

Abstract
SteA is a protein that can be translocated into host cells through the two virulence-related type III secretion systems that are present in Salmonella enterica. We used the T-POP system to carry out general screens for loci that exhibited activation or repression of a steA::lacZ fusion. These screens identified the histidine kinase PhoQ and the response regulator PhoP as positive regulators of steA. Transcription of this gene is σ70 dependent, and the promoter of steA contains a PhoP-binding site that mediates direct regulation by PhoP. Our screens also detected MgrB (also known as YobG) as a negative regulator of the expression of steA. Disruption of the gene encoding the periplasmic disulfide oxidoreductase DsbA or addition of the reducing agent dithiothreitol increases transcription of steA. The effects of MgrB and DsbA on steA are mediated by PhoP. These results suggest that the cellular redox status is a factor contributing to regulation of steA and, probably, other virulence genes regulated by the PhoQ/PhoP two-component system.
INTRODUCTION
Salmonella enterica is a facultative intracellular pathogen responsible for gastroenteritis and systemic infections in humans and many other animals. S. enterica virulence factors include two type III secretion systems, T3SS1 and T3SS2, encoded by genes within Salmonella pathogenicity island 1 (SPI1) and 2 (SPI2), respectively (1–3). T3SSs are complex devices, evolutionary related to flagella, present in many Gram-negative bacteria that are pathogens or symbionts of animal and plants, including members of the genera Salmonella, Shigella, Yersinia, Escherichia, Pseudomonas, and Rhizobium. A T3SS consists of at least 20 different subunits forming a structure that spans bacterial membranes and delivers effector proteins into host cells through a needlelike structure and a translocon pore that is formed in a target host membrane (4).
Collectively, S. enterica T3SS1 and T3SS2 are able to translocate more than 40 effectors into eukaryotic host cells. However, only nine of them, GtgE, PipB2, SlrP, SopD, SpvC, SpvD, SspH1, SteA, and SteE, have been shown to be secreted through both systems (5). Therefore, in most cases, each effector appears to be a specific substrate of a particular T3SS. Although secretion signals or chaperones can participate, this specificity is probably achieved through coregulation of each effector with its cognate T3SS. T3SS1 should be expressed extracellularly to mediate invasion into the host cell (6). T3SS2 is expressed after internalization to facilitate survival of Salmonella inside macrophages and other host cells (2, 7, 8). Regulation of SPI1 and SPI2 gene expression, however, is a complex issue and, although conditions for optimal expression of each island are different, they are not exclusive, and some overlap exists (9–12). The central regulator in the overall scheme of SPI1 regulation is HilA, a transcriptional activator encoded in SPI1 that contains a DNA-binding motif belonging to the OmpR/ToxR family (13). Expression of hilA is controlled by the combined action of three AraC-like transcriptional activators: HilC, HilD, and RtsA (14–16). The expression of SPI2, and hence T3SS2, is directly controlled by the SsrA/SsrB two-component system, which is encoded by the ssrA and ssrB genes located within SPI2. SsrB is a response regulator that, when phosphorylated, binds to several promoters in SPI2, and SsrA is its cognate histidine kinase. The system is activated when Salmonella is inside macrophages but also in Salmonella cells grown in minimal medium at acidic pH (17). SPI1 and SPI2 share some regulators, including the PhoQ/PhoP two-component system, an ancestral regulatory system that is conserved among Salmonella and related species. PhoQ/PhoP have opposite effects on both islands: activation of the system decreases SPI1 expression through reduction in hilA transcription (11) but increases SPI2 expression through binding to the ssrB promoter and posttranscriptional regulation of SsrA (18).
SteA is a Salmonella effector that can be translocated into epithelial cells and macrophages through T3SS1 and T3SS2 (19, 20). This protein localizes to the trans-Golgi network in both transfected and infected human epithelial HeLa cells and to Salmonella-induced membrane tubules containing trans-Golgi markers (20, 21). We have previously studied the conditions that modulate synthesis, secretion to culture media, and translocation to eukaryotic cells of SteA and have identified sequences of this effector that are important for secretion and translocation through T3SS1 and T3SS2 (19). We found that steA is expressed under a wide range of conditions. However, the highest expression was obtained in low-phosphate, low-magnesium minimal medium (LPM) at pH 5.8, a medium that imitates the conditions found in the Salmonella-containing vacuole and that is optimal for the synthesis of T3SS2. Acidic pH is one of the factors contributing to expression of T3SS2, but this is not the case for steA. This gene, in contrast, is repressed by butyric acid, an organic acid found in the gut that is known to repress expression of T3SS1. These data suggest that synthesis of SteA responds to different stimuli, some typical of T3SS1 effectors and others typical of T3SS2 substrates, and are consistent with the ability of this effector to be secreted through two systems expressed under different conditions.
This work was undertaken to identify genetic factors that regulate the expression of steA. We show that the PhoQ/PhoP two-component regulatory system is the main direct activator of the expression of this gene. We also identify the magnesium responsive protein MgrB, also known as YobG, and the periplasmic disulfide bond isomerase DsbA, as negative regulators of steA that act through modulation of the PhoQ/PhoP system. We propose that environmental redox conditions can be sensed by Salmonella to fine-tune expression of steA.
MATERIALS AND METHODS
Bacterial strains, bacteriophages, and strain construction.
Escherichia coli and S. enterica serovar Typhimurium strains used in this study are described in Table 1. Salmonella strains derive from the mouse-virulent strain ATCC 14028. Transductional crosses using phage P22 HT 105/1 int201 (30) were used for strain construction (31). To obtain phage-free isolates, transductants were purified by streaking on green plates. Green plates were prepared as described previously (32), except that methyl blue (Sigma) was substituted for aniline blue. Phage sensitivity was tested by cross-streaking with the clear-plaque mutant P22 H5.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s) | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | supE44 ΔlacU169 (ϕ80 lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | 22 |
| UQ285 | lacZ4 argG75 rpoD285(ts) | 23 |
| XL1-Blue | recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 Δlac-pro/F′ proAB lacIq lacZΔM15 Tn10; Tetr | 24 |
| M15 | lac ara gal mtl | Qiagen |
| S. enterica serovar Typhimurium | ||
| 14028 | Wild type | ATCC |
| 55130a | 14028 pho-24 (PhoP constitutive) | E. A. Groisman |
| SV4699a | 14028 phoP7953::Tn10; Tcr | 25, 26 |
| SV5452a | 14028 ΔssrB::Cmr | 27 |
| SV5846a | 14028 steA::3×FLAG; Kmr | 19 |
| SV6152a | 14028 steA::lacZ; Kmr | 19 |
| SV6712 | 14028 steA::lacZ phoP::T-POP | This study |
| SV6713 | 14028 steA::lacZ phoQ::T-POP | This study |
| SV6973 | 14028 steA::lacZ mgrB::T-POP | This study |
| SV7001a | 14028 ΔdsbA | Laboratory stock |
| SV7329a | 14028 ΔmgrB::Cmr | This study |
| Plasmids | ||
| pIC552 | Parent for lacZ transcriptional fusions; Apr | 28 |
| pIZ1949 | pQE30-phoP | This study |
| pIZ1964 | pIC552-steA(−27/−1) | This study |
| pIZ1965 | pIC552-steA(−49/−1) | This study |
| pIZ1968 | pIC552-steA(−104/−1) | This study |
| pIZ1995 | pIZ1968-(G-30C) | This study |
| pIZ1996 | pIZ1968-(A-26G) | This study |
| pKD3 | bla FRT cat FRT PS1 PS2 oriR6K | 29 |
| pKD46 | bla PBAD gam bet exo pSC101 oriTS | 29 |
| pQE30 | Parent for N-ter His6 fusions; Apr | Qiagen |
| pREP4 | lacI; Kmr | Qiagen |
Derivatives of these strains were used as indicated in the text.
Bacterial culture.
Culture media for S. enterica were Luria-Bertani (LB) broth and low-phosphate, low-magnesium minimal medium (LPM) at pH 5.8. LPM contained 80 mM 2-(N-morpholino) ethanesulfonic acid (pH 5.8), 5 mM KCl, 7.5 mM (NH4)2SO4, 0.5 mM K2SO4, 0.1% Casamino Acids, 38 mM glycerol, 337.5 μM K2HPO4-KH2PO4 (pH 7.4), and 8 μM MgCl2. Solid media contained agar at 1.5% final concentration. Antibiotics were used at the following final concentrations in rich medium: kanamycin (Km), 50 μg ml−1; chloramphenicol (Cm), 20 μg ml−1; ampicillin (Ap), 100 μg ml−1; tetracycline (Tc), 20 μg ml−1. In minimal medium, antibiotics were used at the following concentrations: Km, 125 μg ml−1; Cm, 5 μg ml−1; Ap, 50 μg ml−1; Tc, 10 μg ml−1. Plates for monitoring β-galactosidase activity contained 5-bromo-4-chloro-indolyl-β-d-galactopyranoside (X-Gal; final concentration, 40 μg ml−1).
