

Abstract
Recently, the S-layer protein of Sulfolobus acidocaldarius was shown to be N-linked with a tribranched hexasaccharide, composed of Man2Glc1GlcNAc2 and a sulfated sugar called sulfoquinovose. To identify genes involved in the biosynthesis and attachment of this glycan, markerless in-frame deletions of genes coding for predicted glycosyltransferases were created. The successful deletion of agl16, coding for a glycosyltransferase, resulted in the S-layer protein and archaellins having reduced molecular weights, as visualized by Coomassie staining or immunoblotting. This analysis indicated a change in the N-glycan composition. Nano-liquid chromatography-tandem mass spectrometry (LC-MS/MS) analyses confirmed that the glycan of the S-layer protein from the agl16 deletion mutant was a pentasaccharide, which was missing a terminal hexose residue. High-performance liquid chromatography (HPLC) analyses of the hydrolyzed N-glycan indicated that the missing hexose is a glucose residue. A physiological characterization of the agl16 deletion mutant revealed a significant effect on the growth at elevated salt concentrations. At 300 mM NaCl, the doubling time of the Δagl16 mutant was increased 2-fold compared to that of the background strain. Furthermore, the incomplete glycan structure of the Δagl16 deletion strain affected the assembly and function of the archaellum, as exemplified by semisolid Gelrite plate analysis, in which the motility is decreased according to the N-glycan size.
INTRODUCTION
The majority of the Archaea possess a proteinaceous surface layer (S-layer) as the sole cell wall structure (1), which contributes to cell stability and maintenance of the cell morphology. Archaeal S-layer proteins (2–10), as well as other surface-exposed proteins, like pilins (11), sugar-binding proteins (12), and the archaeal flagellins (13, 14), now termed archaellins (15), are posttranslationally modified via glycosylation. This posttranslational modification affects protein stability (16) and influences the assembly of surface-exposed proteins (17).
Protein glycosylation, in which sugar residues or oligosaccharides are covalently linked mainly to either selected asparagine (N-glycosylation) or serine and threonine residues (O-glycosylation) within a peptide sequence, is probably the most predominant posttranslational modification. In contrast to Bacteria, where N-glycosylation is a rare process, found only in a few species (18), almost all sequenced Archaea, with just a few exceptions, possess the gene encoding the key enzyme of the N-glycosylation process, the oligosaccharyltransferase AglB (19, 20). In the past few years, research on archaeal N-glycosylation performed in three members of the Euryarchaea revealed that Archaea exhibit features of both eukaryal and bacterial glycosylation pathways (21). Genes involved in this N-glycosylation pathway are designated agl (archaeal glycosylation) (21, 22).
Surface-exposed proteins of the crenarchaeon Sulfolobus acidocaldarius, like cytochrome b558/566 (23), sugar-binding proteins (24), the large S-layer protein SlaA (7), and the archaellin (14), are modified by the attachment of glycans. The cytochrome b558/566, which possesses O- and N-linked glycans, was the first crenarchaeal glycoprotein in which the composition and structure of the N-glycan were determined (25). Analysis of O-linked sugars detected only mannose residues (23), whereas the detailed analyses of the N-linked glycans revealed a tribranched hexasaccharide composed of Glc1Man2GlcNAc2 and a sulfated sugar called sulfoquinovose. Analysis of the S-layer glycoprotein SlaA confirmed the same N-glycan and revealed an astonishingly high glycosylation density (7). Beside the high glycosylation density, which is more similar to the highly glycosylated eukaryal cell wall protein ECM33 of Saccharomyces cerevisiae, the presence of the chitobiose core is a further eukaryal characteristic of the N-glycans which has exclusively been found in Sulfolobus so far. Furthermore, incorporation of the rare sulfated sugar sulfoquinovose (QuiS) into the N-glycan has not been detected in any other Archaea. Sulfoquinose has so far been found only as the head group of the nonphosphorous lipid sulfoquinovosyldiacylglycerol, which is incorporated into the photosynthetic membrane of all higher plants, mosses, ferns, and algae, as well as in most photosynthetic bacteria (26, 27). Deletion of the UDP-sulfoquinovose synthesis gene in S. acidocaldarius resulted in the loss of incorporation of sulfoquinovose in the N-glycan as well as one mannose and the glucose residues, implying the highly ordered assembly process of the N-glycans (14). However, unlike some Archaea, in which glycosyltransferases (GTases) are clustered directly next to the oligosaccharyltransferase AglB (19), no aglB-based gene clusters of GTases are present in Sulfolobus, which makes the identification of GTases participating in the N-glycosylation more difficult. Identification of GTases on the basis of their homologies to known N-glycosylation components in Eukarya, Bacteria, or Archaea led to no specific hits for S. acidocaldarius, and deletion studies of several genes coding for predicted GTases in the genome of S. acidocaldarius revealed no alteration of the N-glycan linked to the S-layer protein SlaA except for saci0807.
