

Abstract
The Pel polysaccharide serves as an intercellular adhesin for the formation and maintenance of biofilms in the opportunistic pathogen Pseudomonas aeruginosa. Pel biosynthesis requires the products of a seven-gene operon, pelA-pelG, all of which are necessary for Pel-dependent biofilm formation and Pel-related phenotypes. One of the genes, pelA, encodes a protein with a predicted polysaccharide deacetylase domain. In this work, the role of the putative deacetylase domain in Pel production was examined. We first established that purified recombinant PelA hydrolyzed the pseudosubstrate p-nitrophenyl acetate in vitro, and site-specific mutations of predicted deacetylase active-site residues reduced activity greater than 10-fold. Additionally, these mutants were deficient in Pel-dependent biofilm formation and wrinkly colony morphology in vivo. Subcellular fractionation experiments demonstrate that PelA localizes to both the membrane and periplasmic fractions. Finally, antiserum against the Pel polysaccharide was generated, and PelA deacetylase mutants do not produce Pel-reactive material. Taken together, these results suggest that the deacetylase activity of PelA is important for the production of the Pel polysaccharide.
INTRODUCTION
Biofilms are a prominent mode of bacterial growth in the environment and in disease (1). Biofilm development involves specific stages, including surface adherence, proliferation, cell-cell cohesion, and dispersion (2). The bacterial cells residing within the biofilm are encased in extracellular matrix that plays a key role in each of these developmental steps (2). An important component of the biofilm matrix is extracellular polysaccharides. Extracellular polysaccharides carry out a range of matrix functions, including promoting attachment to surfaces and other cells, building and providing a scaffold to help maintain biofilm structure, and protecting cells from antimicrobials and host defenses (2, 3).
Pseudomonas aeruginosa is a model organism for studying the process of biofilm development, as the bacterium is capable of producing at least three unique extracellular polysaccharides implicated in biofilm development, alginate, Psl, and Pel (4). Previous studies have demonstrated that large strain-to-strain variation exists for polymer production (5). In the current study, we focused on the poorly understood process of Pel biosynthesis. The pel operon was originally identified in a transposon screen for strains defective in biofilm formation at the air-liquid interface of a standing culture in P. aeruginosa strain PA14 (6). This strain lacks the genetic capacity necessary to synthesize Psl and is therefore a useful strain to specifically examine Pel-dependent phenotypes. Biofilms occurring at an air-liquid interface are referred to as pellicles. Pellicles formed by PA14 are rigid and resistant to extensive vortexing, boiling, and enzymatic treatments, including DNase I, RNase A, and proteinase K (6). Additionally, the pel operon is necessary for wrinkly colony morphology, Congo red binding, and mature biofilm development in PA14 (6, 7). Recently, Coulon et al. suggested that the pel operon is involved in controlling the relative amounts of cell-associated versus secreted extracellular 3-deoxy-d-manno-octulosonic acid sugar-containing polysaccharides (LPS). The pel mutant strain had much more Kdo sugar-containing material in the supernatant than the PA14 wild type, suggesting that the pel gene products play a role in maintaining association of the core oligosaccharide of LPS to the cell (8).
Although the structure of the Pel polysaccharide remains unknown, all seven genes in the operon are required for Pel-dependent biofilm phenotypes, and each of the Pel proteins are predicted to have functions involved in polysaccharide synthesis, transport, and processing (4, 9). Biosynthesis of extracellular polysaccharides can be roughly divided into five different stages. Initially the precursor substrate, a nucleotide-activated sugar, is synthesized in the cytoplasm, followed by the polymerization of the precursor substrate onto the growing polysaccharide (10). The polysaccharide is subsequently transported across the inner membrane to the periplasm, where it can be enzymatically modified and finally exported through the outer membrane (10).
Pel polymerization is suggested to begin with the single predicted glycosyltransferase in the pel operon, PelF (4). The polymer is hypothesized to be transferred across the inner membrane by two proteins, PelE and PelG, and following translocation through the periplasm, it is believed to be exported across the outer membrane by PelB (4). The role of the outer membrane lipoprotein, PelC, is currently not well characterized, and its function remains unknown (4, 11, 12). Synthesis of the polymer is allosterically regulated by the secondary messenger c-di-GMP (13, 14), as binding of c-di-GMP to the inner membrane protein PelD is essential for Pel production (13). Based on initial computational analysis, PelA, the first gene in the operon, is speculated to serve a dual function, as it is predicted to contain an N-terminal glycoside hydrolase domain and a C-terminal carbohydrate esterase domain (4). Based on these predictions, it was proposed that PelA modifies the polymer after polymerization by catalyzing the removal of O- or N-acetyl groups from the polymer (4). In addition, PelA may also function as a hydrolase and be required for determining Pel polymer size and/or clearing the polysaccharide from the periplasm. Degradation activity has been previously described for alginate biosynthesis. In the absence of scaffolding proteins necessary for alginate synthesis and transport, the alginate lyase AlgL degrades the alginate polymer via β-elimination into smaller uronic acids (15).
In this study, we show that PelA localizes to both the periplasm and membrane and have examined the role of its predicted polysaccharide deacetylase domain in Pel production. Structural modeling and sequence alignment analysis suggest that the deacetylase domain is a member of the metal-dependent carbohydrate esterase family 4 (CE4) superfamily. We demonstrate that PelA exhibits deacetylase activity in vitro and identify four conserved residues in the putative active site required for this activity. Furthermore, mutation of these residues in vivo abrogates biofilm formation and wrinkly colony morphology, suggesting that deacetylase activity is required for Pel synthesis. Our results suggest that Pel deacetylation in the periplasm is required for mature polymer production and secretion.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains and primers used in this study are listed in Table S1 in the supplemental material (7, 16–22). For routine culture, P. aeruginosa strains were grown at 37°C in Luria-Bertani (LB) medium unless otherwise specified (Difco). Plasmids were selected with 100 μg/ml gentamicin for P. aeruginosa strains and 10 μg/ml gentamicin for Escherichia coli. Twenty-five μg/ml irgasan was used to counterselect for E. coli.
Strain construction.
DNA manipulations were performed using standard techniques. For the pelA deletion mutant, ΔpelA, allelic replacement strains were constructed using an unmarked, nonpolar deletion strategy (23). Flanking regions of pelA were amplified using pelA upstream and pelA downstream primer sets (see Table S1 in the supplemental material). The PCR product was ligated into the suicide vector pEX18Gm via its HindIII restriction site. The plasmid pEX18Gm::ΔpelA was verified by sequence analysis. Single recombinant mutants were selected on LB agar plates containing 30 μg/ml gentamicin and 25 μg/ml irgasan. Double recombination mutants were selected on LB agar plates without NaCl but containing 5% (wt/vol) sucrose and confirmed by PCR.
Chromosomal point mutations were constructed using an unmarked, nonpolar allelic replacement strategy similar to that described above. Full-length pelA was amplified with the pelA F2 and R2 primer set. The plasmid pEX18Gm::pelA was generated by ligating the PCR product of pelA F2 and R2 into pEX18Gm via its HindIII restriction site. The D528A, D530A, H600A, and H604A point mutants were generated from pEX18Gm::pelA by site-directed mutagenesis (Stratagene). Each plasmid was verified by sequence analysis. Single recombinant mutants were selected on LB plates containing 30 μg/ml gentamicin and 25 μg/ml irgasan. Double recombination mutants were selected on LB plates without NaCl but containing 5% sucrose and were confirmed by sequence analysis.
Bioinformatics analysis of PelA.
The amino acid sequence of PelA from Pseudomonas aeruginosa strain PAO1 was obtained from the Pseudomonas Genome database (http://www.pseudomonas.com) and analyzed using a number of different web-based servers, primarily Phyre2, SignalP v3.0, and PRED-TAT (24–27). The full-length sequence of PelA was initially used for the bioinformatics analysis. As analysis of full-length PelA using Phyre2 failed to predict a model for residues 410 to 519 and 803 to 948, these residues were submitted separately to the server. The predicted deacetylase domain, residues 520 to 800, was aligned with other predicted or known polysaccharide deacetylase sequences. Sequences were initially aligned with T-coffee (28) and manually adjusted based on structural comparisons using the multiple DNA sequence alignment feature in MacVector.
Cloning and expression of recombinant wild-type PelA and point mutants.
