

Abstract
Neuropeptide tachykinins (substance P, neurokinin A, and neurokinin B) are present in peripheral terminals of sensory nerve fibers within the respiratory tract and cause airway contractile responses and hyperresponsiveness in humans and most mammalian species. Three subtypes of neurokinin receptors (NK1R, NK2R, and NK3R) classically couple to Gq protein-mediated inositol 1,4,5-trisphosphate (IP3) synthesis and liberation of intracellular Ca2+, which initiates contraction, but their expression and calcium signaling mechanisms are incompletely understood in airway smooth muscle. All three subtypes were identified in native and cultured human airway smooth muscle (HASM) and were subsequently overexpressed in HASM cells using a human immunodeficiency virus-1-based lentivirus transduction system. Specific NKR agonists {NK1R, [Sar9,Met(O2)11]-substance P; NK2R, [β-Ala8]-neurokinin A(4–10); NK3R, senktide} stimulated inositol phosphate synthesis and increased intracellular Ca2+ concentration ([Ca2+]i) in native HASM cells and in HASM cells transfected with each NKR subtype. These effects were blocked by NKR-selective antagonists (NK1R, L-732138; NK2R, GR-159897; NK3R, SB-222200). The initial transient and sustained phases of increased [Ca2+]i were predominantly inhibited by the IP3 receptor antagonist 2-aminoethoxydiphenyl borate (2-APB) or the store-operated Ca2+ channel antagonist SKF-96365, respectively. These results show that all three subtypes of NKRs are expressed in native HASM cells and that IP3 levels are the primary mediators of NKR-stimulated initial [Ca2+]i increases, whereas store-operated Ca2+ channels mediate the sustained phase of the [Ca2+]i increase.
Keywords: intracellular calcium, ryanodine, actin, myosin, lentivirus
Tachykinins are stored and released from unmyelinated pulmonary C fibers in airways (8, 30, 31, 44) and evoke inflammatory peripheral effects including vasodilation, plasma extravasation, leukocyte adhesion, facilitation of cholinergic neurotransmission, and epithelial cell secretion in the airways (1, 12, 16), which are referred to as neurogenic inflammation. Local release of tachykinins from peripheral sensory nerves may also lead to airway contractile responses and hyperresponsiveness in human and most mammalian species (32, 42). At least three subtypes of neurokinin receptors (NK1R, NK2R, and NK3R) have been characterized, which exhibit preferential affinity for the endogenous neuropeptides substance P, neurokinin A, and neurokinin B, respectively (33). All three receptor subtypes are members of the seven-transmembrane domain receptor superfamily that couple to Gq proteins (50).
Classically, the expression of NK1R and NK2R has been described in both the nervous system and peripheral target organs, including human airways, whereas the expression of NK3R was believed to be more limited to the central and peripheral nervous system (31, 34). Furthermore, it has traditionally been thought that only the NK2R mediated direct contraction of airway smooth muscle, especially in human airways (3). However, recently it was reported that in small-diameter human bronchi, tachykinins also cause contraction via NK1R stimulation (4). Moreover, functional evidence for the presence of NK3R exists in both animal and human airways (24, 38, 40, 43). Although most functional studies suggest that the NK3R subtype is not directly involved in airway smooth muscle constriction (2, 13, 14, 53), an NK3R antagonist was mildly potent at inhibiting a neurokinin A-induced contraction in human bronchus (51).
Early studies using RNase protection assays detected mRNA encoding the NK1R and NK2R but not the NK3R in human bronchi (6). However, more recently, mRNA encoding the NK3R was identified by RT-PCR in human bronchi (48), although the specific lung cell type on which the NK3R is expressed is unclear. The NK1R and NK2R proteins were immunohistochemically detected in human bronchial smooth muscle of central airways, but the presence of the NK3R protein was not evaluated (34). Thus the expression and functional coupling of the NK3R on human airway smooth muscle is unclear.
In airway smooth muscle, increases in intracellular Ca2+ concentration ([Ca2+]i) are mediated by intracellular release from sarcoplasmic reticulum (SR) via both ryanodine and inositol 1,4,5-trisphosphate (IP3) receptors (7) and by influx of extracellular Ca2+, which can occur via voltage-gated and receptor-gated channels, as well as in response to SR Ca2+ depletion (store-operated Ca2+ entry; SOCE) (11, 35, 49). Although NKR agonist-induced IP3 accumulation is thought to play a predominant role in the initiation of increases in [Ca2+]i and the initiation of contraction in airway smooth muscle (4, 17), detailed intracellular Ca2+ signaling mechanisms induced by activation of NKR subtypes are not fully understood in HASM.
In the present study, we questioned whether all three known subtypes of tachykinin receptors are expressed in airway smooth muscle cells and whether each of these subtypes is capable of coupling to the activation of inositol phosphate synthesis and elevation of intracellular calcium, and we hypothesized that the predominant mechanism of increased intracellular Ca2+ occurred via activation of IP3 receptors.
METHODS
Materials
Cells were cultured in SmGM-2 smooth muscle medium (Lonza, Walkersville, MD). Fluo-4 AM and Pluronic F-127 were obtained from Molecular Probes (Eugene, OR). Myo-[3H]inositol (20 Ci/mmol) was obtained from MP Biomedicals (Irvine, CA). [Sar9,Met(O2)11]-substance P (SM-SP), [β-Ala8]-neurokinin A(4–10) (β-ala-NKA), 2-aminoethoxydiphenyl borate (2-APB), and ryanodine were obtained from Calbiochem (San Diego, CA). GR-159897 was obtained from Tocris Bioscience (Ellisville, MO). All other chemicals were obtained from Sigma (St. Louis, MO) unless otherwise stated.
Cell culture
Primary cultures of human tracheal smooth muscle cells were obtained from lung transplant donors in accordance with procedures approved by the University of Pennsylvania Committee on Studies Involving Human Beings as previously described (47). For the measurement of [Ca2+]i, the cells were grown to confluence on 96-well plates in culture medium (SmGM-2 supplemented with 5% FBS, 5 μg/ml insulin, 1 ng/ml human fibroblast growth factor, 500 pg/ml human epidermal growth factor, 30 μg/ml gentamicin, and 15 ng/ml amphotericin B; Lonza) at 37°C in an atmosphere of 5% CO2-95% air.
RT-PCR of NKRs in native HASM
Studies were approved by Columbia University’s Institutional Review Board and deemed not human subjects research under 45 CFR 46. Human trachea came from two sources. Snap-frozen tracheas obtained at autopsy from nonasthmatic adults within 8 h of death were obtained from the National Disease Research Exchange (Philadelphia, PA). Additional tracheas were obtained from discarded regions of healthy donor lungs harvested for lung transplantation at Columbia University. The exteriors of human tracheas were dissected free of adherent connective tissue under a dissecting microscope. Tracheas were then cut open longitudinally along the anterior border. The tracheal epithelium was removed, and the airway smooth muscle between the noncontiguous ends of the cartilaginous tracheal rings was dissected. Total RNA was extracted from freshly dissected native human smooth muscle or cultured HASM cells using TRI reagent (Ambion, Austin, TX) according to the manufacturer’s recommendations. Total RNA from whole human brain (Clontech, Mountain View, CA) was used as a positive control. With the use of the Advantage RT-for-PCR kit (Clontech), 1 μg of total RNA was reverse transcribed at 42°C for 1 h in 20 μl including 200 units of Moloney murine leukemia virus reverse transcriptase, 20 units of RNase inhibitor, 20 pmol of oligo(dT) primer, and 0.5 mM each of dNTP mix in reaction buffer (50 mM Tris•HCl, pH 8.3, 75 mM KCl, and 3 mM MgCl2).