In this study, we identified the first thermophilic GTase, Agl16 (Saci0807), that participates in the crenarchaeal N-glycosylation process. Recently developed genetic tools for S. acidocaldarius as well as biochemical and mass-spectrometric methods were used to verify the predicted function of the Agl16 GTase in the biosynthesis of the S-layer and archaellin N-glycan.
MATERIALS AND METHODS
Strains and growth conditions.
The strain S. acidocaldarius MW001 (ΔpyrE) (28) and the Δagl16 mutant were grown in Brock medium at 75°C and pH 3, adjusted using sulfuric acid. The medium was supplemented with 0.1% (wt/vol) NZ-amine and 0.1% (wt/vol) dextrin as carbon and energy sources (29). Gelrite (0.6%) selection plates were supplemented with the same nutrients (as listed above), with the addition of 10 mM MgCl2 and 3 mM CaCl2. For second selection, plates containing 10 μg ml−1 uracil and 100 μg ml−1 5-fluoroorotic acid (5-FOA) were added. For the growth of the uracil auxotrophic mutant MW001 and Δagl16 strains, 10 μg ml−1 uracil was added to the medium. The cell growth was monitored by measuring the optical density at 600 nm. For propagation of plasmids, Escherichia coli DH5α cells were used.
Construction of a deletion plasmid.
Plasmids used in this study are listed in Table 1. To verify whether the gene saci0807, encoding the GTase Agl16, is indeed important for the N-glycosylation pathway, a markerless deletion mutant of this gene was created as previously described (28, 31). Briefly, the uracil-auxotrophic strain MW001, in which the pyrE gene is disrupted, was transformed with plasmid pSVA1233. To construct the plasmid, 800 to 1,000 bp of the up- and downstream fragments of agl16 were PCR amplified. At the 5′ end of the upstream and at the 3′ end of the downstream fragment, the restriction sites for ApaI and BamHI were introduced by the forward and reverse primers, respectively. The upstream reverse primer and the downstream forward primer were designed to each incorporate 15 bp of the reverse complement strand of the other primer, resulting in a 30-bp overlapping stretch. The up- and downstream fragments were fused by an overlapping PCR, using the 3′ ends of the up- and downstream fragments as primers. The amplified overlapping PCR fragment was purified by electrophoresis in 0.8% agarose gel using a Nucleospin extract kit (Macherey-Nagel, Düren, Germany) and digested with ApaI and BamHI. After repurification, the overlap fragment was ligated into an ApaI- and BamHI-predigested plasmid, pSVA407, containing a pyrEF cassette (28). The constructed plasmid, pSVA1233, was transformed into E. coli DH5α and selected on LB plates containing 50 μg ml−1 ampicillin. The accuracy of the plasmid was ascertained by sequencing. In order to avoid restriction in S. acidocaldarius, the plasmid was methylated by transformation in E. coli ER1821 cells containing pM.EsaBC4I (available from NEB), which expresses a methylase.
Table 1.
Plasmids used in this study
| Plasmid | Insert | Reference |
|---|---|---|
| pSAV407 | Gene targeting plasmid, pGEM-T Easy backbone, pyrEF cassette of S. solfataricusa | 28 |
| pSAV1233 | In-frame deletion of agl16 cloned into pSVA407 with ApaI and BamHI | This study |
| pMZ1 | Gene targeting plasmid with a C-terminal Strep tag and 10× His tag | 30 |
| pSAV1450 | pRN1-based shuttle vector with lacS reporter gene of S. solfataricusb | 28 |
| pSVA1273 | Full agl16 gene insert cloned into pMZ1 with NcoI and BamHI | This study |
| pSAV1274 | agl16 insert from pSVA1273 cloned into pSVA1450 with NcoI and EagI | This study |
pyrEF encode the enzymes orotate phosphoribosyltransferase and orotidine-5-mono-phosphate decarboxylase, respectively.
lacS encodes β-d-galactosidase of S. solfataricus.
Transformation and selection of the deletion mutant in S. acidocaldarius.
