

Abstract
Bone marrow-derived endothelial progenitor cells (EPCs) contribute to angiogenesis-mediated pathological neovascularization and recent studies have begun to recognize the biological significance of this contribution. This review will discuss the ability of EPCs to contribute to neovascularization in both physiological and pathological conditions. Circulating EPCs were originally identified in 1997 by Asahara as CD34+ VEGFR2+ mononuclear cells. These cells differentiated into an endothelial phenotype, expressed endothelial markers, and incorporated into neovessels at sites of ischemia (Asahara et al., 1997). EPCs provide both instructive (release of pro-angiogenic cytokines) and structural (vessel incorporation and stabilization) functions that contribute to the initiation of neo-angiogenesis. EPC populations can be characterized based on surface markers of freshly isolated cells or they can be described by their in vitro characteristics once placed in culture. However, a major stumbling block to progress in the field has been the lack of consensus among investigators as to the optimal characterization of EPCs. This review intends to address the role of both EPC classes and evaluate how they interact in the setting of pathological angiogenesis. Since the EPCs may be responsible for turning on the “angiogenic switch,” strategies have been employed to keep this switch in the “off” position for diseases like cancer, retinopathy and wet AMD. The expectation is that EPCs will evolve into clinically useful prognostic and predictive tools in cancer and in ocular diseases associated with pathological neovascularization and that targeting this cell type is a key to successful management of patients suffering from diseases associated with pathological neovascularization.
The significance of the EPC
EPCs, a minor subpopulation of the mononuclear cell fraction in peripheral blood, are believed to be derived from hematopoietic stem cells, (HSCs, Figure 1) or alternatively from the endothelium itself (Yoder et al., 2007). In the last decade, it has been established that EPCs are recruited to sites requiring vascular repair and that these cells contribute to the viability of the vasculature (Asahara et al., 1997). EPCs leave the bone marrow following gradients of growth factors and cytokines that are released into the circulation by injured endothelium and injured tissues (Schatteman et al., 2007). Once in the circulation, EPCs home to sites of damage and promote vascular integrity. They not only mediate repair of injured tissue but lead to reperfusion of ischemic regions within the tissue (Schatteman et al., 2007). Following the discovery of EPCs by Asahara and coworkers, numerous studies demonstrated that EPCs contribute to such repair processes, including myocardial ischemia/infarction, limb ischemia, wound healing, atherosclerosis, endogenous endothelial repair, and tumor neovascularization in mice and humans (Asahara et al., 1997; Spring et al., 2005). This repair occurs as a series of carefully orchestrated steps. EPCs are first mobilized from bone marrow; then circulate to remote sites of vascular injury where they interact with the local endothelium. EPCs comprise 0.02% of the total bone marrow (BM) contribution (compared to 4% by GR1+ myeloid cells); GR1+ myeloid cells represent a heterogeneous population of myeloid cells that comprises immature macrophages, granulocytes, dendridic cells and myeloid cells at early stages of differentiation. GR1+CD11b+ cells (Bronte et al., 2001; Kusmartsev and Gabrilovich, 2002) are present in the bone marrow and spleen of healthy mice and differentiate into mature myeloid cells — that is, granulocytes, macrophages and DCs — in the presence of GM-CSF in vitro or after adoptive transfer to healthy, naïve recipients in vivo (Kusmartsev and Gabrilovich, 2003) and their incorporation into vessels varies dramatically with an engraftment efficiency of up to 95% in some vascular beds (Rafii and Lyden, 2003). Minami et al. has shown that circulating EPCs engraft luminally into 15% to 29% of the vessels of the transplanted human heart (Minami et al., 2005). Bone marrow derived endothelial cells have also been shown to give rise to up to 16% of the neovasculature in spontaneous tumors growing in transgenic mice, and also contribute to human tumor vessels (Peters et al., 2005).
Figure 1. Adult stem cells of the bone marrow.
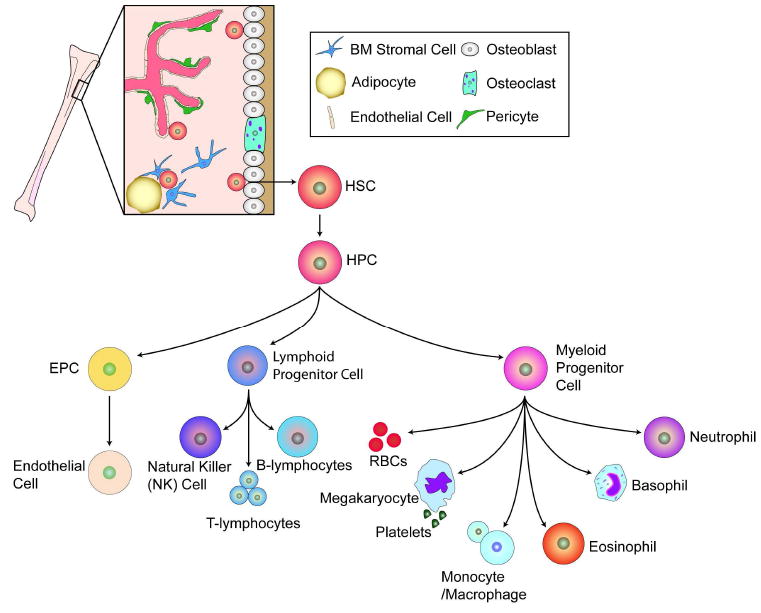
The bone marrow hosts at least two known types of adult stem cells, the mesenchymal stem cells (MSCs) and the hematopoietic stem cells (HSCs); the most prominent adult stem cell in the bone marrow, together with the EPC, is the focus of this review. The HSC can give rise to the hematopoietic progenitor cells (HPCs) which in turn give rise to the lymphoid progenitor cell, the myeloid progenitor cells, and likely the EPC. The precise origin of the EPC is under debate as this cell may directly arise from the HSC or from the HPC. The bone marrow microenvironment is composed of bone marrow stromal cells (which are the source of SDF-1), adipocytes, and cells of the bone matrix, osteoblasts and osteoclasts. The vessels within the bone marrow, composed of pericytes and endothelium, function to provide a barrier between the hematopoietic compartment and the circulatory system discussed in greater detail in Figure 2. Figure adapted from Domen, et al. (Domen et al., 2006).
Our group has observed very high numbers of bone marrow-derived cells contributing to both repair and pathological neovascularization in the eye (Caballero et al., 2007; Grant et al., 2002). We interpreted this finding to be secondary to the highly quiescent nature of the resident retinal vasculature (typical retinal endothelial cell turnover occurs every 4 years) (Engerman et al., 1967), thus facilitating the contribution of circulating EPCs to the newly forming vessel.
EPCs may be isolated from peripheral blood, cord blood or bone marrow; however, an accurate definition and characterization of the various EPC sub-populations is still lacking. Thus, the first controversy is how to best identify these cells, the second is how they precisely function to perform repair, and finally, what makes these beneficial cells participate in pathological repair processes? Many studies have questioned their functional significance as they typically represent a relatively low contribution to vasculature. Extensive variability ranging from a major contribution (Grant et al., 2002; Lyden et al., 2001) to a minor contribution (Peters et al., 2005), and in some cases no contribution (Purhonen et al., 2008) has been reported. These conflicting reports can be, in part, attributed to a limited analysis of the EPC phenotype in each study and the lack of definitive methods for distinguishing vessel incorporated bone marrow-derived endothelial cells from intimately associated perivascular cells.