PCR was performed by adding 5 μl of newly synthesized cDNA to a 45-μl reaction mixture, yielding final concentrations of 0.2 mM of each dNTP, 1× Advantage 2 polymerase mix, PCR buffer (Clontech), and 0.4 μM of both sense and antisense primers corresponding to the three human neurokinin receptors [NK1R: sense primer, 5′-TCC ACA TCT TCT TCC TCC TGC CCT ACA T-3′; antisense primer, 5′-GCT TGA AGC CCA GAC GGA ACC TGT-3′, amplicon size, 162 bp; NK2R: sense primer, 5′-CCA TCT GCT GGC TGC CCT ACC ACC TCT A-3′; antisense primer, 5′-GTG TCC CCA GCC ATG AAC AAA GTC TCC T-3′; amplicon size, 310 bp; NK3R: sense primer, 5′-GGC TGG CAA TGA GCT CAA CCA TGT ACA A-3′; antisense primer, 5′-CAT CCA CAG AGG TAT AGG GTG AGC TTA TGA AAC TT-3′; amplicon size, 360 bp]. Two-step PCR (annealing and extension at the same temperature) was performed with a PTC-200 Peltier thermal cycler (MJ Research, Waltham, MA). Primer sets were designed to span at least one gene intron to avoid the confounding effect of amplifying contaminating genomic DNA within the total RNA samples. PCR conditions for all reactions included an initial denaturation step at 94°C for 1 min, 40 cycles of a denaturation step at 94°C for 10 s, and an annealing/extension step at 72°C for 1 min. PCR products were electrophoresed on 5% nondenaturing polyacryl-amide gel in 1× Tris-acetate-EDTA buffer. The gel was stained with ethidium bromide (Molecular Probes), visualized using ultraviolet illumination, and analyzed using Quantity One software (Bio-Rad, Hercules, CA).
RT-PCR of ryanodine receptors
Total RNA was extracted from primary cultures and NKR-transduced HASM cells as described above. Total RNA from human heart and skeletal muscle were components of the human total RNA Master Panel II (Clontech) and were used as positive controls. The Advantage RT-for-PCR kit was used as described, utilizing primers that transversed at least one intron for each ryanodine receptor (Ryr) subtype: [Ryr1: sense primer, 5′-ATC TGC AGA AGG ACA TGG TGG TGA TGT TGC T-3′; antisense primer, 5′-AAC ATG TCG AAG AAC TTG AGG ATC ATC TCC ACA TT-3′; Ryr2: forward primer, 5′-GGC GTT CAA TTT CTT CCG AAA ATT CTA CAA TAA AAG-3′; reverse primer, 5′-CCC TCC TCC AGC ACG AAC TCC AAC AT-3′; Ryr3: forward primer, 5′-CTG TCA TCT GTA ACT CAC AAT GGC AAA CAG-3′; reverse primer, 5′-ATG GCC AGC AAG ATG ACA ATG ACG AAG AAG-3′]. PCR products were analyzed as described above.
Production of lentiviral vectors and transduction of cultured HASM cells
Plasmids (pcDNA 3.1+) containing the coding sequence for the human (hu) NK1R, NK2R, and NK3R were purchased from the UMR cDNA Resource Center (University of Missouri at Rolla). Coding regions were excised (EcoRI and XbaI for NK1R and NK2R) and subcloned into these same restriction sites in the lentiviral vector Δu6-pLL-IRES-EGFP. Because of the presence of an internal EcoRI restriction endonuclease recognition site, huNK3R was excised from pcDNA 3.1+ using HindIII and XbaI and subcloned into pBluescript. Subsequently, it was excised using HindIII (blunt) and NotI and subcloned into EcoRI (blunt) and NotI in Δu6-pLL-IRES-EGFP. When expressed, this vector yields two independent proteins, enhanced green fluorescent protein (EGFP) and the human tachykinin receptor subtype of interest. Vesicular stomatitis virus G (VSVG)-pseudotyped human immunodeficiency virus (HIV) vectors were generated by cotransfecting the lentivirus shuttle vector (Δu6-pLL-IRES-EGFP-huNK1R, -huNK2R, or -huNK3R) with the HIV-1 packaging vector pCMVΔR8.91 (57) and the pMD.G plasmid (45) encoding the VSVG envelope glycoprotein into HEK-293FT cells. Briefly, 10 μg of Δu6-pLL-IRES-EGFP-huNKxR, 5 μg of pCMVΔR8.91, and 7 μg of pMD.G were cotransfected into 80–90% confluent HEK-293FT cells in 10-cm tissue culture plates using 20 μl of Lipofectamine 2000 (Invitrogen, Carlsbad, CA) in serum-free OptiMEM medium according to the manufacturer’s recommendations. After 6 h, the medium was replaced with DMEM supplemented with 10% FBS and penicillin/streptomycin (Pen-Strep). The cell culture medium from the HEK-293FT cells was collected after 72 h and passed through a 0.45-μm filter, and the virus titer was increased ~10-fold by ultrafiltration (Amicon Ultra 100,000 MW cutoff; Millipore, Bedford, MA). Polybrene was added to a final concentration of 8 μg/ml. Cell culture medium (SmGM-2) was removed from passage 3 HASM cells growing on six-well plates and replaced with 1.5 ml of lentivirus medium (i.e., DMEM-10% FBS-Pen-Strep) and incubated at 37°C. After 24 h, the lentivirus medium was replaced with cell culture medium (SmGM-2). Infected HASM cells were propagated and used for studies between passages 6 and 8.
Inositol phosphate assays
Synthesis of total [3H]inositol phosphates was measured in confluent native HASM cells and in HASM cells stably transfected with the NK1R, NK2R, or NK3R in 24-well tissue culture plates as described previously (21, 25). Briefly, after overnight loading with myo-[3H]inositol (10 μCi/ml, 20 Ci/mmol) in inositol-free and serum-free DMEM (Chemicon, Temecula, CA), plates were washed three times [37°C, 500 μl of Hanks’ balanced salt solution (HBSS) with 10 mM LiCl]. Incubation of cells with NKR-selective agonists (0.01 nM-10 ΔM; NK1R, SM-SP; NK2R, β-ala-NKA; NK3R, senktide) in a final volume of 300 μl at 37°C for 30 min was performed in the absence and presence of NKR antagonists (100 μM) (NK1R, L-732138; NK2R, GR-159897; NK3R, SB-222200). Reactions were terminated, and [3H]inositol and total [3H]inositol phosphates were recovered using chromatography (21).
Measurement of [Ca2+]i
Confluent native HASM cells or HASM cells stably transfected with the NK1R, NK2R, or NK3R in 96-well plates were incubated in modified HBSS (in mM: 138 NaCl, 5.3 KCl, 2.5 CaCl2, 0.4 MgSO4, 0.49 MgCl2, 0.34 Na2HPO4, 4.2 NaHCO3, 0.44 KH2PO4, 5.5 dextrose, and 20 HEPES, pH 7.4) at 100 μl/well containing 5 μM fluo-4 AM (DMSO vehicle final concentration: 0.5%), 0.05% Pluronic F-127 (DMSO vehicle final concentration: 0.25%), and 2.5 mM probenecid for 30 min at 37°C. Once the cells were loaded, the cells were washed twice with modified HBSS containing 2.5 mM probenecid and left for an additional 30 min at room temperature to allow complete deesterification of the intracellular acetoxy methyl esters. This buffer was exchanged (100 μl/well) just before the measurement of fluorescence was started. The fluorescence was then continuously recorded every 5 s at excitation wavelengths of 485 nm and emission wavelengths of 528 nm using a microplate reader (Synergy HT; BioTek Instruments, Winooski, VT). Triplicate wells were simultaneously measured, and values were averaged for each data point. After a stable baseline was established for the first 10 min, the cells were pretreated with inhibitors [10 μM verapamil, an L-type voltage-gated Ca2+ channel blocker; 10 μM SKF-96365, a SOCC blocker; 10 μM 2-APB, an IP3 receptor antagonist; 100 μM ryanodine, a ryanodine receptor antagonist; or 10 mM caffeine, a ryanodine receptor agonist that leads to Ca2+ depletion in the SR] or vehicle (modified HBSS) for 10 min. Baseline was acquired for 2 min just before addition of NKR-selective agonists. The cells were then incubated with 1 μM NKR-selective agonist (SM-SP, β-ala-NKA, or senktide), and the fluorescence was recorded for 10 min. In all studies, the fluorescence after treatments is presented as the percent change from baseline fluorescence.