Preparation of competent cells was performed according to the protocol of Kurosawa and Grogan (32). Briefly, S. acidocaldarius strain MW001 was grown in Brock medium supplemented with 0.1% (wt/vol) NZ-amine and 0.1% dextrin until an optical density at 600 nm (OD600) between 0.1 and 0.3 was reached. Cooled cells were harvested by centrifugation (2,000 × g at 4°C for 20 min). The cell pellet was washed once each in 50 ml, 10 ml, and 1 ml of ice-cold 20 mM sucrose (dissolved in demineralized water) after mild centrifugation (2,000 × g at 4°C for 20 min). The final cell pellet was resuspended in 20 mM sucrose to attain an OD600 of 10 and stored in 50-μl aliquots at −80°C. Methylated pSVA1233 plasmid (400 to 600 ng) was added to the 50-μl aliquot of competent MW001 cells and incubated for 5 min on ice before transformation in a 1-mm-gap electroporation cuvette at 1,250 V, 1,000 Ω, and 25 mF using a Bio-Rad gene pulser II. Directly after transformation, 50 μl of a 2×-concentrated recovery solution (1% sucrose, 20 mM beta-alanine, 20 mM malate buffer [pH 4.5], 10 mM MgSO4) was added to the sample and incubated at 75°C for 30 min under mild shaking conditions (150 rpm). Before plating, the sample was mixed with 100 μl of heated 2×-concentrated recovery solution, and two 100-μl portions were spread on Gelrite plates containing Brock medium supplemented with 0.1% NZ-amine and 0.1% dextrin. After incubation for 5 to 7 days at 78°C, large brownish colonies were used to inoculate 50 μl of Brock medium containing 0.1% NZ-amine and 0.1% dextrin, each culture was incubated for 3 days of 78°C. Each culture, which was confirmed by PCR to contain genomic incorporated plasmid pSVA1233, was grown in Brock medium supplemented with 0.1% NZ-amine and 0.1% dextrin until an OD of 0.4. Aliquots of 40 μl were spread on second-selection plates supplemented with 0.1% NZ-amine, 0.1% dextrin, and 10 mg ml−1 uracil and incubated for 5 to 7 days at 78°C. Newly formed colonies were streaked onto new second-selection plates to ensure that they were formed from single colonies, before each colony was screened for the genomic absence or presence of the agl16 gene by PCR. The deletion of agl16 was further confirmed by sequencing and reverse transcription-PCR (RT-PCR).
RT-PCR.
Primers used in this study are listed in Table 2.The RNA isolation was performed using an RNeasy Plus mini-extraction kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions. The concentration of RNA was analyzed by spectrophotometrically. To eliminate the genomic DNA, two repetitive 2 h of incubation with DNase I (Fermentas, St. Leon-Rot, Germany) followed by repurification steps with the RNeasy Plus mini-extraction kit were performed. Purified total RNA was checked by PCR for the lack of contaminating chromosomal DNA, before cDNA synthesis with a First Strand cDNA synthesis kit (Fermentas) was used, according to the manufacturer's instruction. To verify the deletion of agl16 by RT-PCR, a specific forward primer (4101) and reverse primer (4106) amplifying the internal region of agl16 and, as a control, primers specific for the internal region of aglB (1727 and 1730) were designed and used in a PCR with single-stranded cDNA as a template.
Table 2.
Oligonucleotide primers used in this study
| Primer | Sequence (5′–3′)a | Restriction site | Description and target gene |
|---|---|---|---|
| 1848 | CTCTACGGGCCCACTTCTGTCTCCCCACTGG | ApaI | Up-for, Δagl16 |
| 1849 | AAGGTCTGGATGTTGTAAAAATTATGTCCATGACATCACAGGACA | Up-rev, Δagl16 | |
| 1850 | TGGACATAATTTTTACAACATCCAGACCTTATACATGAATCATCC | Down-for, Δagl16 | |
| 1851 | CGCCGAGGATCCAACCATGGAATTGTAGCC | BamHI | Down-rev, Δagl16 |
| 4101 | GTCCTCAAGACCCAATTCACTAAC | RT-PCR-for, agl16 | |
| 4106 | GCCGACTGCAGCATTAGTAATGACTCCACACCAC | RT-PCR-rev, agl16 | |
| 2799 | CATGCGGATCCTTAACTTAAACTTTTATAGAGCTCAAGC | BamHI | Forward primer complementing agl16 |
| 4100 | CATGCCCATGGATGTATAAGGTCTGGATGTTGACCCCAC | NcoI | Reverse primer complementing agl16 |
| 1729 | CTGCTGCAATTACAGCGTTC | RT-PCR-for aglB | |
| 1730 | AACCGTGAGCTACTTCAGAC | RT-PCR-rev aglB |
Underlining indicates restriction sites.
S-layer isolation.
Cell pellets of frozen or fresh cells from a 50-ml culture were incubated under shaking condition for 45 min at 37°C in 40 ml of buffer A (10 mM NaCl, 1 mM phenylmethylsulfonyl fluoride [PMSF], 0.5% sodium lauroylsarcosine) with the addition of a small amount of DNase. Samples were centrifuged for 40 min in an Optima Max-XU ultracentrifuge (Beckman Coulter) at 21,000 rpm, yielding a brownish tan pellet. The pellet was resuspended and incubated for 30 min at 37°C in 1.5 ml of buffer A. Repetitive steps of washes in buffer B (10 mM NaCl, 0.5 mM MgSO4, 0.5% SDS), incubation for 20 min at 37°C, and subsequent centrifugation (tabletop centrifuge at 14,000 rpm), yielded in a translucent tan pellet of S-layer proteins. Purified S-layer proteins were washed once with water and then stored in water at 4°C.
Induction and immunostaining of the archaellin FlaB.