Vectors for expression of recombinant PelA and its variants were generated as follows. The nucleotide sequence of pelA from Pseudomonas aeruginosa PAO1 was used to design primers for PCR (see Table S1 in the supplemental material) (24). NdeI and XhoI restriction sites were included in the forward and reverse primers, respectively. The amplified region excluded the first 46 amino acids, which correspond to the predicted transmembrane region and signal sequence. The PCR product was cloned into the pET-28a vector following digestion with NdeI and XhoI (Novagen). The resulting expression plasmid, pNApelAΔ46, was then used to generate the point mutants of the conserved residues utilizing the QuikChange site-directed mutagenesis lightning kit (Stratagene). The wild-type and mutant expression plasmids, pNApelAΔ46, pNApelAΔ46 D528A, pNApelAΔ46 D530A, pNApelAΔ46 H600A, and pNApelAΔ46 H604A, include residues 47 to 948 of PelA fused to a thrombin-cleavable N-terminal 6-histidine tag used for purification purposes. The fidelity of each construct was verified using DNA sequencing (ACGT Inc., Toronto, Canada).
Expression was achieved by transforming each vector individually into E. coli BL21(DE3) competent cells, which were grown in 2 liters of LB broth containing 50 μg/ml kanamycin at 37°C. The cells were grown to an optical density at 600 nm (OD600) of 0.6 to 0.7, whereupon protein expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. Following induction, the cells were left to grow overnight at 18°C for 22 h prior to being harvested via centrifugation at 7,300 × g for 20 min at 4°C. The resulting cell pellets were stored at −20°C until needed.
Protein purification of wild-type PelA and point mutants.
The following purification protocol was used to purify all of the recombinant proteins utilized in the functional in vitro assays described in this study. A cell pellet from a 2-liter bacterial culture was thawed and resuspended in 30 ml of buffer A (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 10% [vol/vol] glycerol, and 1 Sigmafast protease inhibitor tablet [Sigma]). The resuspended cells were then subjected to five 60-s sonication pulses at 50% amplitude, alternated with 60 s of cooling on ice in order to lyse the cells without heating up the sample. The soluble cell lysate was separated via centrifugation at 25,000 × g for 30 min at 4°C prior to loading onto a 5-ml Ni2+-nitrilotriacetic acid (NTA) superflow cartridge (Qiagen) preequilibrated with five column volumes of buffer B (20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 10% [vol/vol] glycerol, and 5 mM imidazole). Ten column volumes of buffer B containing 20 mM imidazole was used to wash any contaminants off the column, and the bound protein was eluted with three column volumes of buffer B containing 250 mM imidazole. The eluted protein was further purified and buffer exchanged into buffer C (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 10% [vol/vol] glycerol) by size-exclusion chromatography using a HiLoad 16/60 Superdex 200 gel filtration column (GE Healthcare). SDS-PAGE analysis revealed that the purity of each of the proteins was ∼85 to 90%. Each protein was concentrated to 2 mg/ml and used within 1 week for an in vitro enzymatic assay. The concentration of each protein was determined using the Pierce bicinchoninic acid (BCA) protein assay kit from Thermo Scientific (Rockford, IL).
Antibody production and absorption.
PelAΔ46 protein was purified as described above. Purified PelAΔ46 was used to generate antiserum from rabbits using a 70-day standard protocol (Open Biosystems). Antiserum was absorbed using P. aeruginosa PA14ΔpelA lysates. Lysates were generated as previously described (14). The cell lysate was used for absorption by mixing 30 μl α-PelA antisera, 75 μl P. aeruginosa PA14ΔpelA lysate in 1 ml of 5% (wt/vol) nonfat milk in 50 mM Tris, 150 mM NaCl, and 0.05% (vol/vol) Tween 20 (TBST). The antiserum was absorbed for 4 h at room temperature.
To generate a crude Pel polysaccharide sample, 0.5 ml of P. aeruginosa PA14PBADpel overnight culture was added to 50 ml of LB medium supplemented with 0.5% arabinose and incubated for 20 h at 30°C. Cells were harvested by centrifugation at 5,000 × g for 20 min. The supernatant was precipitated overnight at −20°C with cold ethanol to a final concentration of 70% (vol/vol). The precipitate was resuspended in 2 ml of buffer (50 mM Tris, pH 7.5, 1 mM CaCl2, and 2 mM MgCl2) and treated with 5 mg DNase I, 5 mg RNase A for 2 h at 37°C, followed by 5 mg proteinase K treatment overnight at 37°C. This sample was lyophilized and used to generate antiserum from rabbits using a 70-day standard protocol (Open Biosystems). Antiserum was absorbed using PA14ΔpelA and PAO1ΔwspFΔpelΔpsl lysates. Lysates were generated as previously described (14). The cell lysate was used for absorption by mixing 30 μl α-PelA antiserum, 50 μl PA14ΔpelA lysate, and 50 μl PAO1ΔwspFΔpelΔpsl lysate in 1 ml of 5% (wt/vol) nonfat milk in TBST. The antiserum was absorbed for 4 h at room temperature.
Enzyme assay.
All enzyme assays were performed at least in triplicate, in a 96-well microtiter plate, using a SpectraMax M2 from Molecular Devices (Sunnyvale, CA). Standard assays contained 2.5 mM p-nitrophenyl acetate (pNPA), dissolved in ethanol, and ∼40 μg of PelA in buffer A in a total volume of 200 μl of 50 mM sodium HEPES buffer (pH 8.0) at 25°C. To remove the predicted metal ion, PelA (2 mg/ml in buffer A) was incubated with shaking for 90 min in the same buffer supplemented with 200 mM EDTA. Reactions were initiated by the addition of pNPA and were allowed to proceed for 10 min. Reaction progress was monitored in real time at 405 nm for the appearance of p-nitrophenyl. Per the manufacturer's instructions, the extinction coefficient was taken to be 18,300 M−1 cm−1. The background hydrolysis rate was monitored and subtracted from the enzyme-catalyzed reactions. The protein concentration of each enzyme variant was determined using the Pierce BCA protein assay kit from Thermo Scientific (Rockford, IL).
Colony morphology.
Overnight cultures were diluted 1:100 in phosphate-buffered saline (PBS). Five μl of cells was spotted onto tryptone plates containing 10 g/liter tryptone (Difco), 10 g/liter Bacto agar (Difco), 40 μg/ml Congo red (Sigma-Aldrich), and 15 μg/ml brilliant blue R (Sigma-Aldrich) and then incubated at room temperature for 5 days.
Microtiter dish biofilm.
A 96-well microtiter dish assay was preformed as described previously (29). Briefly, 100 μl of mid-log-phase cells (OD600 of ∼0.5) was added to the wells of a 96-well polypropylene plate (Nunc) and incubated statically for 20 h at room temperature. Following incubation, nonattached cells were removed and the plate was rinsed thoroughly with water. Plates were stained with 150 μl 0.1% (wt/vol) crystal violet for 10 min. The plate was rinsed and adhered crystal violet was solubilized in 200 μl 95% (vol/vol) ethanol for 10 min, and then 100 μl was transferred to a new 96-well plate to measure the absorbance at OD595.
Subcellular fractionation.
Subcellular fractionations were preformed as described previously (30, 31). Briefly, 2 ml of mid-log-phase cells (OD600 of ∼0.6) were harvested and either resuspended in 100 μl PBS for whole-cell (WC) analysis or resuspended in 100 μl of lysis buffer (30 mM Tris-HCl, pH 8.0, 20% [wt/vol] sucrose, 4 mM EDTA, 0.5 mg/ml lysozyme, 1 mM phenylmethylsulfonyl fluoride [PMSF]). Cells resuspended in lysis buffer were incubated at room temperature for 2 min. MgCl2 was added to a final concentration of 10 mM and incubated at 30°C for 1 h with occasional inversion. The sample was centrifuged at 4°C, and the supernatant fraction was removed for periplasmic analysis. The pellet consisting of spheroplasts and unlysed cells was washed once in resuspension buffer (30 mM Tris-HCl, pH 8.0, 20% [wt/vol] sucrose). The pellet was resuspended in 100 μl of 1 mM EDTA and lysed by three freeze/thaw cycles in a dry ice/ethanol bath. Unlysed cells were removed by centrifugation at 5,000 × g for 5 min. The supernatant was further centrifuged at 338,800 × g for 1 h. The supernatant consisted of the cytoplasmic fraction, and the pellet contained the membrane fraction.
To analyze Pel polysaccharide subcellular localization, a strategy similar to that described above, with the following modifications, was used. One ml of overnight culture was harvested and resuspended in 100 μl 0.5 M EDTA (EDTA cell pellet) or 100 μl lysis buffer to extract the periplasmic fraction as described above. The pellet containing the spheroplasts was resuspended in 100 μl of 0.5 M EDTA. Each sample was boiled, centrifuged, treated with proteinase K, and probed for Pel polysaccharide reactivity as described in the Pel immunoblotting section below.