Measurement of store-operated Ca2+ entry
These experiments were designed to examine the effects of 2-APB, caffeine, or ryanodine on store-operated Ca2+ entry (SOCE) in HASM. SOCE was evaluated using previously described protocols (5, 46, 49). Extracellular Ca2+ was removed by exposure to 0-Ca2+ modified HBSS containing 1 mM EGTA for 10 min. Cells were exposed to 1 μM verapamil and 10 mM KCl to inhibit L-type Ca2+ channels and depolarize cells, respectively. Cells were then exposed to 10 μM cyclopiazonic acid [CPA; an inhibitor of SR Ca2+-ATPase (SERCA)] in 0-Ca2+ modified HBSS with 1 mM EGTA, resulting in passive SR depletion with continued SR Ca2+ leak (likely from both IP3- and ryanodine-sensitive SR stores). As in previous studies (5, 46), a gradual elevation of [Ca2+]i was typically noted that eventually reached a plateau or started trending downward (because plasma membrane Ca2+ efflux was not inhibited). Pretreatment with SKF-96365 (10 μM; positive control inhibitor), 2-APB (10 μM), ryanodine (100 μM), or caffeine (10 mM) was performed, and then CaCl2 (to achieve a final extracellular Ca2+ concentration of 2.5 mM) was rapidly reintroduced (in the continued presence of verapamil, KCl, and CPA) and the peak fluorescence was measured.
Western blot analysis of the heavy chain of smooth muscle-specific myosin
Confluent HASM cells either nontransduced or stably transduced with the NK1R, NK2R, or NK3R were scraped from T75 flasks in ice-cold lysis buffer [50 mM Tris•HCl, pH 8.0, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 1 mM EDTA, 1:200 dilution of protease inhibitor cocktail III (Calbiochem), 1 mM Na3VO4, and 1 mM NaF]. Freshly dissected human tracheal airway smooth muscle was homogenized on ice using a Tekmar homogenizer set at top speed for 30 s in the same lysis buffer. After centrifugation (15,000 g, 15 min, 4°C) of the whole cell lysate, the supernatant was saved and protein concentrations were determined. Aliquots of the supernatants were solubilized by heating at 95°C for 5 min in sample buffer (final concentrations: 50 mM Tris•HCl, pH 6.8, 2.5% SDS, 6% glycerol, 2.5% 2-mercaptoethanol, and bromphenol blue) and stored at −20°C. The supernatants of solubilized whole cell or tissue lysates were electrophoresed through a discontinuous SDS-PAGE gel (3% stacking over 5% separating) and transferred to polyvinylidene difluoride (PVDF) membranes. The PVDF membranes were blocked for 2 h at room temperature with 5% milk in Tris-buffered saline (TBS) with 0.1% Tween 20 (TBS-T) and then probed overnight while shaking at 4°C with a 1:1,000 dilution of mouse monoclonal antibody in 1% milk in TBS-T directed against the heavy chain of smooth muscle-specific myosin (MAB3570; Chemicon). After being washed three times, PVDF membranes were incubated for 2 h at room temperature with horseradish peroxidase-conjugated secondary anti-mouse antibodies (1:5,000 dilution in 1% milk in TBS-T; Amersham Biosciences NA931V). The signal from the immunoreactive bands was detected using enhanced chemiluminescence (ECL Plus; Amersham Biosciences) according to the manufacturer’s recommendations, developed on film (Kodak Biomax light film; Kodak, Rochester, NY), and detected using Quantity One software (Bio-Rad).
Filamentous-to-globular actin ratio measurements
Basal and agonist-induced ratios of filamentous (F) and globular (G) actin were determined as previously described (55), except that a blue fluorescence-tagged phalloidin was used instead of a green FITC-tagged phalloidin because of the coexpression of green fluorescent protein (GFP) in these NKR subtype transduced cells. Cells at 70% confluence on eight-well microscope slides were exposed to vehicle (water) or NKR subtype-specific agonists (10 μM for 5 min) before being fixed by the addition of an equal volume of 7.4% paraformaldehyde in PBS to achieve a final concentration of 3.7% paraformaldehyde. After permeabilization with Triton X-100 (0.5% in PBS, 5 min) and blocking (1% BSA in 0.1% Triton-X100 in PBS, 10 min), cells were stained with Alexa Fluor 350-phalloidin (3 U/ml; Invitrogen) and Alexa Fluor 594-DNase I (10 μg/ml; Invitrogen) in 300 μl of blocking solution in the dark for 20 min. After being washed (twice in 0.1% Triton X-100 in PBS and twice in PBS), a coverslip was mounted and visualized with an inverted fluorescent microscope (Olympus IX-70). Digitized images were quantified as described previously (20).
Statistical analysis
Statistical analysis was performed using repeated measures of ANOVA, followed by Bonferroni posttest comparison using Prism 4.0 software (GraphPad, San Diego, CA). Data are means ± SE. P < 0.05 was considered significant.
RESULTS
Expression of native and overexpressed NKRs in HASM cells
Messenger RNA encoding the NK1R, NK2R, and NK3R was detected in total RNA extracted from freshly dissected airway smooth muscle from human bronchi and in primary cell cultures of HASM cells by RT-PCR (Fig. 1). Bands of expected size (162, 310, and 360 bp, respectively) were detected in at least three independent RNA samples from each source and in total RNA from whole human brain used as a positive control.
Fig. 1.
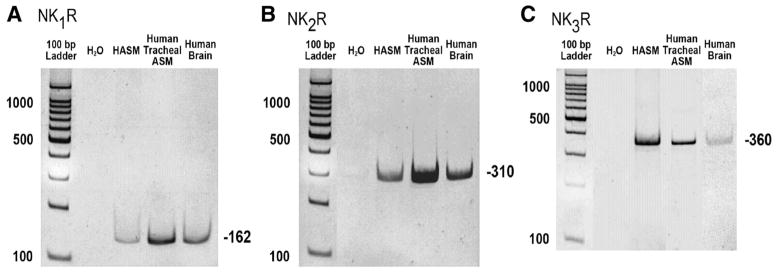
Endogenous neurokinin receptor (NKR) expression in human airway smooth muscle (HASM) cells. Representative RT-PCR analysis of total RNA using primers that specially recognize mRNA for each NKR subtype [NK1R (A), NK2R (B), and NK3R (C)] in freshly dissected human tracheal airway smooth muscle (ASM) and in primary cultures of HASM cells. Human brain cDNA was used as a positive control.
We first established functional coupling of each tachykinin receptor subtype in native cultured airway smooth muscle cells by using receptor subtype-specific agonist stimulation of inositol phosphate synthesis. In native HASM cells, all three NKR-selective agonists (10 μM SM-SP, 10 μM β-ala-NKA, and 10 μM senktide) significantly increased inositol phosphate synthesis (SM-SP: P < 0.05, n = 7; β-ala-NKA: P < 0.05, n = 7; senktide: P < 0.05, n = 10) (Fig. 2). We next demonstrated in native cultured airway smooth muscle cells that subtype-selective agonists selective for the NK1R, NK2R, and NK3R increased [Ca2+]i (Fig. 3).
Fig. 2.
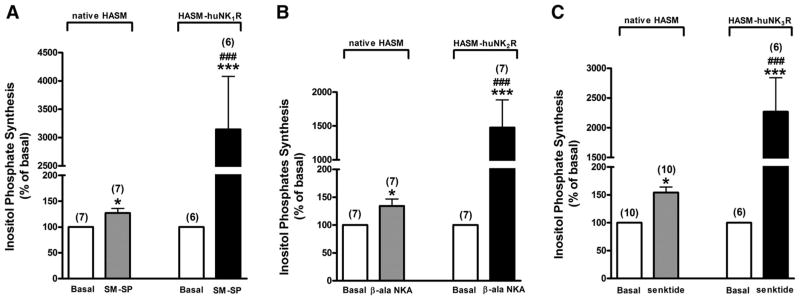
NKR subtype-selective agonist effects on inositol phosphate synthesis. Effects of selective NKR agonists [Sar9,Met(O2)11]-substance P (SM-SP; 10 μM) for NK1R (A), [β-Ala8]-neurokinin A(4–10) (β-ala-NKA; 10 μM) for NK2R (B), and senktide (10 μM) for NK3R (C) on total inositol phosphate synthesis in both native cultured HASM and NKR-transduced cultured HASM (HASM-huNK1R, -huNK2R, -huNK3R) cells. Data are means ± SE, presented as percentages of basal values. *P < 0.05; ***P < 0.001 compared with basal. ###P < 0.001 compared with NKR agonist in native HASM. Numbers in parentheses indicate the number of experiments.
Fig. 3.