Pellets of fresh 50-ml cultures of MW001 and the Δagl16 strain were resuspended and grown for 4 h at 79°C in 40 ml of Brock medium lacking NZ-amine as a carbon and energy source to induce the production of archaella (33). Centrifuged cells were resolved in 1 ml of 20 mM Tris HCl and 500 mM NaCl buffer. Aliquots of 30 μl were loaded on a 11% SDS-PAGE gel, electrophoresed (100 V), and blotted onto a polyvinylidene difluoride (PVDF) membrane. For detection, the primary antibody raised in rabbits against a FlaB peptide (Eurogentec) and an alkaline phosphatase-coupled anti-rabbit IgG antibody (Sigma-Aldrich, St. Louis, MO) were used. The chemifluorescence signal was measured in a Fujifilm LAS-4000 luminescent image analyzer (Fujifilm, Düsseldorf, Germany).
Motility assay.
Swimming motility was analyzed on semisolid plates consisting of 0.15% Gelrite supplemented with Brock medium with a reduced amount of NZ-amine (0.0015%) and either 0 mM or 300 mM NaCl. To inoculate the plates, aliquots of 5 to 7 μl, corresponding to the OD600 of MW001 or Δagl16 cultures (grown to an OD600 of 0.5), were dropped on soft Gelrite plates. Plates were incubated 4 to 9 days in a humid chamber at 75°C. Swimming behavior of the different Sulfolobus strains was analyzed by measuring the swimming radius.
Complementation of agl16 by cloning of agl16 into the S. acidocaldarius expression vector.
To complement the defect in the biosynthesis of the N-glycan in the Δagl16 strain, the gene saci0807 (agl16) was amplified from genomic DNA of S. acidocaldarius, introducing BamHI and NcoI restriction site at the 5′ ends of primers 2799 and 4100, respectively. The PCR fragment was cloned into pMZ1 (30), yielding vector pSVA1273, the plasmid was digested with NcoI and EagI and the insert containing the agl16 gene was ligated into pSVA1450, an improved expression vector (M. Wagner and S. V. Albers, unpublished data) based on pCmalLacS (34), yielding pSVA1274. The pCmalLacS plasmid contains an inducible maltose promoter that is used for the expression of agl16 in S. acidocaldarius. Before transformation into the Δagl16 deletion strain, the plasmid was methylated (see “Transformation and selection of the deletion mutant in S. acidocaldarius” above). Transformed cells were plated on first-selection Gelrite plates, and single colonies were picked and induced in Brock medium with 0.4% maltose. Pellets of a culture grown to an OD of 0.8 were used for the S-layer isolation.
Tryptic digestion for the mutant S-layer samples.
Purified S-layer mutants were run on a 2 to 8% precast gel (Invitrogen, Paisley, United Kingdom) and stained with Novotex colloidal blue stain (Invitrogen). The stained band was excised from the gel and cut into relatively small pieces and destained, using 400 ml of 50% (vol/vol) acetonitrile in 0.1 M ammonium bicarbonate (pH 8.4) and dried in a SpeedVac. Dried gel pieces were typically reswollen in 20 μl of 50 mM ammonium bicarbonate (pH 8.4) containing 0.5 ng ml−1 trypsin (Promega catalog no. V5111) and incubated at 37°C, overnight. The supernatant was transferred to a clean Eppendorf tube. In order to stop the tryptic reaction, the gel pieces were incubated (37°C for 10 min) in 0.1% trifluoroacetic acid (TFA) (50 ml) followed by incubation (37°C for 10 min) in acetonitrile (100 ml) to shrink the gel pieces. The supernatant was then added to the previously collected glycopeptides and the final volume reduced to ∼20 to 40 μl for nano-LC-ES-MS analysis.
Nano-LC-ES-MS/MS analysis.
Tryptic digests were analyzed by nano-LC-ES-MS/MS using a reverse-phase nano-HPLC system (Dionex, Sunnyvale, CA) connected to a quadrupole time-of-flight (TOF) mass spectrometer (Q-STAR Pulsar I; MDS Sciex). The digests were separated by a binary nano-high-performance liquid chromatography (nano-HPLC) gradient generated by an Ultimate pump fitted with a Famos autosampler and a Switchos microcolumn switching module (LC Packings, Amsterdam, The Netherlands). An analytical C18 nanocapillary (75 μm [inside diameter] by 15 cm; PepMap) and a micro-precolumn C18 cartridge were employed for on-line peptide separation. The digest was first loaded onto the precolumn and eluted with 0.1% formic acid (Sigma) in water (HPLC grade; Purite) for 4 min. The eluant was then transferred onto an analytical C18 nanocapillary HPLC column and eluted at a flow rate of 150 nl/min using the following gradient of solvent A [0.05% (vol/vol) formic acid in a 95:5 (vol/vol) water/acetonitrile mixture] and solvent B [0.04% formic acid in a 95:5 (vol/vol) acetonitrile/water mixture]: 99% A from 0 to 5 min, 99 to 90% A from 5 to 10 min, 90 to 60% A from 10 to 70 min, 60 to 50% A from 70 to 71 min, 50 to 5% A from 71 to 75 min, 5% A from 75 to 85 min, 5 to 95% A from 85 to 86 min, and 95% A from 86 to 90 min. Data acquisition was performed using Analyst QS software with an automatic information-dependent-acquisition (IDA) function.
