

Abstract
The periplasmic binding protein (PBP) IbpA mediates the uptake of myo-inositol by the IatP-IatA ATP-binding cassette transmembrane transporter. We report a crystal structure of Caulobacter crescentus IbpA bound to myo-inositol at 1.45 Å resolution. This constitutes the first structure of a PBP bound to inositol. IbpA adopts a type I PBP fold consisting of two α-β lobes that surround a central hinge. A pocket positioned between the lobes contains the myo-inositol ligand, which binds with submicromolar affinity (0.76 ± 0.08 μM). IbpA is homologous to ribose-binding proteins and binds d-ribose with low affinity (50.8 ± 3.4 μM). On the basis of IbpA and ribose-binding protein structures, we have designed variants of IbpA with inverted binding specificity for myo-inositol and d-ribose. Five mutations in the ligand-binding pocket are sufficient to increase the affinity of IbpA for d-ribose by 10-fold while completely abolishing binding to myo-inositol. Replacement of ibpA with these mutant alleles unable to bind myo-inositol abolishes C. crescentus growth in medium containing myo-inositol as the sole carbon source. Neither deletion of ibpA nor replacement of ibpA with the high-affinity ribose binding allele affected C. crescentus growth on d-ribose as a carbon source, providing evidence that the IatP-IatA transporter is specific for myo-inositol. This study outlines the evolutionary relationship between ribose- and inositol-binding proteins and provides insight into the molecular basis upon which these two related, but functionally distinct, classes of periplasmic proteins specifically bind carbohydrate ligands.
INTRODUCTION
Cellular growth in chemically complex environments requires multiple systems for the transport of metabolic substrates. In the bacterial kingdom, specific transmembrane transporters facilitate the uptake of diverse molecular substrates as energy sources and anabolic building blocks. Three families of transporters have been broadly characterized: (i) the ATP-binding cassette (ABC) transporters (1, 2), (ii) the tripartite ATP-independent periplasmic (TRAP) transporters (3, 4), and (iii) the tripartite tricarboxylate (TTT) transporters (5). These transporters are known to recruit periplasmic binding proteins (PBPs) that bind a specific ligand(s) with high affinity (5–7). PBPs are widespread in Gram-negative bacteria but vary significantly both at the primary sequence level and in the types of ligands they bind. The primary functional roles of PBPs are nutrient transport and environmental sensing (6, 8–12). In the case of nutrient transport, the PBP undergoes a transition upon substrate binding that typically involves open-to-closed conformational changes. The PBP then docks with the transmembrane transporter and releases the substrate for carriage across the membrane and into the cell. Depending on the transporter class, the substrate translocation process is driven by proton motive force, electrochemical gradients, or ATP hydrolysis (7, 13).
The freshwater alphaproteobacterium Caulobacter crescentus transports and metabolizes a range of organic nutrients, including the abundant environmental carbohydrate myo-inositol (14). Inositol is an important molecule in many microbial ecosystems; inositol and its derivatives can serve as antioxidants, as cell membrane components, as osmolytes, and as carbon storage units in bacteria and archaea (15, 16). It is also a carbon and energy source for many bacterial species (17–21); in its phosphorylated forms, inositol can serve as a phosphorus source (22, 23).
In Caulobacter, cellular utilization of myo-inositol requires an ABC transporter that consists of the PBP IbpA, the transmembrane permease IatP, and the ABC IatA (14). Once myo-inositol has entered the cell, enzymes encoded from a conserved gene cluster (iolCDEBA-idhA) catalyze its stepwise degradation, yielding dihydroxyacetone phosphate, acetyl coenzyme A, NADH, and NADPH (17, 24, 25, 26, 27). The transcriptional regulatory protein IolR inhibits the expression of the transporter operon and the catabolic enzymes in C. crescentus (14) (Fig. 1) and in related alphaproteobacteria (14, 28).
Fig 1.

Model of myo-inositol uptake and catabolism in Caulobacter. When present in the environment, myo-inositol is bound by the PBP IbpA, which contacts the transmembrane transporter IatP and delivers the sugar to the cytoplasmic space. Translocation of myo-inositol (red circle) from the periplasm to the cytoplasm is energized by the hydrolysis of ATP by the ABC protein IatA. myo-Inositol in the cytoplasm is then catabolized, and the late pathway intermediate 2-keto-5-deoxy-d-gluconate-6-phosphate (orange circle) functions as the inducer of the iol genes by inhibiting the IolR response regulator through a direct interaction with its sugar-binding domain (SBD). Genes (idhA, iolC, and ibpA) normally repressed in the absence of myo-inositol are transcribed, facilitating the metabolism of myo-inositol. OM, outer membrane; IM, inner membrane.
In an effort to define the molecular basis of myo-inositol binding specificity in the C. crescentus ABC transporter system, we have solved the crystal structure of IbpA bound to myo-inositol at 1.45 Å resolution. To our knowledge, this constitutes the first structure of a PBP bound to any form of inositol. IbpA adopts a classic type I clam-like PBP fold (6, 29), with α-β globular lobes separated by a central three-segment hinge. The two lobes and the hinge surround a central ligand-binding cavity, where the six-carbon carbohydrate myo-inositol (cis-1,2,3,5-trans-4,6-cyclohexanehexol) is bound with high affinity (Kd = 760 ± 80 nM). IbpA has sequence and structural features in common with the ribose-binding proteins (RBPs) (30–33) and binds the 5-carbon carbohydrate α-d-ribose [(3R,4S,5R)-5-(hydroxymethyl)oxolane-2,3,4-triol], with low affinity (Kd = 50.8 ± 3.4 μM). A phylogenetic analysis of IbpA and ribose-binding PBP sequences reveals that C. crescentus IbpA occupies an intermediate evolutionary position between myo-inositol-binding PBPs and ribose-binding PBPs. Combining primary sequence data with our IbpA crystal structure, we have engineered a variant cavity mutant (CM), IbpACM2, that binds ribose with high affinity (4.9 ± 1.2 μM) but does not bind myo-inositol. This switch in carbohydrate binding specificity required a total of five mutations that modify specific polar contacts with the carbohydrate and alleviate the steric clash between carbohydrate-interacting side chains. Replacement of the wild-type ibpA allele on the C. crescentus chromosome with ibpACM2 completely blocked cellular growth in myo-inositol minimum medium and did not confer improved growth on d-ribose. Moreover, strains in which the permease (iatP) or ATPase (iatA) transporter components were deleted exhibited wild-type growth rates on d-ribose as the sole carbon source. Thus, the IbpA-IatP-IatA ABC transporter system specifically transports myo-inositol and not d-ribose under the culture conditions tested.
MATERIALS AND METHODS
Construction of expression plasmids.