NKR subtype-selective agonist effects on intracellular Ca2+ concentration ([Ca2+]i). A: representative trace of time plotted against fluorescence intensity (ΔF/Fo) illustrating the characteristics of the NKR agonist-induced [Ca2+]i increase in HASM-NKxR cells. The responses to NKR agonists were “biphasic,” with a transient rise in [Ca2+]i followed by a sustained [Ca2+]i increase lasting many minutes. Int., interruption of fluorescence measurement due to drug addition. Dotted line indicates the estimated fluorescence intensity during the interruption. B: effects of selective NKR agonists [NK1R, 1 μM SM-SP; NK2R, 1 μM β-ala-NKA; NK3R, 1 μM senktide] and vehicle control (modified HBSS) on [Ca2+]i in native cultured HASM cells. Peak fluorescence increase was measured and expressed as a maximum compared with basal values (F/Fo). Data are means ± SE. *P < 0.05; **P < 0.01 compared with vehicle control. Numbers in parentheses indicate the number of experiments.
To enhance the expression of each receptor subtype to facilitate signaling studies, we transduced primary cell lines of HASM cells with lentivirus encoding each of the human NKR subtypes along with coexpressed GFP. Transfected HASM cells exhibited bright green fluorescence 3–5 days after transduction (Fig. 4). Approximately 70–80% of the HASM cells routinely expressed GFP at 3–5 days.
Fig. 4.

Lentivirus-mediated overexpression of NKR subtypes in HASM cells. Representative images of fluorescent microscopy in native cultured HASM cells (negative control; A) and lentivirus-mediated transduction of huNK1R (B), huNK2R (C), and huNK3R (D) in cultured HASM cells. HASM cells expressed green fluorescent protein (GFP) 3–5 days after lentivirus vector exposure. Original magnifications, ×200.
Confirmation of smooth muscle cell phenotype in transduced cells
Two independent studies were performed to confirm that both native and NKR-transduced HASM cells retained a smooth muscle phenotype. We confirmed the expression of myosin by immunoblotting and confirmed activation of the actin cytoskeleton (19, 20, 54) by NKR agonists. An antibody specific for the heavy chain of smooth muscle myosin detected a 204-kDa protein in solubilized lysates of freshly dissected HASM and in native cultured HASM cells as well as cultured HASM cells transduced with the NK1R subtype (Fig. 5A).
Fig. 5.

Confirmation of smooth muscle cell phenotype of NKR-transduced HASM cells. A: representative immunoblot identifying expression of the 204-kDa smooth muscle-specific heavy chain of myosin in freshly dissected human tracheal ASM and in primary (HASM) and transduced (HASM-huNK1R) cultures of HASM cells. B: fluorescent staining ratios of filamentous and globular actin (F/G actin) in NKR-transduced HASM cells (HASM-huNK1R, HASM-huNK2R, and HASM-huNK3R) under untreated (control) or subtype-specific agonist (10 μM)-treated conditions [NK1 agonist, SM-SP (n = 11); NK2 agonist, β-ala-NKA (n = 12); NK3 agonist, senktide (n = 8)]. In each cell type, subtype-specific activation of the NKR increased F actin, which is a component of smooth muscle cell contraction.
Similarly, activation of the actin cytoskeleton, a process required for smooth muscle contraction, was induced by the respective specific agonists for each NKR subtype (Fig. 5B). In NK1R-transduced HASM cells, 10 μM SM-SP for 5 min increased the basal F/G actin ratio from 2.6 ± 0.13 to 3.6 ± 0.25 (n = 11, P = 0.003).
In NK2R-transduced HASM cells, 10 μM β-ala-NKA for 5 min increased the basal F/G actin ratio from 2.3 ± 0.11 to 3.5 ± 0.26 (n = 12, P = 0.0004). In NK3R-transduced HASM cells, 10 μM senktide for 5 min increased the basal F/G actin ratio from 2.3 ± 0.08 to 3.0 ± 0.24 (n = 8, P = 0.016).
Effect of NKR-selective agonists on inositol phosphate synthesis
In these HASM cell lines individually transfected with each of the huNKRs, NKR-selective agonists significantly increased inositol phosphate synthesis (SM-SP: P < 0.001, n = 6; β-ala-NKA: P < 0.001, n = 7; and senktide: P < 0.001, n = 6 in HASM-huNK1R, -huNK2R, and -huNK3R cells, respectively), which was significantly greater than that in native cells (SM-SP: P < 0.001; β-ala-NKA: P < 0.001; and senktide: P < 0.001) (Fig. 2). These increases were virtually abolished by pretreatment with 100 μM NKR-selective antagonists (L-732138, GR-159897, and SB-222200) (P < 0.001, n = 6; P < 0.001, n = 4; and P < 0.001, n = 6 in HASM-huNK1R, -huNK2R, and -huNK3R cells, respectively) (Fig. 6).
Fig. 6.
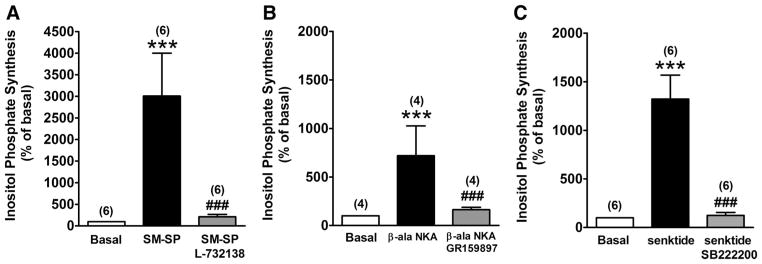
NKR subtype-selective antagonists block effects of selective agonists on inositol phosphate synthesis. Effects of selective NKR antagonists L-732138 (100 μM), GR-159897 (100 μM), and SB-222200 (100 μM) on selective NKR agonist [SM-SP (1 μM) for NK1R, β-ala-NKA (1 μM) for NK2R, and senktide (1 μM) for NK3R]-stimulated total inositol phosphate synthesis in HASM-huNK1R (A), HASM-huNK2R (B), and HASM-huNK3R cells (C), respectively. Data are means ± SE, presented as percentages of basal values. ***P < 0.001 compared with basal. ###P < 0.001 compared with selective NKR-selective agonists. Numbers in parentheses indicate the number of experiments.
In all three HASM-huNKR cell lines, NKR-selective agonists (SM-SP, β-ala-NKA, and senktide, 0.01 nM–10 μM) increased both inositol phosphate synthesis and peak [Ca2+]i mobilization in a concentration-dependent manner. At higher concentrations (10 μM), all relatively selective NKR agonists activated all three NKR subtypes but had markedly lower affinities compared with that to their preferred receptor subtype (Fig. 7). Thus each of the subtype-specific agonists displayed expected selectivity for their respective human NKR subtype.
Fig. 7.

Concentration-dependent effects of agonists on inositol phosphate. Concentration-dependent effects of NKR-selective agonists SM-SP (●; NK1R), β-ala-NKA (▲; NK2R), and senktide (■; NK3R) on total inositol phosphate synthesis (a) and peak (transient) fluorescence increase (b) in HASM-huNK1R (A), HASM-huNK2R (B), and HASM-huNK3R cells (C), respectively. Data are means ± SE; for inositol phosphate assays, data are presented as percentages of basal values, and for the measurement of Ca2+, data are presented as percent change (ΔF) from baseline fluorescence (Fo). Numbers in parentheses indicate the number of experiments.