HPLC analyses.
The N-glycans of purified S-layer derived from a 25-ml culture were hydrolyzed in 2 N TFA for 20 h at 80°C. After the treatment, the S-layer was removed from the sample by centrifugation in a tabletop centrifuge at 14,000 rpm for 20 min. An 80-μl portion of the clarified supernatant was acidified with H2SO4 (4 μl, 1 M). Subsequently this solution was analyzed by high-performance liquid chromatography (HPLC) using an 8-μm H+ IEX resin column (250 by 8 mm [inside diameter]; Grom, Rottenburg, Germany) at 80°C and a mobile phase of 5 mM H2SO4 (0.35 ml min−1).
Transmission electron microscopy (TEM).
For TEM, cells were fixed with 2.5% glutaraldehyde for 15 min at room temperature. Cell suspensions were placed directly on glow-discharged Formvar-coated 200-mesh copper grids (Plano, Wetzlar, Germany). The samples were washed once with water, negatively stained for 15 s with 2% uranyl acetate, and air dried. Grids were investigated on a JEOL 3010 300-kV transmission electron microscope (JEOL, Eching, Germany).
RESULTS
Identification of saci0807, its genomic localization, and protein characterization.
To elucidate the N-glycosylation pathway in S. acidocaldarius, the genome was screened for the presence of genes coding for predicted GTases. However, none of these identified GTases (Table 3) possesses a high sequence similarity to known eukaryal, bacterial, or archaeal GTases involved in the N-glycosylation pathway. Deletion studies of a variety of putative GTases in the genome of S. acidocaldarius were fruitless, except that deletion of saci0807, encoding a predicted GTase, resulted in an alteration in the biosynthesis of the N-linked hexasaccharide. The gene agl16 (saci0807) encodes a soluble 40.6-kDa protein with a glycosyltransferases 1 domain (PF00534) spanning residues 167 to 330, whereas outside this region the protein contains no other known domain. However, the broad range of functions related to the glycosyltransferase 1 domain did not allow a precise prediction of the function of this GTase. The CAZy database grouped Agl16, based on similarity of short sequence motifs, in GTase family 4, with a GT-B fold and a retaining enzyme according to the stereochemistry of the substrates and reaction products. Besides the vast number of different GTase sequence families, only a limited number of structural folds have been determined, in which the majority of GTases are found to be either GT-A or GT-B folded. In GT-A-folded proteins, only a single catalytic domain is found, whereas GT-B-folded enzymes possess two catalytically distinct domains, separated by a stretch which binds the acceptor. All domains possess structural elements which are similar to the Rossmann fold. The C-terminal domain of the GT-B-folded GTases mediates the binding of the nucleotide-activated sugar. In contrast to the GT-A GTases, the catalysis of GT-B GTases is independent of the metal ion cofactor, and these enzymes do not possess the metal ion-binding DxD motif. However, in Agl16 we could identify a DxD motif; whether this motif is indeed important for the catalysis by binding a cofactor is still unclear.
Table 3.
Overview of genes encoding predicted GTase in S. acidocaldarius
| GTase family | 3D fold, mechanism of sugar transfer | S. acidocaldarius gene(s) encoding GTase | No. of GTase genes |
|---|---|---|---|
| 2 | GT-A, inverting | saci0124, saci0275, saci0956, saci1011, saci1051, saci1499, saci1909, saci1911, saci1915, saci1927 | 10 |
| 4 | GT-B, retaining | saci0807, saci0869, saci1249, saci1827, saci1904, saci1907, saci1914, saci1916, saci1921, saci1922, saci1923 | 11 |
| 5 | GT-B, retaining | saci1201 | 1 |
| 35 | GT-B, retaining | saci0294 | 1 |
| 39 | GT-C, inverting | saci0421 | 1 |
| 66 | GT-C, inverting | saci1274 (aglB) | 2 |
| Not classified | saci0201, saci0870, saci1865, saci2027 | 4 |
BLAST analysis against known protein structures of GT-B-folded GTases revealed a crystal structure (residues 172 to 338) of an archaeal GTase from Archaeoglobus fulgidus (W. Zhou, F. Forouhar, K. Conover, R. Xiao, T. B. Acton, G. T. Montelione, L. Tong, J. F. Hunt, PDB no. O30192, 2005) which showed 26% sequence identity to Agl16. This GTase is homologous to the bacterial mannosyltransferase WbaZ, which catalyzes an α-Manp(1 → 3)-β-d-Galp linkage in the O-antigens from Salmonella enterica (35) and from E. coli K30 (36). Modeling of the Agl16 to the archaeal WbaZ homolog (PDB no. 2f9f) showed the two-domain structure, including the Rossmann-like fold.