The sequence encoding residues 39 to 327 of myo-inositol-binding protein (IBP; ibpA gene number CC_0859) was amplified by PCR from C. crescentus NA1000 genomic DNA. The PCR product was isolated by agarose gel extraction (Omega Biotek), cloned into the PCR-Blunt II-TOPO vector (Invitrogen), and sequenced. ibpA was cut as an NdeI-HindIII fragment from the TOPO vector and ligated into the corresponding sites of pET28c (Novagen). The resulting plasmid encodes IbpA(39-327) with an N-terminal 6×His tag. Mutations in the IbpA ligand-binding cavity were successively introduced by overlapping PCR (for the sequences of the primers used, see Table S1 in the supplemental material). Two different sets of mutations were introduced, corresponding to IbpA CM1 (IbpACM1: Q49N, D169I, S174A, and S203F) and IbpA CM2 (IbpACM2: Q49N, N168G, D169I, S174A, and S203F).
IbpA expression and purification.
Recombinant IbpA proteins (wild-type IbpA, IbpACM1, and IbpACM2) were expressed in E. coli Rosetta(DE3)pLysS (Novagen) (for strain numbers and characteristics, see Table S2 in the supplemental material). A 50-ml overnight LB medium culture supplemented with 50 μg/ml kanamycin (LB-Kan50) was used to inoculate 1 liter of LB-Kan50; this culture was incubated at 37°C in a rotary shaker at 220 rpm. Transcription of recombinant ibpA was induced at an optical density at 600 nm (OD600) of 0.8 by adding 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). After 4 h of induction, the cells were harvested by centrifugation at 12,000 × g for 20 min at 4°C. Cell pellets were resuspended in 30 ml of lysing/binding buffer (10 mM Tris [pH 7.4], 150 mM NaCl, 10 mM imidazole) supplemented with DNase I (Sigma-Aldrich) and phenylmethylsulfonyl fluoride (Sigma-Aldrich).
Cells were disrupted by three passages in a French pressure cell, and the cell debris was removed by centrifugation for 20 min at 25,000 × g. The supernatant was loaded onto a Ni2+ Sepharose affinity column (GE Life Sciences) pre-equilibrated with the binding buffer. Two washing steps were performed with 10 and 75 mM imidazole, followed by two elution steps with 200 mM and 1 M imidazole in the binding buffer. The protein solution was then dialyzed against 10 mM Tris (pH 7.4) and 150 mM NaCl buffer to remove the imidazole. All purification steps were carried out at 4°C. All concentrations were measured with a NanoDrop 1000 spectrophotometer (Thermo Scientific).
Crystallization of IbpA bound to myo-inositol.
Initial attempts to phase the IbpA structure by molecular replacement failed because of problems with crystal pseudosymmetry. We produced selenium-labeled protein for experimental phase determination. IbpA was expressed in a defined medium containing selenomethionine as previously described (34). Purified wild-type SeMet-labeled IbpA was purified and concentrated with a centrifugal filter (3-kDa molecular mass cutoff; Amicon-Millipore). Protein purity was estimated to be 95% as assessed by 12% SDS-PAGE and staining with Coomassie brilliant blue. Initial crystallization screening was carried out by the sitting-drop vapor diffusion technique in 96-well microplates (Nunc). Trays were set up with a Mosquito robot (TTP LabTech) and commercial crystallization kits (Nextal-Qiagen). The drops were set up by mixing equal volumes (0.1 μl) of the protein and the precipitant solutions equilibrated against 75 μl of the precipitant solution. In all trials, the protein concentration was ∼50 mg/ml (∼1.6 mM) supplemented with 5 mM myo-inositol (Calbiochem, EMD Millipore). In approximately 1 week, small crystals appeared in condition 44 of the Classics Suite II crystallization kit (Qiagen). After manual refinement of the crystallization condition, the best crystals were obtained at 19°C with a crystallization solution containing 75 mM HEPES (pH 7.4), 25% (wt/vol) polyethylene glycol 3350, and 5 mM myo-inositol. All manual crystallization attempts were carried out by the hanging-drop vapor diffusion technique in 24-well plates (Hampton). Microseeding of pre-equilibrated drops with a cat whisker improved the shape, size, and quality of the crystals. The drops were set up by mixing equal volumes (3 μl) of the protein and the precipitant solutions equilibrated against 500 μl of the precipitant solution. Crystals grew to their final size in 7 to 10 days. Before flash freezing with liquid nitrogen, crystals were cryoprotected by soaking in a crystallization solution containing 30% glycerol and 5 mM β-mercaptoethanol.
Crystallographic data collection and data processing.
Crystal diffraction was measured at a temperature of 100 K with a 1° oscillation range on beamline 21-ID-D (Life Sciences Collaborative Access Team, Advanced Photon Source, Argonne, IL); diffraction images were collected on a MAR Mosaic 300 detector. Diffraction images were processed with the HKL 2000 suite (35). Geometric refinement and examination of the scaled amplitudes revealed that the Se-Met IbpA crystals belong to monoclinic space group C2, with cell dimensions a = 83.1 Å, b = 34.9 Å, and c = 181.8 Å (β = 102.6°) (see Table 1).
Table 1.
Crystallographic data and refinement statistics
| Parameter | Value(s)e |
|---|---|
| Data collection statistics | |
| Energy (keV) | 12.66 |
| Resolution range (Å) | 19.9–1.45 (1.47–1.45) |
| No. of unique reflections | 77,441 |
| Rmergea | 0.08 |
| 〈I〉/〈sI〉 ratio | 14.8 (2.0) |
| Avg % redundancy | 4.2 (3.8) |
| Avg % completeness | 99.0 (98.9) |
| Phasing statistics (19.9–1.45 Å),b figures of merit: | |
| Acentric | 0.47 |
| Centric | 0.17 |
| Overall | 0.35 |
| Refinement statistics | |
| Space group | C2 |
| a, b, c (Å) | 83.1, 34.9, 181.8 |
| β (°) | 102.6 |
| Rcrystc | 0.166 |
| Rfreed | 0.188 |
| 〈B〉 (Å2) | 16.9 |
| RMSD of bond lengths (Å) | 0.010 |
| RMSD of bond angles (°) | 1.321 |
| Ramachandran analysis | |
| % Preferred | 98.5 |
| % Allowed | 0.8 |
| % Disallowed | 0.7 |
Rmerge = ΣhklΣi |Ii − 〈I〉|/ΣhklΣiIi. For all data, I/σ(I) = >3.
Initial phases were determined by selenium Autosol SAD (see Materials and Methods).
Rcryst = Σhkl ||Fobs| − |Fcalc||/Σhkl |Fobs| (includes all data).
Rfree uses 1,945 total reflections for cross-validation.
The values in parentheses represent the highest-resolution shell.
Diffraction from a single Se-Met IbpA protein crystal was measured at an energy of 12.66 keV (0.979 Å), and the structure was phased from the resulting 1.45-Å data set by single-wavelength anomalous dispersion (36). Two IbpA monomers are in the asymmetric unit. Eighteen selenium sites (nine in each monomer) were located within the asymmetric unit by the Autosol SAD routine within the Phenix software suite (37). A preliminary IbpA structural model (Rwork of 20%, Rfree of 23%) was built de novo from the initial experimental, solvent-flattened maps by the AutoBuild routine and phenix.refine. This initial model was then manually examined and corrected; solvent addition and refinement of the structure were conducted iteratively with Coot (38) and phenix.refine (37). The final structural model was refined to an Rwork of 16.6% and an Rfree of 18.8%. Crystallographic data and refined model statistics are presented in Table 1.