Effect of NKR-selective receptor agonists on [Ca2+]i response
In HASM cells, exposure to G protein-coupled receptor (GPCR) agonist such as bradykinin, histamine, and acetylcholine typically resulted in a characteristic “biphasic” [Ca2+]i response, with an initial, significantly higher peak (transient phase) followed by a sustained elevation significantly above the baseline (sustained phase) (39). Likewise, NKR-selective agonists (1 μM SM-SP, 1 μM β-ala-NKA, and 1 μM senktide) produced the biphasic [Ca2+]i response in HASM-huNKR cells (Fig. 3, top). To characterize the mechanisms of NKR-mediated [Ca2+]i mobilization in both the transient and sustained phases, HASM-huNKR cells were pretreated for 5 min with 1) verapamil (10 μM), 2) SKF-96365 (10 μM), 3) 2-APB (10 μM), 4) ryanodine (100 μM), or 5) caffeine (10 mM) and then stimulated with NKR agonists. In the transient phase, 2-APB predominantly inhibited the [Ca2+]i mobilization (57.39 ± 9.65%, P < 0.001, n = 11; 44.07 ± 5.84%, P < 0.001, n = 8; and 69.49 ± 8.27%, P < 0.001, n = 8 in HASM-huNK1R, -huNK2R, and -huNK3R cells, respectively), whereas verapamil (9.65 ± 3.61%, P < 0.05, n = 8; 11.62 ± 7.28%, P < 0.05, n = 10; and 13.35 ± 4.74%, P < 0.01, n = 9 in HASM-huNK1R, -huNK2R, and -huNK3R cells, respectively), SKF-96365 (7.61 ± 3.25%, P < 0.05, n = 9; 18.17 ± 3.34%, P < 0.001, n = 11; and 9.53 ± 4.72%, P < 0.05, n = 8 in HASM-huNK1R, -huNK2R, and -huNK3R cells, respectively), and caffeine (16.07 ± 4.89%, P < 0.01, n = 10; 30.9 ± 5.13%, P < 0.01, n = 9; and 35.49 ± 5.73%, P < 0.01, n = 11 in HASM-huNK1R, -huNK2R, and -huNK3R cells, respectively) inhibited less in all three HASM-huNKR cells (Fig. 8). Ryanodine did not inhibit NKR-selective agonist-induced [Ca2+]i mobilization [1.78 ± 1.27%, not significant (NS), n = 11; 3.27 ± 1.71%, NS, n = 11; and 4.58 ± 2.12%, NS, n = 15 in HASM-huNK1R, -huNK2R, and -huNK3R cells, respectively].
Fig. 8.

Initial transient phase of increased [Ca2+]i. Effect of NKR-selective agonist [SM-SP (1 μM) for NK1R, β-ala-NKA (1 μM) for NK2R, and senktide (1 μM) for NK3R]-stimulated peak fluorescence increase pretreated with verapamil (10 μM), SKF-96365 (10 μM), 2-aminoethoxydiphenyl borate (2-APB; 10 μM), ryanodine (100 μM), or caffeine (10 mM) on HASM-huNK1R (A), HASM-huNK2R (B), and HASM-huNK3R cells (C). Data are means ± SE, presented as a percentage of the NKR-selective agonist (1 μM)-stimulated fluorescence increase (ΔF/Fo). *P < 0.05; **P < 0.01; ***P < 0.001 compared with NKR-selective agonist. Numbers in parentheses indicate the number of experiments.
During the sustained phase of the increase in intracellular Ca2+, 2-APB (75.16 ± 12.39%, P < 0.001, n = 11; 60.23 ± 6.67%, P < 0.01, n = 8; and 60.94 ± 13.56%, P < 0.001, n = 8 in HASM-huNK1R, -huNK2R, and -huNK3R cells, respectively), SKF-96365 (76.63 ± 4.50%, P < 0.001, n = 9; 84.19 ± 14.42%, P < 0.001, n = 11; and 60.67 ± 9.11%, P < 0.01, n = 8 in HASM-huNK1R, -huNK2R, and -huNK3R cells, respectively), and caffeine (76.13 ± 8.71%, P < 0.001, n = 10; 83.70 ± 14.74%, P < 0.01, n = 9; and 60.34 ± 9.85%, P < 0.01, n = 11 in HASM-huNK1R, -huNK2R, and -huNK3R cells, respectively) inhibited the NKR-selective agonist-induced [Ca2+]i mobilization, whereas verapamil inhibited less (17.57 ± 6.58%, P < 0.05, n = 8; 20.10 ± 7.91%, P < 0.05, n = 10; and 20.98 ± 6.26%, P < 0.01, n = 9 in HASM-huNK1R, -huNK2R, and -huNK3R cells, respectively) (Fig. 9). Ryanodine also partially inhibited the NKR-selective agonist-induced [Ca2+]i mobilization in the sustained phase (13.34 ± 1.73%, P < 0.001, n = 11; 11.78 ± 3.06%, P < 0.01, n = 11; and 15.44 ± 5.25%, P < 0.05, n = 15 in HASM-huNK1R, -huNK2R, and -huNK3R cells, respectively) (Fig. 9).
Fig. 9.

Sustained phase of increased [Ca2+]i. Effect of NKR-selective agonist [SM-SP (1 μM) for NK1R, β-ala-NKA (1 μM) for NK2R, and senktide (1 μM) for NK3R]-stimulated sustained fluorescence increase pretreated with verapamil (10 μM), SKF-96365 (10 μM), 2-APB (10 μM), ryanodine (100 μM), or caffeine (10 mM) on HASM-huNK1R (A), HASM-huNK2R (B), and HASM-huNK3R cells (C). Data are means ± SE, presented as a percentage of the NKR-selective agonist (1 μM)-stimulated fluorescence increase (ΔF/Fo). *P < 0.05; **P < 0.01; ***P < 0.001 compared with NKR-selective agonist. Numbers in parentheses indicate the number of experiments.
Effect of SKF-96365, 2-APB, ryanodine, and caffeine on SOCE in HASM
The sustained phase is thought to be primarily mediated by the extracellular Ca2+ influx through SOCC (35). SKF-96365, 2-APB, ryanodine, and caffeine also inhibited the sustained phase in the present study. To examine whether SOCC contributes to the SKF-96365-, 2-APB-, ryanodine-, or caffeine-induced inhibition of the sustained phase, we examined the effect of SKF-96365 (10 μM), 2-APB (10 μM), ryanodine (100 μM), and caffeine (10 mM) on SOCE in native HASM cells. Exposure to SKF-96365, ryanodine, and caffeine resulted in partial inhibition of SOCE (27.49 ± 2.09%, P < 0.001, n = 7; 10.07 ± 2.60%, P < 0.01, n = 9; and 18.51 ± 2.85%, P < 0.01, n = 6, respectively), whereas exposure to 2-APB did not exert any effect on SOCE (− 0.60 ± 4.72%, NS, n = 5) (Fig. 10).
Fig. 10.

Store-operated calcium entry (SOCE). Effect of SKF-96365 (10 μM), 2-APB (10 μM), ryanodine (100 μM), or caffeine (10 mM) on SOCE in native HASM cells. Data are means ± SE, presented as a percentage of control (ΔF/Fo). Data represent means ± SEM. **P < 0.01 compared with control. Numbers in parentheses indicate the number of experiments.
Confirmation of expression of ryanodine receptor subtypes in cultured transduced HASM
Messenger RNA encoding all three known isoforms of ryanodine receptors was identified in both native and NKR-transduced HASM cells (Fig. 11). PCR products of expected sizes (142, 127, and 314 bp) were also detected in total RNA isolated from respective positive control tissues (human and mouse skeletal muscle for the Ryr1 and Ryr3 and human and mouse heart for the Ryr2) (Fig. 11).
Fig. 11.

Ryanodine receptor isoform expression. Representative RT-PCR analysis of total RNA using primers that specially recognize mRNA for each ryanodine receptor isoform (Ryr1, Ryr2, Ryr3) in primary cultures of native (HASM) and NK receptor-transduced (HASM-huNK1R, HASM-huNK2R, HASM-huNK3R) HASM cells. Human and mouse skeletal muscle RNA were used as a positive control for Ryr1 and Ryr3 isoforms, and human and mouse heart were used as a positive control for Ryr2.
DISCUSSION
We have demonstrated that all three known subtypes of NKRs are expressed at the mRNA level in native HASM and that all three receptor subtypes couple to the synthesis of inositol phosphates and increase [Ca2+]i. Utilizing independent lentivirus-mediated overexpression of each receptor subtype in primary cultures of HASM cells, we have demonstrated agonist and antagonist specificity of each receptor subtype to inositol phosphate generation and have dissected the cellular compartmental source of intracellular Ca2+.
The expression and functional significance of all three NKR subtypes in native HASM cells has been under some debate. Most functional studies in both human and animal models have suggested that the NK2R subtype is the primary receptor subtype that mediates direct smooth muscle constriction (3, 51, 53). Some studies also have suggested a role for the NK1R in HASM constriction (4). However, the role of the NK3R in airways has traditionally been attributed to modulation of peripheral nerve function (9).
To characterize the coupling of NKRs to pivotal intracellular signaling pathways in HASM cells, we individually overexpressed the three subtypes of wild-type huNKRs in primary cultures of HASM cells (HASM-NK1R, -NK2R, and -NK3R) using an HIV-1-based lentivirus transduction system (29). A three-plasmid expression system was used to generate the lentiviruses encoding each subtype of the human NKRs. This system has three advantages over other systems: it can be used on relatively slowly dividing cells such as airway smooth muscle (41), does not require a cell surface receptor for entry into the target cell but mediates viral entry through lipid binding and plasma membrane fusion (45), and allows for a higher transduction efficiency than most other transfection methods in smooth muscle cells.