The analysis of the genetic localization of alg16 revealed two genes which might also participate in the N-glycosylation pathway by activating the sugar precursor. Directly upstream, the gene saci0806 encodes a predicted phosphoglucomutase/phosphomannomutase and the gene saci0802 encodes a predicted 3-hexulose-6-phosphate synthase (Fig. 1). In our proposed model, Saci0802 would activate the sugar by transferring a phosphate from nucleotide triphosphate onto the C6 atom of the sugar. The phosphoglucomutase/phosphomannomutase is predicted to transfer the phosphate to create sugar-3-phosphate. This phosphorylated sugar would then be nucleotide activated by a yet-unknown enzyme. The nucleotide-activated sugar acts as a donor for the GTase Agl16, which either directly transfers the sugar to the premature N-glycan or to the dolichyl phosphate. However, the direct transfer to the N-glycan is more likely, because all eukaryal GTase transferring sugars onto dolichyl phosphate, like Dpm1 or Alg5, are membrane bound.
Fig 1.
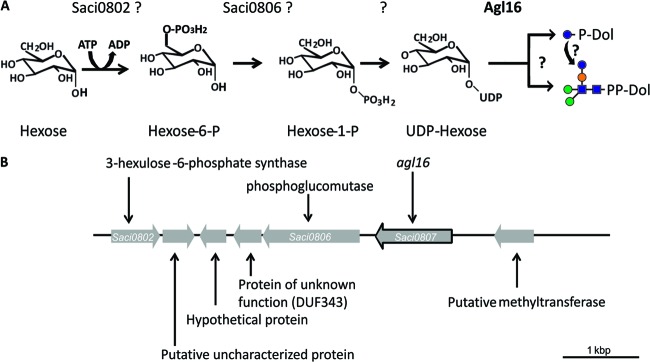
Proposed activation and transfer pathway of the last hexose residue of the N-glycan in S. acidocaldarius. (A) Proposed biosynthesis and transferase pathway of the last hexose. The structure of the unidentified hexose is shown as glucose. saci0802 encodes a proposed 3-hexulose-6-phosphate synthase; saci0806 is annotated as a phosphoglucomutase, and agl16 codes for a soluble glycosyltransferase. Glycan symbols: blue sphere, glucose; green sphere, mannose; orange sphere, sulfoquinovose; blue square, GlcNAc. (B) Physical map of the gene region of S. acidocaldarius, where the gene coding for the glycosyltransferase Agl16 is located. The illustrated region encompasses saci0802 to saci0808. The outlined gene encodes the glycosyltransferase Agl16. The genes saci0802 and saci0806 are most likely also involved in the activation of the hexose.
Markerless in-frame deletion of agl16.
To delete agl16, 800 to 1,000 bp stretches flanking the gene were amplified and fused by PCR. The resulting overlapping fragment was inserted into the plasmid pSAV0407, including the pyrEF genes for selection. The obtained vector was transformed in the background strain S. acidocaldarius MW001 and selected first for the integration and second for the segregation of the plasmid, yielding either the background strain or the in-frame deletion mutant. The deletion of agl16 was first confirmed by PCR amplification of the gene area with upstream forward and downstream reverse primers, resulting in either a 3,125-bp or a 2,090-bp fragment for the background strain MW001 or the Δagl16 mutant, respectively (Fig. 2). To verify the Δagl16 mutant, a reverse transcription-PCR (RT-PCR) was performed. RNA of the background strain MW001 and the Δagl16 strain, grown to mid-exponential growth phase, was isolated and used as a template for the synthesis of single-stranded cDNA. The control PCR used the genomic DNA, cDNA, and RNA as templates, as well as primers against the internal region of either agl16 or aglB, encoding the oligosaccharyl transferase (Fig. 2). The PCR confirmed the lack of genomic DNA in the RNA sample as well as the functional synthesis of the cDNA for the background strain MW001 and for the mutant. PCR with primers against the internal region of agl16 confirmed the absence of agl16 in the deletion mutant strain.
Fig 2.
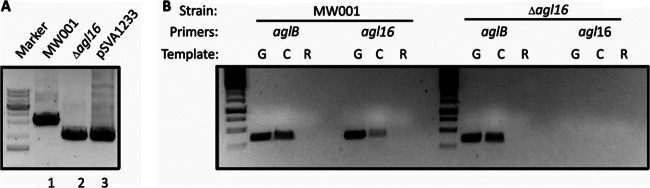
Confirmation of the in-frame Δagl16 mutant. (A) Gene deletion was confirmed by PCR using the outside primers against the flanking regions of agl16 and template DNA isolated from the background strain MW001, the mutant lacking agl16, or the plasmid pSVA1233, used for the homologous recombination incorporating the up- and downstream region of deleted agl16. (B) RT-PCR confirmed the deletion of the gene agl16, encoding a glycosyltransferase. The cDNA (C) served as a template in PCR amplification using primers against the internal region of either aglB or agl16. In each case, PCR amplifications were also performed using genomic DNA (G) as a positive control and total RNA (R) as a negative control.
Deletion of agl16 reduced the molecular weight of the S-layer protein SlaA.