Genetic manipulations and engineering of C. crescentus allelic-replacement strains.
The chromosomal copy of the wild-type ibpA allele was replaced in the C. crescentus NA1000 (39) genetic background with two ibpA CM alleles (ibpACM1 and ibpACM2) by a double-recombination gene replacement strategy (40). Briefly, ibpA mutant alleles were cloned into suicide plasmid pNPTS138 (M. R. K. Alley, unpublished data), which carries the nptI gene for initial selection and the sacB gene for counterselection on sucrose. A C. crescentus NA1000 ΔibpA strain (FC488) was transformed with pNPTS138-ibpA plasmids by electroporation, and single-crossover integrants were selected on peptone-yeast extract (PYE) kanamycin (25 μg/ml) agar plates. Counterselection for the second crossover and PCR screening to identify strains in which the wild-type allele was replaced with a mutant allele were carried out as described previously (41). See Tables S1 and S2 in the supplemental material for primer, plasmid, and strain information.
Cell growth assays.
Wild-type and mutant strains of C. crescentus NA1000 were grown overnight in culture tubes containing 5 ml of PYE medium. After overnight growth, OD660s were assessed. Bacteria were then harvested by centrifugation (20,000 × g for 1 min), and the pellets were washed three times with minimum medium (M2) and reharvested by centrifugation. The densities of cultures for growth rate assays were adjusted by resuspending the washed pellets in the appropriate volume of M2 medium. Equal volumes of the density-adjusted cultures were then used to inoculate (to an OD660 of 0.05) tubes containing 3 ml of M2 minimum medium supplemented with 0.2% (wt/vol) d-ribose (Sigma-Aldrich) or 0.2% (wt/vol) myo-inositol (Calbiochem). The growth kinetics of the different strains were measured by tracking the OD660 each day for 1 week. Each growth experiment was repeated three times.
ITC ligand biding assays.
To ensure that the IbpA protein sample lacked ligand prior to the isothermal titration calorimetry (ITC) carbohydrate binding assays, purified wild-type and mutant IbpA proteins were extensively dialyzed against 2 liters of 10 mM Tris (pH 7.4)–150 mM NaCl buffer over 12 h; this dialysis procedure was serially repeated four times. We assume that after affinity chromatography and four serial 2-liter dialysis steps carried out across 4 days, random stray ligand obtained from E. coli lysate will be mostly lost. This assumption is based on typical microscopic dissociation rate constants of 1 to 100 s−1 for ligands bound to PBPs (42) and a low micromolar concentration of IbpA in the dialysis bag. However, it is possible that a small fraction of our sample had unknown ligand bound prior to the ITC measurements.
All samples (proteins and ligands) were degassed for 20 min prior to ITC measurements, and final sample dilutions were carried out by using the final dialysis buffer. Ligands were injected into a 200-μl sample cell containing 50 μM protein for the myo-inositol titrations and 100 μM protein for the d-ribose titrations. Concentrations of 1 and 10 mM myo-inositol were tested on wild-type IbpA (40 injections) and CM1 and CM2 IbpA mutants (20 injections), respectively. For d-ribose, a concentration of 10 mM was used for wild-type IbpA and IbpACM1 (40 and 20 injections, respectively). A concentration of 5 mM d-ribose was used for IbpACM2 titrations (40 injections). ITC was performed at 25°C with a 0.5-μl injection volume every 200 s. All ligands were titrated into ITC buffer alone, and the resulting heat of dilution was subtracted from each experimental curve. ITC was performed with an iTC200 microcalorimeter (MicroCal; GE Healthcare). Data were analyzed and fitted with Microcal Origin software. Each titration was performed three times.
CD spectroscopy.
Overall folding and secondary structure of IbpA and mutant variants were assessed by circular dichroism (CD) spectroscopy. Prior to CD measurements, purified protein samples were dialyzed in a buffer containing 50 mM sodium phosphate (pH 7.4) and 50 mM NaCl. CD spectra from 180 to 260 nm were measured on an AVIV 202 CD spectrometer. Mean ellipticity units per residue (after buffer subtraction) were calculated for each spectrum, and data were analyzed with the K2D3 software (http://www.ogic.ca/projects/k2d3) to estimate protein secondary structure (43).
Sequence alignment and protein visualization methods.
The Dali server (http://ekhidna.biocenter.helsinki.fi/dali_server/) (44) was used to align the IbpA structure with structures in the Protein Data Bank (PDB). Protein sequence alignments were carried out in ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/) (45) and shaded in Boxshade (http://www.ch.embnet.org/software/BOX_form.html). IbpA ribbon rendering, IbpA/RBPTt structural alignment, and ligand binding comparisons were carried out in PyMOL (version 1.3; Schrödinger, LLC). The PDBsum server (http://www.ebi.ac.uk/thornton-srv/databases/pdbsum/Generate.html) was used to define the molecular interaction map between the myo-inositol ligand and atoms in the IbpA protein.
Phylogenetic tree construction.
Six sequences from proteins involved in myo-inositol uptake, including C. crescentus IbpA (UniProt accession no. B8H228), Sinorhizobium meliloti Q926E6, Mesorhizobium loti Q98CU7, Agrobacterium tumefaciens A9CF36, Brucella melitensis Q8YIQ0 (14), and Pseudomonas sp. strain GM48 J2ZWP7 (46), and five sequences corresponding to PBPs that mediate ribose uptake, including Escherichia coli P02925 (47), Salmonella enterica serovar Typhimurium F5ZWH0 (48), Bacillus subtilis P36949 (49), Thermoanaerobacter tengcongensis Q8RD41 (32), and Thermotoga maritima Q9X053 (30), were used to construct a BIONJ neighbor-joining phylogenetic tree with the phylogeny.fr suite (50). Xylose-binding proteins from E. coli (UniProt accession no. P37387) (51) and Thermoanaerobacter ethanolicus (O68456) (52) were used as an outgroup. Evolutionary distances for tree construction were calculated with protdist (53) and the WAG amino acid replacement matrix (54). Bootstrapping was conducted with Seqboot and Consense (number of bootstrap replicates = 100) (53).
Protein structure accession number.
Coordinates of C. crescentus IbpA have been deposited in the PDB (code 4IRX).
RESULTS
Crystal structure of IBP IbpA.
An N-terminally His6-tagged fusion of C. crescentus IbpA from which the first 38 signal peptide amino acids were removed was expressed in E. coli and purified by affinity chromatography. His6-IbpA(39-326), here referred to simply as IbpA, was crystallized in the presence of the myo isomer of inositol (cis-1,2,3,5-trans-4,6-cyclohexanehexol) and formed monoclinic crystals of space group C2 with cell dimensions of 83.1, 34.9, and 181.8 Å (β = 102.6°). There are two molecules of IbpA in the crystal asymmetric unit. Statistics relating to the final refined model are summarized in Table 1. The Ramachandran plot of each monomer in the asymmetric unit reveals two outliers, D169 and D258. The side chain carboxyl of D169 makes direct contact with a hydroxyl group of myo-inositol; residue D258 is a Ramachandran outlier across several members of the periplasmic carbohydrate-binding protein family (55). It is not known if atypical backbone geometry at the D258 position has general functional significance in carbohydrate-binding PBPs.