DNA amplification with the PCR.
Amplification reactions were carried out in a PerkinElmer Gene-Amp PCR System 2400 (PerkinElmer Cetus). The final volume of reactions was 50 μl, and the final concentration of MgCl2 was 1.5 mM. Reagents were used at the following concentrations: deoxynucleoside triphosphates (dNTPs), 300 μM; primers, 0.3 μM; and Taq polymerase (KAPA HiFi DNA polymerase; Kapa Biosystems), 1 unit per reaction. The thermal program included the following steps: (i) initial denaturation, 5 min at 95°C; (ii) 30 cycles of denaturation (98°C, 20 s), annealing (55°C, 20 s), and extension (72°C, 30 s per kb); and (iii) final incubation at 72°C for 5 min, to complete extension. To generate point mutations in the steA promoter cloned in pIC552, the thermal program included the following steps: (i) initial denaturation, 30 s at 95°C; (ii) 12 cycles of denaturation (95°C, 30 s), annealing (42°C, 1 min), and extension (68°C, 5 min). Primers are listed in Table 2. PCR constructs were sequenced with an automated DNA sequencer (Stab Vida, Oeiras, Portugal).
Table 2.
Oligonucleotides used in this study
| Oligonucleotide by use | 5′→3′ sequence |
|---|---|
| ST-PCR | |
| tpop1 | GCCTTCTTATTCGGCCTTGAATTGATCATATGCGG |
| stACGCC | GGCCACGCGTCGACTAGTACNNNNNNNNNNACGCC |
| stGATAT | GGCCACGCGTCGACTAGTACNNNNNNNNNNGATAT |
| tpop2 | CTTTTTCCGTGATGGTAACC |
| st1 | GGCCACGCGTCGACTAGTAC |
| 5′ RACE | |
| RNAsteA1 | TAGGCCAGTCCTGCGGTAAGCAC |
| RNAsteA2 | CTCCTGCAGCAACATGAGGTAGC |
| GeneRacer RNA oligo | CGACUGGAGCACGAGGACACUGACAUGGACUGAAGGAGUAGAAA |
| GeneRacer 5′ nested primer | GGACACTGACATGGACTGAAGGAGTA |
| mgrB deletion | |
| mgrBP1 | ATTTAGCGACATAAGATGTAAGATCGGAGAGTGGAGTGAAGTGTAGGCTGGAGCTGCTTC |
| mgrBP2 | GGGAAGGAAATCTCTGGTGTAAAACGTTTACCAGGGAATACATATGAATATCCTCCTTAG |
| mgrB ext5 | TTGCGCGAATGATCAAACGC |
| mgrB ext3 | TTGACAAAGAACCAGAGCGC |
| steA qRT-PCR | |
| steA left | TGTTAACATTCGAACAACTCTCG |
| steA right | TCCGATATCCTGACGATTGG |
| rfaH left | TTCAGGATCGACAACGCCTT |
| rfaH right | TCAGCCATTTTGTGCGCTT |
| Construction of pIZ1949 | |
| phoPbampqe5′ | GCATGGATCCATGATGCGCGTACTGGTTGTAG |
| phoPsalpqe3′ | GCATGTCGACGAGCAAATTTATTCATTAGCGC |
| Construction of pIZ1964 | |
| steA(−27) 5′ | GATCAGATCTGATTGACATATCGTCATAATG |
| steA(−1) 3′ | GATCCTCGAGCTCTCATTATGACGATATGTC |
| Construction of pIZ1965 | |
| steA(−49) 5′ | GATCAGATCTTATGCCTTTGAGCAATTTTG |
| steA(−1) 3′ | As above |
| Construction of pIZ1968 | |
| steA(−104) 5′ | GATCAGATCTAAGCAGCATAAGATCAGGCC |
| steA(−1) 3′ | As above |
| Construction of pIZ1995 | |
| G30C 5′ | CCTTTGAGCAATTTTCTTGATTGACATATCG |
| G30C 3′ | GGAAACTCGTTAAAAGAACTAACTGTATAGC |
| Construction of pIZ1996 | |
| A26G 5′ | AGCAATTTTGTTGGTTGACATATCGTC |
| A26G 3′ | TCGTTAAAACAACCAACTGTATAGCAG |
| steA promoter | |
| promsteAdir | GAGCATCCTACCTGCAATTG |
| promsteArev | GGCATAGGTAGAAACTGATG |
| slyB promoter | |
| promslyBdir | AGACTTGCCTGTTGCGCAAC |
| promslyBrev | AAACGCTATTTCAGCATCCC |
| phoN promoter | |
| promphoNdir | AATGCGTGTCAGTCAGGCAC |
| promphoNrev | TTAGCTACGATCAGTGGTAG |
Plasmids.
Plasmids used in this study are listed in Table 1. Plasmids expressing transcriptional lacZ fusions were derivatives of pIC552. To construct these plasmids, DNA from strain 14028 was used as a template for PCR amplification with the primers listed in Table 2. The amplified fragments were digested with BglII and XhoI and ligated with BglII/XhoI-digested pIC552. To generate point mutations in the steA promoter, pIZ1968 was used as a template for PCR amplification. Products were digested with 1 μl of DpnI (10 U μl−1) for 1 h at 37°C, and E. coli DH5α was directly transformed with these digested plasmids. To obtain the His6-PhoP recombinant protein, DNA from strain 14028 was used as a template for PCR amplification with primers listed in Table 2. The amplified fragment was digested with BamHI and SalI and ligated with BamHI/SalI-digested pQE30. The product of this ligation was then transformed into E. coli XL1-Blue. All constructs were confirmed by DNA sequencing.
Mutagenesis with T-POP.
Strain 14028 of S. enterica serovar Typhimurium was mutagenized with T-POP, a derivative of Tn10dTc (33). Pools of 5,000 colonies, each carrying an independent T-POP insertion, were then prepared and lysed with phage P22 HT. The lysates were used to transduce strain SV6152 (14028 steA::lacZ), selecting Tc-resistant transductans on LB and LPM plates supplemented with X-Gal, Tc, and Km.
Molecular characterization of T-POP insertions.
A semirandom, two-step PCR protocol (34) was used to amplify genomic regions adjacent to the T-POP insertions. The reactions were carried out as previously described (35) by using primers listed in Table 2. The final products were sequenced using primers tpop2 and st1. Sequence analysis was performed with molecular biology algorithms from the National Center for Biotechnology Information at www.ncbi.nlm.nih.gov.
Construction of an mgrB mutant.
Disruption and replacement of mgrB with a Cm resistance cassette were performed as described previously (29). Briefly, the Cm resistance cassette from plasmid pKD3 was PCR amplified with primers mgrBP1 and mgrBP2 (Table 2). The PCR product was used to transform the wild-type strain carrying the Red recombinase expression plasmid pKD46. Transformants were selected in LB with Cm.
β-Galactosidase assays.
Levels of β-galactosidase were assayed as described previously (36), using the CHCl3/SDS permeabilization procedure. Bacteria were grown overnight in LB medium at 37°C with shaking. Cells were then diluted 1:50 into LB medium or washed and diluted 1:50 into LPM and incubated at 37°C with shaking, for LB and LPM, or without shaking, only for LB. Cultures were incubated for 1.5 to 2 h to reach exponential phase or 8 h to reach stationary phase.
RNA extraction.
Two protocols were used, one for real-time PCR (RT-PCR) and a different one for 5′ rapid amplification of cDNA ends (RACE). In the first protocol, bacterial strains were grown overnight in LB in capped standing tubes at 37°C. After this, 4 ml of three independent replicates for each strain was pelleted and resuspended in 100 μl of TE buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA) containing 3 mg ml−1 lysozyme. RNA from these lysates was isolated with 1 ml of TRIzol reagent (Invitrogen) by using the protocol supplied by the manufacturer. In the second protocol, three independent colonies of Salmonella 14028 were grown overnight in LB with shaking and then diluted in fresh LB and grown until an optical density at 600 nm of ∼2. RNA extraction was carried out as described above, with an additional step of phenolization to obtain pure samples.
Quantitative RT-PCR and calculation of relative expression levels.