Immunoblot analysis. (i) Western blots.
One ml of mid-log-phase cells (OD600 of ∼0.5) grown in LB was harvested and resuspended in 100 μl PBS. A 50-μl sample was mixed with 50 μl 2× Laemmli buffer and boiled for 5 min. Protein concentration was measured using a Pierce 660-nm protein assay with ionic detergent compatibility reagent as described by the manufacturer (Thermo Scientific). Equal amounts of total protein were loaded onto a precast 12.5% Tris-HCl polyacrylamide gel and transferred to a polyvinylidene difluoride (PVDF) membrane for immunoblotting (Bio-Rad). The membrane was blocked in 5% (wt/vol) nonfat milk in TBST for 1 h at room temperature. The membrane was subsequently probed with absorbed α-PelA antisera at a 1:500 dilution, unabsorbed α-PelC antisera (5) at 1:5,000, commercial α-RNA polymerase antibody (RNAP; Neoclone Biotechnologies) at 1:20,000, or α-β-lactamase antibody (QED Biosciences Inc.) at 1:1,000 in 1% (wt/vol) nonfat milk in TBST overnight at 4°C (see the next section for antisera production and absorption). Blots were washed and probed with goat α-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (Thermo Scientific) and developed using the Pierce detection kit.
(ii) Pel immunoblots.
One-ml aliquots of cultures grown overnight were harvested and resuspended in 100 μl 0.5 M EDTA. Cells were boiled for 20 min with periodic vortexing and centrifuged. The supernatant fraction was treated with proteinase K (final concentration, 0.5 mg/ml) for 60 min at 60°C, followed by 30 min at 80°C to inactivate proteinase K. Polysaccharide preparations were normalized to total protein in the PBS resuspension as determined by Bio-Rad protein assay (Bio-Rad). A 5-μl sample was spotted onto a nitrocellulose membrane, allowed to dry, and blocked in 5% (wt/vol) nonfat milk in TBST for 1 h at room temperature. The membrane was probed with absorbed α-Pel antisera at a 1:1,000 dilution in 1% (wt/vol) nonfat milk in TBST for 1 h at room temperature (see the previous section for antiserum production and absorption). Blots were washed, probed with goat α-rabbit HRP-conjugated secondary antibody (Thermo-Scientific), and developed using the Pierce detection kit.
RESULTS
PelA is predicted to contain glycoside hydrolase and polysaccharide deacetylase domains.
Bioinformatics analysis of PelA suggests that it is a multidomain periplasmic protein with three potential catalytic activities (Fig. 1A). The PRED-TAT server predicts a Tat signal peptide with the most likely cleavage site after reside 45. Examination of the N-terminal region of PelA reveals a conserved Tat recognition motif, Z-R-R-X-Φ-Φ (where Z is a polar residue, X is any residue, and Φ is a hydrophobic residue [32]), between residues 15 and 20 (S-R-R-N-I-L). This suggests that PelA is a periplasmic protein that is transported across the cytoplasmic membrane in its folded state via the Tat secretion machinery (33).
Fig 1.

Schematic representation of PelA and structural model of the predicted deacetylase domain. (A) Domain organization of full-length PelA. The approximate boundaries for each domain are indicated on the diagram. The relative size of each domain is proportional to the number of residues in each predicted region. The predicted Tat signal sequence is abbreviated as SS. TIM-barrel, Reductase, and Deacetylase represent the hydrolase, reductase, and deacetylase domains predicted by Phyre2, respectively. β-Jelly Roll represents the low-confidence two-layered β-sheet jelly roll domain predicted for residues 840 to 927. Gray rectangles represent regions of the PelA protein for which Phyre2 was unable to make high-confidence predictions, or for which the entire domain could not be modeled. (B) Homology model of the putative deacetylase domain of PelA based on HpPgdA (PDB entry 3QBU) shown in cartoon representation, with the highly conserved putative catalytic/active site residues D528, D530, H600, and H604 shown in stick representation (purple). (C) A close-up representation of the PelA model showing the putative metal-coordinating residues D530, H600, and H604 and the catalytic residue, D528 (purple), superimposed with the equivalent residues from HpPgdA (gray). The zinc metal ion from the HpPgdA structure is shown as a sphere (cyan). The dotted lines and distances (in Å) represent potential interactions between PelA and the metal ion. For the carboxylate group of D530 to interact with the metal ion, we anticipate that minor structural rearrangements will need to occur. (D) The P. aeruginosa PelA predicted deacetylase domain sequence is compared to known polysaccharide deacetylases, including PgdA from Helicobacter pylori, IcaB from Staphylococcus epidermidis, NodB from Sinorhizobium meliloti, and PdaA from Bacillus subtilis. Conserved residues with greater than 60% identity are highlighted in dark gray. Similar residues with greater than 60% identity are highlighted in light gray. A consensus sequence of conserved and similar residues is located immediately below the PdaA sequence. Alanine replacements made in P. aeruginosa PelA are shown as asterisks located immediately above the PelA amino acid sequence.
Bioinformatics analysis of PelA using Phyre2 predicts at least four, and possibly five, distinct domains, three of which have structural similarities to proteins with known enzymatic activity (25). The first domain, encompassing residues 47 to 303, is predicted to have a canonical (β/α)8 TIM barrel fold (Fig. 1A). The highest confidence model (100%) is based on a putative glycosidase, tm1410 from Thermotoga maritima (PDB code 2AAM). Examination of the highest confidence models reveals that this region of PelA, as for tm1410, has potential structural similarity to proteins that belong to families 13, 36, and 66 of the glycoside hydrolase (GH) superfamily. In the predicted model, the barrel has a canonical TIM barrel fold, with two additional α-helices and two short β-strands. TIM barrel folds have been shown to have many modifications, including domain insertions before, within, or after the barrel, which are essential, in some cases, for the proper functioning of the enzyme (34). The GH family is a large superfamily, and while it is not clear which subfamily of GHs domain 1 of PelA belong to, the predicted TIM barrel fold suggests that its enzymatic function is important for the proper processing of the Pel polysaccharide as it traverses the periplasm. Alternatively, this domain, as is predicted for the C-terminal TIM barrel domain of E. coli PgaB, a protein involved in partial de-N-acetylation and export of the exopolysaccharide poly β-1,6-N-acetyl-d-glucosamine (PNAG), may not exhibit hydrolase activity but instead is involved in polymer binding and is required for the subsequent processing and/or export of Pel (35, 36).
The second domain, encompassing residues 303 to 409, is predicted to be similar (94.5% confidence level) to the flavin mononucleotide-binding domain of Cpr (PDB code 2BPO), a yeast cytochrome p450 reductase (37). At present, it is unclear what function, if any, this domain has; however, the translocation of PelA across the inner membrane as a folded protein via the Tat pathway would enable it to carry along any required cofactors (38). In the context of the full-length protein, Phyre2 was unable to make any predictions for the region immediately following the putative reductase domain, residues 410 to 519. To determine if these residues in isolation had structural homology to a known protein, they were submitted to Phyre2. Phyre2 could not find a high-confidence model for this region, although residues 458 to 496 were predicted with a 45% confidence level to be similar to part of a putative 3-demethylubiquinone-9 3-methyltransferase, PhnB from Bacillus cereus (PDB code 3OMS). Since this prediction covers less than 35% of the residues in this region of the protein, it has not been included in our domain analysis of PelA (Fig. 1A).
Residues 520 to 800 constitute the third predicted domain of PelA. This region of the protein is suggested to be structurally similar to members of carbohydrate esterase family 4 (CE4). Members of this superfamily have been found to exhibit metal-dependent deacetylation of O- and N-acetylated polysaccharides, such as chitin, peptidoglycan, and acetylxylan (39). Proteins belonging to this family have conserved residues that are important for metal coordination (D-H-H triad) and enzymatic activity (Fig. 1). The top three hits include a putative chitooligosaccharide deacetylase (PDB code 2VYO), a probable polysaccharide deacetylase (PDB code 1Z7A), and a putative peptidoglycan deacetylase (PgdA) from Helicobacter pylori, HpPgdA (PDB code 3QBU) (40). The Phyre2 prediction for this domain suggests that the Pel polysaccharide is deacetylated during its passage through the periplasm.
