 16. Luckett S., Garcia R. S., Barker J. J., Konarev A. V., Shewry P. R., Clarke A. R., Brady R. L. (1999) High-resolution structure of a potent, cyclic proteinase inhibitor from sunflower seeds. J. Mol. Biol. 290, 525–533 [DOI] [PubMed] [Google Scholar]
- 17. Swedberg J. E., Nigon L. V., Reid J. C., de Veer S. J., Walpole C. M., Stephens C. R., Walsh T. P., Takayama T. K., Hooper J. D., Clements J. A., Buckle A. M., Harris J. M. (2009) Substrate-guided design of a potent and selective kallikrein-related peptidase inhibitor for kallikrein 4. Chem. Biol. 16, 633–643 [DOI] [PubMed] [Google Scholar]
- 18. Legowska A., Bulak E., Jaśkiewicz A., Małuch I., Sieracki M., Wysocka M., Lesner A., Rolka K. (2010) Analogues of trypsin inhibitor SFTI-1 with disulfide bridge substituted by various length of carbonyl bridges. Protein Pept. Lett. 17, 1223–1227 [DOI] [PubMed] [Google Scholar]
- 19. Legowska A., Dȩbowski D., Lukajtis R., Wysocka M., Czaplewski C., Lesner A., Rolka K. (2010) Implication of the disulfide bridge in trypsin inhibitor SFTI-1 in its interaction with serine proteinases. Bioorg. Med. Chem. 18, 8188–8193 [DOI] [PubMed] [Google Scholar]
- 20. Li P., Jiang S., Lee S.-L., Lin C. Y., Johnson M. D., Dickson R. B., Michejda C. J., Roller P. P. (2007) Design and synthesis of novel and potent inhibitors of the type II transmembrane serine protease, matriptase, based upon the sunflower trypsin inhibitor-1. J. Med. Chem. 50, 5976–5983 [DOI] [PubMed] [Google Scholar]
- 21. Guo X., Shi J., Tang Z., Cui D., Zhang Y. (2006) Synthesis and biological activity of seleno sunflower trypsin inhibitor analog. Chem. Biol. Drug Des. 68, 341–344 [DOI] [PubMed] [Google Scholar]
- 22. Avrutina O., Fittler H., Glotzbach B., Kolmar H., Empting M. (2012) Between two worlds. A comparative study on in vitro and in silico inhibition of trypsin and matriptase by redox-stable SFTI-1 variants at near physiological pH. Org. Biomol. Chem. 10, 7753–7762 [DOI] [PubMed] [Google Scholar]
- 23. Barbeta B. L., Marshall A. T., Gillon A. D., Craik D. J., Anderson M. A. (2008) Plant cyclotides disrupt epithelial cells in the midgut of lepidopteran larvae. Proc. Natl. Acad. Sci. U.S.A. 105, 1221–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gustafson K. R., McKee T. C., Bokesch H. R. (2004) Anti-HIV cyclotides. Curr. Protein Pept. Sci. 5, 331–340 [DOI] [PubMed] [Google Scholar]
- 25. Daly N. L., Love S., Alewood P. F., Craik D. J. (1999) Chemical synthesis and folding pathways of large cyclic polypeptides. Studies of the cystine knot polypeptide kalata B1. Biochemistry 38, 10606–10614 [DOI] [PubMed] [Google Scholar]
- 26. Hernandez J. F., Gagnon J., Chiche L., Nguyen T. M., Andrieu J. P., Heitz A., Trinh Hong T., Pham T. T., Le Nguyen D. (2000) Squash trypsin inhibitors from Momordica cochinchinensis exhibit an atypical macrocyclic structure. Biochemistry 39, 5722–5730 [DOI] [PubMed] [Google Scholar]
- 27. Daly N. L., Chen Y.-K., Foley F. M., Bansal P. S., Bharathi R., Clark R. J., Sommerhoff C. P., Craik D. J. (2006) The absolute structural requirement for a proline in the P3′-position of Bowman-Birk protease inhibitors is surmounted in the minimized SFTI-1 scaffold. J. Biol. Chem. 281, 23668–23675 [DOI] [PubMed] [Google Scholar]
- 28. Chan L. Y., Gunasekera S., Henriques S. T., Worth N. F., Le S. J., Clark R. J., Campbell J. H., Craik D. J., Daly N. L. (2011) Engineering pro-angiogenic peptides using stable, disulfide-rich cyclic scaffolds. Blood 118, 6709–6717 [DOI] [PubMed] [Google Scholar]
- 29. Goddard T. D., Kneller D. G. (YEAR) SPARKY 3. University of California, San Francisco [Google Scholar]
- 30. Wüthrich K. (1986) NMR of Proteins and Nucleic Acids, Wiley-Interscience, New York [Google Scholar]