The bone marrow microenvironment: priming these cells for action
Arterial vessels enter the marrow space through the bone. These vessels branch out into arterioles and then capillaries spread throughout the marrow and supply the bone marrow sinusoids. Sinusoids drain into a central sinus, which is the largest vascular structure found in the bone marrow.
Adult bone marrow is known to contain a population of hematopoietic stem cells (HSCs) that can be divided into lineage positive (Lin+) and lineage negative (Lin−) categories with regard to their potential to differentiate into formed elements of the blood. Lin− HSCs have been shown to contain a population of endothelial precursor cells (EPCs) capable of forming blood vessels. The bone marrow compartment comprises the osteoblastic (or endosteal) and the vascular niches (Figure 2). The vascular niche contains more committed stem and progenitor cells than the relatively quiescent stem cells of the endosteal niche. The osteoblastic niche provides a quiescent microenvironment for stem cell maintenance, and the resident HSCs are anchored to the endosteal surface by calcium sensing receptors present on the HSC. The bone marrow microenvironment switches from a quiescent state to an activated state when local tissue injury results in the release of soluble factors including VEGF, FGF, GM-CSF, or osteopontin into the circulation. This in turn promotes the mobilization of both vascular and hematopoietic progenitors into the peripheral circulation from which these cells are recruited to sites of injury. In the injured tissue, these bone marrow recruited cells and other stromal cells (adipocytes, fibroblasts etc.) constitute a unique environment that can modify the phenotype of the bone marrow cell (Figure 3).
Figure 2. Bone marrow and hematopoietic stem cell niches.
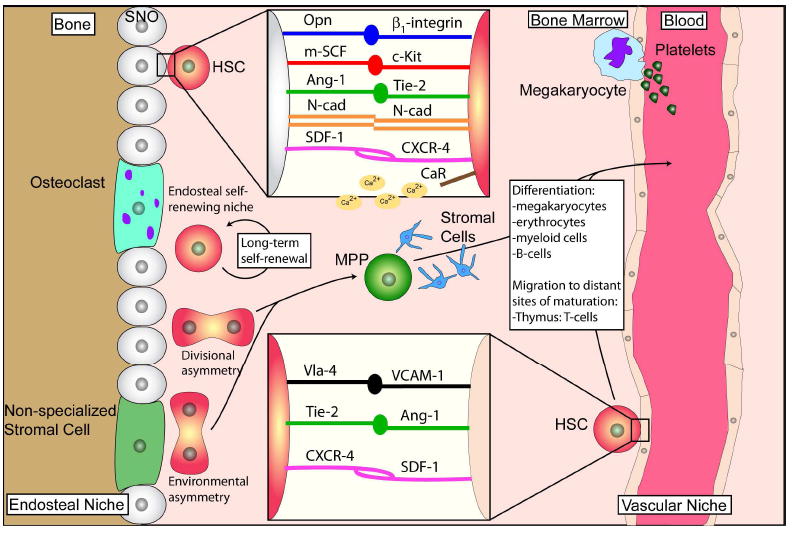
The maintenance of HSC self-renewal and differentiation is dependent on the specific micro-environment in which they reside. These specialized microenvironments or “niches” include the endosteal or osteoblast niche and the vascular niche. In the endosteal niche, stem cells reside in close proximity to endosteal linings of the bone marrow cavities of the trabecular regions of long bones. HSCs dock to spindle-shaped N-cadherin+ CD45- osteoblasts (SNO) (Upper inset) and to endothelial cells (Lower inset) through a variety of ligand/receptor binding (Opn/α1-integrin; m-SCF/c-Kit; Ang-1/Tie-2; N-cad/n-cad; SDF-1/CXCR-4. Upper inset); (Vla-4/VCAM-1; Tie-2/Ang-1; CXCR-4/SDF-1. Lower inset). The vascular niche is where stem and progenitor cells have been identified to reside close to sinusoidal endothelium. The endosteal and vascular niches seem to cooperate and both niches are indispensible in controlling HSC quiescence and self renewal activities. Opn: Osteopontin; m-SCF: membrane-bound stem cell factor; Ang-1: angiopoietin-1; N-cad: neural cadherin; SDF-1: stromal cell-derived factor-1; CXCR-4: chemokine (C-X-C motif) receptor 4; CaR: calcium receptor; Vla-4: very late antigen 4; VCAM-1: vascular cell adhesion molecule 1. Figure adapted from Wilson and Trumpp (Wilson and Trumpp, 2006).
Figure 3. Mobilization and homing are two processes closely related.
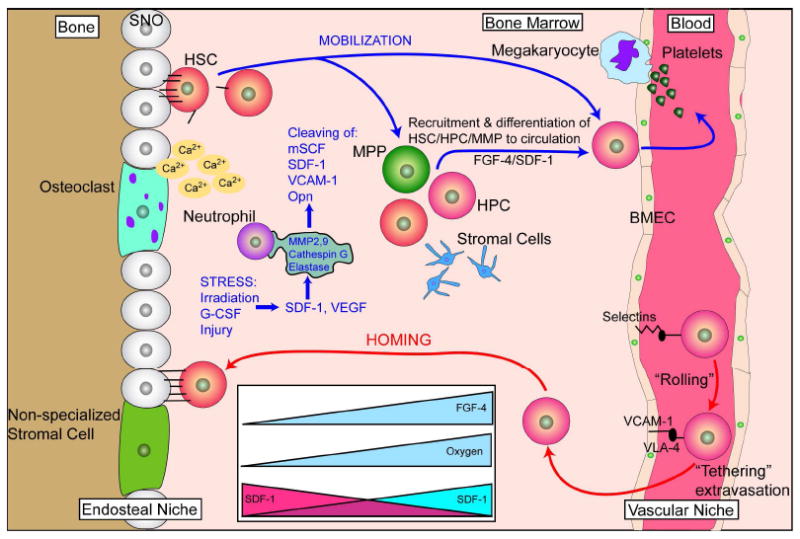
Mobilization involves the exodus of HSC/HPC from the bone marrow into the circulation while homing is the “opposite” of this event. HSC mobilize from the endosteal niche, move to the vascular niche, and ultimately into the circulation. This normally occurs when stress induces changes of SDF-1 levels in the bone marrow. The mechanism of stress-induced mobilization as occurs following irradiation or G-CSF-induced mobilization is not fully known, but is, in part, accomplished by the upregulation of proteases such as MMP-2, MMP-9, cathepsin-G and elastase. These proteases cleave niche retention signals like membrane-bound stem cell factor (mSCF), SDF-1, VCAM-1 and osteopontin (Opn). Gradients of fibroblast growth factor 4 (FGF-4) also regulate mobilization. For homing events, key steps are needed. Upon reaching the bone marrow vasculature, SDF-1-stimulated circulating HSC/HPC express integrins such as very late antigen 4 (VLA-4) and hyaluronan binding–cellular adhesion molecule (CD44). These integrins, in turn, interact with vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), E-and P-selectins expressed on bone marrow endothelial cells which slows down the circulating HSC/HSP in a process known as “rolling.” Following rolling, firm adhesion and subsequent endothelia trans- migration into the hematopoietic compartment is mainly accomplished by VLA-4 interactions. Once extravasated, the cells migrate along extravascular hematopoietic cords toward specific niches through as SDF-1 gradient or receding oxygen gradient originating from the supporting osteoblastic or endothelia niches. BMEC: Bone marrow microvascular endothelial cell. Figure adapted from Wilson and Trumpp (Wilson and Trumpp, 2006) and from Yin, et al. (Yin and Li, 2006).