We demonstrated that NKR subtype-selective agonists significantly increased inositol phosphate synthesis in both native HASM and HASM NKR-transfected cells and that these increases were completely blocked by NKR subtype-selective antagonists. Receptor agonists designed to be selective for one receptor subtype typically demonstrate preferred selectivity for one receptor subtype but at higher concentrations can also activate related receptor subtypes. Moreover, subtype-selective agonists do not always maintain predicted selectivity for receptor subtypes across species because of subtle species differences in receptor protein structure (10, 15, 27). In the current study, we have demonstrated that [Sar9,Met(O2)11]-substance P, [β-Ala8]-neurokinin A(4–10), and senktide display the predicted selectivities for the human NK1R, NK2R, and NK3R, respectively. These results agree with the previous study showing that all tachykinins can act as full agonists on the three different NKRs but with lower affinities than on the preferred receptor (50).
NKRs belong to the seven-transmembrane domain receptor superfamily and classically couple to Gq proteins. In HASM cells, activation of GPCRs, which signal through Gq proteins such as bradykinin, histamine, and acetylcholine, classically result in a biphasic [Ca2+]i response, a rapid initial transient phase followed by a prolonged sustained phase (39). The initial rapid rise in [Ca2+]i levels reaches a maximum within 10–15 s and then rapidly declines and is dependent on the release of Ca2+ from intracellular stores (SR), induced by stimulation of inositol phosphate and/or ryanodine receptors. Classically, the depletion of SR Ca2+ stores triggers SOCE through SOCC.
We analyzed three components of intracellular Ca2+ regulation following NKR activation: 1) the initial transient phase, 2) the more prolonged sustained phase, and 3) SOCE. Pretreatment with 2-APB significantly inhibited the initial transient [Ca2+]i increase induced by NKR-selective agonists in all three types of HASM NKR-transfected cells. These results agree with previous evidence that IP3 plays a predominant role in tachykinin-induced airway contraction (17). In contrast, caffeine but not ryanodine inhibited the transient [Ca2+]i increase induced by NKR-selective agonists. Hyvelin et al. (22) reported that only ryr3 mRNA was expressed in human bronchial smooth muscle cells. The function of the type 3 ryanodine receptor is still poorly understood, whereas type 1 and type 2 ryanodine receptors have been clearly implicated in excitation-contraction coupling in skeletal and cardiac muscle (18). In the present study, caffeine, which depletes the SR via ryanodine activation and prevents it from refilling (52), had a small effect on the initial phase of increased [Ca2+]i but a large effect on the sustained phase in all three types of HASM NKR-transfected cells. Ryanodine had no effect on the initial transient phase of increased [Ca2+]i but a small effect on the sustained phase and SOCE. This result indicates that functional ryanodine receptors exist in HASM, and this was confirmed by demonstrating the mRNA expression of all three ryanodine receptor subtypes in native and NKR-transduced HASM cells. The effect of caffeine on the initial phase can be attributed to a partial depletion of SR stores by the preexposure to caffeine. However, it has been reported that acetylcholine-induced [Ca2+]i increase in human bronchial smooth muscle cell was not impaired by ryanodine (22). Together, these results suggest that ryanodine receptors expressed in HASM cells do not have a significant role in the initial transient phase of NKR-mediated increases in [Ca2+]i.
It has been reported that Ca2+ influx via L-type voltage-gated Ca2+ channels also takes part in airway smooth muscle contraction (23, 26, 36). In the present study, NKR-selective agonist-induced [Ca2+]i increases were partially blocked by verapamil. Likewise, Lin et al. (28) reported that substance P-evoked airway smooth muscle contraction and transient Ca2+ mobilization were partially inhibited by verapamil, indicating that the NKR-mediated [Ca2+]i increases are associated with activation of voltage-gated Ca2+ channels. They further demonstrated that smooth muscle contraction and [Ca2+]i increases activated by substance P were also partially blocked under Ca2+-free external conditions (28). Together, these findings indicate that Ca2+ influx from the extracellular space is a component of the transient initial phase of NKR-mediated transient [Ca2+]i increases. In the present study, SKF-96365 had a small effect on the NKR-mediated transient [Ca2+]i increase but a much greater effect on the sustained phase. This finding raises the possibility that SOCE also takes part in the transient [Ca2+]i increase. However, a limitation of the interpretation of the results of the present study is that SKF-96365 also has a minor inhibitory effect on voltage-gated Ca2+ channels (56).
The sustained Ca2+ response is important for maintaining the contractile response to agonist (18) and requires the continued presence of agonist (11, 39). In the sustained phase, SOCE has been considered as the predominant source of Ca2+ influx (11, 35). SOCC are Ca2+-permeable channels in the plasma membrane that are open following depletion of intra-cellular Ca2+ stores underlying the so-called “store-operated” or “capacitative” Ca2+ entry (35). In the present study, the NKR-selective agonist-induced sustained [Ca2+]i increase was significantly blocked by SKF-96365. These findings agree with the previous study (11, 35). However, verapamil also partially blocked this sustained [Ca2+]i increase. In contrast, it has been known that agonist-induced sustained [Ca2+]i increases in airway smooth muscle are relatively insensitive to inhibitors of voltage-gated Ca2+ channel despite their established presence (23, 26, 36). Our findings indicate that voltage-gated Ca2+ channels partially contribute to the maintenance of the NKR-mediated sustained [Ca2+]i response. We used SKF-96365 as an inhibitor of SOCE and validated this effect in the absence of receptor agonists (Fig. 10). However, SKF-96365 also has been shown to inhibit receptor-operated Ca2+ entry in airway smooth muscle (37), which could confound the interpretation of the studies of the sustained phase of Ca2+ entry in the presence of NKR agonists (Fig. 9).
Interestingly, 2-APB inhibited the NKR-mediated sustained [Ca2+]i increases. Interpretation of these results is confounded by two issues: 1) the dose-dependent specificity of 2-APB, and 2) 2-APB blockade of agonist-mediated SR Ca2+ depletion never allows SOCE to become activated. At low concentrations (1–10 μM), 2-APB inhibits IP3 receptor-mediated Ca2+ release, whereas it also inhibits SOCE at higher concentrations (25–100 μM). In the present study, 10 μM 2-APB was used to examine the contribution of IP3 receptors on NKR-mediated [Ca2+]i increases. In the subsequent focused studies on SOCE, this concentration of 2-APB did not inhibit SOCE, indicating that we were selectively studying inhibition of IP3 receptors using 10 μM 2-APB. The second confounding issue is that when cells are pretreated with 2-APB, preventing NKR-mediated SR Ca2+ release, SOCE never becomes activated. Thus pretreatment with an IP3 receptor antagonist never allows SOCE to be detected. Since inhibition of SOCE with SKF-96365 had a large effect on the sustained phase of Ca2+ increases, this is likely the primary mechanism of sustained elevations in intracellular Ca2+ following NKR activation.
Caffeine or ryanodine inhibited the NKR-mediated sustained [Ca2+]i increase in all three types of HASM NKR-transfected cells. This effect could be explained by at least two mechanisms: 1) caffeine depletion or ryanodine blockade of SR stores may reduce the amount of SR Ca2+ available for release, and 2) we have independently shown that caffeine or ryanodine has a small effect in attenuating SOCE. However, these effects of caffeine or ryanodine on SOCE may be indirect. They may prevent a portion of SR Ca2+ depletion such that less SOCE is necessary to replenish intracellular Ca2+ levels, and thus less Ca2+ enters the cell upon replacement of Ca2+ in the external medium.
In summary, we have demonstrated that native HASM cells express all three known subtypes of neurokinin receptors, which couple to activation of inositol phosphate and increases in [Ca2+]i. Utilizing lentivirus-mediated overexpression of each receptor subtype in primary cultures of HASM cells, we have demonstrated agonist specificity at each receptor subtype and complex regulation of the [Ca2+]i phases following NKR activation. Activation of the SR IP3 receptor primarily mediates the initial transient phase of elevation of [Ca2+]I, whereas the sustained phase is primarily mediated by SOCE. The SR ryanodine receptor plays a minor role in the sustained phase of NKR-mediated increases in [Ca2+]i, whereas the L-type voltage-gated Ca2+ channels have minor contributions to both the initial and sustained phases of increases in [Ca2+]i. These findings suggest that endogenous tachykinins in airways may have effects on airway smooth muscle that contribute to the asthmatic phenotype.