One microgram of total RNA was used as the template for each 20-μl reverse transcription reaction mixture with Quantiscript reverse transcriptase (Qiagen) and an appropriate DNA primer, using the buffer and the protocol supplied by the manufacturer. Negative control without enzyme was included for each sample or DNA primer pair used. Four microliters of each reverse transcription reaction was used as the template for PCR with the DNA primers indicated in Table 2 (primers were designed with the Universal ProbeLibrary Assay Design Center software [Roche]). Real-time PCR was carried out in a LightCycler 480 II (Roche) with SYBR Premix Ex Taq in a total volume of 10 μl on a 384-well optical reaction plate (Roche), using the protocol for LightCycler. The cycling conditions were as follows: (i) 95°C for 10 s, 4.8°C s−1; (ii) 40 cycles at 95°C for 5 s, 4.8°C s−1, 60°C for 20 s, 2.5°C s−1; (iii) 95°C for 1 s, 4.8°C s−1, 65°C for 0 s, 0.11°C s−1, 95°C for 1 s, 4.8°C s−1. Melting curve analysis verified that each reaction mixture contained a single PCR product. Gene expression levels were normalized to transcripts of rfaH (primers sequences obtained from reference 37), a housekeeping gene that served as an internal control.
5′ RACE.
Fifteen micrograms of RNA was used to determine the cDNA 5′ end by using a protocol similar to that described previously (38, 39). RNAs were prepared either with or without tobacco acid pyrophosphatase to distinguish primary transcript 5′ ends from internal 5′ processing sites. DNA primer RNAsteA1 was used for cDNA synthesis. A second DNA primer for subsequent PCR amplification of cDNAs, GeneRacer 5′ nested primer, is homologous to the adaptor RNA, GeneRacer RNA oligo, used for 5′ RACE. PCR products that were detected both with and without tobacco acid pyrophosphatase treatment were purified by using a Wizard SV gel and PCR clean-up system kit (Promega) and cloned by using the pGEM-T Easy kit (Promega), and 19 clones were sequenced.
Protein purification and phosphorylation.
His6-PhoP protein was produced and purified as previously described (40) with some modifications. E. coli M15/pREP4 strain carrying pIZ1949 (pQE30-phoP) was grown overnight in LB medium containing 50 μg ml−1 Km and 100 μg ml−1 Ap at 37°C with vigorous aeration. The culture was diluted 1:200 into 1 liter of fresh LB with antibiotics and grown for 2.5 h. To induce expression of the recombinant protein, isopropyl-β-d-thiogalactopyranoside (IPTG) was added at a final concentration of 1 mM, and the culture was grown for 3 h at 30°C and harvested by centrifugation. The pellet was resuspended in 17 ml lysis buffer (20 mM HEPES [pH 8.0], 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 1 mM MgCl2, 1 mM EDTA, 1 mM dithiothreitol [DTT] in the presence of proteases inhibitors [1:100; Sigma-Aldrich], DNase I [5 μg ml−1], and RNase A [10 μg ml−1]). Cells were disrupted by sonication and then centrifuged at 15,000 × g for 30 min. His6-PhoP was purified by nickel-affinity chromatography (GE Healthcare) at 4°C. The column was washed with 5 ml of washing buffer (20 mM HEPES [pH 8.0], 50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, 1 mM MgCl2, 1 mM EDTA, 1 mM DTT) and washed again with the same buffer containing 50 mM imidazole. His6-PhoP was eluted with 5 ml elution buffer (20 mM HEPES [pH 8.0], 50 mM NaH2PO4, 300 mM NaCl, 300 mM imidazole, 1 mM MgCl2, 1 mM EDTA, 1 mM DTT) and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The selected eluted fractions were then collected and dialyzed against dialysis buffer (20 mM HEPES [pH 8.0], 100 mM KCl, 20% glycerol, 1 mM DTT, and 1 mM MgCl2) in dialysis tubing cellulose membranes (Sigma-Aldrich). Dialyzed fraction was analyzed by UV absorbance at 280 nm and by SDS-PAGE. Protein samples were frozen and stored at −80°C for further use. For slot blot assays, S. enterica His6-PhoP was phosphorylated with acetyl phosphate as previously described (41) with modifications. Briefly, His6-PhoP was incubated in 20 μl of phosphorylation buffer (50 mM Tris-HCl [pH 7.5], 50 mM KCl, 10 mM MgCl2) containing 10 mM acetyl phosphate (Sigma-Aldrich) for 1 h at 37°C.
Slot blot for DNA-protein interaction.
DNA fragments used for the PhoP binding assay were amplified by PCR using Salmonella 14028 as a template. The reverse primers, listed in Table 2, were labeled with 6-carboxyfluorescein (FAM) fluorochrome. PCR amplification rendered fragments of 281, 355, and 412 bp for phoN, slyB, and steA promoters, respectively. The binding assay was carried out as previously described (41) with modifications. Briefly, a solution of 10 nM FAM-labeled DNA and 0, 0.125, 0.25, 0.5, 1, and 2 μM phosphorylated His6-PhoP was prepared in binding buffer (50 mM Tris-HCl [pH 8.0], 50 mM KCl) in a total volume of 20 μl and incubated for 30 min at room temperature. After this, binding reactions were diluted 1:10 with phosphate-buffered saline (PBS) and blotted onto nitrocellulose filters using a PR 600 slot blot manifold (Hoefer Scientific Instruments, San Francisco, CA) connected to a portable vacuum/pressure pump (Millipore). Wells were then washed five times with PBS, and membranes were air dried. Images were acquired using a Fujifilm FLA-5100 system, and the quantification was performed using the Image J software.
Western blotting and antibodies.
Salmonella strains were cultured as explained above for β-galactosidase assays. The bacteria were then pelleted by centrifugation and resuspended in SDS-PAGE sample buffer. Proteins were separated by SDS-PAGE (Mini-PROTEAN TGX precast gels; 12% or 4 to 15%) and electrophoretically transferred to nitrocellulose filters for Western blot analysis using anti-FLAG M2 monoclonal antibodies (1:5,000; Sigma) and anti-DnaK (8E2/2) (1:1,0000; Assay Designs) monoclonal antibodies. Goat anti-mouse horseradish peroxidase (HRP)-conjugated antibodies (1:5,000; Bio-Rad) were used as secondary antibodies.
Statistical analyses.
Student's t test was used to analyze differences in enzymatic activities. P values of 0.05 or less were considered significant.
RESULTS
T-POP-based screens identify PhoP, PhoQ, and MgrB as regulators of steA expression.
The S. enterica gene steA encodes a T3SS effector that can be secreted by the two virulence-related T3SS that are present in these bacteria. In a previous work, we studied the environmental conditions affecting SteA synthesis and we found that, although this protein is significantly produced under a wide range of conditions, expression is 6-fold higher under conditions favoring T3SS2 expression than under conditions optimized for T3SS1 expression. In order to understand the regulation of steA expression, we took advantage of a steA::lacZ translational fusion (19) to carry out a T-POP-based screen (33). This defective transposon was used to mutagenize S. enterica serovar Typhimurium strain SV6152, a derivative of the wild-type strain 14028 carrying a chromosomal steA::lacZ fusion. Two independent screens were carried out.
In the first screen, colonies of mutagenized bacteria were grown on minimal medium with tetracycline, to select for T-POP insertions, and the chromogenic indicator X-Gal, to compare the level of expression of steA::lacZ in different colonies. Most colonies were blue because of the high expression of steA in this medium. Visual inspection, however, detected 28 white colonies out of 93,000 colonies screened. Reconstruction experiments and β-galactosidase assays revealed 7 independent T-POP insertions that consistently resulted in a decrease in steA expression. Since T-POP can disrupt a gene by insertion and/or increase transcription of adjacent genes from internal tetracycline-dependent promoters, the effect of these insertions on the expression of steA::lacZ was measured both in the presence and in the absence of tetracycline. All of them decreased β-galactosidase activity in a tetracycline-independent manner, suggesting that their effect was due to disruption of a gene encoding a positive regulator of steA. The insertions were mapped by sequencing DNA adjacent to the transposon that was amplified using a semirandom, two-step PCR protocol (see Materials and Methods). Five insertions were located in phoP, and two insertions were in phoQ. The products of these genes, PhoP and PhoQ, constitute a two-component regulatory system, with PhoQ acting as a membrane sensor that, in response to environmental signals, phosphorylates and activates the transcriptional regulator PhoP. Figure 1 compares β-galactosidase activities of strains carrying the steA::lacZ fusion and a T-POP insertion in phoP, phoQ, or in a random, unrelated location used as a control.