Finally, Phyre2 predicts that the C-terminal region of PelA, residues 840 to 927, has structural similarity to the two-layered β-sheet jelly roll motif found in the C-terminal region of maltose phosphorylase (PDB code 1H54) (41). This model is predicted with low confidence (45%), and as for residues 458 to 496, it could not be detected when the full-length protein was submitted to the Phyre2 server. This region of maltose phosphorylase has superficial resemblance to the starch binding domain of glucoamylase from Aspergillus niger and the N-terminal domain of the endocellulase CelD (42, 43). The function of the β-rich domain in these proteins is unknown.
Conserved amino acids in the predicted deacetylase domain of PelA are required for in vitro deacetylase activity.
As PelA harbors a putative polysaccharide deacetylase domain, we hypothesized that the Pel polysaccharide is subject to deacetylation, and that this step is required for polymer secretion and/or biofilm formation. Partial deacetylation of PNAG has been shown to be required for the secretion of the polymer in E. coli (36) and for biofilm formation for a number of both Gram-negative and Gram-positive bacteria (44–47). In addition, acetylation of polysaccharides can play a crucial role in modifying their biophysical properties within the biofilm matrix of various species (48, 49). Thus, demonstrating that PelA has deacetylase activity and determining if this activity is required for Pel synthesis is an important step toward characterizing this matrix polymer.
Given that the Pel polysaccharide generally is insoluble in aqueous solution, we reasoned that we could use p-nitrophenyl acetate as a pseudosubstrate to probe the deacetylase activity of PelA. Deacetylation activity in this assay results in the production of p-nitrophenol, which can be monitored spectrophotometrically. This assay has been used extensively to study other deacetylases (50–52). Mature wild-type PelA (PelAΔ46) was purified from E. coli and tested for deacetylation activity. PelAΔ46 was able to hydrolyze p-nitrophenyl acetate in vitro and had an observed specific activity of 0.042 ± 0.004 μmol · min−1 · mg−1 using 2.5 mM substrate (Fig. 2).
Fig 2.
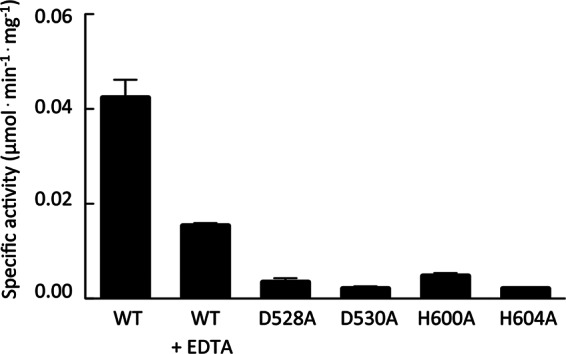
Specific activity of PelA and variants catalyzing the hydrolysis of p-nitrophenyl acetate. Assays contained 2.5 mM p-nitrophenyl acetate, dissolved in ethanol, and ∼40 μg of wild-type PelA (WT) or one of its variants (D528A, D530A, H600A, or H604A) in a total volume of 200 μl of 50 mM sodium HEPES buffer (pH 8.0) at 25°C. To test the metal dependency of the reaction, the wild-type protein was preincubated with EDTA and assayed (WT+EDTA). Error bars represent the standard errors of the means (SEM).
The prediction that PelA is a member of the CE4 family suggests that its enzymatic activity, similar to that of other members of this enzyme family, is metal dependent. CE4 enzymes typically require a divalent Zn2+ or Ni2+ metal ion that is usually coordinated by an aspartate and two histidine residues (53–56). According to the proposed mechanism, an acidic catalytic base extracts a proton from water, creating a nucleophile, while attacks on the carbonyl carbon of the acetate result in the formation of a tretrahedral oxyanion intermediate. A catalytic acid subsequently donates a proton to this intermediate releasing acetate (53). To confirm that the predicted deacetylase domain was responsible for the observed in vitro activity, we modeled this domain using HpPgdA as the model template. Using this model, we identified four residues, D528, D530, H600, and H604, in PelA as being potentially important for deacetylation (Fig. 1B and C). Our analysis suggests that D528 serves as the catalytic base to extract the proton, activating the nucleophilic water, while D530, H600, and H604 are involved in metal ion coordination. While the side chain of D530 in PelA and the analogous residue in HpPgdA do not completely superimpose, due to a slight difference in the predicted secondary structure in this region, rotation by about χ-1 brings the carboxyl oxygen of D530 into an orientation that could allow it to coordinate with a metal ion (Fig. 1C). We anticipate that minor rearrangement(s) of the structure in this region would be necessary to ensure that the metal is bound with the correct coordination geometry. Sequence alignments to other predicted or characterized polysaccharide deacetylases demonstrate that these four amino acids are well conserved (Fig. 1D). Furthermore, previous studies demonstrate that mutation of these conserved residues in other CE4 enzymes results in the abolishment of deacetylase activity (36, 53, 57). We found that purified alanine mutants of D528, D530, H600, and H604 led to a greater than 10-fold reduction in specific activity in vitro (Fig. 2), suggesting that these residues are involved in the catalytic mechanism. This loss of activity is not due to protein misfolding, as the proteins behaved similarly to the wild type during protein expression and purification, and circular dichroism spectroscopy of the point mutants indicated that they were folded (see Fig. S1 in the supplemental material). As members of the CE4 superfamily are known to require a divalent metal ion for activity, we also tested whether the deacetylase activity observed was dependent on the presence of a metal ion by incubating the protein with a large molar excess of EDTA. We observed that addition of this metal chelator reduced catalytic activity by ∼3-fold (Fig. 2). These results suggest that PelA exhibits metal-dependent deacetylase activity in vitro.
Deacetylase activity is required for Pel-dependent biofilm formation in PA14.
To test whether the polysaccharide deacetylase activity of PelA is required for Pel polysaccharide synthesis, transport, and/or structural modifications in the biofilm architecture, each of the four conserved amino acids in PelA was replaced with an alanine residue by site-directed mutagenesis on the chromosome of PA14. An unmarked, nonpolar pelA deletion was used for comparison studies with the site-directed mutants. As demonstrated previously, under specific growth conditions, PA14 develops into a wrinkly colony and forms robust biofilms that are dependent on the Pel polysaccharide (6, 7). As expected, a pelA mutant is deficient in wrinkly colony morphology (Fig. 3A). Similarly, each point mutant was incapable of forming a wrinkly colony and remained smooth, similar to the pelA mutant strain.
Fig 3.

Effects of PelA catalytic mutations on biofilm formation. (A) Colony morphology (top) and pellicle formation (bottom) are shown. (B) Microtiter dish biofilm assay of P. aeruginosa PA14, PA14ΔpelA, and the indicated pelA point mutants are shown. (C) Western blots for PelA (top), PelC (middle) and RNA polymerase (RNAp), a protein loading control (bottom) are shown for PA14, PA14ΔpelA, and the indicated pelA point mutants expressing the diguanylate cyclase PA1120 (pJN1120). The asterisk indicates the PelA protein. (D) Microtiter dish biofilm assay for strains expressing pJN1120 is shown. Error bars represent standard deviations.
We further examined biofilm development by evaluating each strain's ability to form a biofilm in a microtiter dish. PA14ΔpelA formed significantly reduced biofilms compared to the parental strain and, likewise, each point mutant was comparably impaired (Fig. 3B). To confirm that this was not due to differences in protein expression levels or stability, PelA protein expression was measured using PelA-specific antisera. We could not detect PelA from PA14 log-phase cultures due to low expression levels (7). To increase PelA production, a plasmid expressing PA1120, a diguanylate cyclase, was transformed into each strain. Expression of pJN1120 increases intracellular concentrations of the biofilm-inducing signaling molecule c-di-GMP, resulting in increased pel transcription and Pel protein levels (16, 17). Under these conditions, the point mutant alleles produce PelA at levels comparable to those of the parental strain (Fig. 3C). In addition, we also monitored PelC levels to confirm that the pelA in-frame mutation and PelA point mutants did not disrupt downstream gene expression. Moreover, the strains expressing pJN1120 and the point mutant alleles of pelA displayed a biofilm-deficient phenotype trend similar to that of the ΔpelA null strain (Fig. 3D). Combined, these results suggest that PelA deacetylase activity is required for Pel-dependent biofilm formation and colony morphology in PA14.
Deacetylase activity is required for Pel production in PAO1.