Classification of in vitro expanded EPCs
In vitro methods can be used to identify specific subsets of isolated EPCs, expand cell numbers and modify function ex vivo by exposure to certain factors or by modifying gene expression. Based on in vitro characteristics, two distinct EPC phenotypes have been described. The “outgrowth endothelial cells” (OECs) which display a clonal phenotype, are derived from CD34+CD45+ population (Case et al., 2007) and have been called the “late EPC.” The second population is termed the “early EPC” (eEPC) and has a monocyte/macrophage phenotype (CD14+/CD45+/CD68+ or F4/80+ in mice) but display endothelial-like markers and behavior (and participate in “vascular mimicry”, (Figure 4A) (Anghelina et al., 2006). This in vitro characterization inherently means that these cells likely lose some of their intrinsic features when taken out of the individual and placed in culture (Hur et al., 2004).
Figure 4. Tunneling and vascular mimicry by MC/Mph in Matrigel plugs in vivo.
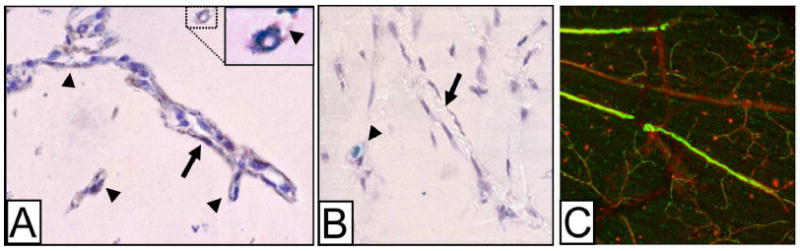
A. Mph-lined, capillary-like structures (arrows) detected at 1 week in a Matrigel plug in a Tie2-βgal mouse (no Tie2-βgal+ cells present). Note the branching (arrowheads), and that the cells in the lumen are F4/80+. Insert: Enlargement of the F4/80+ cell marked with dashed lines, showing a lumen-like vacuole and a low-density tunnel in Matrigel (arrowhead). B. Negative control for the F4/80 immunostaining (omission of the primary antibody). Cell columns line a collagen bundle cut longitudinally, visible in the background of this phase contrast micrograph (arrow). In this field, a Tie2-ßgal+ progenitor cell (light blue) is wrapped by another cell (arrowhead). Counterstaining is with hematoxylin. Original magnifications: A-B, ×120. (From Anghelina et al., AJP, 2006). C. Peripheral blood CD14+ cells (green, visualized by reaction to anti-human nuclear antigen) isolated from a diabetic patient injected within the vitreous of a mouse that had STZ-induced diabetes for 11 months show vascular association with presumably damaged capillaries. Original magnification: ×20.
Freshly isolated EPCs are characterized based on surface markers including CD34+, CD133+, VEGFR-2+ in humans and lin− sca1+ cells in mice. While differences in isolation methodologies have created some confusion in the field, recently there is a widely accepted consensus that bona fide circulating EPCs express surface markers shared by both endothelial and stem cell phenotypes (Barber and Iruela-Arispe, 2006) and must exhibit key behaviors such as eNOS expression, enhancement of HUVEC tube formation, and incorporation with HUVECs in tube formation in vitro (Schatteman et al., 2007).
Finding the bone marrow-derived endothelial cells in vivo is a great challenge, but in our experience the best way to characterize an EPC is to use genetically labeled cells such as GFP (Figure 4C). To properly characterize a cell as an EPC, one may show that, in a single nucleus, GFP and CD31 signals occur in the same cell and then show that VE-cadherin is localized to the new endothelial cell junctions (Caballero et al., 2007).
The angiogenic monocyte paves the way for the EPC
The role of monocyte (MC) and macrophage (Mph) subpopulations in vascular repair/reperfusion and neovascularization responses has been an area of hot debate (Anghelina et al., 2004; Anghelina et al., 2006; Moldovan et al., 2000). Anghelina et al. demonstrate the MC/Mph population drill conduits creating “cell columns.” These columns facilitate other progenitor cell populations to come into the area and eventually these columns are converted into blood vessels (Figure 4B) (Anghelina et al., 2006). These cells typically start out with their distinct MC/Mph characteristics but during the actual engraftment process they may begin expressing endothelial cell markers. This same type of transition (myeloid to endothelial) is believed to occur during the generation of early endothelial progenitor cells (eEPCs) in vitro (Yoder et al., 2007). These MC/Mph cells also participate in angiogenesis by secreting paracrine factors that support new vessel formation and thus create the ideal provisional environment for the “second wave” of cells that are “true” endothelial building blocks for vessels and generate stable vasculature.
The large cadre of MC/Mph cells, which are part of the cells that form eEPCs in vitro, have yet to be fully characterized. The MC/Mph cells clearly participate in repair/reperfusion and neovascularization responses and represent populations of cells that can be used therapeutically. Moreover, their structural presence encompasses the lymphangiogeneis as well (Cursiefen et al., 2006). The key role of these cells is not surprising as circulating MCs are the first cells to be recruited to sites of tissue damage and developing vessels. The presence of Mph in damaged endothelium and the migration of Mph through walls of vessels have been known for decades (Poole and Florey, 1958). See Moldovan and Asahara for a review (Moldovan and Asahara, 2003). Recent studies using parabiosis models (the uniting of two individual organisms or animals anatomically and physiologically, either under experimental or natural conditions) demonstrated that circulating blood Mph (F4/80+) also express the endothelial marker CD31 using three different endpoints: wound healing, gel foam plugs containing angiogenic factors, and a tumor (Kim et al., 2009).
The communication between MC/Mph and resident endothelial cells is bi-directional as these cells each dramatically and distinctly influence each other. For example, endothelial cells stimulate MC to produce MMP-9 that facilitates the angiogenic process. In states of pathological neovascularization, the inflammatory response that is critical to the initiation and maintenance of this aberrant process includes MC/Mph. Such is the case in diabetes and may set the stage for the development of not only early background retinopathy but also proliferative diabetic retinopathy (Joussen et al., 2004; Kern, 2007; Portillo et al., 2008). Inhibiting macrophage colony stimulating factor specifically targets pathological neovascularization in the eye and in tumors (Kubota et al., 2009) which suggests that MC/Mph cells are central players in this type of pathological angiogenesis.