Fig 1.
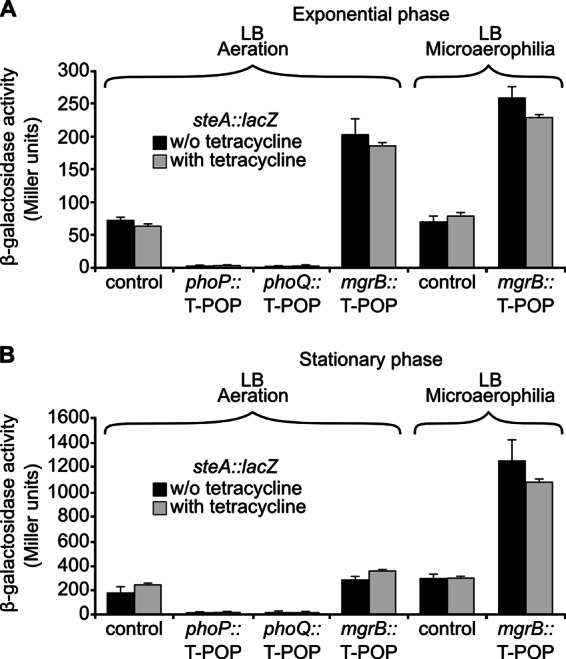
PhoP, PhoQ, and MgrB are regulators of steA expression. A T-POP screen for steA regulators identified three chromosomal loci: phoP, phoQ, and mgrB. Expression levels of a chromosomal steA::lacZ fusion were monitored in four representative strains carrying T-POP insertions in phoP, phoQ, mgrB, and a random nonregulatory locus (control). Gene expression was measured with and without tetracycline, under aeration and microaerophilic conditions, for exponential-phase cultures (A) and for stationary-phase cultures (B). Data presented here are means and standard deviations from two independent β-galactosidase measurements. Similar results were obtained in three independent experiments performed in duplicate.
A second screen was carried out by plating mutagenized bacteria onto LB medium containing 680 mM NaCl and supplemented with tetracycline and X-Gal. Because expression of steA in this medium is relatively low, the background color of most colonies was light blue. However, several dark-blue colonies were detected, although only one of them carried a T-POP insertion that consistently reproduced the same phenotype after reconstruction. This insertion was located in mgrB, also known as yobG, and resulted in an increase in β-galactosidase activity of the strain carrying steA::lacZ in a tetracycline-independent manner (Fig. 1). In the exponential phase (Fig. 1A), this increase was observed in standing (microaerophilia) and shaking (aeration) culture conditions; however, in the stationary phase (Fig. 1B), the effect of the insertion of T-POP in mgrB was detected only in standing cultures.
In summary, our T-POP-based screens detected PhoP and PhoQ as positive regulators and MgrB as a negative regulator of steA.
The PhoQ/PhoP two-component system regulates steA expression in an SsrB-independent manner.
To confirm regulation of steA by the PhoQ/PhoP system, two additional mutations were used: a null phoP mutation due to a Tn10 insertion and a point mutation in phoQ (pho-24) causing constitutive activation of the system (42, 43). The effect of these mutations on steA::lacZ was quantified by measuring β-galactosidase activities in LB medium and in LPM. As seen in Fig. 2A, the level of expression of steA is higher in LPM minimal medium than in LB rich medium. Interestingly, PhoP is absolutely essential for expression in both media. Activation of steA expression by the PhoQ/PhoP system is further confirmed by the pho-24 mutation increasing steA expression in LB. This effect is not observed in LPM, presumably because the PhoQ/PhoP system is already fully activated in this medium, which mimics intracellular conditions (44). The results obtained with steA::lacZ and β-galactosidase activities were confirmed at the protein level using an SteA-3xFLAG fusion and immunoblotting with an anti-FLAG antibody (Fig. 2B).
Fig 2.
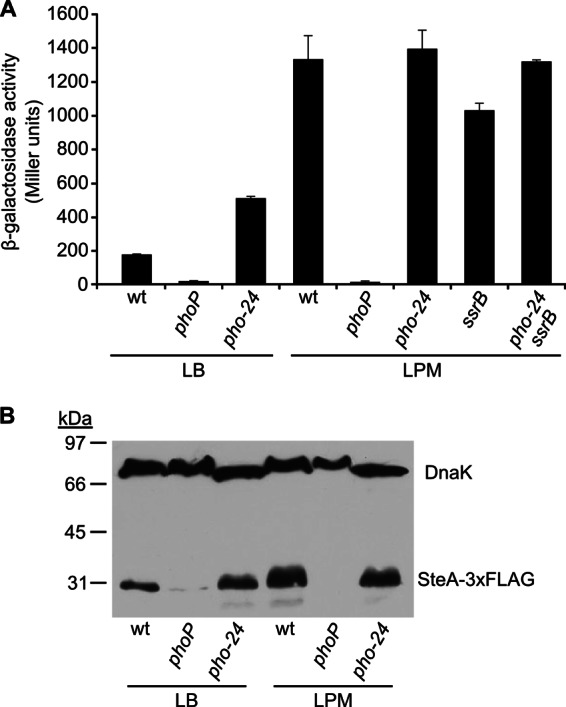
The PhoQ/PhoP two-component system regulates steA expression in an SsrB-independent manner. (A) β-Galactosidase activities were measured from stationary-phase aerated cultures in LB or LPM of several S. enterica serovar Typhimurium strains: wild-type 14028 (wt), null mutants (phoP and ssrB), a mutant with constitutive activation of the PhoQ/PhoP system (pho-24), and a double mutant (ssrB pho-24), all of them carrying a chromosomal steA::lacZ translational fusion. Means and standard deviations from one experiment representative of three independent experiments performed in duplicate are represented. (B) Expression at the protein level was studied by Western blotting using strains expressing SteA-3xFLAG. Extracts from stationary-phase cultures of these strains were resolved by SDS-PAGE (12%), and a monoclonal anti-FLAG antibody was used for immunoblotting (lower bands). A polyclonal anti-DnaK antibody was used to get a loading control (upper bands). Sizes of molecular mass markers are indicated on the left.
The PhoQ/PhoP two-component system regulates SPI2 genes and some T3SS2-related genes located outside SPI2 through another two-component system, SsrA/SsrB (18, 45). To test whether the same regulatory cascade affected steA, expression of steA::lacZ was measured in an ssrB-null background. Only a slight effect was observed (Fig. 2A). In addition, the double mutant ssrB pho-24 exhibited the same level of expression of steA as the pho-24 single mutant or the wild type in LPM (Fig. 2A). These results establish that regulation of steA by PhoQ/PhoP does not occur through activation of the SsrA/SsrB system and suggest that PhoP could be a direct activator of steA.
MgrB regulates expression of steA in a PhoP-dependent manner.
One of the T-POP insertions detected in the screens described above was located in mgrB. To confirm that disruption of this gene caused an increase in steA expression, an mgrB-null mutant was constructed using a method based on λ Red recombinase, and the expression of steA::lacZ was measured in wild-type and mgrB backgrounds. The mgrB mutation caused a 2-fold increase in β-galactosidase activities of aerated LB cultures in exponential phase and of standing LB cultures in exponential and stationary growth phases (Fig. 3A).
Fig 3.

MgrB and DsbA regulate expression of steA through the PhoQ/PhoP system. (A) β-Galactosidase activities were measured from aerated (upper panel) or standing (lower panel) cultures in LB of wild-type 14028 (wt) and mgrB- and dsbA-null mutants. Means and standard deviations from triplicated experiments are represented. Similar results were obtained in two independent experiments performed in triplicate. The asterisks indicate values that are significantly higher than wt values obtained under the same conditions. (B) β-Galactosidase activities were measured from stationary-phase standing cultures in LB, in the presence or in the absence of 1 mM DTT, of several S. enterica serovar Typhimurium strains: wild-type 14028 (wt), null mutants (phoP, mgrB, and dsbA mutants), a mutant with constitutive activation of the PhoQ/PhoP system (pho-24 mutant), double mutants (mgrB phoP, mgrB dsbA, and dsbA phoP mutants), and a triple mutant (mgrB dsbA phoP mutant), all of them carrying a chromosomal steA::lacZ translational fusion. Means and standard deviations from duplicated experiments are represented. Similar results were obtained in three independent experiments performed in duplicate. (C) Total RNA was isolated from S. enterica serovar Typhimurium strain 14028 (wt) and the indicated mutants cultured under microaerophilic conditions (standing cultures in capped tubes) in LB. Levels of steA mRNA were measured by real-time PCR. For the wt, mRNA level was also measured from cultures in LB containing 1 mM DTT. Represented data are means and standard deviations from triplicated experiments and are relativized to the levels detected in the wild type. (D) Expression at the protein level was studied by Western blotting using strains expressing SteA-3xFLAG. Extracts from these strains were resolved by SDS-PAGE (4 to 15% gradient), and a monoclonal anti-FLAG antibody was used for immunoblotting (bottom). A polyclonal anti-DnaK antibody was used to get a loading control (top).