Acknowledgments
This work was supported by National Institute of General Medical Sciences Grant GM-065281.
References
- 1.Advenier C, Lagente V, Boichot E. The role of tachykinin receptor antagonists in the prevention of bronchial hyperresponsiveness, airway inflammation and cough. Eur Respir J. 1997;10:1892–1906. doi: 10.1183/09031936.97.10081892. [DOI] [PubMed] [Google Scholar]
- 2.Advenier C, Naline E, Drapeau G, Regoli D. Relative potencies of neurokinins in guinea pig trachea and human bronchus. Eur J Pharmacol. 1987;139:133–137. doi: 10.1016/0014-2999(87)90244-5. [DOI] [PubMed] [Google Scholar]
- 3.Advenier C, Naline E, Toty L, Bakdach H, Emonds-Alt X, Vilain P, Breliere JC, Le FG. Effects on the isolated human bronchus of SR 48968, a potent and selective nonpeptide antagonist of the neurokinin A (NK2) receptors. Am Rev Respir Dis. 1992;146:1177–1181. doi: 10.1164/ajrccm/146.5_Pt_1.1177. [DOI] [PubMed] [Google Scholar]
- 4.Amadesi S, Moreau J, Tognetto M, Springer J, Trevisani M, Naline E, Advenier C, Fisher A, Vinci D, Mapp C, Miotto D, Cavallesco G, Geppetti P. NK1 receptor stimulation causes contraction and inositol phosphate increase in medium-size human isolated bronchi. Am J Respir Crit Care Med. 2001;163:1206–1211. doi: 10.1164/ajrccm.163.5.2002079. [DOI] [PubMed] [Google Scholar]
- 5.Ay B, Prakash YS, Pabelick CM, Sieck GC. Store-operated Ca2+ entry in porcine airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2004;286:L909–L917. doi: 10.1152/ajplung.00317.2003. [DOI] [PubMed] [Google Scholar]
- 6.Bai TR, Zhou D, Weir T, Walker B, Hegele R, Hayashi S, McKay K, Bondy GP, Fong T. Substance P (NK1)- and neurokinin A (NK2)-receptor gene expression in inflammatory airway diseases. Am J Physiol Lung Cell Mol Physiol. 1995;269:L309–L317. doi: 10.1152/ajplung.1995.269.3.L309. [DOI] [PubMed] [Google Scholar]
- 7.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 8.Bertrand C, Geppetti P. Tachykinin and kinin receptor antagonists: therapeutic perspectives in allergic airway disease. Trends Pharmacol Sci. 1996;17:255–259. doi: 10.1016/0165-6147(96)10027-4. [DOI] [PubMed] [Google Scholar]
- 9.Canning BJ. Neurokinin3 receptor regulation of the airways. Vascul Pharmacol. 2006;45:227–234. doi: 10.1016/j.vph.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 10.Chung FZ, Wu LH, Vartanian MA, Watling KJ, Guard S, Woodruff GN, Oxender DL. The non-peptide tachykinin NK2 receptor antagonist SR 48968 interacts with human, but not rat, cloned tachykinin NK3 receptors. Biochem Biophys Res Commun. 1994;198:967–972. doi: 10.1006/bbrc.1994.1138. [DOI] [PubMed] [Google Scholar]
- 11.Corteling RL, Li S, Giddings J, Westwick J, Poll C, Hall IP. Expression of transient receptor potential C6 and related transient receptor potential family members in HASM and lung tissue. Am J Respir Cell Mol Biol. 2004;30:145–154. doi: 10.1165/rcmb.2003-0134OC. [DOI] [PubMed] [Google Scholar]
- 12.Di Maria GU, Bellofiore S, Geppetti P. Regulation of airway neurogenic inflammation by neutral endopeptidase. Eur Respir J. 1998;12:1454–1462. doi: 10.1183/09031936.98.12061454. [DOI] [PubMed] [Google Scholar]
- 13.Ellis JL, Undem BJ, Kays JS, Ghanekar SV, Barthlow HG, Buckner CK. Pharmacological examination of receptors mediating contractile responses to tachykinins in airways isolated from human, guinea pig and hamster. J Pharmacol Exp Ther. 1993;267:95–101. [PubMed] [Google Scholar]
- 14.Faisy C, Naline E, Diehl JL, Emonds-Alt X, Chinet T, Advenier C. In vitro sensitization of human bronchus by beta2-adrenergic agonists. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1033–L1042. doi: 10.1152/ajplung.00063.2002. [DOI] [PubMed] [Google Scholar]
- 15.Gao ZG, Blaustein JB, Gross AS, Melman N, Jacobson KA. N6-substituted adenosine derivatives: selectivity, efficacy, and species differences at A3 adenosine receptors. Biochem Pharmacol. 2003;65:1675–1684. doi: 10.1016/s0006-2952(03)00153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geppetti P, Tognetto M, Trevisani M, Amadesi S, Bertrand C. Tachykinins and kinins in airway allergy. Expert Opin Investig Drugs. 1999;8:947–956. doi: 10.1517/13543784.8.7.947. [DOI] [PubMed] [Google Scholar]
- 17.Grandordy BM, Frossard N, Rhoden KJ, Barnes PJ. Tachykinin-induced phosphoinositide breakdown in airway smooth muscle and epithelium: relationship to contraction. Mol Pharmacol. 1988;33:515–519. [PubMed] [Google Scholar]
- 18.Hall IP. Second messengers, ion channels and pharmacology of airway smooth muscle. Eur Respir J. 2000;15:1120–1127. doi: 10.1034/j.1399-3003.2000.01523.x. [DOI] [PubMed] [Google Scholar]
- 19.Hirshman CA, Emala CW. Actin reorganization in airway smooth muscle cells involves Gq and Gi-2 activation of Rho. Am J Physiol Lung Cell Mol Physiol. 1999;277:L653–L661. doi: 10.1152/ajplung.1999.277.3.L653. [DOI] [PubMed] [Google Scholar]
- 20.Hirshman CA, Zhu D, Pertel T, Panettieri RA, Emala CW. Isoproterenol induces actin depolymerization in HASM cells via activation of an Src kinase and GS. Am J Physiol Lung Cell Mol Physiol. 2005;288:L924–L931. doi: 10.1152/ajplung.00463.2004. [DOI] [PubMed] [Google Scholar]
- 21.Hotta K, Emala CW, Hirshman CA. TNF-α upregulates Giα and Gqα protein expression and function in HASM cells. Am J Physiol Lung Cell Mol Physiol. 1999;276:L405–L411. doi: 10.1152/ajplung.1999.276.3.L405. [DOI] [PubMed] [Google Scholar]
- 22.Hyvelin JM, Martin C, Roux E, Marthan R, Savineau JP. Human isolated bronchial smooth muscle contains functional ryanodine/caffeine-sensitive Ca-release channels. Am J Respir Crit Care Med. 2000;162:687–694. doi: 10.1164/ajrccm.162.2.9911025. [DOI] [PubMed] [Google Scholar]
- 23.Janssen LJ. Ionic mechanisms and Ca2+ regulation in airway smooth muscle contraction: do the data contradict dogma? Am J Physiol Lung Cell Mol Physiol. 2002;282:L1161–L1178. doi: 10.1152/ajplung.00452.2001. [DOI] [PubMed] [Google Scholar]
- 24.Joos GF, De Swert KO, Pauwels RA. Airway inflammation and tachykinins: prospects for the development of tachykinin receptor antagonists. Eur J Pharmacol. 2001;429:239–250. doi: 10.1016/s0014-2999(01)01323-1. [DOI] [PubMed] [Google Scholar]
- 25.Jooste E, Zhang Y, Emala CW. Rapacuronium preferentially antagonizes the function of M2 versus M3 muscarinic receptors in guinea pig airway smooth muscle. Anesthesiology. 2005;102:117–124. doi: 10.1097/00000542-200501000-00020. [DOI] [PubMed] [Google Scholar]
- 26.Kotlikoff MI. Calcium currents in isolated canine airway smooth muscle. Am J Physiol Cell Physiol. 1988;254:C793–C801. doi: 10.1152/ajpcell.1988.254.6.C793. [DOI] [PubMed] [Google Scholar]