EPCs in ocular angiogenesis
Retinopathy is the most common diabetic complication with almost all diabetic patients developing background retinopathy (Klein et al., 2008). Both type 1 and type 2 diabetes is associated with widespread EPC dysfunction (Fadini et al., 2005). Several studies have shown that circulating EPC number is reduced in patients with nonproliferative diabetic retinopathy (NPDR) (Kusuyama et al., 2006) but increased in patients with proliferative diabetic retinopathy (PDR ) (Lee et al., 2006). These observations were confirmed by Brunner et al. who conducted a case-control study that compared 90 patients with type 1 diabetes with and without retinopathy. They also demonstrated that in type 1 patients with retinopathy, EPCs underwent stage-related regulation. In NPDR, a reduction of EPCs was observed, while in proliferative retinopathy, a dramatic increase of mature EPCs was observed (Brunner et al., 2009). Typically in diabetes CD34+ EPCs are not only reduced in number but also in their function, demonstrating decreased migration, decreased colony formation in vitro, decreased differentiation into endothelial cells and decreased tube formation in vitro (Schatteman et al., 2007). We determined that the mechanism for reduced migration was due to reduced levels of bioavailable nitric oxide (NO). These reduced levels are secondary to increased levels of reactive oxygen species (ROS) which scavenge the NO and convert it to toxic peroxynitrite, increasing levels of NO or reducing ROS improved diabetic EPC migratory function in vitro (Segal et al., 2006). To further explore this mechanism, we examined the effect of (NO) on the phosphorylation and intracellular redistribution of vasodilator-stimulated phosphoprotein (VASP), a critical actin motor protein required for cell migration. We found that NO promotes cytoskeletal changes through site- and cell type-specific VASP phosphorylation and that in diabetes there was a markedly blunted response to NO associated with reduced cell migration. This migratory defect, when corrected, results in enhanced vascular repair and tissue perfusion in diabetic mice (Jarajapu Y et al unpublished data). These findings are consistent with the hypothesis that the vasodegenerative phase (acellular capillaries) of diabetic retinopathy is likely associated with not only reduced numbers of EPCs but EPCs with reduced reparative function. In contrast with PDR, these cells may be responsible for pathological neovascularization and serving as a nidus for new vessel growth in the diabetic retina (Figure 5).
Figure 5. Schematic of the hypotheses for the pathogenesis of PDR and the involvement of EPCs.
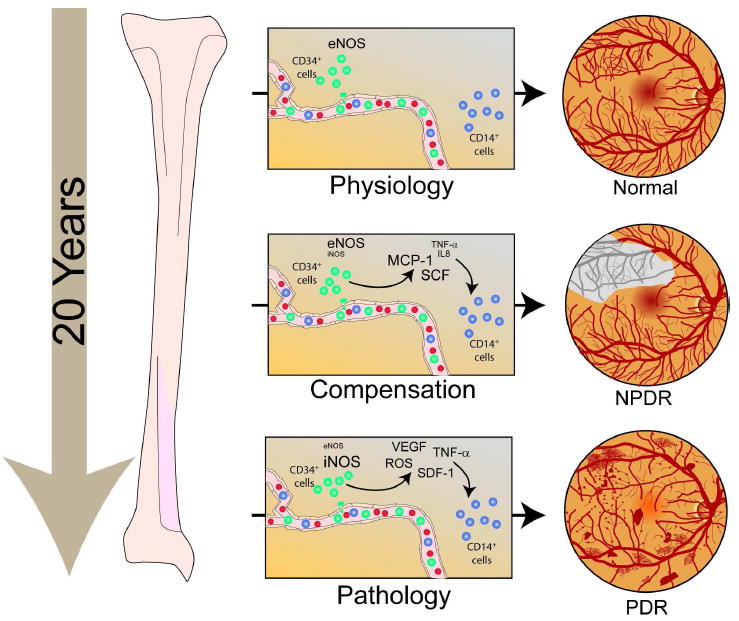
In physiological conditions, CD34+ EPCs contribute to routine blood vessel maintenance through eNOS activation. In diabetes, initially, cytokines like stem cell factor (SCF), monocyte chemoattractant protein-1 (MCP-1), interleukin-8 (IL-8), tumor necrosis factor alpha (TNF-α released by dysfunctional CD34+ EPCs) and iNOS activation initiate CD14+ EPC-mediated aberrant vascular repair and CD34+ EPC dysfunction which ultimately results in retinal ischemia. This phase is referred to as non proliferative diabetic retinopathy (NPDR). The vasodegenerative phase of diabetic retinopathy associated with reduced reparative function of EPCs evolves in the proliferative diabetic retinopathy (PDR). PDR is associated with marked iNOS activation in the CD14+ EPC population. Increased levels of NO generated by a change of NOS isoform expression (from eNOS in health to iNOS in late diabetes) is associated with a phenotypic switch in these cells to a more inflammatory cell with secretion of proangiogenic growth factors and cytokines. This phase is characterized by pathological neovascularization seen in the diabetic retina.
Astrocytes, as mentioned earlier, are known to play a pivotal role in normal developmental retinal vascularization. Otani et al. showed that intravitreally injected Lin− BM cells selectively target retinal astrocytes, cells that serve as a template for both developmental and injury-associated retinal angiogenesis. When Lin− HSCs were injected into neonatal mouse eyes, they participated in normal developmental angiogenesis. When EPC-enriched HSCs were injected into the eyes of neonatal rd/rd mice, whose vasculature ordinarily degenerates with age, they prevented the retinal vascular degeneration and this vascular rescue was associated with neuronal rescue (Otani et al., 2002). Normal developmental vessels and exogenous progenitor cell targeting to astrocytes and the normal vascular plexus were both found to be dependent on the functional adhesion of R-cadherin as confirmed by disruption of HSCs targeting to the three distinct retinal vascular plexuses following R-cadherin blockade (Dorrell et al., 2004).
The role of glial cells during pathological retinal neovascularization is still under investigation. Loss of astrocytes and microglia directly correlates with the development of pathological NV in a mouse model of oxygen-induced retinopathy (OIR) (Smith et al., 1994). These two distinct glial cell populations were found to have cooperative survival effects in vitro and in vivo. The intravitreal injection of CD44hi myeloid progenitor cells (Ritter et al., 2006), astrocytes, or astrocyte-conditioned media (Dorrell et al.) rescued endogenous astrocytes from degeneration that normally occurs within the hypoxic, hyperoxia-induced vaso-obliterated retina following return to normoxia. Protection of the retinal astrocytes and microglia was directly correlated with accelerated revascularization of the normal retinal plexuses and reduction of pathological intravitreal neovascularization normally associated with OIR. Using astrocyte-conditioned media, several factors were identified that may contribute to the observed astrocytic protection and subsequent normalization of the retinal vasculature, including vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF). Injection of VEGF or bFGF at specific doses rescued the retinas from developing OIR-associated pathology, an effect that was also preceded by protection of endogenous glia from hypoxia-induced degeneration. These data suggest that vascular-associated glia are also required for normalized revascularization of the hypoxic retina and protecting glial cells may provide a novel strategy by which normalized revascularization can be promoted and the consequences of abnormal neovascularization in retinal vascular diseases can be prevented (Dorrell and Friedlander, 2006).