mgrB was identified in Escherichia coli as a gene whose expression was induced by magnesium limitation and depended on PhoP and PhoQ (46). The product of this gene, MgrB, is a broadly conserved protein of just 47 amino acids. Interestingly, mgrB is a PhoP-regulated gene that has been found to mediate feedback in this system, since deletion of this gene results in a potent increase in PhoP-regulated transcription in E. coli. In addition, overexpression of mgrB decreases PhoP-regulated transcription in E. coli, S. enterica, and Yersinia pestis (47). To investigate the relationships between MgrB and the PhoQ/PhoP system in the regulation of steA, we carried out epistasis analysis. Figure 3 shows that the mgrB phoP double mutant has the same level of steA expression as the phoP mutant, suggesting that PhoP is downstream of MgrB in this regulatory pathway.
A very recent report has shown that DsbA acts as a periplasmic oxidant and that deletion of dsbA leads to increased transcription of PhoP-regulated genes in E. coli. Interestingly, this effect appears to be mediated by MgrB and can be mimicked by the addition of the reducing agent DTT to the culture media (48). To test whether DsbA could affect expression of steA in S. enterica, we used an S. enterica dsbA-null mutant to measure the expression of steA::lacZ in a dsbA background. As seen in Fig. 3A and B, this mutation caused an increase of steA expression under microaerophilic conditions similar to the increase caused by an mgrB mutation. Epistasis analysis showed that the effect of DsbA on steA expression was mediated by the PhoQ/PhoP system (Fig. 3B). In addition, the effects of mutations in dsbA and mgrB were not additive, a result which is consistent with the notion that the products of both genes act in the same regulatory pathway. We also found similar levels of increase of steA expression after addition of the reducing agent DTT, suggesting that conditions that perturb the oxidizing environment of the periplasm can lead to expression of steA through stimulation of the PhoQ/PhoP system.
The role of MgrB, DsbA, and the oxidizing environment on steA expression were further explored at two additional levels: (i) the amount of steA mRNA was measured in different genetic backgrounds and in the presence of DTT using real-time PCR (Fig. 3C); and (ii) the influence of the same genetic backgrounds and environmental conditions was investigated at the protein level by Western blotting using an anti-FLAG antibody to detect SteA-3xFLAG (Fig. 3D). The results were totally consistent with the idea that DsbA, MgrB, and oxidizing conditions have a negative impact on the expression of steA through inactivation of the PhoQ/PhoP system.
Characterization of the steA promoter.
Results shown in previous sections suggest that PhoP could be a direct activator of steA transcription. To analyze this issue, we decided to characterize the promoter region of this gene. 5′ RACE was employed for the identification of the transcriptional start site (Fig. 4A). It was experimentally determined to be located 46 bp upstream of the steA coding region. Visual inspection revealed the presence of −10 and −35 consensus sequences typical of σ70-dependent promoters. To test whether σ70 was necessary for steA transcription, we studied the effect of a thermosensitive rpoD mutation on the expression of a steA::lacZ fusion. A derivative of plasmid pIC552 containing the promoter region of steA (from −49 to −1) in fusion with lacZ was introduced into E. coli strain UQ285, which carries a thermosensitive rpoD allele. Exponential-phase cultures grown in LB with Ap at 37°C (permissive temperature) were maintained at the permissive temperature or shifted to 42°C (restrictive temperature). At different time points, aliquots were extracted and β-galactosidase activities were measured. As shown in Fig. 4B, activities decreased over time at the restrictive temperature, indicating that transcription of steA requires the rpoD-encoded σ70 factor. Moreover, a putative PhoP-binding site was found to be located in the promoter region of steA (Fig. 4A). As shown in Fig. 4C, this motif is also observed in a similar position and in the same orientation in a subset of PhoP-activated genes, including phoP, mgtA, pmrD, yrbL, slyB, and orgB (49).
Fig 4.

Transcription of steA is σ70 dependent. (A) 5′ RACE was carried out to map the transcriptional start site of steA. The PCR products obtained with and without TAP treatment are shown. The sequence surrounding the transcriptional start site (+1) is shown. Putative σ70 consensus sequences are underlined, and a putative PhoP-binding site is boxed. The ribosome binding site and the start of the coding region are also indicated. (B) Exponential-phase cultures of E. coli strain UQ285 (rpoD thermosensitive)/pIZ1965 (steA::lacZ transcriptional fusion) were either maintained at 37°C (permissive temperature) or shifted to 42°C (restrictive temperature), and β-galactosidase activities were measured at the indicated times (in minutes). Similar results were obtained in two independent experiments performed in duplicate. (C) Alignment of the promoter regions of steA and six PhoP-activated genes with a similar architecture (49). Predicted −10 hexamers used as a reference for alignment and putative PhoP boxes are indicated.
Direct binding of PhoP to the steA promoter.
A slot blot method was used to analyze the binding of PhoP to the promoter of steA. Two additional PhoP-activated genes, slyB and phoN, were used as positive and negative controls, respectively, since previous experiments have shown direct interaction of PhoP with the promoter of slyB but not with the promoter of phoN (50). Purified His6-PhoP was used for these assays together with PCR-amplified fragments derived from the promoter regions of the three genes. As seen in Fig. 5, phosphorylated PhoP was able to bind to the promoter of steA and slyB with similar kinetics, whereas no binding was detected to the promoter of phoN.
Fig 5.
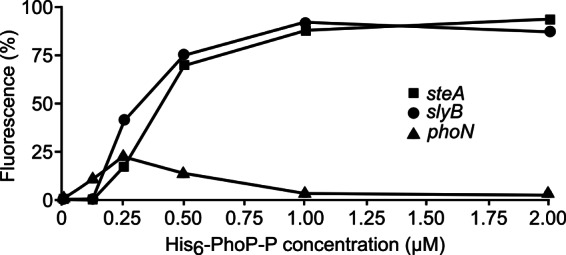
PhoP binds directly to the promoter of steA. Purified His6-PhoP was phosphorylated in vitro using acetyl phosphate. DNA fragments containing the promoter region of steA (−337/+76), slyB, and phoN were PCR amplified using fluorochrome-labeled primers and incubated with the indicated concentrations of phosphorylated His6-PhoP (His6-PhoP-P). Slot blot was used to quantify binding. Results from an experiment, representative of three independent experiments, are shown.
Determination of the PhoP-binding site in the promoter of steA.
Data presented in Fig. 4 and 5 suggest that PhoP could bind to a putative PhoP-binding site located at positions −31 to −25 relative to the transcriptional start site of steA. We carried out a mutational analysis to test this hypothesis. First, fragments −104/−1 and −49/−1 were cloned into pIC552 to generate lacZ transcriptional fusions. These plasmids were transformed into two strains of S. enterica serovar Typhimurium, a wild type and a phoP mutant, and the level of expression of the fusions was measured. PhoP regulation was observed for fragments −104/−1 and −49/−1 (Fig. 6A), consistent with the idea that a region located between positions −49 and −1 is needed for PhoP binding. Then, we obtained two independent point mutants. One resulted from a transversion at position −30 and the other from a transition at position −26 in the fragment −104/−1 of the steA promoter cloned in pIC552. These nucleotides are part of the putative PhoP-binding sites but do not overlap −10 or −35 consensus sequences. Activities of the fusions were compared in wild-type and phoP-null backgrounds. Both mutations resulted in a significant reduction (or even a complete abrogation) of PhoP regulation (Fig. 6B). Finally, slot blot analysis was carried out to test the effect of these mutations in the binding of PhoP to steA promoter. Binding was totally prevented by both mutations (Fig. 6C). Results shown in this section strongly support the hypothesis that this site is necessary for PhoP binding.
Fig 6.