Each IbpA monomer consists of two α-β lobes that surround a three-segment hinge (segment 1, A140 to N142; segment 2, E277 to P280; segment 3, L306 to V309) (Fig. 2A and 3A). The hinged lobes form a deep cleft that constitutes the myo-inositol binding cavity (Fig. 2B). Lobe 1 begins with the amino terminus and consists of β-strands 1 to 5 and 12 and α-helices 1 to 3 and 9. Lobe 2 consists of β-strands 6 to 11 and 13 and α-helices 4 to 8, 10, and 11, and ends at the carboxy terminus (Fig. 2A and 3A). The binding cavity for myo-inositol is acidic; 11 residues in the ligand-binding cavity make direct contact with the hydroxyl groups of myo-inositol, including Q49, D125, and R126 from lobe 1; N168, D169, S174, R178, S203, N231, and D258 from lobe 2; and Q278 of the hinge. myo-inositol is flanked by two phenylalanines (F51 and F52) that make van der Waals contact with the ligand (Fig. 2C).
Fig 2.

Structure of the IbpA protein bound to myo-inositol. (A) Ribbon structure of IbpA. α-Helices (light gray) and β-strands (light pink) are numbered. (B) Simulated annealing composite omit map (contoured at 2σ) of bound myo-inositol in the IbpA ligand-binding cavity. (C) Interaction map of the IbpA side chain-ligand interactions in the IbpA/myo-inositol structure. Measured distances from side chain nitrogen and oxygen atoms to the hydroxyl oxygens of myo-inositol are shown in green. myo-Inositol carbons are numbered in panels B and C.
Fig 3.

Structural comparison of C. crescentus IbpA and T. tengcongensis RBP. (A) Amino acid sequence alignment of IbpA (upper sequence) and RBPTt (lower sequence). Solid and open circles highlight residues involved in polar interactions and hydrophobic interactions, respectively, between IbpA and myo-inositol (blue) and between RBPTt and ribose (orange). Red boxes highlight residues N168 and S203 from IbpA and G169 and F203 from RBPTt (see Fig. 5). Red lines highlight residues present in the three-segment hinge. α-Helices are represented by cylinders, and β-strands are represented by arrows. The residue at the beginning of each line is numbered. (B) Structural alignment between IbpA (light blue) and RBPTt (light orange, PDB code 2IOY). RMSD = 1.03 Å. (C) Structural alignment of the binding cavity residues of IbpA (light blue) and RBPTt (light orange). myo-Inositol and d-ribose are in blue and orange, respectively. To improve the visibility of the side chains presented, F51 and F52 in IbpA and F52 and F53 in RBPTt are not shown. The carbons of each ligand are been numbered in blue for myo-inositol and in red for d-ribose.
IbpA is structurally homologous to ribose-binding PBPs.
We compared the C. crescentus IbpA structure to available structures in the PDB by using the Dali server (44). IbpA shares the highest structural similarity (root mean square deviation [RMSD] = 1.03 Å) with the RBP from T. tengcongensis (RBPTt) (32) (PDB code 2IOY; Z score = 38.7); the next highest hit (RMSD = 1.02 Å) is the structure of E. coli RBP (33) (PDB code 2DRI; Z score = 37.5). Amino acid sequence alignment and structural alignment of RBPTt and IbpA revealed a large set of identical residues in the carbohydrate-binding cavity (identity, 32%). Five residues that make direct contact with myo-inositol in IbpA differ from residues at the same positions in RBPTt: Q49 versus N51, N168 versus G169, D169 versus I170, S174 versus A175, and S203 versus F203 (Fig. 3).
Phylogenetic analysis of RBPs and IBPs.
To further assess the relationship between RBPs and IBPs, we conducted a phylogenetic analysis of these carbohydrate-binding proteins. Primary sequences of PBPs that have been experimentally classified as ribose or myo-inositol binders were used as our analysis set. Six sequences corresponding to PBPs involved in myo-inositol uptake, including C. crescentus IbpA (UniProt accession no. B8H228), S. meliloti (Q926E6), M. loti (Q98CU7), A. tumefaciens (A9CF36), B. melitensis (Q8YIQ0) (14), and Pseudomonas sp. strain GM48 (J2ZWP7) (46), and five sequences corresponding to PBPs involved in ribose uptake in E. coli (UniProt accession no. P02925) (47), S. enterica serovar Typhimurium (F5ZWH0) (48), B. subtilis (P36949) (49), T. tengcongensis (Q8RD41) (32), and T. maritima (Q9X053) (30), were aligned in order to calculate evolutionary protein distances and construct a neighbor-joining phylogenetic tree (see Materials and Methods). Xylose-binding proteins from E. coli (UniProt accession no. P37387) (51) and T. ethanolicus (O68456) (52) were used as an outgroup. C. crescentus IbpA occupies an intermediate position on the tree, between RBPs and IBPs (Fig. 4).
Fig 4.

Neighbor-joining phylogenetic tree of IBPs and RBPs. The tree is constructed from six IBP sequences (C. crescentus [Cc], S. meliloti [Sm], M. loti [Ml], A. tumefaciens [At], B. melitensis [Bm], and Pseudomonas sp. strain GM48 [Ps]) and five RBP sequences (E. coli [Ec], S. enterica serovar Typhimurium [St], B. subtilis [Bs], T. tengcongensis [Tt], and T. maritima [Tm]). Xylose-binding protein sequences from E. coli (Ec) and T. ethanolicus (Te) are the outgroup. Bootstrap values for 100 replicates are presented at the nodes on the tree.
IbpA has slightly higher homology to RBP sequences (31% ± 2% identity) than to IBP sequences (27% ± 2% identity). Within their ligand-binding cavities, RBPs and IBPs are similar but can be distinguished at selected positions. The ligand-binding cavity of IbpA has characteristics of an RBP-IBP hybrid. Eleven residues make polar contacts with myo-inositol in IbpA, while two phenylalanine residues form packing interactions with ligand-interacting side chains. Certain IbpA residues are more closely related to RBPs in our alignment (e.g., F52 and D125), while others (Q49, N168, D169, and S203) are distinct from the RBP and IBP lineages (Fig. 5A and B). As outlined in the section above, five residues distinguish C. crescentus IbpA from RBPs. We note a pair of residues in the ligand-binding cavity (positions 168 and 203) that distinguish IbpA from both RBPs and other IBPs in our alignment (Fig. 5A and B; see Fig. S1 in the supplemental material). IbpA contains a serine at position 203; this serine residue is flanked by asparagine 168. In all other proteins in the alignment, a glycine is present at the position corresponding to N168 and flanks a phenylalanine or a tryptophan at position 203. The Phe residue forms stacking interactions on top of the ribose in RBPs (Fig. 5C). Experiments in which we investigated the functional roles of positions 168 and 203 in ligand binding are described below.