Another condition associated with pathological angiogenesis is age related macular degeneration. Csaky’s group demonstrated that EPCs, in the form of OECs, can be isolated and expanded successfully from the peripheral blood of elderly control and AMD-affected patients. They demonstrated significantly higher numbers of initial OEC clusters and expansion potential of OECs in patients at risk for or already affected by neovascular age-related macular degeneration (nvAMD). The group postulated that OECs may be used for further phenotypic, genetic, and functional analyses in patients with nvAMD (Thill et al., 2008).
Proliferative sickle cell retinopathy (PSR) is an uncommon complication in individuals with sickle cell trait, also called AS hemoglobinopathy (HAS), occurring more frequently in patients with SC hemoglobinopathy, S-thalassemia and SS hemoglobinopathy (Livrea et al., 1996). Interestingly, KDR+/CD34+/Cd45dim cells were found to be significantly higher during painful crisis (van Beem et al., 2009). Both PDR and PSR are associated with retinal ischemia, just in different locations. PSR is usually described as a peripheral neovascular tuft in a “sea fan” like configuration, while neovascularization of the disc (NVD) is a common finding in PDR. In PSR, the mechanism of the ischemia is retinal obliteration due to RBC plugging whereas in diabetic retinopathy the acellular capillaries are the initial source of ischemia. The possibility exists that mobilization of EPCs in sickle crisis could be due to injury to vessels throughout the body and even the bone marrow. It is highly likely that injured marrow is likely to result in “leakage” of these progenitor populations into the circulation rather than their orchestrated release. Also because sickle cell crisis is so painful, it is likely to initiate a surge of norepinephrine that will result in the release of these cells from the bone marrow into the circulation (Mendez-Ferrer and Frenette, 2009). Interestingly, we showed that in diabetes there is a defect in the release of these cells from the bone marrow due to peripheral neuropathy due to diabetes.
While van Beem et al. showed that there were higher counts of circulating endothelial progenitor cells during painful crisis, their studies did not find a direct relationship of EPCs levels with the established mobilizing growth factors (VEGF and IL-8) and concluded that the mechanism of EPC mobilization in sickle cell crisis is unknown.
EPCs in tumor angiogenesis
The existence of the bone marrow reservoir of progenitor cells and their involvement in neovascularization has attracted the interest of tumor biologists. The initial demonstration that EPCs contribute to tumor angiogenesis was demonstrated by Lyden et al. (Lyden et al., 2001). Transplantation of donor β-galactosidase+ (α-gal+) bone marrow from Rosa-26 mice into lethally irradiated angiogenesis-defective Id1-mutant mice revealed the presence of donor-derived LacZ+ BM cells in tumor vessels. Similarly, in humans previously transplanted with bone marrow cells from a sex-mismatched donor, the examination of secondary tumors revealed that 0.5–12% of tumor endothelial cells were donor-derived as determined using sex chromosome FISH analysis (Peters et al., 2005).
Tumor-derived paracrine signals activate the bone marrow compartment resulting in the mobilization and recruitment of discrete subsets of bone marrow-derived cells to the tumor bed (Figure 6). Recruited pro-angiogenic bone marrow-derived cells contribute significantly to neovasculature formation and tumor growth in adults (Kopp et al., 2008). Bone marrow-derived hematopoietic cells contribute to endothelial cells that contribute to neovessel formation directly and perivascular cells (Khakoo and Finkel, 2005). In response to tumor cytokines, including VEGF, putative VEGFR2+ EPCs mobilize into the peripheral circulation, and move to the tumor bed where they incorporate into sprouting neovessels (Lyden et al., 2001). Among the bone marrow derived cells, much attention has been given to the pro-angiogenic mural cells that are recruited to the tumor bed where they exert their functions perivascularly via paracrine release of pro-angiogenic cytokines (Kopp et al., 2006).
Figure 6. EPCs in tumor angiogenesis.
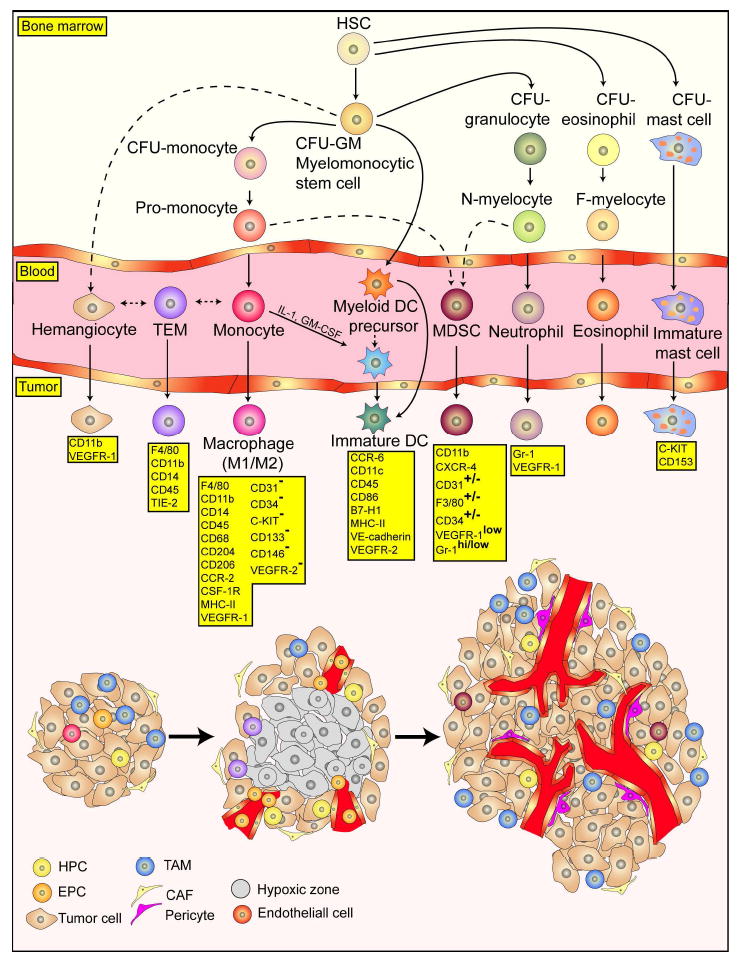
Bone marrow-derived hematopoietic stem cells supply endothelial cells that directly contribute to neovessel formation and perivascular cells. Putative VEGFR2+ EPCs mobilize into the peripheral circulation in response to tumor cytokines, including VEGF, and move to the tumor bed where they incorporate into sprouting neovessels. Several specific populations of bone marrow derived hematopoietic cells have been reported to contribute to tumor angiogenesis and invasion including GR1+CD11b+ myeloid progenitors, F4/80+ CD11b+ tumor-associated macrophages (TAMs), Tie2-expressing monocytes (TEMs), CXCR4+VEGFR1+ hemangiocytes, VE-cadherin+ CD45+ vascular leukocytes, and infiltrating mast cells and neutrophils. However, exactly how each of these cells contributes remains a topic of great debate. Figure adapted from Murdoch, et al. (Murdoch et al., 2008).