We next sought to determine whether the biofilm deficiencies of the point mutants were due to a reduction in Pel polysaccharide levels. To measure Pel polysaccharide levels more directly, we generated antisera against a crude polysaccharide preparation from the supernatant of PA14PBADpel cultures. PA14PBADpel overexpresses the pel operon upon addition of the inducer arabinose (7). The antiserum generated was highly cross-reactive against the O-specific antigen of PA14 (data not shown), making Pel measurements in PA14 difficult. However, since PAO1 and PA14 express two distinct O-specific antigens (serotype O5 and O10, respectively [58]), we reasoned that we could effectively reduce the amount of cross-reactivity by analyzing Pel production in a PAO1 background. To confirm the specificity of the Pel antisera, we evaluated PAO1ΔwspF, PAO1ΔwspFΔpel, PAO1ΔwspFΔpsl, and PAO1ΔwspFΔpelΔpsl for reactivity to the Pel antiserum (Fig. 4D). As expected, strains incapable of synthesizing Pel, PAO1ΔwspFΔpel and PAO1ΔwspFΔpelΔpsl, were much less reactive.
Fig 4.

PelA catalytic mutation in PAO1 arrests colony morphology, biofilm formation, and Pel synthesis. PAO1ΔwspFΔpsl PBADpel and the catalytic mutant D528A in pelA were assessed for colony morphology in the presence of 0.5% arabinose (A) and microtiter dish biofilm formation in the presence and absence of the arabinose inducer (B). (C) Strains were probed for PelA, PelC, and RNA polymerase (RNAp) protein expression by Western blotting and Pel polysaccharide by dot blotting. The asterisk indicates the PelA protein. The bottom panel represents an anti-Pel blot of these strains. (D) To verify the Pel antisera, Pel polysaccharide dot blots were analyzed for PAO1ΔwspF, PAO1ΔwspFΔpel, PAO1ΔwspFΔpsl, and PAO1ΔwspFΔpelΔpsl.
We generated a pel-inducible overexpression strain in a high c-di-GMP background to augment Pel production. The native promoter region of pelA was replaced with the araC-PBAD promoter on the PAO1 chromosome as previously described, allowing arabinose-dependent expression of the pel operon (7). By introducing an in-frame mutation in wspF, a negative regulator of the diguanylate cyclase WspR, a high c-di-GMP background was generated. This results in elevated pel and psl transcription, elevated biofilm formation, and wrinkly colony morphology (17). In addition, we introduced a polar mutation in the psl operon in this strain to allow us to study the Pel polysaccharide in the absence of any potential cross-reactivity with the Psl polysaccharide. This inducible strain is referred to as PAO1ΔwspFΔpsl PBADpel.
To test if PelA deacetylase activity is also required for Pel-dependent phenotypes in PAO1, we introduced the D528A point mutation into pelA. Similar to PA14, the wrinkly colony morphology seen in the induced strain, PAO1ΔwspFΔpsl PBADpel, was absent from the isogenic PelAD528A mutant (Fig. 4A). To assess the effect of the point mutation on biofilm development, we grew both strains in the presence and absence of the inducer arabinose. In the absence of arabinose, neither strain produced biofilms. In contrast, the addition of arabinose resulted in a dramatic increase in biofilm formation for PAO1ΔwspFΔpsl PBADpel but not the PelAD528A mutant (Fig. 4B). We reasoned that the observed phenotypes could be due to a reduction in polysaccharide production, a failure to export the polysaccharide, or a structural change affecting the physical properties of the polymer. To explore these possibilities, Pel polysaccharide levels were measured semiquantitatively using Pel-specific antiserum. Addition of arabinose dramatically increased Pel reactivity in PAO1ΔwspFΔpsl PBADpel (Fig. 4C). In contrast, the PelAD528A mutant strain was not reactive in either the presence or absence of arabinose (Fig. 4C). Western blot analysis clearly shows that the lack of Pel production is not due to loss of protein expression, as comparable amounts of PelA and the product of the downstream gene, PelC, are produced (Fig. 4C).
PelA localizes to the periplasmic and membrane fractions.
Bioinformatics analysis suggests that PelA is a periplasmic protein, as it contains a Tat signal sequence. This would place PelA in position to modify Pel as the polymer is transferred from the cytoplasm to the extracellular surface. To determine subcellular localization of PelA, bacterial cells overexpressing Pel containing a plasmid carrying the β-lactamase gene (bla), pPSV18, were fractionated into membrane (M), cytoplasmic (C), and periplasmic (P) fractions. Controls for the membrane (PelC), cytoplasm (RNAp), and periplasm (Bla) were predominantly found in the expected fractions (Fig. 5). We found that PelA localized to both the periplasmic and membrane fractions but not to the cytoplasm.
Fig 5.
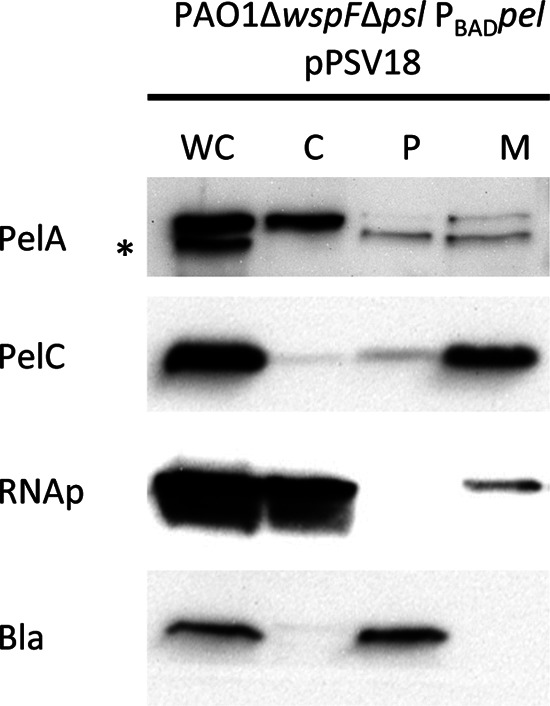
PelA localizes primarily to the periplasmic and membrane fractions. Western blots of whole-cell (WC), cytoplasmic (C), periplasmic (P), and membrane (M) fractions are shown for PAO1ΔwspFΔpsl PBADpel expressing pPSV18, a plasmid that synthesizes β-lactamase. Subcellular fractions were probed for PelA, PelC (membrane protein), RNA polymerase (RNAp; cytosolic protein), and β-lactamase (Bla; periplasmic protein). The asterisk indicates that the band for PelA protein is the lower of the two immunoreactive bands.
PelA deacetylase activity is required for the synthesis of Pel-reactive material.
Previous studies in E. coli have demonstrated that deacetylation of the PNAG polymer is required for transport out of the periplasm (36). Mutants deficient in deacetylase activity appeared to retain the polymer in the periplasmic space, as demonstrated by transmission electron microscopy (TEM) (36). To test if the PelAD528A mutant is deficient in polymer export, we purified the periplasm and tested for Pel reactivity. The periplasm did not contain Pel-reactive material in either PAO1ΔwspFΔpsl PBADpel or the PelAD528A mutant (Fig. 6A). EDTA extraction of both the untreated cell pellet and the spheroplasts successfully extracted Pel-reactive material from only the induced PAO1ΔwspFΔpsl PBADpel strain. To confirm that the periplasm was being properly fractionated, we probed for the periplasmic protein Bla (Fig. 6B), which was observed to be present. Combined, these results suggest that the Pel-reactive material is not being trapped in the periplasm; rather, it is not being synthesized or is being degraded, or the altered structure of Pel is not being recognized by the Pel antisera.
Fig 6.
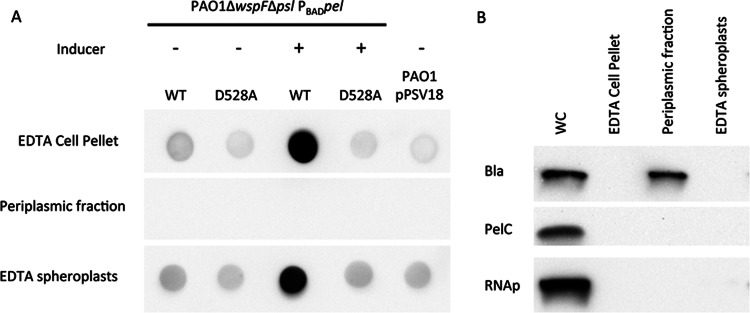
Pel-reactive material is not detected in the periplasmic fraction. (A) PAO1ΔwspFΔpsl PBADpel, the pelA catalytic mutant D528A, and PAO1 expressing pPSV18 were assessed for Pel production by dot blotting. PAO1ΔwspFΔpsl PBADpel strains were grown in the presence or absence of 0.5% arabinose as indicated. Pel polysaccharide expression was analyzed for the EDTA-extracted whole-cell pellet, periplasmic fraction, and EDTA-extracted spheroplasts. (B) The whole-cell (WC) fraction, EDTA-extracted whole-cell pellet, periplasmic fraction, and EDTA-extracted spheroplasts were probed for PelC (membrane protein), RNA polymerase (RNAp; cytosolic protein), and β-lactamase (Bla; periplasmic protein).