As described above, the GTase Agl16 was first identified by screening a variety of GTase deletion mutants for defects in the N-glycan biosynthesis. To detect differences in the N-glycan size, the S-layer glycoprotein SlaA was used as a reporter protein. Isolated S-layer proteins from the background as well as from the agl16 deletion strain were separated on an 11% SDS-PAGE gel (Fig. 3). Staining with Coomassie brilliant blue revealed that the samples contained similar amounts of the S-layer glycoprotein. Furthermore, the SlaA from the Δagl16 deletion mutant migrated faster than the wild-type protein (Fig. 3). This was the first indication that the Agl16 is involved in the N-glycosylation pathway. To determine whether the defect in the N-glycosylation was indeed caused by the deletion of agl16 and was not a result of polar effects, the deletion strain was complemented with an empty plasmid (control) or with a plasmid containing the full-length agl16 gene. The S-layer of the complemented strain (Δagl16 + pSVA1274) showed no alteration in the protein size, implying the successful restoration of the full N-glycan biosynthesis pathway by the expression of the agl16 gene, excluding any polar effects in the deletion mutant (Fig. 3).
Fig 3.
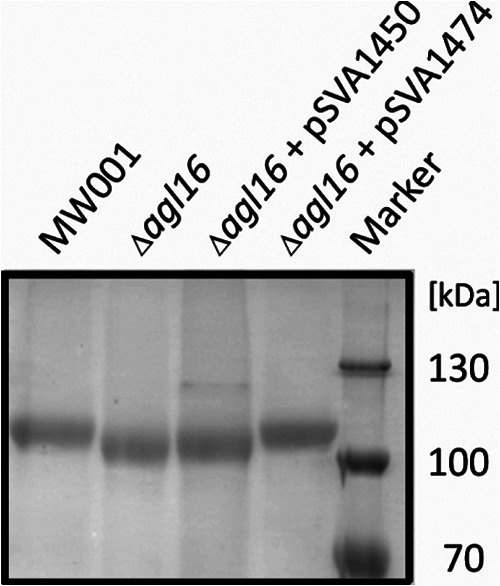
Effects of the agl16 deletion on S. acidocaldarius S-layer glycoprotein. Equivalent amounts of the S-layer protein SlaA from S. acidocaldarius MW001, the Δagl16 strain, the Δagl16 strain with control plasmid pSVA1450, and the Δagl16 strain complemented with pSVA1274 containing agl16 were separated by 11% SDS-PAGE and Coomassie blue stained.
Deletion of the agl16 resulted in a reduced FlaB protein size.
As it had been previously shown that deletion of agl3, encoding a UDP-sulfoquinovose synthase, resulted in an altered N-glycan on SlaA and the archaellin FlaB (14), we reasoned that the deletion of agl16 might also lead to a different migration behavior of the FlaB protein in SDS-PAGE. Therefore, cell pellets from the background strain S. acidocaldarius MW001, the Δagl3 strain (14), the Δagl3 strain complemented with pSVA1266 (14), the Δagl16 strain, and the Δagl16 strain complemented with pSVA1274 were separated by 11% SDS-PAGE and immunoblotted with antibodies raised against FlaB. Both FlaB proteins from the Δagl16 and from the Δagl3 deletion strain migrated faster than the wild-type protein (Fig. 4), whereas in the complementation strains, the original FlaB wild-type size was restored. As was shown for the S-layer glycoprotein, FlaB in the Δagl16 strain was small compared to FlaB in the wild-type and Δagl3 strains. This difference most likely is due to the fact that only one sugar residue is lost within the N-glycan in the Δagl16 strain, whereas in the Δagl3 strain, the N-glycan is reduced to three instead of six sugar residues (14). Although equivalent amounts of cells were used, as confirmed by Coomassie staining (data not shown), the immunoblot signal was stronger in the wild-type and complemented strains, implying a higher FlaB protein concentration present in these cells.
Fig 4.
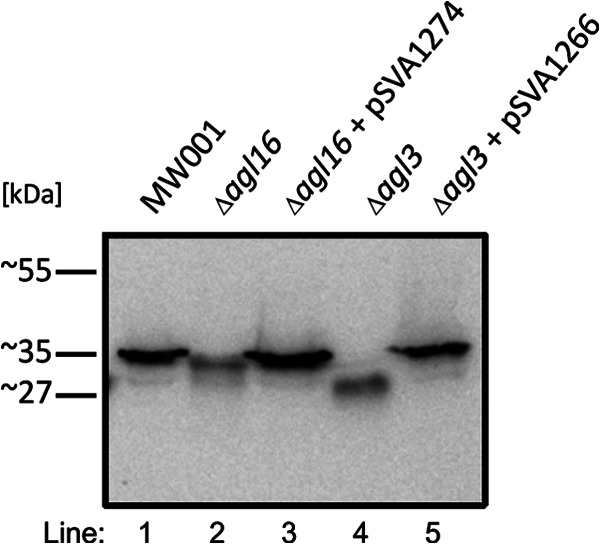
Glycosylation defects on the archaellin FlaB from S. acidocaldarius. Equivalent amounts of cells from S. acidocaldarius MW001 (lane 1), the Δagl16 strain (lane 2), the Δagl16 strain complemented with pSVA1274 (lane 3), the Δagl3 strain (lane 4), and the Δagl3 strain complemented with pSVA1266 (lane 5) were separated by 11% SDS-PAGE and immunoblotted with antibodies raised against FlaB.