Fig 5.

Comparative residue frequencies in the IBP and RBP ligand-binding cavities. Residue numbering is based on the IbpA sequence. For the amino acid sequence alignment used to produce this figure, see Fig. S1 in the supplemental material. (A) Residue frequency in the ligand-binding cavities of IBPs. The residue at each of these positions in Caulobacter IbpA is in red. (B) Residue frequency in the ligand-binding cavities of RBPs. The residue at each of these positions in Caulobacter IbpA is in red. The residues at positions 169 and 174 are not involved in ligand interaction and are in gray. (C) Spatial organization of the N168-S203 pair in IbpA and the G169-F203 pair in RBPTt (PDB code 2IOY). myo-Inositol and ribose carbons are numbered.
Structure-function analysis of myo-inositol and ribose binding to IbpA: an engineered switch in carbohydrate binding specificity.
To test the functional roles of specific IbpA residues in binding to carbohydrate ligands, we measured the myo-inositol and ribose binding affinities of wild-type and mutant variants of purified IbpA protein by ITC (Fig. 6). Consistent with a role in myo-inositol transport, wild-type C. crescentus IbpA binds myo-inositol with high affinity (0.76 ± 0.08 μM) (Fig. 6A). As discussed above, the binding cavity of IbpA is structurally homologous to RBPs. To assess whether structural homology to RBPs in the ligand-binding cavity is reflected in actual ligand binding function, we also measured the binding of d-ribose to IbpA. IbpA binds d-ribose, albeit with ∼70-fold lower affinity (50.8 ± 3.4 μM) than myo-inositol (Fig. 6D).
Fig 6.

ITC carbohydrate binding assays. In each panel, the raw heat signal is displayed above the integrated and fitted data (below). Equilibrium dissociation constants (Kd values) were calculated for each titration after subtraction of the buffer heat of dilution. (A) Binding interaction between IbpA and myo-inositol. WT, wild type. (B) Binding interaction between IbpACM1 and myo-inositol. (C) Binding interaction between IbpACM2 and myo-inositol. (D) Binding interaction between IbpA and d-ribose. (E) Binding interaction between IbpACM1 and d-ribose. (F) Binding interaction between IbpACM2 and d-ribose.
Given that IbpA binds both ribose and myo-inositol, we sought to assess the function of residues within the ligand-binding cavity of IbpA in the binding of these carbohydrate ligands. We first introduced mutations into the ligand-binding cavity at four positions that distinguish IbpA from known RBPs: Q49, D169, S174, and S203 (Fig. 5B). These residues were mutated to the most conserved residues in RBPs at the corresponding structural positions (Q49N, D169I, S174A, and S203F; Fig. 5B; see Fig. S1 in the supplemental material) to produce IbpA CM1 (IbpACM1). Though the ligand-binding cavity of mutant IbpACM1 is significantly more closely related to bona fide RBPs, its affinity for ribose is ∼6-fold lower (Kd = 289 ± 32 μM) than that of wild-type IbpA. Binding to myo-inositol is completely abolished in mutant IbpACM1 (Fig. 6B and E).
We constructed a homology model of IbpACM1 that predicted steric side chain clash between the phenylalanine introduced at position 203 and asparagine 168. All sequences within the RBP clade encode a glycine at position 168 (Fig. 5B; see Fig. S1 in the supplemental material), which allows F203 to enter a stacked conformation with the ribose ring (Fig. 5C). On the basis of these structures, we predicted that the addition of an N168G mutation would alleviate the steric clash with the F203 position and increase the affinity of the mutant protein for a ribose ligand. We thus generated IbpA CM2 (IbpACM2), which contained the mutations Q49N, N168G, D169I, S174A, and S203F. IbpACM2 binds d-ribose with high affinity (4.9 ± 1.2 μM) (Fig. 6F), approximately 60-fold higher than ribose binding to IbpACM1 and 10-fold higher than ribose binding to wild-type IbpA. Like IbpACM1, IbpACM2 does not bind myo-inositol (Fig. 6C). Thus, the introduction of five mutations into the ligand-binding cavity of C. crescentus IbpA is sufficient to confer a switch in binding specificity from myo-inositol to ribose.
As a control to ensure that the changes in ligand binding affinities between wild-type and mutant IbpA proteins were not a result of changes in global protein folding and stability, we measured the UV CD spectra of purified IbpA, IbpACM1, and IbpACM2. Estimation of the α-helix and β-strand contents of each mutant from the UV CD spectra demonstrated that all three proteins have similar α-helix and β-strand contents (average α-helix content, ∼34%; average β-strand content, ∼17%). These values are consistent with the helix and strand contents of the IbpA crystal structure (α-helix content, 44%; β-strand content, 18%) (Fig. 7A).
Fig 7.
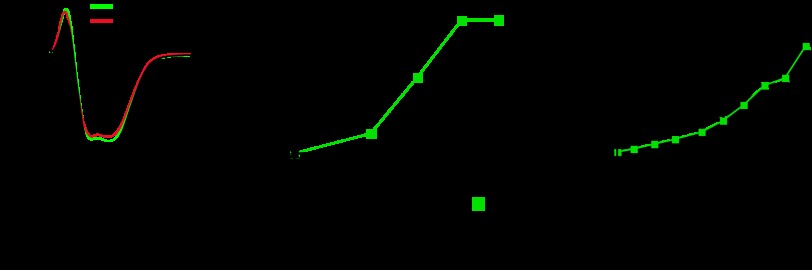
Effects of ABC transporter mutations and IbpA cavity mutations on C. crescentus growth on myo-inositol and d-ribose. (A) Folding of the different IbpA CMs (CM1 and CM2) was compared to that of the wild-type (WT) protein by UV CD spectroscopy. The α-helix and β-strand contents of each protein were calculated from the CD spectra and compared to the IbpA crystal structure. mdeg, millidegrees. (B) Growth curves of wild-type C. crescentus, ABC transporter mutants, and IbpA CMs in M2 defined medium supplemented with 0.2% (wt/vol) myo-inositol. (C) Growth curves of wild-type C. crescentus, ABC transporter mutants, and IbpA CMs in M2 defined medium supplemented with 0.2% (wt/vol) d-ribose.
Functional analysis of wild-type and mutant IbpA in C. crescentus cells.
We next measured the effect of IbpA ligand-binding cavity mutations on C. crescentus cell growth in medium containing either myo-inositol or ribose as the sole carbon source. As previously described (14), the growth of C. crescentus in M2 defined medium (56) supplemented with 0.2% (wt/vol) myo-inositol (M2-inositol) requires each gene of the myo-inositol transport system (i.e., ibpA, iatP, and iatA) (Fig. 7B). We performed this same cell growth experiment with M2 defined medium supplemented with 0.2% (wt/vol) d-ribose (M2-ribose). Wild-type C. crescentus grows slowly in M2-ribose, with a doubling time of 62 ± 1 h (Fig. 7C). Deletion of ibpA, iatP, or iatA has no effect on cell growth in M2-ribose (Fig. 7C).