DISCUSSION
This study examined the role of PelA putative deacetylase activity on Pel production and Pel-dependent phenotypes. Bioinformatics analysis predicts that PelA contains a Tat-dependent signal sequence, suggesting the protein is localized to the periplasm and three putative distinct enzymatic domains. The first predicted enzymatic domain is a glycoside hydrolase (GH), which hydrolyzes a glycosidic bond between two or more carbohydrates or between a carbohydrate and a noncarbohydrate moiety (59). It remains to be seen if this domain is important in determining chain length and/or hydrolyzing improperly synthesized Pel. The second is a flavin mononucleotide-binding domain, of unknown function, while the third domain is a predicted carbohydrate esterase family 4 (CE4) deacetylase domain. In this study, we demonstrate that PelA exhibits deacetylase activity, and that this activity is required for Pel-dependent biofilm formation. Additionally, we generated Pel-specific antisera from PA14 that is reactive to the synthesis of Pel from both PAO1 and PA14 but is not reactive to Pel produced from a PelA deacetylase mutant.
Using an in vitro assay, we established that purified PelA hydrolyzes the pseudosubstrate p-nitrophenyl acetate, providing direct evidence that the enzyme acts as a deacetylase in P. aeruginosa. Furthermore, the specific activity of PelA was reduced when incubated with EDTA, providing additional evidence that PelA is a member of the metal-dependent CE4 superfamily. The metal ion in the CE4 reaction is thought to play a dual role: initiating the reaction by binding a water molecule and helping to stabilize the tetrahedral oxyanion intermediate that is produced after the aspartate residue extracts a proton from the bound water, creating the nucleophile that attacks the carbonyl carbon of the substrate (53). As seen for other CE4 proteins, incubation with EDTA or other chelators does not always completely ablate activity (50, 55), suggesting that the metal is tightly associated with the protein and/or that EDTA is a poor chelator of the metal for members of this superfamily. CE4 enzymes depend on Zn2+ and Ni2+ and, in some cases, Co2+ and Fe2+ for activity (39, 50, 60). While our results suggest that optimal deacetylation activity of PelA requires a metal ion, the identity of the bound metal remains to be determined.
Through sequence analysis and structural homology modeling, we were able to identify four conserved amino acids predicted to be important for catalytic function in the deacetylase domain. D528 is predicted to serve as the catalytic base, while D530, H600, and H604 are proposed to be involved in metal ion coordination. Mutation of these residues in vitro resulted in a ≥10-fold reduction in specific activity, while replacing each conserved residue with an alanine on the chromosome abolished biofilm formation and wrinkly colony morphology, two phenotypes dependent on the production of the Pel polysaccharide. In addition, using Pel-specific antisera, we demonstrated that the Pel-reactive polymer is not synthesized in a PelAD528A mutant. Pel-reactive material was not detected in the periplasm of the PelAD528A mutant, suggesting that the lack of Pel detection is not due to a failure to export the polymer across the outer membrane. These data suggest that PelA deacetylase activity is required for Pel polysaccharide production.
We generated antisera against PA14PBADpel, a Pel overexpression strain. The antisera were reactive to both PA14 and PAO1 strains overexpressing the pel operon but not the corresponding pel mutant strains, suggesting that the structure of the Pel polysaccharide, or parts of it, is conserved between PA14 and PAO1. Recent data suggest that the pel-encoded functions are important in maintaining cell-associated forms of LPS (8). The results that the Pel-specific antisera recognized Pel produced by two strains that have structurally distinct O-antigens (8, 61), and that the pel mutant strain showed very little reactivity to the antisera, suggest that the pel genes do not influence LPS localization. We propose that our data support the view that the pel operon is involved in synthesizing a unique polysaccharide. However, we cannot rule out the possibility that the pel gene cluster influences both the production of a novel exopolysaccharide and LPS localization.
Acetylation is a common modification of polysaccharides that can impact the physical and chemical properties of polymers, including hydrophobicity, viscosity, and solubility (10, 62). One example is seen by comparing chitin to chitosan. Chitin is predominantly composed of large amounts of the acetylated form of glucosamine (GlcNAc). The increased proportion of acetyl groups increases hydrophobicity, making the chitin polymer highly insoluble, as evident in that chitin is commonly found as part of invertebrate exoskeletons, such as shrimp and cicada (63). In contrast, chitosan is predominantly composed of the nonacetylated glucosamine polymer and is soluble under conditions in which chitin is not. Interestingly, the degree of deacetylation (%DD) is an important variable in solubility and is commonly reported with commercial chitosan synthesis (64).
Acetylation impacts a number of extracellular polysaccharides implicated in biofilm formation, including alginate, cellulose, and glycopeptidolipids (48, 65, 66). For example, alginate is a high-molecular-weight linear polysaccharide composed of β-1,4-linked d-mannuronic acid (M) and l-glucuronic acid (G) residues, which can be selectively O-acetylated on the C-2 and/or C-3 hydroxyl groups of M residues by the concerted action of AlgI, AlgJ, and AlgF and, potentially, AlgX (4, 67). Interestingly, the structure of alginate is made up of a random arrangement of M and G residues, and this arrangement varies depending on the organism or strain producing the polymer. Alginate is first synthesized as a linear homopolymer of d-mannuronic acid and transported into the periplasm. Once there, the M residue can be selectively acetylated, while nonacetylated d-mannuronic acids can be epimerized to l-glucuronic acid by the enzyme AlgG (4). A mutation in algJ synthesizes alginate lacking O-acetyl groups, and this strain is significantly impaired in surface attachment and biofilm development (48). A separate study demonstrated that alginate acetylation was necessary for the aggregation of bacteria into microcolonies (68). Thus, it appears that acetyl groups help mediate interactions between neighboring cells and surface colonization, and these factors are important in the ability of strains to form robust biofilms. P. aeruginosa mucoid strains isolated from cystic fibrosis patients reportedly have differing levels of acetylation (69). It remains to be determined if the degree of alginate acetylation correlates with disease severity.
A second example of biofilm formation being linked to acetylation is seen in Pseudomonas fluorescens SBW25. This strain forms a robust pellicle that is dependent on the synthesis of a cellulosic polymer from a 10-gene operon, wssA-wssJ (65). The cellulose-like polymer is believed to be acetylated by the concerted action of wssGHI, which are thought to be analogous to the algFIJ genes, respectively, in the alginate system (49). Disruption of these genes in P. fluorescens SBW25 results in a weaker pellicle and bacteria that fail to rapidly spread across solid surfaces despite production of similar levels of the core polysaccharide (65). A third example is the glycopeptidolipids (GPLs) found in the outermost layer of the cell wall in mycobacterial species (66). The GPLs are necessary for sliding motility, colony morphology, and biofilm formation. A mutation in the atf1 gene prevents the GPLs from being acetylated in Mycobacterium smegmatis and impairs sliding motility, colony morphology, and biofilm formation (66). These studies demonstrate that acetyl modifications can greatly alter biofilm development.
While we currently do not know the biosynthetic mechanism of Pel production, the observation that Pel does not appear to be synthesized in a catalytically inactive deacetylase mutant is interesting, and to our knowledge, this is the first report suggesting that deacetylation is necessary for polymer synthesis. In the examples described above, the core polymer is still synthesized and can be detected (65). However, in other cases, deacetylation can be required for either polysaccharide export or attachment to the cell surface. For example, the deacetylase IcaB in Staphylococcus epidermidis converts 15 to 20% of GlcNAc residues from PNAG to glucosamine (70). Deletion of icaB leads to shedding of the polymer from the cell surface and an inability to form biofilms (70). In contrast, the deacetylase PgaB in E. coli is required for polysaccharide export across the outer membrane (36). In both cases, the polymer is still made in the absence of deacetylase activity. Additional structural studies will be required to determine if changes in acetylation affect recognition by the Pel-specific antisera. For PNAG, the antisera can recognize both deacetylated and acetylated forms (35).