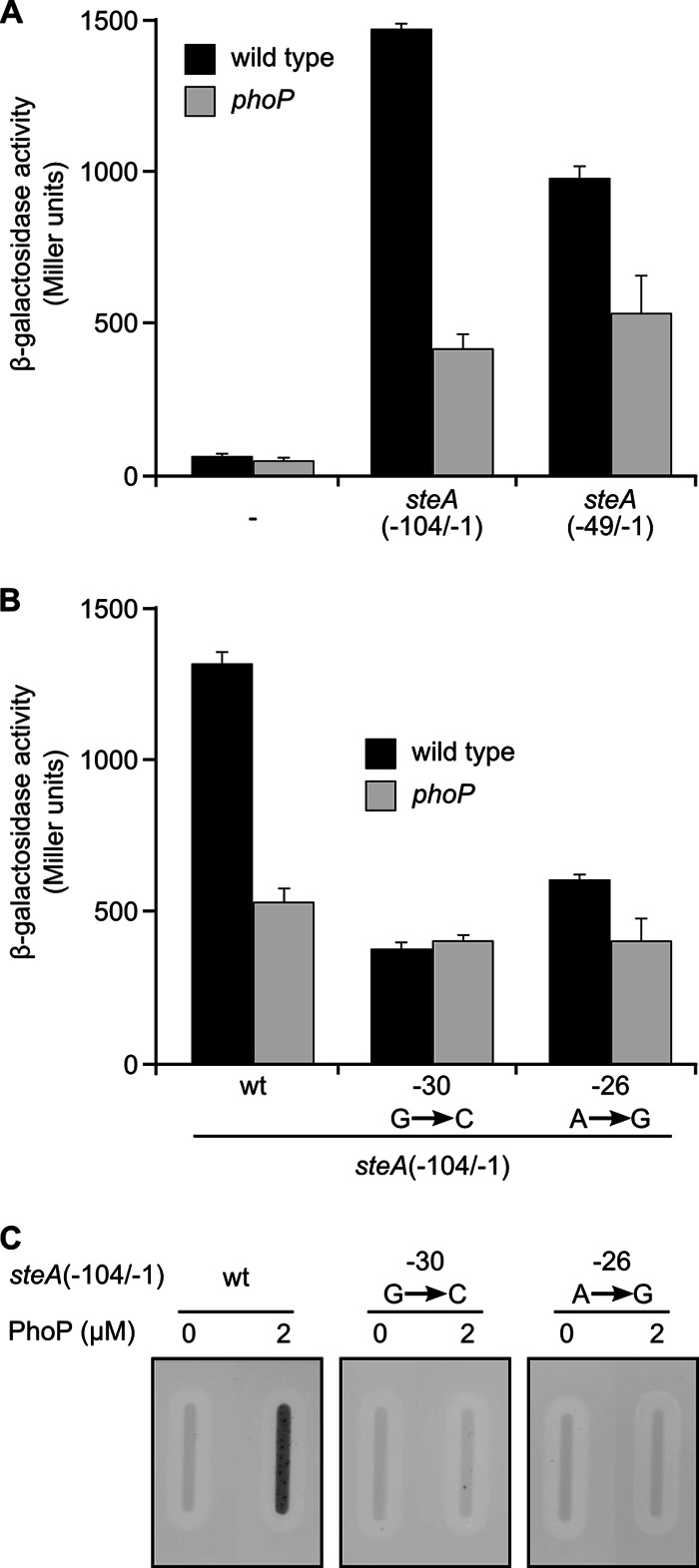
The integrity of a putative PhoP box is essential for binding of PhoP and regulation of steA expression by PhoP. (A) Two fragments of the steA promoter, −104/−1 and −49/−1, were inserted into plasmid pIC552 to generate transcriptional fusions to the lacZ gene. These plasmids were introduced in S. enterica serovar Typhimurium strain 14028 (wild type) or a phoP-null mutant, and β-galactosidase activities were measured in cultures grown to stationary phase in liquid LB. Means and standard deviations from two independent β-galactosidase measurements are shown. (B) A derivative of pIC552 carrying fragment −104/−1 of steA or derivatives of this plasmid with the indicated point mutations were introduced in S. enterica serovar Typhimurium strain 14028 (wild type) or a phoP-null mutant. β-Galactosidase activities were measured from stationary-phase LB broth cultures. Means and standard deviations from two independent β-galactosidase measurements are shown. Similar results were obtained in three independent experiments performed in duplicate. (C) Slot blot was used to detect binding of phosphorylated His6-PhoP (PhoP), at the indicated concentrations, to the promoter region of steA (−104/−1) (wt) or to the same region with the indicated point mutations. Similar results were obtained in three independent experiments.
DISCUSSION
In a previous work, we defined the environmental factors controlling the expression of steA (19). In this work, we studied the genetic factors involved in regulating the expression of this gene. Our results indicate that the PhoQ/PhoP two-component regulatory system has an essential role in the activation of steA expression, since null mutations in phoP or phoQ completely abolished expression. Binding of PhoP to its targets requires phosphorylation, a process controlled by PhoQ, a membrane protein that senses extracellular Mg2+. Thus, disruption of either phoP or phoQ should have similar effects, as is the case for steA regulation. PhoP regulates expression of more than 100 genes in S. enterica serovar Typhimurium (49, 51). Regulation can be direct, by binding of PhoP to the corresponding promoter regions, or indirect, by regulating the expression of other regulatory proteins or RNAs. A previous study, combining computational and experimental methods, identified 50 genes with PhoP binding sites with detectable effects upon transcription (51). A recent report investigated the number of PhoP binding sites, their orientation, location, and sequences, in 23 PhoP-activated promoters (49). This analysis identified five distinct promoter architectures composed of specific combinations of cis-acting regulatory elements. Architecture I and III harbor a single PhoP box in the direct orientation located 11 or 12 and 21 to 23 nucleotides, respectively, upstream of the predicted −10 hexamer. Architecture II, IV, and V contain two PhoP boxes that differ in their locations and orientations. Here, we show that steA possesses a PhoP-activated promoter with a PhoP box located 12 nucleotides upstream of the −10 hexamer, resembling promoters with architecture I. The promoter proximal half of this PhoP box is very similar to the consensus (T/G)GTTTA. The distal half is not so similar, which could explain why this PhoP-activated promoter was not detected in previous computational searches. Several lines of evidence suggest that PhoP directly activates steA and that the proposed PhoP binding site is functional. First, an in vitro slot blot-based assay indicated binding of PhoP to the region −337/+76. Second, PhoP regulation and binding was observed for the fragment −104/−1. Third, point mutations in the putative PhoP box abolished PhoP regulation and binding. In addition, consensus sequences for σ70 promoters are present upstream of the steA transcriptional start site, and we have shown that expression of steA is σ70 dependent. This is in agreement with previous reports indicating that this factor is responsible for transcription of PhoP-regulated genes (52).
A significant fraction of the PhoP-regulated genes in S. enterica are involved in virulence. In fact, strains lacking the PhoQ/PhoP system are highly attenuated for virulence. This attenuation is probably due, at least in part, to their inability to sense the transition from an extracellular environment to an intravacuolar location (low Mg2+ environment) and to activate a set of virulence factors (53, 54). As mentioned above, PhoP activates the entire SPI2 by activating the SsrA/SsrB system (18). This pathway also operates for many T3SS2 substrates that are encoded outside SPI2, including GogB, PipB, PipB2, SifA, SifB, SopD2, SrfJ, SseI (also known as SrfH), SseJ, SseK1, SseK2, SseL, SspH1, SspH2, and SteC (45, 55, 56). SteA is an exception since, as shown here, it is regulated by PhoP in an SsrB-independent manner. SlrP is another T3SS2 effector that is not regulated by SsrB (56, 57). These effectors have in common their ability to be secreted by T3SS1 and T3SS2. Therefore, it makes sense that their expression is not strictly dependent on SsrB since they need to be expressed under conditions that prevent activation of SsrB and SPI2 expression. The situation can be more complex, as exemplified by sseL, a gene located outside SPI2 that encodes a T3SS2 effector. This gene is indirectly regulated by PhoP through SsrB but also directly through binding to the sseL promoter (40).
Our screens also identified MgrB as a negative regulator of steA. A previous study showed that MgrB is associated with the inner membrane, interacts with PhoQ, and represses expression of PhoP-activated genes in E. coli (47). A gene homologous to mgrB in S. enterica has been shown to be activated by PhoP, suggesting that MgrB is part of a negative feedback loop in the PhoQ/PhoP signaling circuit (58). Consistent with these previous results, here, we show that PhoP mediates the effect of MgrB on steA expression. We also explored whether an additional upstream regulatory component recently described for the E. coli PhoQ/PhoP circuit, the periplasmic oxidant DsbA (48), also modulates expression of steA in S. enterica. Our results show that the effect of a dsbA mutation on steA expression is similar to the effect of an mgrB mutation, and both are PhoP dependent. In addition, the effects of dsbA and mgrB mutations are not additive, suggesting that they are in the same pathway. Addition of DTT had a similar effect, indicating that disruption of the oxidizing environment in the periplasm stimulates the PhoQ/PhoP system in S. enterica. It is interesting to note that regulation of steA by DsbA is more clearly observed under low-oxygen conditions (standing cultures) than under aerobic conditions (shaken cultures). These observations are consistent with a previous report showing that DsbA was absolutely required for disulfide bond formation in the E. coli periplasm under low-oxygen conditions but not under aerobic conditions (59).