To assess the effects of mutations in the IbpA ligand-binding cavity on C. crescentus growth in defined medium, we replaced the wild-type chromosomal copy of ibpA with either the ibpACM1 or the ibpACM2 allele by double-crossover recombination. Neither the ibpACM1 nor the ibpACM2 allele replacement strain was able to grow in M2-inositol medium (Fig. 7B). This result provides evidence that C. crescentus growth on myo-inositol as the sole carbon source requires an IbpA allele that is competent to bind myo-inositol. We further tested the growth of the ibpACM1 and ibpACM2 allele replacement strains in M2-ribose medium. As expected from the ΔibpA mutant growth experiment, neither the ibpACM1 nor the ibpACM2 mutant had defects in growth on ribose (Fig. 7C).
DISCUSSION
We have solved a high-resolution crystal structure of IbpA bound to myo-inositol, which constitutes the first structure of a PBP bound to this class of carbohydrate. A comparison of IbpA to PBP sequences in the nonredundant database of GenBank (57) and to structures in the PDB (58) revealed significant homology between IbpA and RBPs (Fig. 3 to 5). The crystal structure of IbpA thus provides a foundation for comparative analysis of PBPs that bind myo-inositol and d-ribose. The phylogenetic, structural, and biochemical analyses presented herein have yielded molecular-level insight into inositol- and ribose-binding specificity in these related classes of ABC transporter PBPs.
IbpA binds myo-inositol in vitro with an affinity of 0.76 μM, which is consistent with the measured affinities of carbohydrate-binding PBPs, including the myo-inositol-binding PBP of Pseudomonas sp. (46). As previously described, ibpA and the adjacent membrane permease (iatP) and ATPase (iatA) genes are required for C. crescentus growth on myo-inositol as the sole carbon and energy source (14). We have shown that IbpA also binds d-ribose with 51 μM affinity and demonstrated that C. crescentus will grow slowly on ribose as the sole carbon and energy source. However, genetic perturbation of the IbpA-IatP-IatA myo-inositol transporter has no effect on C. crescentus growth on d-ribose as the sole carbon and energy source at the assessed ribose concentration of 0.2% (13.3 mM) (Fig. 7C). Thus, the IbpA-IatP-IatA transporter system is apparently not involved in ribose uptake, even though IbpA binds ribose. This result is consistent with the known properties of ABC carbohydrate transporters, which exhibit a high degree of specificity for their transported substrates (1, 2, 59). To date, the system responsible for d-ribose uptake in C. crescentus remains undefined.
IbpA binds myo-inositol with an affinity approximately 70-fold higher than that for d-ribose (Fig. 6). Previous studies have shown that the specificity and affinity of PBPs for a ligand can be modified by cavity mutagenesis (60–62). Given the sequence and structural homology between IbpA and RBPs, we sought to design a variant of IbpA in which the specificities for myo-inositol and d-ribose are switched (i.e., an IbpA mutant that binds ribose with an affinity higher than that for inositol). To this end, we engineered two distinct IbpA CMs, IbpACM1 and IbpACM2. The CM1 mutant protein contains four mutations, Q49N, D169I, S174A, and S203F, that were predicted to increase the affinity of IbpA for d-ribose by modifying the cavity to an RBP-like cavity (32, 33). However, the introduction of these residues did not result in increased affinity of the protein for a ribose ligand. Rather, the affinity of IbpACM1 for ribose was significantly lower than that of the wild-type protein (Kd = 289 μM). We detected no binding interaction between IbpACM1 and myo-inositol, providing evidence that these four mutations increase the specificity for ribose relative to that for inositol (though ribose binding affinity was very low).
On the basis of homology modeling, we attributed the lowered ribose binding affinity of IbpACM1 to a steric clash between the introduced side chain of F203 and the side chain of N168 (Fig. 3C and 5C). Substitution of glycine for N168 was predicted to alleviate this steric clash and allow the F203 phenyl ring to align in a parallel conformation with the ribose ring, as observed in RBP crystal structures (Fig. 3C and 5C). We thus engineered CM2 (IbpACM2), which contained the same four mutations described in IbpACM1 plus an N168G mutation. Introduction of N168G increased the affinity of IbpACM2 for d-ribose by ∼60-fold relative to that of IbpACM1 and 10-fold relative to that of wild-type IbpA (Kd = 5 μM) (Fig. 6). Like IbpACM1, IbpACM2 could be stably expressed and purified but failed to bind myo-inositol in vitro (Fig. 6). Thus, a combination of five mutations in the ligand-binding cavity was sufficient to create a variant of IbpA that binds d-ribose with high affinity but does not bind myo-inositol. We further assessed the loss of myo-inositol binding function in IbpACM1 and IbpACM2 in vivo; C. crescentus ibpACM1 and ibpACM2 chromosomal allelic-replacement mutants failed to grow on myo-inositol as the sole carbon and energy source (Fig. 7B).
The role of residues at IbpA positions 168 and 203 merits further discussion. Classic carbohydrate-binding proteins typically have an aromatic residue (Phe or Trp) at position 203 (Fig. 3 and 5) that makes van der Waals contact with the carbohydrate (30, 32, 51, 63, 64). When Phe or Trp is present at this position, a glycine residue is systematically present at position 168 (Fig. 3 and 5; see Fig. S1 in the supplemental material). Presumably, the absence of a Cβ at position 168 is required for the proper packing of Phe/Trp within the carbohydrate-binding cavity (32, 33). Indeed, experimental data presented in this study reveal a key role for G168 in the designed high-affinity ribose-binding variant IbpACM2, which contains a Phe at position 203 (Fig. 6F). Compared to other IBP and RBP proteins (Fig. 5; see also alignment in Fig. S1), C. crescentus IbpA is atypical at positions 203 and 168 as it contains neither an aromatic residue at position 203 nor a glycine at position 168. Rather, the polar S203 and N168 side chains in wild-type IbpA make close hydrogen bonds with adjacent hydroxyl groups of the bound myo-inositol ligand (Fig. 2C).
Though the designed IbpACM2 variant matches known RBPs in terms of predicted structural contacts with the ribose ligand (Fig. 3 and 5), the affinity of this mutant with a “ribose-like” cavity is not equivalent to that of bona fide RBP proteins, which have measured ribose binding affinities in the 0.1-to-0.2 μM range (33, 47). This result is consistent with a model in which carbohydrate binding affinity is determined by the additive effects of residues that make direct ligand contacts, as well as residues that indirectly affect ligand cavity side chain conformations and, perhaps, global protein conformational transitions between the open and closed states, as described in maltose-binding protein (65).
In conclusion, C. crescentus IbpA presents a case study of a PBP that is positioned at the phylogenetic intersection of IBPs and RBPs (Fig. 4; see Fig. S1 in the supplemental material). Though IbpA binds both myo-inositol and d-ribose in vitro, it has no apparent role in ribose utilization in the C. crescentus cell, where it specifically functions as the PBP component of an ABC transporter that is required for the utilization of myo-inositol as a carbon and energy source (Fig. 7). We have solved a crystal structure of IbpA to 1.45 Å resolution and used the resulting structural model to design an IbpA variant with strongly inverted specificities for inositol and ribose binding. This protein design effort has defined structural determinants of carbohydrate-binding specificity in the related ribose- and myo-inositol-binding classes of ABC transporter PBPs.