Although the structure of the Pel polysaccharide remains unknown, we provide evidence that this polymer can be deacetylated. This raises a number of important questions for future studies. Can Pel be differentially acetylated, and does this impact its function on the matrix? Is deacetylation involved in Pel transport? Can targeting deacetylation activity prove to be an effective intervention for anti-Pseudomonas biofilm therapy? In addition, our data suggest that acetyl sugars comprise the building blocks for Pel synthesis, which should help efforts to identify the chemical structure of Pel. Finally, examination of the other putative catalytic domain(s) in PelA may further define the role of this important protein in Pel biosynthesis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by research grants from the NIH (R01 AI077628-01A1) and NSF (MCB0822405) to M.R.P. and the Canadian Institutes of Health Research (CIHR MT 43998) to P.L.H. P.L.H. was the recipient of a Canada Research Chair. P.B. was supported by a postdoctoral fellowship from Cystic Fibrosis Canada. J.C.W. was supported by graduate scholarships from the Natural Sciences and Engineering Research Council of Canada, Cystic Fibrosis Canada, the Ontario Graduate Scholarship Program, the Ontario Student Opportunities Trust Fund, and The Hospital for Sick Children Foundation Student Scholarship Program.
Footnotes
Published ahead of print 15 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02150-12.
REFERENCES
- 1. Costerton JW, Lewandowski Z, Caldwell DE, Korber DR, Lappin-Scott HM. 1995. Microbial biofilms. Annu. Rev. Microbiol. 49:711–745 [DOI] [PubMed] [Google Scholar]
- 2. Stoodley P, Sauer K, Davies DG, Costerton JW. 2002. Biofilms as complex differentiated communities. Annu. Rev. Microbiol. 56:187–209 [DOI] [PubMed] [Google Scholar]
- 3. Stewart PS, Costerton JW. 2001. Antibiotic resistance of bacteria in biofilms. Lancet 358:135–138 [DOI] [PubMed] [Google Scholar]
- 4. Franklin MJ, Nivens DE, Weadge JT, Howell PL. 2011. Biosynthesis of the Pseudomonas aeruginosa extracellular polysaccharides, alginate, Pel, and Psl. Front. Microbiol. 2:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Colvin KM, Irie Y, Tart CS, Urbano R, Whitney JC, Ryder C, Howell PL, Wozniak DJ, Parsek MR. 2011. The Pel and Psl polysaccharides provide Pseudomonas aeruginosa structural redundancy within the biofilm matrix. Environ. Microbiol. 14:1913–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Friedman L, Kolter R. 2004. Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol. Microbiol. 51:675–690 [DOI] [PubMed] [Google Scholar]
- 7. Colvin KM, Gordon VD, Murakami K, Borlee BR, Wozniak DJ, Wong GC, Parsek MR. 2011. The Pel polysaccharide can serve a structural and protective role in the biofilm matrix of Pseudomonas aeruginosa. PLoS Pathog. 7:e1001264 doi:10.1371/journal.ppat.1001264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coulon C, Vinogradov E, Filloux A, Sadovskaya I. 2010. Chemical analysis of cellular and extracellular carbohydrates of a biofilm-forming strain Pseudomonas aeruginosa PA14. PLoS One 5:e14220 doi:10.1371/journal.pone.0014220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vasseur P, Vallet-Gely I, Soscia C, Genin S, Filloux A. 2005. The pel genes of the Pseudomonas aeruginosa PAK strain are involved at early and late stages of biofilm formation. Microbiology 151:985–997 [DOI] [PubMed] [Google Scholar]
- 10. Varki A, Cummings R, Esko J, Freeze H, Hart G, Marth J. 1999. Essentials of glycobiology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 11. Vasseur P, Soscia C, Voulhoux R, Filloux A. 2007. PelC is a Pseudomonas aeruginosa outer membrane lipoprotein of the OMA family of proteins involved in exopolysaccharide transport. Biochimie 89:903–915 [DOI] [PubMed] [Google Scholar]
- 12. Kowalska K, Soscia C, Combe H, Vasseur P, Voulhoux R, Filloux A. 2010. The C-terminal amphipathic alpha-helix of Pseudomonas aeruginosa PelC outer membrane protein is required for its function. Biochimie 92:33–40 [DOI] [PubMed] [Google Scholar]
- 13. Lee VT, Matewish JM, Kessler JL, Hyodo M, Hayakawa Y, Lory S. 2007. A cyclic-di-GMP receptor required for bacterial exopolysaccharide production. Mol. Microbiol. 65:1474–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Whitney JC, Colvin KM, Marmont LS, Robinson H, Parsek MR, Howell PL. 2012. Structure of the cytoplasmic region of PelD, a degenerate diguanylate cyclase receptor that regulates exopolysaccharide production in Pseudomonas aeruginosa. J. Biol. Chem. 287:23582–23593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jain S, Ohman DE. 2005. Role of an alginate lyase for alginate transport in mucoid Pseudomonas aeruginosa. Infect. Immun. 73:6429–6436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Borlee BR, Goldman AD, Murakami K, Samudrala R, Wozniak DJ, Parsek MR. 2010. Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol. Microbiol. 75:827–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hickman JW, Tifrea DF, Harwood CS. 2005. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc. Natl. Acad. Sci. U. S. A. 102:14422–14427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86 [DOI] [PubMed] [Google Scholar]
- 19. Irie Y, Starkey M, Edwards AN, Wozniak DJ, Romeo T, Parsek MR. 2010. Pseudomonas aeruginosa biofilm matrix polysaccharide Psl is regulated transcriptionally by RpoS and post-transcriptionally by RsmA. Mol. Microbiol. 78:158–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Newman JR, Fuqua C. 1999. Broad-host-range expression vectors that carry the L-arabinose-inducible Escherichia coli araBAD promoter and the araC regulator. Gene 227:197–203 [DOI] [PubMed] [Google Scholar]
- 21. Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, Ausubel FM. 1995. Common virulence factors for bacterial pathogenicity in plants and animals. Science 268:1899–1902 [DOI] [PubMed] [Google Scholar]
- 22. Rietsch A, Wolfgang MC, Mekalanos JJ. 2004. Effect of metabolic imbalance on expression of type III secretion genes in Pseudomonas aeruginosa. Infect. Immun. 72:1383–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Choi KH, Schweizer HP. 2005. An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol. 5:30 doi:10.1186/1471-2180-5-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock RE, Brinkman FS. 2011. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 39:D596–D600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371 [DOI] [PubMed] [Google Scholar]
- 26. Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8:785–786 [DOI] [PubMed] [Google Scholar]
- 27. Bagos PG, Nikolaou EP, Liakopoulos TD, Tsirigos KD. 2010. Combined prediction of Tat and Sec signal peptides with hidden Markov models. Bioinformatics 26:2811–2817 [DOI] [PubMed] [Google Scholar]
- 28. Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205–217 [DOI] [PubMed] [Google Scholar]
- 29. O'Toole GA, Pratt LA, Watnick PI, Newman DK, Weaver VB, Kolter R. 1999. Genetic approaches to study of biofilms. Methods Enzymol. 310:91–109 [DOI] [PubMed] [Google Scholar]
- 30. Liu J, Walsh CT. 1990. Peptidyl-prolyl cis-trans-isomerase from Escherichia coli: a periplasmic homolog of cyclophilin that is not inhibited by cyclosporin A. Proc. Natl. Acad. Sci. U. S. A. 87:4028–4032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Russell AB, Hood RD, Bui NK, LeRoux M, Vollmer W, Mougous JD. 2011. Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475:343–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Berks BC. 1996. A common export pathway for proteins binding complex redox cofactors? Mol. Microbiol. 22:393–404 [DOI] [PubMed] [Google Scholar]
- 33. Lee PA, Tullman-Ercek D, Georgiou G. 2006. The bacterial twin-arginine translocation pathway. Annu. Rev. Microbiol. 60:373–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nagano N, Orengo CA, Thornton JM. 2002. One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J. Mol. Biol. 321:741–765 [DOI] [PubMed] [Google Scholar]