DsbA plays an important role in the biogenesis of bacterial toxins and virulence factors in many bacteria, including Haemophilus influenzae, Shigella flexneri, Vibrio cholerae, Erwinia carotovora, Pseudomonas aeruginosa, and S. enterica. As a consequence, mutants defective in DsbA have reduced fitness and attenuated virulence in infection models (reviewed in reference 60). In S. enterica serovar Typhimurium, DsbA is required for secretion of effector proteins via T3SS1 and T3SS2 (61, 62). DsbA positively regulates transcription of SPI1 and of genes encoding T3SS1 effectors SopA and SopB, which are located outside SPI1 (61, 63). This transcriptional response is dependent on both the flagellar protein FliZ and the RcsCDB system (63). Independently of the transcriptional effect on the SPI1 regulatory circuit, loss of DsbA affects either the assembly or the function of T3SS1 (63), but the target is unknown. A direct target of DsbA is SpiA (also known as SsaC), one of the components of the T3SS2 apparatus. DsbA catalyzes disulfide bond formation in SpiA, a posttranslational modification that is necessary for proper folding and function of this protein (62). In addition, SlrP, an effector that can be translocated by T3SS1 and T3SS2, is positively regulated by DsbA at the posttranscriptional level (61). We now add a new way for DsbA to contribute to the control of Salmonella virulence negatively regulating the expression of the effector SteA. Our data, together with previous data in E. coli, suggest a model in which DsbA is a negative regulator of the PhoQ/PhoP system probably through disulfide bond formation in MgrB: reducing conditions disrupting disulfide bonds increase PhoQ/PhoP activity, and phosphorylated PhoP binds then to the steA promoter to directly activate transcription of this gene. Since PhoP is a global transcriptional regulator, DsbA is expected to have an indirect effect on transcription of many genes, including genes in SPI1 and SPI2, through this pathway.
ACKNOWLEDGMENTS
This work was supported by grant SAF2010-15015, from the Spanish Ministry of Economy and Competitiveness and the European Regional Development Fund, and grant P08-CVI-03487, from the Consejería de Economía, Innovación y Ciencia, Junta de Andalucía, Spain.
We are grateful to Ana I. Prieto (University of Cambridge) for critical reading of the manuscript.
Footnotes
Published ahead of print 15 March 2013
REFERENCES
- 1. Galán JE, Curtiss R., III 1989. Cloning and molecular characterization of genes whose products allow Salmonella Typhimurium to penetrate tissue culture cells. Proc. Natl. Acad. Sci. U. S. A. 86:6383–6387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ochman H, Soncini FC, Solomon F, Groisman EA. 1996. Identification of a pathogenicity island required for Salmonella survival in host cells. Proc. Natl. Acad. Sci. U. S. A. 93:7800–7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shea JE, Hensel M, Gleeson C, Holden DW. 1996. Identification of a virulence locus encoding a second type III secretion system in Salmonella Typhimurium. Proc. Natl. Acad. Sci. U. S. A. 93:2593–2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buttner D. 2012. Protein export according to schedule: architecture, assembly, and regulation of type III secretion systems from plant- and animal-pathogenic bacteria. Microbiol. Mol. Biol. Rev. 76:262–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ramos-Morales F. 2012. Impact of Salmonella enterica type III secretion system effectors on the eukaryotic host cell. ISRN Cell Biol. doi:10.5402/2012/787934 [Google Scholar]
- 6. Galán JE. 1999. Interaction of Salmonella with host cells through the centisome 63 type III secretion system. Curr. Opin. Microbiol. 2:46–50 [DOI] [PubMed] [Google Scholar]
- 7. Cirillo DM, Valdivia RH, Monack DM, Falkow S. 1998. Macrophage-dependent induction of the Salmonella pathogenicity island 2 type III secretion system and its role in intracellular survival. Mol. Microbiol. 30:175–188 [DOI] [PubMed] [Google Scholar]
- 8. Hensel M, Shea JE, Waterman SR, Mundy R, Nikolaus T, Banks G, Vazquez-Torres A, Gleeson C, Fang FC, Holden DW. 1998. Genes encoding putative effector proteins of the type III secretion system of Salmonella pathogenicity island 2 are required for bacterial virulence and proliferation in macrophages. Mol. Microbiol. 30:163–174 [DOI] [PubMed] [Google Scholar]
- 9. Ellermeier JR, Slauch JM. 2007. Adaptation to the host environment: regulation of the SPI1 type III secretion system in Salmonella enterica serovar Typhimurium. Curr. Opin. Microbiol. 10:24–29 [DOI] [PubMed] [Google Scholar]
- 10. Fass E, Groisman EA. 2009. Control of Salmonella pathogenicity island-2 gene expression. Curr. Opin. Microbiol. 12:199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Golubeva YA, Sadik AY, Ellermeier JR, Slauch JM. 2012. Integrating global regulatory input into the Salmonella pathogenicity island 1 type III secretion system. Genetics 190:79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hautefort I, Thompson A, Eriksson-Ygberg S, Parker ML, Lucchini S, Danino V, Bongaerts RJ, Ahmad N, Rhen M, Hinton JC. 2008. During infection of epithelial cells Salmonella enterica serovar Typhimurium undergoes a time-dependent transcriptional adaptation that results in simultaneous expression of three type 3 secretion systems. Cell Microbiol. 10:958–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee CA, Jones BD, Falkow S. 1992. Identification of a Salmonella Typhimurium invasion locus by selection for hyperinvasive mutants. Proc. Natl. Acad. Sci. U. S. A. 89:1847–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ellermeier CD, Slauch JM. 2003. RtsA and RtsB coordinately regulate expression of the invasion and flagellar genes in Salmonella enterica serovar Typhimurium. J. Bacteriol. 185:5096–5108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Olekhnovich IN, Kadner RJ. 2002. DNA-binding activities of the HilC and HilD virulence regulatory proteins of Salmonella enterica serovar Typhimurium. J. Bacteriol. 184:4148–4160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schechter LM, Lee CA. 2001. AraC/XylS family members, HilC and HilD, directly bind and derepress the Salmonella Typhimurium hilA promoter. Mol. Microbiol. 40:1289–1299 [DOI] [PubMed] [Google Scholar]
- 17. Miao EA, Freeman JA, Miller SI. 2002. Transcription of the SsrAB regulon is repressed by alkaline pH and is independent of PhoPQ and magnesium concentration. J. Bacteriol. 184:1493–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bijlsma JJ, Groisman EA. 2005. The PhoP/PhoQ system controls the intramacrophage type three secretion system of Salmonella enterica. Mol. Microbiol. 57:85–96 [DOI] [PubMed] [Google Scholar]
- 19. Cardenal-Muñoz E, Ramos-Morales F. 2011. Analysis of the expression, secretion and translocation of the Salmonella enterica type III secretion system effector SteA. PLoS One 6:e26930 doi:10.1371/journal.pone.0026930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Geddes K, Worley M, Niemann G, Heffron F. 2005. Identification of new secreted effectors in Salmonella enterica serovar Typhimurium. Infect. Immun. 73:6260–6271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Engelenburg SB, Palmer AE. 2010. Imaging type-III secretion reveals dynamics and spatial segregation of Salmonella effectors. Nat. Methods 7:325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557–580 [DOI] [PubMed] [Google Scholar]
- 23. Isaksson LA, Skold SE, Skjoldebrand J, Takata R. 1977. A procedure for isolation of spontaneous mutants with temperature sensitive of RNA and/or protein. Mol. Gen. Genet. 156:233–237 [DOI] [PubMed] [Google Scholar]
- 24. Bullock WO, Fernandez JM, Short JM. 1987. XL1-BLUE: a high efficiency plasmid transforming RecA Escherichia coli strain with beta-galactosidase selection. Biotechniques 5:376–379 [Google Scholar]
- 25. Groisman EA, Chiao E, Lipps CJ, Heffron F. 1989. Salmonella Typhimurium phoP virulence gene is a transcriptional regulator. Proc. Natl. Acad. Sci. U. S. A. 86:7077–7081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Segura I, Casadesús J, Ramos-Morales F. 2004. Use of mixed infections to study cell invasion and intracellular proliferation of Salmonella enterica in eukaryotic cell cultures. J. Microbiol. Methods 56:83–91 [DOI] [PubMed] [Google Scholar]