Several specific populations of bone marrow derived hematopoietic cells have been reported to contribute to tumor angiogenesis and invasion including GR1+CD11b+ myeloid progenitors (Yang et al., 2004), F4/80+ CD11b+ tumor-associated macrophages (TAMs) (Pollard, 2004), Tie2-expressing monocytes (TEMs) (De Palma et al., 2005), CXCR4+VEGFR1+ hemangiocytes (Jin et al., 2006), CD45+/CD11b+ myeloid cells (Grunewald et al., 2006), PDGFR+ pericyte progenitors (Song et al., 2005), VE-cadherin+ CD45+ vascular leukocytes (Soucek et al., 2007), and infiltrating mast cells and neutrophils (Soucek et al., 2007). However, exactly how each of these cells contributes remains a topic of great debate. Moreover, the general plasticity of these bone marrow cells and the rapidity with which they change surface markers makes these cells particularly elusive. Confusion in the field is further intensified by the wide variety of tumor models used, reviewed by Gao D, 2009 (Gao et al., 2009).
Recent studies have begun to set forward a notion that paracrine signaling by specific populations of cells that comprise a pathological microenvironment has significant biological effects. For example, depletion of myeloid cell-derived VEGF caused vasculature normalization even when abundant sources of VEGF were present in the tumor microenvironment (Stockmann et al., 2008). Blocking recruitment of specific subsets of bone marrow-derived cells such as neutrophils, macrophages, pericyte progenitors, and Tie2+ monocytes had dramatic effects in tumor progression (De Palma et al., 2003; Nozawa et al., 2006). Recombinant granulocyte colony-stimulating factor (G-CSF) is used for cancer patients with myelosuppression induced by chemotherapy. However, G-CSF promotes tumor angiogenesis by increasing circulating EPCs and Gr1+CD11b+ cells (Shaked et al., 2008). Thus, caution must be exercised in using G-CSF in cancer patients with residual tumors, diabetic patients, or patients with choroidal neo-vascularization (CNV).
Since most studies now agree that bone marrow-derived components of the tumor microenvironment are not merely passive bystanders, but serve critical roles in regulating tumor growth and metastasis (McAllister et al., 2008), these cells represent a novel target for therapeutic intervention. For example, the polyphenol epigallocatechin-3 gallate (EGCG) in green tea suppresses tumor growth and inhibits angiogenesis. Moreover, it reduces expression of MMP-9 mRNA in bone marrow stromal cells resulting in decreased EPC mobilization and increasing its therapeutic efficacy (Ohga et al., 2009).
Key regulators of the generation of endothelial progenitor cells
One key regulator appears to be Inhibitor of DNA binding 1 (Id1) which belongs to the helix–loop–helix (HLH) family of transcription factors (Perk et al., 2005). The Id1 knockout mice were critical in demonstrating that bone marrow-derived progenitors are the source of tumor endothelium, as Id1 knockout mice failed to mobilize these progenitors while BM transplantation of Id1 knockout mice with wild type BM was shown to rescue the observed vascular defects (Lyden et al., 2001). The importance of Id1+ progenitor cells was confirmed recently in “vascular rebound” that results from vascular disrupting therapies (Shaked et al., 2006). Id1 is expressed in the long term repopulating hematopoietic stem cells (lin− Sca+ kit+ CD34−) (Jankovic et al., 2007) and loss of Id 1 increases cyclin-dependent kinase inhibitor p21 expression which drives the stem cells towards a more committed myeloid state. This in turn is associated with the depletion of cells capable of endothelial cell fate commitment. These results suggest that Id1 is required in early hematopoietic stem cells to restrain the commitment to the myeloid lineage and protect the pool of cells that give rise to endothelial progenitors.
EPCs in aberrant angiogenesis: clinical translation
Selective targeting of EPCs has been heralded as a promising avenue for anti-angiogenic therapy whether in the eye or in tumors. EPCs are being considered as useful surrogate markers for monitoring the activity of PDR, cancer progression, and for optimizing the efficacy of anti-angiogenic therapies, such as anti-VEGFR2 antibody therapy (Bertolini et al., 2006). As clinical specimens need to be processed in “real time” and the timing of these specimens can be unpredictable, subpar testing conditions may result. Since the turning “on” of the angiogenic switch is probably not an event that happens over a prolonged period of time, observational studies in high risk cohorts are needed in order to better define the precise role of this and other cells in disease progression. Moreover, it must be kept in mind that circulating levels of these cells are not as important as the much harder to assess tissue levels.
Based on human and mouse work (Blinder and Fisher, 2008), there appears to be a peak in VEGFR2+ EPCs as tumors transition from micrometastatic to macrometastatic and recent observations suggest that EPCs are the main regulators of the “angiogenic switch” in progression from micrometastases to macrometastases. This suggests that their selective targeting may be a promising approach in cancer patients representing a paradigm shift in the field and a foundation for very exciting clinical applications (Gao et al., 2008).
Given that in many cancer patients metastatic spread has already occurred by the time of primary diagnosis, selectively targeting the angiogenic switch via endothelial progenitor cells may provide a clinically feasible approach to block metastatic progression and prevent death in cancer patients. Thus, combinations of adjuvant chemotherapy with anti-angiogenic agents to block recruitment of the endothelial progenitor cells may provide a highly effective strategy to impair the establishment of metastatic lesions. Indeed, EPC targeting will enhance the efficacy of other anti-cancer therapies, such as vascular disruptive agents and select chemotherapy drugs (Shaked et al., 2006). Similarly the concept of targeting these cells for treatment of PDR and CNV is likely to provide a new and viable approach for the management of ocular diseases associated with pathological neovascularization.
Emerging concepts and future directions
Understanding the mechanisms governing EPC activation and expansion in the BM compartment and their subsequent mobilization and recruitment to the areas of injury in the eye or tumor bed is critical to recognize the initiation of the angiogenesis program. Parallel technological advancements in BM transplantation systems (Figure 7), informative mouse genetic models of disease progression and cell-specific reporters have facilitated a more detailed investigation of these cells. Conspicuously, EPC ablation resulted in severe angiogenesis inhibition and impaired tumor growth and metastasis. Considering that cancer is the most aggressive of all the disease processes associated with pathological angiogenesis, it is likely that targeting these cells in ocular neovascularization may be even easier. Indeed, once in the incorporated state, recruited EPCs secrete numerous pro-angiogenic factors, suggesting that in addition to providing structural support to nascent vessels, EPCs have paracrine roles that may be essential in resident endothelial cell migration and proliferation at even the early stages of pathological neovascularization. One of many key questions remaining to be addressed is whether EPCs are necessary only for initiating neovessel formation or also for their maintenance. Finally, to move forward, it will be critical to identify EPC-specific markers and gene promoters so that direct single cell lineage tracing can be performed to unravel their true identity in both physiological and pathological settings.
Figure 7. Laser injury promotes recruitment of bone-marrow derived GFP+HSCs into the adult retinal vasculature.