- 35. Wang X, Preston JF, III, Romeo T. 2004. The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J. Bacteriol. 186:2724–2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Itoh Y, Rice JD, Goller C, Pannuri A, Taylor J, Meisner J, Beveridge TJ, Preston JF, III, Romeo T. 2008. Roles of pgaABCD genes in synthesis, modification, and export of the Escherichia coli biofilm adhesin poly-beta-1,6-N-acetyl-d-glucosamine. J. Bacteriol. 190:3670–3680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lamb DC, Kim Y, Yermalitskaya LV, Yermalitsky VN, Lepesheva GI, Kelly SL, Waterman MR, Podust LM. 2006. A second FMN binding site in yeast NADPH-cytochrome P450 reductase suggests a mechanism of electron transfer by diflavin reductases. Structure 14:51–61 [DOI] [PubMed] [Google Scholar]
- 38. Natale P, Bruser T, Driessen AJ. 2008. Sec- and Tat-mediated protein secretion across the bacterial cytoplasmic membrane–distinct translocases and mechanisms. Biochim. Biophys. Acta 1778:1735–1756 [DOI] [PubMed] [Google Scholar]
- 39. Caufrier F, Martinou A, Dupont C, Bouriotis V. 2003. Carbohydrate esterase family 4 enzymes: substrate specificity. Carbohydr. Res. 338:687–692 [DOI] [PubMed] [Google Scholar]
- 40. Shaik MM, Cendron L, Percudani R, Zanotti G. 2011. The structure of Helicobacter pylori HP0310 reveals an atypical peptidoglycan deacetylase. PLoS One 6:e19207 doi:10.1371/journal.pone.0019207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Egloff MP, Uppenberg J, Haalck L, van Tilbeurgh H. 2001. Crystal structure of maltose phosphorylase from Lactobacillus brevis: unexpected evolutionary relationship with glucoamylases. Structure 9:689–697 [DOI] [PubMed] [Google Scholar]
- 42. Sorimachi K, Jacks AJ, Le Gal-Coeffet MF, Williamson G, Archer DB, Williamson MP. 1996. Solution structure of the granular starch binding domain of glucoamylase from Aspergillus niger by nuclear magnetic resonance spectroscopy. J. Mol. Biol. 259:970–987 [DOI] [PubMed] [Google Scholar]
- 43. Juy M, Amrit AG, Alzari PM, Poljak RJ, Mclaeyssens Beguin P, Aubert J-P. 1992. Three-dimensional structure of a thermostable bacterial cellulase. Nature 357:89–91 [Google Scholar]
- 44. Rupp ME, Ulphani JS, Fey PD, Mack D. 1999. Characterization of Staphylococcus epidermidis polysaccharide intercellular adhesin/hemagglutinin in the pathogenesis of intravascular catheter-associated infection in a rat model. Infect. Immun. 67:2656–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rupp ME, Ulphani JS, Fey PD, Bartscht K, Mack D. 1999. Characterization of the importance of polysaccharide intercellular adhesin/hemagglutinin of Staphylococcus epidermidis in the pathogenesis of biomaterial-based infection in a mouse foreign body infection model. Infect. Immun. 67:2627–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rupp ME, Fey PD, Heilmann C, Gotz F. 2001. Characterization of the importance of Staphylococcus epidermidis autolysin and polysaccharide intercellular adhesin in the pathogenesis of intravascular catheter-associated infection in a rat model. J. Infect. Dis. 183:1038–1042 [DOI] [PubMed] [Google Scholar]
- 47. Li H, Xu L, Wang J, Wen Y, Vuong C, Otto M, Gao Q. 2005. Conversion of Staphylococcus epidermidis strains from commensal to invasive by expression of the ica locus encoding production of biofilm exopolysaccharide. Infect. Immun. 73:3188–3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nivens DE, Ohman DE, Williams J, Franklin MJ. 2001. Role of alginate and its O acetylation in formation of Pseudomonas aeruginosa microcolonies and biofilms. J. Bacteriol. 183:1047–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Whitney JC, Howell PL. Synthase-dependent exopolysaccharide secretion in Gram-negative bacteria. Trends Microbiol., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Little DJ, Poloczek J, Whitney JC, Robinson H, Nitz M, Howell PL. 2012. The structure- and metal-dependent activity of Escherichia coli PgaB provides insight into the partial de-N-acetylation of poly-beta-1,6-N-acetyl-d-glucosamine. J. Biol. Chem. 287:31126–31137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gupta N, Rathi P, Gupta R. 2002. Simplified para-nitrophenyl palmitate assay for lipases and esterases. Anal. Biochem. 311:98–99 [DOI] [PubMed] [Google Scholar]
- 52. Tiwari R, Koffel R, Schneiter R. 2007. An acetylation/deacetylation cycle controls the export of sterols and steroids from S. cerevisiae. EMBO J. 26:5109–5119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Blair DE, Schuttelkopf AW, MacRae JI, van Aalten DM. 2005. Structure and metal-dependent mechanism of peptidoglycan deacetylase, a Streptococcal virulence factor. Proc. Natl. Acad. Sci. U. S. A. 102:15429–15434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Taylor EJ, Gloster TM, Turkenburg JP, Vincent F, Brzozowski AM, Dupont C, Shareck F, Centeno MS, Prates JA, Puchart V, Ferreira LM, Fontes CM, Biely P, Davies GJ. 2006. Structure and activity of two metal ion-dependent acetylxylan esterases involved in plant cell wall degradation reveals a close similarity to peptidoglycan deacetylases. J. Biol. Chem. 281:10968–10975 [DOI] [PubMed] [Google Scholar]
- 55. Blair DE, Hekmat O, Schuttelkopf AW, Shrestha B, Tokuyasu K, Withers SG, van Aalten DM. 2006. Structure and mechanism of chitin deacetylase from the fungal pathogen Colletotrichum lindemuthianum. Biochemistry 45:9416–9426 [DOI] [PubMed] [Google Scholar]
- 56. Oberbarnscheidt L, Taylor EJ, Davies GJ, Gloster TM. 2007. Structure of a carbohydrate esterase from Bacillus anthracis. Proteins 66:250–252 [DOI] [PubMed] [Google Scholar]
- 57. Forman S, Bobrov AG, Kirillina O, Craig SK, Abney J, Fetherston JD, Perry RD. 2006. Identification of critical amino acid residues in the plague biofilm Hms proteins. Microbiology 152:3399–3410 [DOI] [PubMed] [Google Scholar]
- 58. Lam JS, Taylor VL, Islam ST, Hao Y, Kocíncová D. 2011. Genetic and functional diversity of Pseudomonas aeruginosa lipopolysaccharide. Front. Microbiol. 2:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Henrissat B, Davies G. 1997. Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7:637–644 [DOI] [PubMed] [Google Scholar]
- 60. Deng DM, Urch JE, ten Cate JM, Rao VA, van Aalten DM, Crielaard W. 2009. Streptococcus mutans SMU. 623c codes for a functional, metal-dependent polysaccharide deacetylase that modulates interactions with salivary agglutinin. J. Bacteriol. 191:394–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rocchetta HL, Burrows LL, Lam JS. 1999. Genetics of O-antigen biosynthesis in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 63:523–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Flemming HC, Wingender J. 2010. The biofilm matrix. Nat. Rev. Microbiol. 8:623–633 [DOI] [PubMed] [Google Scholar]
- 63. Aranaz I, Mengibar M, Harris R, Panos I, Miralles B, Acosta N, Galed G, Heras A. 2009. Functional characterization of chitin and chitosan. Curr. Chem. Biol. 3:203–230 [Google Scholar]
- 64. Khan TA, Peh KK, Ch'ng HS. 2002. Reporting degree of deacetylation values of chitosan: the influence of analytical methods. J. Pharm. Pharm. Sci. 5:205–212 [PubMed] [Google Scholar]
- 65. Spiers AJ, Bohannon J, Gehrig SM, Rainey PB. 2003. Biofilm formation at the air-liquid interface by the Pseudomonas fluorescens SBW25 wrinkly spreader requires an acetylated form of cellulose. Mol. Microbiol. 50:15–27 [DOI] [PubMed] [Google Scholar]
- 66. Recht J, Kolter R. 2001. Glycopeptidolipid acetylation affects sliding motility and biofilm formation in Mycobacterium smegmatis. J. Bacteriol. 183:5718–5724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Skjåk-Braek G, Grasdalen H, Larsen B. 1986. Monomer sequence and acetylation pattern in some bacterial alginates. Carbohydr. Res. 154:239–250 [DOI] [PubMed] [Google Scholar]
- 68. Tielen P, Strathmann M, Jaeger KE, Flemming HC, Wingender J. 2005. Alginate acetylation influences initial surface colonization by mucoid Pseudomonas aeruginosa. Microbiol. Res. 160:165–176 [DOI] [PubMed] [Google Scholar]
- 69. McAvoy MJ, Newton V, Paull A, Morgan J, Gacesa P, Russell NJ. 1989. Isolation of mucoid strains of Pseudomonas aeruginosa from non-cystic-fibrosis patients and characterisation of the structure of their secreted alginate. J. Med. Microbiol. 28:183–189 [DOI] [PubMed] [Google Scholar]
- 70. Vuong C, Kocianova S, Voyich JM, Yao Y, Fischer ER, DeLeo FR, Otto M. 2004. A crucial role for exopolysaccharide modification in bacterial biofilm formation, immune evasion, and virulence. J. Biol. Chem. 279:54881–54886 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.