Supplementary Material
ACKNOWLEDGMENTS
We thank Elena Solomaha (Chicago Biophysics Core) for assistance with ITC and Heather Pinkett (Northwestern University) for helpful discussion. We thank Jon Henry for providing a whisker from Midnight the cat.
The Advanced Photon Source is supported by the Department of Energy Office of Basic Energy Sciences (contract DE-AC02-06CH11357). The Life Sciences Collaborative Access Team is supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (grant 085P1000817).
Footnotes
Published ahead of print 15 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00116-13.
REFERENCES
- 1. Nikaido H, Hall JA. 1998. Overview of bacterial ABC transporters. Methods Enzymol. 292:3–20 [DOI] [PubMed] [Google Scholar]
- 2. Rees DC, Johnson E, Lewinson O. 2009. ABC transporters: the power to change. Nat. Rev. Mol. Cell Biol. 10:218–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fischer M, Zhang QY, Hubbard RE, Thomas GH. 2010. Caught in a TRAP: substrate-binding proteins in secondary transport. Trends Microbiol. 18:471–478 [DOI] [PubMed] [Google Scholar]
- 4. Rabus R, Jack DL, Kelly DJ, Saier MH., Jr 1999. TRAP transporters: an ancient family of extracytoplasmic solute-receptor-dependent secondary active transporters. Microbiology 145(Pt 12):3431–3445 [DOI] [PubMed] [Google Scholar]
- 5. Winnen B, Hvorup RN, Saier MH., Jr 2003. The tripartite tricarboxylate transporter (TTT) family. Res. Microbiol. 154:457–465 [DOI] [PubMed] [Google Scholar]
- 6. Berntsson RP, Smits SH, Schmitt L, Slotboom DJ, Poolman B. 2010. A structural classification of substrate-binding proteins. FEBS Lett. 584:2606–2617 [DOI] [PubMed] [Google Scholar]
- 7. Saier MH., Jr 2000. A functional-phylogenetic classification system for transmembrane solute transporters. Microbiol. Mol. Biol. Rev. 64:354–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Felder CB, Graul RC, Lee AY, Merkle HP, Sadee W. 1999. The Venus flytrap of periplasmic binding proteins: an ancient protein module present in multiple drug receptors. AAPS PharmSci. 1:E2 doi:10.1208/ps010202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tam R, Saier MH., Jr 1993. Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol. Rev. 57:320–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wadhams GH, Armitage JP. 2004. Making sense of it all: bacterial chemotaxis. Nat. Rev. Mol. Cell Biol. 5:1024–1037 [DOI] [PubMed] [Google Scholar]
- 11. Ng WL, Bassler BL. 2009. Bacterial quorum-sensing network architectures. Annu. Rev. Genet. 43:197–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jacob-Dubuisson F, Wintjens R, Herrou J, Dupré E, Antoine R. 2012. BvgS of pathogenic Bordetellae: a paradigm for sensor-kinases with Venus flytrap perception domains. In Gross R, Beier D. (ed), Two-component systems in bacteria. Caister Academic Press, Würzburg, Germany [Google Scholar]
- 13. Lolkema JS, Poolman B, Konings WN. 1998. Bacterial solute uptake and efflux systems. Curr. Opin. Microbiol. 1:248–253 [DOI] [PubMed] [Google Scholar]
- 14. Boutte CC, Srinivasan BS, Flannick JA, Novak AF, Martens AT, Batzoglou S, Viollier PH, Crosson S. 2008. Genetic and computational identification of a conserved bacterial metabolic module. PLoS Genet. 4:e1000310 doi:10.1371/journal.pgen.1000310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Galbraith MP, Feng SF, Borneman J, Triplett EW, de Bruijn FJ, Rossbach S. 1998. A functional myo-inositol catabolism pathway is essential for rhizopine utilization by Sinorhizobium meliloti. Microbiology 144:2915–2924 [DOI] [PubMed] [Google Scholar]
- 16. Roberts MF. 2006. Inositol in bacteria and archaea, p 103–104 In Majumder AL, Biswas BB. (ed), Biology of inositols and phosphoinositides, vol 39 Springer, New York, NY [Google Scholar]
- 17. Berman T, Magasanik B. 1966. The pathway of myo-inositol degradation in Aerobacter aerogenes. Ring scission. J. Biol. Chem. 241:807–813 [PubMed] [Google Scholar]
- 18. Kawsar HI, Ohtani K, Okumura K, Hayashi H, Shimizu T. 2004. Organization and transcriptional regulation of myo-inositol operon in Clostridium perfringens. FEMS Microbiol. Lett. 235:289–295 [DOI] [PubMed] [Google Scholar]
- 19. Krings E, Krumbach K, Bathe B, Kelle R, Wendisch VF, Sahm H, Eggeling L. 2006. Characterization of myo-inositol utilization by Corynebacterium glutamicum: the stimulon, identification of transporters, and influence on l-lysine formation. J. Bacteriol. 188:8054–8061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poole PS, Blyth A, Reid CJ, Walters K. 1994. myo-Inositol catabolism and catabolite regulation in Rhizobium leguminosarum bv. viciae. Microbiology 140:2787–2795 [Google Scholar]
- 21. Yoshida KI, Aoyama D, Ishio I, Shibayama T, Fujita Y. 1997. Organization and transcription of the myo-inositol operon, iol, of Bacillus subtilis. J. Bacteriol. 179:4591–4598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mullaney EJ, Daly CB, Ullah AHJ. 2000. Advances in phytase research. Adv. Appl. Microbiol. 47:157–199 [DOI] [PubMed] [Google Scholar]
- 23. Turner BL, Paphazy MJ, Haygarth PM, McKelvie ID. 2002. Inositol phosphates in the environment. Philos. Trans. R. Soc. Lond. B Biol. Sci. 357:449–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anderson WA, Magasanik B. 1971. The pathway of myo-inositol degradation in Aerobacter aerogenes. Conversion of 2-deoxy-5-keto-d-gluconic acid to glycolytic intermediates. J. Biol. Chem. 246:5662–5675 [PubMed] [Google Scholar]
- 25. Anderson WA, Magasanik B. 1971. The pathway of myo-inositol degradation in Aerobacter aerogenes. Identification of the intermediate 2-deoxy-5-keto-d-gluconic acid. J. Biol. Chem. 246:5653–5661 [PubMed] [Google Scholar]
- 26. Berman T, Magasanik B. 1966. The pathway of myo-inositol degradation in Aerobacter aerogenes. Dehydrogenation and dehydration. J. Biol. Chem. 241:800–806 [PubMed] [Google Scholar]
- 27. Yoshida K, Yamaguchi M, Morinaga T, Kinehara M, Ikeuchi M, Ashida H, Fujita Y. 2008. myo-Inositol catabolism in Bacillus subtilis. J. Biol. Chem. 283:10415–10424 [DOI] [PubMed] [Google Scholar]