- 27.Kull B, Arslan G, Nilsson C, Owman C, Lorenzen A, Schwabe U, Fredholm BB. Differences in the order of potency for agonists but not antagonists at human and rat adenosine A2A receptors. Biochem Pharmacol. 1999;57:65–75. doi: 10.1016/s0006-2952(98)00298-6. [DOI] [PubMed] [Google Scholar]
- 28.Lin YR, Kao PC, Chan MH. Involvement of Ca2+ signaling in tachy-kinin-mediated contractile responses in swine trachea. J Biomed Sci. 2005;12:547–558. doi: 10.1007/s11373-005-6796-0. [DOI] [PubMed] [Google Scholar]
- 29.Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 30.Lundberg JM, Hokfelt T, Martling CR, Saria A, Cuello C. Substance P-immunoreactive sensory nerves in the lower respiratory tract of various mammals including man. Cell Tissue Res. 1984;235:251–261. doi: 10.1007/BF00217848. [DOI] [PubMed] [Google Scholar]
- 31.Maggi CA. Tachykinin receptors and airway pathophysiology. Eur Respir J. 1993;6:735–742. [PubMed] [Google Scholar]
- 32.Maggi CA, Patacchini R, Quartara L, Rovero P, Santicioli P. Tachykinin receptors in the guinea-pig isolated bronchi. Eur J Pharmacol. 1991;197:167–174. doi: 10.1016/0014-2999(91)90517-t. [DOI] [PubMed] [Google Scholar]
- 33.Maggi CA, Patacchini R, Rovero P, Giachetti A. Tachykinin receptors and tachykinin receptor antagonists. J Auton Pharmacol. 1993;13:23–93. doi: 10.1111/j.1474-8673.1993.tb00396.x. [DOI] [PubMed] [Google Scholar]
- 34.Mapp CE, Miotto D, Braccioni F, Saetta M, Turato G, Maestrelli P, Krause JE, Karpitskiy V, Boyd N, Geppetti P, Fabbri LM. The distribution of neurokinin-1 and neurokinin-2 receptors in human central airways. Am J Respir Crit Care Med. 2000;161:207–215. doi: 10.1164/ajrccm.161.1.9903137. [DOI] [PubMed] [Google Scholar]
- 35.Marthan R. Store-operated calcium entry and intracellular calcium release channels in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2004;286:L907–L908. doi: 10.1152/ajplung.00410.2003. [DOI] [PubMed] [Google Scholar]
- 36.Marthan R, Martin C, Amedee T, Mironneau C. Calcium channel currents in isolated smooth muscle cells from human bronchus. J Appl Physiol. 1989;66:1706–1714. doi: 10.1152/jappl.1989.66.4.1706. [DOI] [PubMed] [Google Scholar]
- 37.Montano LM, Bazan-Perkins B. Resting calcium influx in airway smooth muscle. Can J Physiol Pharmacol. 2005;83:717–723. doi: 10.1139/y05-063. [DOI] [PubMed] [Google Scholar]
- 38.Mukaiyama O, Morimoto K, Nosaka E, Takahashi S, Yamashita M. Involvement of enhanced neurokinin NK3 receptor expression in the severe asthma guinea pig model. Eur J Pharmacol. 2004;498:287–294. doi: 10.1016/j.ejphar.2004.07.071. [DOI] [PubMed] [Google Scholar]
- 39.Murray RK, Kotlikoff MI. Receptor activated calcium influx in HASM cells. J Physiol. 1991;45:123–144. doi: 10.1113/jphysiol.1991.sp018501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Myers AC, Goldie RG, Hay DW. A novel role for tachykinin neurokinin-3 receptors in regulation of human bronchial Ganglia neurons. Am J Respir Crit Care Med. 2005;171:212–216. doi: 10.1164/rccm.200405-600OC. [DOI] [PubMed] [Google Scholar]
- 41.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 42.Naline E, Devillier P, Drapeau G, Toty L, Bakdach H, Regoli D, Advenier C. Characterization of neurokinin effects and receptor selectivity in human isolated bronchi. Am Rev Respir Dis. 1989;140:679–686. doi: 10.1164/ajrccm/140.3.679. [DOI] [PubMed] [Google Scholar]
- 43.Naline E, Hoglund CO, Vincent F, Emonds-Alt X, Lagente V, Advenier C, Frossard N. Role of tachykinin NK3 receptors in the release and effects of nerve growth factor in human isolated bronchi. Eur J Pharmacol. 2007;560:206–211. doi: 10.1016/j.ejphar.2006.10.068. [DOI] [PubMed] [Google Scholar]
- 44.Ollerenshaw SL, Jarvis D, Sullivan CE, Woolcock AJ. Substance P immunoreactive nerves in airways from asthmatics and nonasthmatics. Eur Respir J. 1991;4:673–682. [PubMed] [Google Scholar]
- 45.Ory DS, Neugeboren BA, Mulligan RC. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc Natl Acad Sci USA. 1996;93:11400–11406. doi: 10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pabelick CM, Ay B, Prakash YS, Sieck GC. Effects of volatile anesthetics on store-operated Ca2+ influx in airway smooth muscle. Anesthesiology. 2004;101:373–380. doi: 10.1097/00000542-200408000-00018. [DOI] [PubMed] [Google Scholar]
- 47.Panettieri RA, Murray RK, DePalo LR, Yadvish PA, Kotlikoff MI. A HASM cell line that retains physiological responsiveness. Am J Physiol Cell Physiol. 1989;256:C329–C335. doi: 10.1152/ajpcell.1989.256.2.C329. [DOI] [PubMed] [Google Scholar]
- 48.Pinto FM, Almeida TA, Hernandez M, Devillier P, Advenier C, Candenas ML. mRNA expression of tachykinins and tachykinin receptors in different human tissues. Eur J Pharmacol. 2004;494:233–239. doi: 10.1016/j.ejphar.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 49.Prakash YS, Iyanoye A, Ay B, Sieck GC, Pabelick CM. Store-operated Ca2+ influx in airway smooth muscle: interactions between volatile anesthetic and cyclic nucleotide effects. Anesthesiology. 2006;105:976–983. doi: 10.1097/00000542-200611000-00019. [DOI] [PubMed] [Google Scholar]
- 50.Regoli D, Boudon A, Fauchere JL. Receptors and antagonists for substance P and related peptides. Pharmacol Rev. 1994;46:551–599. [PubMed] [Google Scholar]
- 51.Rizzo CA, Valentine AF, Egan RW, Kreutner W, Hey JA. NK2-receptor mediated contraction in monkey, guinea-pig and HASM. Neuropeptides. 1999;33:27–34. doi: 10.1054/npep.1999.0027. [DOI] [PubMed] [Google Scholar]
- 52.Rousseau E, Meissner G. Single cardiac sarcoplasmic reticulum Ca2+-release channel: activation by caffeine. Am J Physiol Heart Circ Physiol. 1989;256:H328–H333. doi: 10.1152/ajpheart.1989.256.2.H328. [DOI] [PubMed] [Google Scholar]
- 53.Sheldrick RL, Rabe KF, Fischer A, Magnussen H, Coleman RA. Further evidence that tachykinin-induced contraction of human isolated bronchus is mediated only by NK2-receptors. Neuropeptides. 1995;29:281–292. doi: 10.1016/0143-4179(95)90037-3. [DOI] [PubMed] [Google Scholar]
- 54.Togashi H, Emala CW, Hall IP, Hirshman CA. Carbachol-induced actin reorganization involves Gi activation of Rho in HASM cells. Am J Physiol Lung Cell Mol Physiol. 1998;274:L803–L809. doi: 10.1152/ajplung.1998.274.5.L803. [DOI] [PubMed] [Google Scholar]
- 55.Togashi H, Emala CW, Hall IP, Hirshman CA. Carbachol-induced actin reorganization involves Gi activation of Rho in HASM cells. Am J Physiol Lung Cell Mol Physiol. 1998;274:L803–L809. doi: 10.1152/ajplung.1998.274.5.L803. [DOI] [PubMed] [Google Scholar]
- 56.Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia W, Tesco G, Saunders AJ, Ding K, Frosch MP, Tanzi RE, Kim TW. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27:561–572. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 57.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. 1997;15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]