- 27. García-Calderón CB, Casadesús J, Ramos-Morales F. 2007. Rcs and PhoPQ regulatory overlap in the control of Salmonella enterica virulence. J. Bacteriol. 189:6635–6644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Macián F, Pérez-Roger I, Armengod ME. 1994. An improved vector system for constructing transcriptional lacZ fusions: analysis of regulation of the dnaA, dnaN, recF and gyrB genes of Escherichia coli. Gene 145:17–24 [DOI] [PubMed] [Google Scholar]
- 29. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmieger H. 1972. Phage P22-mutants with increased or decreased transduction abilities. Mol. Gen. Genet. 119:75–88 [DOI] [PubMed] [Google Scholar]
- 31. Maloy SR. 1990. Experimental techniques in bacterial genetics. Jones & Barlett, Boston, MA [Google Scholar]
- 32. Chan RK, Botstein D, Watanabe T, Ogata Y. 1972. Specialized transduction of tetracycline resistance by phage P22 in Salmonella Typhimurium. II. Properties of a high-frequency-transducing lysate. Virology 50:883–898 [DOI] [PubMed] [Google Scholar]
- 33. Rappleye CA, Roth JR. 1997. A Tn10 derivative (T-POP) for isolation of insertions with conditional (tetracycline-dependent) phenotypes. J. Bacteriol. 179:5827–5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chun KT, Edenberg HJ, Kelley MR, Goebl MG. 1997. Rapid amplification of uncharacterized transposon-tagged DNA sequences from genomic DNA. Yeast 13:233–240 [DOI] [PubMed] [Google Scholar]
- 35. Baisón-Olmo F, Cardenal-Muñoz E, Ramos-Morales F. 2012. PipB2 is a substrate of the Salmonella pathogenicity island 1-encoded type III secretion system. Biochem. Biophys. Res. Commun. 423:240–246 [DOI] [PubMed] [Google Scholar]
- 36. Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 37. Sittka A, Lucchini S, Papenfort K, Sharma CM, Rolle K, Binnewies TT, Hinton JC, Vogel J. 2008. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 4:e1000163 doi:10.1371/journal.pgen.1000163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EG, Margalit H, Altuvia S. 2001. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr. Biol. 11:941–950 [DOI] [PubMed] [Google Scholar]
- 39. Bensing BA, Meyer BJ, Dunny GM. 1996. Sensitive detection of bacterial transcription initiation sites and differentiation from RNA processing sites in the pheromone-induced plasmid transfer system of Enterococcus faecalis. Proc. Natl. Acad. Sci. U. S. A. 93:7794–7799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gal-Mor O, Elhadad D, Deng W, Rahav G, Finlay BB. 2011. The Salmonella enterica PhoP directly activates the horizontally acquired SPI-2 gene sseL and is functionally different from a S. bongori ortholog. PLoS One 6:e20024 doi:10.1371/journal.pone.0020024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tang YT, Gao R, Havranek JJ, Groisman EA, Stock AM, Marshall GR. 2012. Inhibition of bacterial virulence: drug-like molecules targeting the Salmonella enterica PhoP response regulator. Chem. Biol. Drug Des. 79:1007–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gunn JS, Hohmann EL, Miller SI. 1996. Transcriptional regulation of Salmonella virulence: a PhoQ periplasmic domain mutation results in increased net phosphotransfer to PhoP. J. Bacteriol. 178:6369–6373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miller SI, Mekalanos JJ. 1990. Constitutive expression of the phoP regulon attenuates Salmonella virulence and survival within macrophages. J. Bacteriol. 172:2485–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Coombes BK, Brown NF, Valdez Y, Brumell JH, Finlay BB. 2004. Expression and secretion of Salmonella pathogenicity island-2 virulence genes in response to acidification exhibit differential requirements of a functional type III secretion apparatus and SsaL. J. Biol. Chem. 279:49804–49815 [DOI] [PubMed] [Google Scholar]
- 45. Cordero-Alba M, Bernal-Bayard J, Ramos-Morales F. 2012. SrfJ, a Salmonella type III secretion system effector regulated by PhoP, RcsB, and IolR. J. Bacteriol. 194:4226–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kato A, Tanabe H, Utsumi R. 1999. Molecular characterization of the PhoP-PhoQ two-component system in Escherichia coli K-12: identification of extracellular Mg2+-responsive promoters. J. Bacteriol. 181:5516–5520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lippa AM, Goulian M. 2009. Feedback inhibition in the PhoQ/PhoP signaling system by a membrane peptide. PLoS Genet. 5:e1000788 doi:10.1371/journal.pgen.1000788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lippa AM, Goulian M. 2012. Perturbation of the oxidizing environment of the periplasm stimulates the PhoQ/PhoP system in Escherichia coli. J. Bacteriol. 194:1457–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zwir I, Latifi T, Pérez JC, Huang H, Groisman EA. 2012. The promoter architectural landscape of the Salmonella PhoP regulon. Mol. Microbiol. 84:463–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lejona S, Aguirre A, Cabeza ML, García Vescovi E, Soncini FC. 2003. Molecular characterization of the Mg2+-responsive PhoP-PhoQ regulon in Salmonella enterica. J. Bacteriol. 185:6287–6294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harari O, Park SY, Huang H, Groisman EA, Zwir I. 2010. Defining the plasticity of transcription factor binding sites by deconstructing DNA consensus sequences: the PhoP-binding sites among gamma/enterobacteria. PLoS Comput. Biol. 6:e1000862 doi:10.1371/journal.pcbi.1000862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yamamoto K, Ogasawara H, Fujita N, Utsumi R, Ishihama A. 2002. Novel mode of transcription regulation of divergently overlapping promoters by PhoP, the regulator of two-component system sensing external magnesium availability. Mol. Microbiol. 45:423–438 [DOI] [PubMed] [Google Scholar]
- 53. Galán JE, Curtiss R., III 1989. Virulence and vaccine potential of phoP mutants of Salmonella Typhimurium. Microb. Pathog. 6:433–443 [DOI] [PubMed] [Google Scholar]
- 54. Miller SI, Kukral AM, Mekalanos JJ. 1989. A two-component regulatory system (phoP phoQ) controls Salmonella Typhimurium virulence. Proc. Natl. Acad. Sci. U. S. A. 86:5054–5058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Worley MJ, Ching KH, Heffron F. 2000. Salmonella SsrB activates a global regulon of horizontally acquired genes. Mol. Microbiol. 36:749–761 [DOI] [PubMed] [Google Scholar]
- 56. Xu X, Hensel M. 2010. Systematic analysis of the SsrAB virulon of Salmonella enterica. Infect. Immun. 78:49–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Miao EA, Miller SI. 2000. A conserved amino acid sequence directing intracellular type III secretion by Salmonella Typhimurium. Proc. Natl. Acad. Sci. U. S. A. 97:7539–7544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zwir I, Shin D, Kato A, Nishino K, Latifi T, Solomon F, Hare JM, Huang H, Groisman EA. 2005. Dissecting the PhoP regulatory network of Escherichia coli and Salmonella enterica. Proc. Natl. Acad. Sci. U. S. A. 102:2862–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Leichert LI, Jakob U. 2004. Protein thiol modifications visualized in vivo. PLoS Biol. 2:e333 doi:10.1371/journal.pbio.0020333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Heras B, Shouldice SR, Totsika M, Scanlon MJ, Schembri MA, Martin JL. 2009. DSB proteins and bacterial pathogenicity. Nat. Rev. Microbiol. 7:215–225 [DOI] [PubMed] [Google Scholar]
- 61. Ellermeier CD, Slauch JM. 2004. RtsA coordinately regulates DsbA and the Salmonella pathogenicity island 1 type III secretion system. J. Bacteriol. 186:68–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Miki T, Okada N, Danbara H. 2004. Two periplasmic disulfide oxidoreductases, DsbA and SrgA, target outer membrane protein SpiA, a component of the Salmonella pathogenicity island 2 type III secretion system. J. Biol. Chem. 279:34631–34642 [DOI] [PubMed] [Google Scholar]
- 63. Lin D, Rao CV, Slauch JM. 2008. The Salmonella SPI1 type three secretion system responds to periplasmic disulfide bond status via the flagellar apparatus and the RcsCDB system. J. Bacteriol. 190:87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]