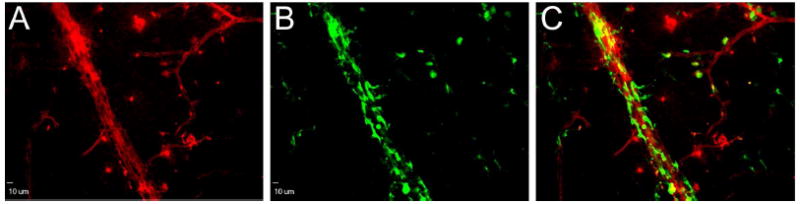
Confocal image of a retinal flatmount from a chimeric mouse that underwent laser retinal vessel occlusion injury, double stained with rhodamine-agglutinin (A) to depict retinal blood vessels and anti-GFP (B) to reveal GFP+ c-Kit+, Sca-1+ HSCs. C: Composite image of red (A) and green (B) channels. Scale bar: 10μm.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anghelina M, et al. Monocytes and macrophages form branched cell columns in matrigel: implications for a role in neovascularization. Stem Cells Dev. 2004;13:665–76. doi: 10.1089/scd.2004.13.665. [DOI] [PubMed] [Google Scholar]
- Anghelina M, et al. A subpopulation of peritoneal macrophages form capillarylike lumens and branching patterns in vitro. J Cell Mol Med. 2006;10:708–15. doi: 10.1111/j.1582-4934.2006.tb00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahara T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- Barber CL, Iruela-Arispe ML. The ever-elusive endothelial progenitor cell: identities, functions and clinical implications. Pediatr Res. 2006;59:26R–32R. doi: 10.1203/01.pdr.0000203553.46471.18. [DOI] [PubMed] [Google Scholar]
- Bertolini F, et al. The multifaceted circulating endothelial cell in cancer: towards marker and target identification. Nat Rev Cancer. 2006;6:835–45. doi: 10.1038/nrc1971. [DOI] [PubMed] [Google Scholar]
- Blinder V, Fisher SG. The role of environmental factors in the etiology of lymphoma. Cancer Invest. 2008;26:306–16. doi: 10.1080/07357900701805686. [DOI] [PubMed] [Google Scholar]
- Bronte V, et al. Tumor-induced immune dysfunctions caused by myeloid suppressor cells. J Immunother. 2001;24:431–46. doi: 10.1097/00002371-200111000-00001. [DOI] [PubMed] [Google Scholar]
- Brunner S, et al. Correlation of different circulating endothelial progenitor cells to stages of diabetic retinopathy: first in vivo data. Invest Ophthalmol Vis Sci. 2009;50:392–8. doi: 10.1167/iovs.08-1748. [DOI] [PubMed] [Google Scholar]
- Caballero S, et al. Ischemic vascular damage can be repaired by healthy, but not diabetic, endothelial progenitor cells. Diabetes. 2007;56:960–7. doi: 10.2337/db06-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case J, et al. Human CD34+AC133+VEGFR-2+ cells are not endothelial progenitor cells but distinct, primitive hematopoietic progenitors. Exp Hematol. 2007;35:1109–18. doi: 10.1016/j.exphem.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Cursiefen C, et al. Time course of angiogenesis and lymphangiogenesis after brief corneal inflammation. Cornea. 2006;25:443–7. doi: 10.1097/01.ico.0000183485.85636.ff. [DOI] [PubMed] [Google Scholar]
- De Palma M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211–26. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]
- De Palma M, et al. Targeting exogenous genes to tumor angiogenesis by transplantation of genetically modified hematopoietic stem cells. Nat Med. 2003;9:789–95. doi: 10.1038/nm871. [DOI] [PubMed] [Google Scholar]
- Domen J, et al. Bone Marrow (hematiopoietic) Stem Cells. Stem Cell Information: The National Institutes of Health resource for stem cell research. 2006 [Google Scholar]
- Dorrell MI, et al. Maintaining retinal astrocytes normalizes revascularization and prevents vascular pathology associated with oxygen-induced retinopathy. Glia. 58:43–54. doi: 10.1002/glia.20900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell MI, Friedlander M. Mechanisms of endothelial cell guidance and vascular patterning in the developing mouse retina. Prog Retin Eye Res. 2006;25:277–95. doi: 10.1016/j.preteyeres.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Dorrell MI, et al. Adult bone marrow-derived stem cells use R-cadherin to target sites of neovascularization in the developing retina. Blood. 2004;103:3420–7. doi: 10.1182/blood-2003-09-3012. [DOI] [PubMed] [Google Scholar]
- Engerman RL, et al. Cell turnover of capillaries. Lab Invest. 1967;17:738–43. [PubMed] [Google Scholar]
- Fadini GP, et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol. 2005;45:1449–57. doi: 10.1016/j.jacc.2004.11.067. [DOI] [PubMed] [Google Scholar]
- Gao D, et al. Bone marrow-derived endothelial progenitor cells contribute to the angiogenic switch in tumor growth and metastatic progression. Biochim Biophys Acta. 2009;1796:33–40. doi: 10.1016/j.bbcan.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, et al. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science. 2008;319:195–8. doi: 10.1126/science.1150224. [DOI] [PubMed] [Google Scholar]
- Grant MB, et al. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nat Med. 2002;8:607–12. doi: 10.1038/nm0602-607. [DOI] [PubMed] [Google Scholar]
- Grunewald M, et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell. 2006;124:175–89. doi: 10.1016/j.cell.2005.10.036. [DOI] [PubMed] [Google Scholar]
- Hur J, et al. Characterization of two types of endothelial progenitor cells and their different contributions to neovasculogenesis. Arterioscler Thromb Vasc Biol. 2004;24:288–93. doi: 10.1161/01.ATV.0000114236.77009.06. [DOI] [PubMed] [Google Scholar]
- Jankovic V, et al. Id1 restrains myeloid commitment, maintaining the self-renewal capacity of hematopoietic stem cells. Proc Natl Acad Sci USA. 2007;104:1260–5. doi: 10.1073/pnas.0607894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin DK, et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med. 2006;12:557–67. doi: 10.1038/nm1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joussen AM, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. Faseb J. 2004;18:1450–2. doi: 10.1096/fj.03-1476fje. [DOI] [PubMed] [Google Scholar]
- Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007;2007 doi: 10.1155/2007/95103. 95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakoo AY, Finkel T. Endothelial progenitor cells. Annu Rev Med. 2005;56:79–101. doi: 10.1146/annurev.med.56.090203.104149. [DOI] [PubMed] [Google Scholar]
- Kim SJ, et al. Circulating monocytes expressing CD31: implications for acute and chronic angiogenesis. Am J Pathol. 2009;174:1972–80. doi: 10.2353/ajpath.2009.080819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, et al. The Wisconsin Epidemiologic Study of Diabetic Retinopathy: XXII the twenty-five-year progression of retinopathy in persons with type 1 diabetes. Ophthalmology. 2008;115:1859–68. doi: 10.1016/j.ophtha.2008.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp HG, et al. Potential combination chemotherapy approaches for advanced adult-type soft-tissue sarcoma. Am J Clin Dermatol. 2008;9:207–17. doi: 10.2165/00128071-200809040-00001. [DOI] [PubMed] [Google Scholar]
- Kopp HG, et al. Contribution of endothelial progenitors and proangiogenic hematopoietic cells to vascularization of tumor and ischemic tissue. Curr Opin Hematol. 2006;13:175–81. doi: 10.1097/01.moh.0000219664.26528.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, et al. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J Exp Med. 2009;206:1089–102. doi: 10.1084/jem.20081605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusmartsev S, Gabrilovich DI. Immature myeloid cells and cancer-associated immune suppression. Cancer Immunol Immunother. 2002;51:293–8. doi: 10.1007/s00262-002-0280-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusmartsev S, Gabrilovich DI. Inhibition of myeloid cell differentiation in cancer: the role of reactive oxygen species. J Leukoc Biol. 2003;74:186–96. doi: 10.1189/jlb.0103010. [DOI] [PubMed] [Google Scholar]