- 31. Morrison J. F. (1969) Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta 185, 269–286 [DOI] [PubMed] [Google Scholar]
- 32. Güntert P. (2009) Automated structure determination from NMR spectra. Eur. Biophys. J. 38, 129–143 [DOI] [PubMed] [Google Scholar]
- 33. Koradi R., Billeter M., Wüthrich K. (1996) MOLMOL. A program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 29–32 [DOI] [PubMed] [Google Scholar]
- 34. Yuan C., Chen L., Meehan E. J., Daly N., Craik D. J., Huang M., Ngo J. C. (2011) Structure of catalytic domain of Matriptase in complex with Sunflower trypsin inhibitor-1. BMC Struct. Biol. 11, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 36. Shen M. Y., Sali A. (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Case D. A., Cheatham T. E., 3rd, Darden T., Gohlke H., Luo R., Merz K. M., Jr., Onufriev A., Simmerling C., Wang B., Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cerutti D. S., Le Trong I., Stenkamp R. E., Lybrand T. P. (2009) Dynamics of the streptavidin-biotin complex in solution and in its crystal lattice. Distinct behavior revealed by molecular simulations. J. Phys. Chem. B 113, 6971–6985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ryckaert J. P., Ciccotti G., Berendsen H. J. (1977) Numerical integration of the cartesian equations of motion of a system with constraints. Molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 [Google Scholar]
- 40. Darden T., York D., Pedersen L. (1993) Particle mesh Ewald. An N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 41. Tam J. P., Lu Y.-A. (1998) A biomimetic strategy in the synthesis and fragmentation of cyclic protein. Protein Sci. 7, 1583–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Avrutina O., Schmoldt H. U., Gabrijelcic-Geiger D., Le Nguyen D., Sommerhoff C. P., Diederichsen U., Kolmar H. (2005) Trypsin inhibition by macrocyclic and open-chain variants of the squash inhibitor MCoTI-II. Biol. Chem. 386, 1301–1306 [DOI] [PubMed] [Google Scholar]
- 43. Wong C. T., Rowlands D. K., Wong C. H., Lo T. W., Nguyen G. K., Li H. Y., Tam J. P. (2012) Orally active peptidic bradykinin B1 receptor antagonists engineered from a cyclotide scaffold for inflammatory pain treatment. Angew. Chem. Int. Ed. Engl. 51, 5620–5624 [DOI] [PubMed] [Google Scholar]
- 44. Takeuchi T., Harris J. L., Huang W., Yan K. W., Coughlin S. R., Craik C. S. (2000) Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J. Biol. Chem. 275, 26333–26342 [DOI] [PubMed] [Google Scholar]
- 45. Rafał Ł., Anna L., Magdalena W., Dawid D., Adam L., Krzysztof R. (2011) Analogues of trypsin inhibitor SFTI-1 modified in the conserved P(1)′ position by synthetic or non-proteinogenic amino acids retain their inhibitory activity. J. Pept. Sci. 17, 281–287 [DOI] [PubMed] [Google Scholar]
- 46. Fittler H., Avrutina O., Glotzbach B., Empting M., Kolmar H. (2013) Combinatorial tuning of peptidic drug candidates. High-affinity matriptase inhibitors through incremental structure-guided optimization. Org. Biomol. Chem. 11, 1848–1857 [DOI] [PubMed] [Google Scholar]
- 47. Mylne J. S., Colgrave M. L., Daly N. L., Chanson A. H., Elliott A. G., McCallum E. J., Jones A., Craik D. J. (2011) Albumins and their processing machinery are hijacked for cyclic peptides in sunflower. Nat. Chem. Biol. 7, 257–259 [DOI] [PubMed] [Google Scholar]
- 48. Clark R. J., Jensen J., Nevin S. T., Callaghan B. P., Adams D. J., Craik D. J. (2010) The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. Engl. 49, 6545–6548 [DOI] [PubMed] [Google Scholar]
- 49. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems. Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wishart D. S., Bigam C. G., Holm A., Hodges R. S., Sykes B. D. (1995) 1H, 13C, and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 5, 67–81 [DOI] [PubMed] [Google Scholar]