- 28. Kohler PR, Choong EL, Rossbach S. 2011. The RpiR-like repressor IolR regulates inositol catabolism in Sinorhizobium meliloti. J. Bacteriol. 193:5155–5163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fukami-Kobayashi K, Tateno Y, Nishikawa K. 1999. Domain dislocation: a change of core structure in periplasmic binding proteins in their evolutionary history. J. Mol. Biol. 286:279–290 [DOI] [PubMed] [Google Scholar]
- 30. Cuneo MJ, Beese LS, Hellinga HW. 2008. Ligand-induced conformational changes in a thermophilic ribose-binding protein. BMC Struct. Biol. 8:50 doi:10.1186/1472-6807-8-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Björkman AJ, Mowbray SL. 1998. Multiple open forms of ribose-binding protein trace the path of its conformational change. J. Mol. Biol. 279:651–664 [DOI] [PubMed] [Google Scholar]
- 32. Cuneo MJ, Tian Y, Allert M, Hellinga HW. 2008. The backbone structure of the thermophilic Thermoanaerobacter tengcongensis ribose binding protein is essentially identical to its mesophilic E. coli homolog. BMC Struct. Biol. 8:20 doi:10.1186/1472-6807-8-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Björkman AJ, Binnie RA, Zhang H, Cole LB, Hermodson MA, Mowbray SL. 1994. Probing protein-protein interactions. The ribose-binding protein in bacterial transport and chemotaxis. J. Biol. Chem. 269:30206–30211 [PubMed] [Google Scholar]
- 34. Doublié S. 2007. Production of selenomethionyl proteins in prokaryotic and eukaryotic expression systems. Methods Mol. Biol. 363:91–108 [DOI] [PubMed] [Google Scholar]
- 35. Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326 [DOI] [PubMed] [Google Scholar]
- 36. Dauter Z. 2002. One-and-a-half wavelength approach. Acta Crystallogr. D Biol. Crystallogr. 58:1958–1967 [DOI] [PubMed] [Google Scholar]
- 37. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 39. Marks ME, Castro-Rojas CM, Teiling C, Du L, Kapatral V, Walunas TL, Crosson S. 2010. The genetic basis of laboratory adaptation in Caulobacter crescentus. J. Bacteriol. 192:3678–3688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ried JL, Collmer A. 1987. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene 57:239–246 [DOI] [PubMed] [Google Scholar]
- 41. Fiebig A, Castro Rojas CM, Siegal-Gaskins D, Crosson S. 2010. Interaction specificity, toxicity and regulation of a paralogous set of ParE/RelE-family toxin-antitoxin systems. Mol. Microbiol. 77:236–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Miller DM, III, Olson JS, Pflugrath JW, Quiocho FA. 1983. Rates of ligand binding to periplasmic proteins involved in bacterial transport and chemotaxis. J. Biol. Chem. 258:13665–13672 [PubMed] [Google Scholar]
- 43. Louis-Jeune C, Andrade-Navarro MA, Perez-Iratxeta C. 2012. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins 80:374–381 [DOI] [PubMed] [Google Scholar]
- 44. Holm L, Rosenstrom P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38:W545–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 46. Deshusses J, Belet M. 1984. Purification and properties of the myo-inositol-binding protein from a Pseudomonas sp. J. Bacteriol. 159:179–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Willis RC, Furlong CE. 1974. Purification and properties of a ribose-binding protein from Escherichia coli. J. Biol. Chem. 249:6926–6929 [PubMed] [Google Scholar]
- 48. Aksamit RR, Koshland DE., Jr 1974. Identification of the ribose binding protein as the receptor for ribose chemotaxis in Salmonella typhimurium. Biochemistry 13:4473–4478 [DOI] [PubMed] [Google Scholar]
- 49. Woodson K, Devine KM. 1994. Analysis of a ribose transport operon from Bacillus subtilis. Microbiology 140(Pt 8):1829–1838 [DOI] [PubMed] [Google Scholar]
- 50. Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36:W465–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sooriyaarachchi S, Ubhayasekera W, Park C, Mowbray SL. 2010. Conformational changes and ligand recognition of Escherichia coli d-xylose binding protein revealed. J. Mol. Biol. 402:657–668 [DOI] [PubMed] [Google Scholar]
- 52. Erbeznik M, Strobel HJ, Dawson KA, Jones CR. 1998. The d-xylose-binding protein, XylF, from Thermoanaerobacter ethanolicus 39E: cloning, molecular analysis, and expression of the structural gene. J. Bacteriol. 180:3570–3577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Felsenstein J. 1989. PHYLIP—phylogeny inference package (version 3.2). Cladistics 5:164–166 [Google Scholar]
- 54. Whelan S, Goldman N. 2001. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18:691–699 [DOI] [PubMed] [Google Scholar]
- 55. Magnusson U, Chaudhuri BN, Ko J, Park C, Jones TA, Mowbray SL. 2002. Hinge-bending motion of d-allose-binding protein from Escherichia coli: three open conformations. J. Biol. Chem. 277:14077–14084 [DOI] [PubMed] [Google Scholar]
- 56. Ely B. 1991. Genetics of Caulobacter crescentus. Methods Enzymol. 204:372–384 [DOI] [PubMed] [Google Scholar]
- 57. Benson DA, Karsch-Mizrachi I, Clark K, Lipman DJ, Ostell J, Sayers EW. 2012. GenBank. Nucleic Acids Res. 40:D48–D53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Saier MH., Jr 2000. Families of transmembrane sugar transport proteins. Mol. Microbiol. 35:699–710 [DOI] [PubMed] [Google Scholar]
- 60. Sakaguchi-Mikami A, Taneoka A, Yamoto R, Ferri S, Sode K. 2008. Engineering of ligand specificity of periplasmic binding protein for glucose sensing. Biotechnol. Lett. 30:1453–1460 [DOI] [PubMed] [Google Scholar]
- 61. Looger LL, Dwyer MA, Smith JJ, Hellinga HW. 2003. Computational design of receptor and sensor proteins with novel functions. Nature 423:185–190 [DOI] [PubMed] [Google Scholar]
- 62. Jeffery CJ. 2011. Engineering periplasmic ligand binding proteins as glucose nanosensors. Nano Rev. 2:5743 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3215197/pdf/NANO-2-5743.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chaudhuri BN, Ko J, Park C, Jones TA, Mowbray SL. 1999. Structure of d-allose binding protein from Escherichia coli bound to d-allose at 1.8 A resolution. J. Mol. Biol. 286:1519–1531 [DOI] [PubMed] [Google Scholar]
- 64. Zou JY, Flocco MM, Mowbray SL. 1993. The 1.7 Å refined X-ray structure of the periplasmic glucose/galactose receptor from Salmonella typhimurium. J. Mol. Biol. 233:739–752 [DOI] [PubMed] [Google Scholar]
- 65. Marvin JS, Hellinga HW. 2001. Manipulation of ligand binding affinity by exploitation of conformational coupling. Nat. Struct. Biol. 8:795–798 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.