- Kusuyama T, et al. Effects of treatment for diabetes mellitus on circulating vascular progenitor cells. J Pharmacol Sci. 2006;102:96–102. doi: 10.1254/jphs.fp0060256. [DOI] [PubMed] [Google Scholar]
- Lee IG, et al. Involvement of circulating endothelial progenitor cells and vasculogenic factors in the pathogenesis of diabetic retinopathy. Eye (Lond) 2006;20:546–52. doi: 10.1038/sj.eye.6701920. [DOI] [PubMed] [Google Scholar]
- Livrea MA, et al. Oxidative stress and antioxidant status in beta-thalassemia major: iron overload and depletion of lipid-soluble antioxidants. Blood. 1996;88:3608–14. [PubMed] [Google Scholar]
- Lyden D, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194–201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- McAllister SS, et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell. 2008;133:994–1005. doi: 10.1016/j.cell.2008.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Ferrer S, Frenette PS. Galpha(s) uncouples hematopoietic stem cell homing and mobilization. Cell Stem Cell. 2009;4:379–80. doi: 10.1016/j.stem.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Minami E, et al. Extracardiac progenitor cells repopulate most major cell types in the transplanted human heart. Circulation. 2005;112:2951–8. doi: 10.1161/CIRCULATIONAHA.105.576017. [DOI] [PubMed] [Google Scholar]
- Moldovan NI, Asahara T. Role of blood mononuclear cells in recanalization and vascularization of thrombi: past, present, and future. Trends Cardiovasc Med. 2003;13:265–9. doi: 10.1016/s1050-1738(03)00108-7. [DOI] [PubMed] [Google Scholar]
- Moldovan NI, et al. Contribution of monocytes/macrophages to compensatory neovascularization: the drilling of metalloelastase-positive tunnels in ischemic myocardium. Circ Res. 2000;87:378–84. doi: 10.1161/01.res.87.5.378. [DOI] [PubMed] [Google Scholar]
- Murdoch C, et al. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–31. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- Nozawa H, et al. Small interfering RNA targeting epidermal growth factor receptor enhances chemosensitivity to cisplatin, 5-fluorouracil and docetaxel in head and neck squamous cell carcinoma. Cancer Sci. 2006;97:1115–24. doi: 10.1111/j.1349-7006.2006.00287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohga N, et al. Inhibitory effects of epigallocatechin-3 gallate, a polyphenol in green tea, on tumor-associated endothelial cells and endothelial progenitor cells. Cancer Sci. 2009;100:1963–70. doi: 10.1111/j.1349-7006.2009.01255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani A, et al. Bone marrow-derived stem cells target retinal astrocytes and can promote or inhibit retinal angiogenesis. Nat Med. 2002;8:1004–10. doi: 10.1038/nm744. [DOI] [PubMed] [Google Scholar]
- Perk J, et al. Id family of helix-loop-helix proteins in cancer. Nat Rev Cancer. 2005;5:603–14. doi: 10.1038/nrc1673. [DOI] [PubMed] [Google Scholar]
- Peters BA, et al. Contribution of bone marrow-derived endothelial cells to human tumor vasculature. Nat Med. 2005;11:261–2. doi: 10.1038/nm1200. [DOI] [PubMed] [Google Scholar]
- Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–8. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- Poole JC, Florey HW. Changes in the endothelium of the aorta and the behaviour of macrophages in experimental atheroma of rabbits. J Pathol Bacteriol. 1958;75:245–51. doi: 10.1002/path.1700750202. [DOI] [PubMed] [Google Scholar]
- Portillo JA, et al. CD40 mediates retinal inflammation and neurovascular degeneration. J Immunol. 2008;181:8719–26. doi: 10.4049/jimmunol.181.12.8719. [DOI] [PubMed] [Google Scholar]
- Purhonen S, et al. Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proc Natl Acad Sci USA. 2008;105:6620–5. doi: 10.1073/pnas.0710516105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafii S, Lyden D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat Med. 2003;9:702–12. doi: 10.1038/nm0603-702. [DOI] [PubMed] [Google Scholar]
- Ritter MR, et al. Myeloid progenitors differentiate into microglia and promote vascular repair in a model of ischemic retinopathy. J Clin Invest. 2006;116:3266–76. doi: 10.1172/JCI29683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatteman GC, et al. Biology of bone marrow-derived endothelial cell precursors. Am J Physiol Heart Circ Physiol. 2007;292:H1–18. doi: 10.1152/ajpheart.00662.2006. [DOI] [PubMed] [Google Scholar]
- Segal MS, et al. Nitric oxide cytoskeletal-induced alterations reverse the endothelial progenitor cell migratory defect associated with diabetes. Diabetes. 2006;55:102–9. [PubMed] [Google Scholar]
- Shaked Y, et al. Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science. 2006;313:1785–7. doi: 10.1126/science.1127592. [DOI] [PubMed] [Google Scholar]
- Shaked Y, et al. Rapid chemotherapy-induced acute endothelial progenitor cell mobilization: implications for antiangiogenic drugs as chemosensitizing agents. Cancer Cell. 2008;14:263–73. doi: 10.1016/j.ccr.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LE, et al. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35:101–11. [PubMed] [Google Scholar]
- Song S, et al. PDGFRbeta+ perivascular progenitor cells in tumours regulate pericyte differentiation and vascular survival. Nat Cell Biol. 2005;7:870–9. doi: 10.1038/ncb1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L, et al. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat Med. 2007;13:1211–8. doi: 10.1038/nm1649. [DOI] [PubMed] [Google Scholar]
- Spring H, et al. Chemokines direct endothelial progenitors into tumor neovessels. Proc Natl Acad Sci USA. 2005;102:18111–6. doi: 10.1073/pnas.0507158102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockmann C, et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature. 2008;456:814–8. doi: 10.1038/nature07445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thill M, et al. Late outgrowth endothelial progenitor cells in patients with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:2696–708. doi: 10.1167/iovs.07-0955. [DOI] [PubMed] [Google Scholar]
- van Beem RT, et al. Elevated endothelial progenitor cells during painful sickle cell crisis. Exp Hematol. 2009;37:1054–9. doi: 10.1016/j.exphem.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6:93–106. doi: 10.1038/nri1779. [DOI] [PubMed] [Google Scholar]
- Yang L, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–21. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Yin T, Li L. The stem cell niches in bone. J Clin Invest. 2006;116:1195–201. doi: 10.1172/JCI28568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder MC, et al. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007;109:1801–9. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]