The SlaA N-glycan composition is changed in the Δagl16 deletion mutant.
In order to verify that the observed changes in the size of the S-layer glycoprotein and the archaellin FlaB (Fig. 3 and 4) are indeed caused by the lack of one sugar residue of the N-glycan within the Δagl16 mutant, the N-glycan composition was analyzed. Thus, glycopeptides of the trypsin-digested S-layer proteins from wild-type S. acidocaldarius MW001 and the Δagl16 mutant were analyzed by nano-LC-ES-MS/MS. The glycopeptides of the wild-type S. acidocaldarius were previously shown to carry a hexasaccharide with the composition Glc1Man2GlcNAc2QuiS, with some glycoforms being truncated at presumed intermediate steps of biosynthesis and with the trisaccharide Man1GlcNAc2 being the most dominant truncated structure (7). As shown in Fig. 5, for the tryptic peptides T26 and T28 (for designations of the tryptic peptides, see reference 7), the intact hexasaccharide is no longer present in the Δagl16 mutant, as the corresponding glycopeptides of m/z [1561.5]3+ for the T24 and m/z [1151.6]4+ for the T26 were missing (7). Instead, the largest molecular ions were 1507.373+ for T24 glycopeptides and 1111.594+ for T26 glycopeptides, which correspond to pentasaccharide-linked peptides. These data confirmed that indeed a single hexose is missing in the N-glycan composition in the Δagl16 mutant. The identity of the hexose is either mannose or glucose, as both are present as terminal sugars. Since mannose and glucose have the same mass, the identity of the missing hexose cannot be unequivocally assigned from the MS data. Therefore, the N-glycan of the Δagl16 mutant has a composition of either Glc1Man1GlcNAc2QuiS or Man2GlcNAc2QuiS, as shown in Fig. 5.
Fig 5.
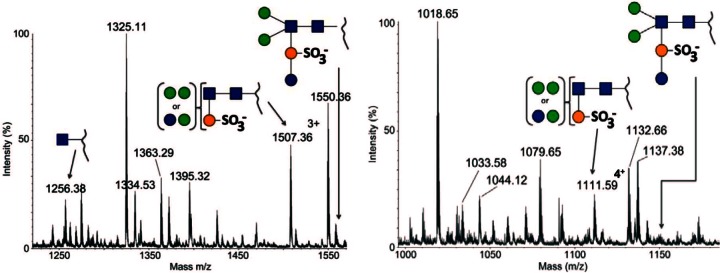
Detailed analysis of the S. acidocaldarius Δagl16 mutant background S-layer glycopeptide. The total ion chromatograms of the glycan profiles for the tryptic peptides T24 (A) and T26 (B) are shown. The annotated peaks show that the truncated Hex2QuiS1HexNAc2 glycoform is present on the observed glycopeptides, whereas the full glycan is missing. For reference, a cartoon depicting the full glycan is provided for each spectrum. Glycan symbols: blue sphere, glucose; green sphere, mannose; orange sphere, sulfoquinovose; blue square, GlcNAc.
HPLC analyses lead to identification of the missing hexose as glucose.
To identify the nature of the missing hexose in the N-glycan of the Δagl16 deletion mutant, the N-glycans of isolated SlaA from the background strain MW001 and the Δagl16 mutant strain were hydrolyzed with TFA, and the supernatant containing the hydrolyzed monosaccharides were analyzed by HPLC. Based on the composition of the fully assembled N-glycan tree, the Glc/Man ratio of the N-glycan from the MW001 background strain should be 1:2 (7, 25). However, HPLC analyses revealed a higher Glc ratio in the background strain than expected. The increase of the Glc peak within the HPLC analyses is due to the presence of GlcN derived from GlcNAc hydrolysis, which runs at the same retention time in the HPLC analysis. Furthermore, in analyses of the N-glycan composition of the Δagl3 strain, which includes only Man1GlcNAc2 and lacks the terminal glucose residue, a peak with the same retention time as Glc was apparent. Nevertheless, as the GlcNAc and QuiS concentrations are equal in MW001 and the Δagl16 strain, the changes in the Glc/Man concentration in the mutant and background strain made identifying the missing hexose possible. HPLC analyses of three independent isolated S-layers that were TFA hydrolyzed revealed average Glc/Man ratios of 9.2:1 in the MW001 background strain and 4.4:1 in the Δagl16 deletion strain, showing that the Glc concentration in the Δagl16 deletion strain is 2-fold reduced (Table 4). Based on the changes in the Glc concentration, it is proposed that Agl16 is a glycosyltransferases transferring the terminal Glc residue to the N-glycan.
Table 4.
HPLC analyses of TFA treatzed isolated S-layer from the background strain MW001 and the Δagl16 deletion mutant
| Sample | Retention time (min) |
Area (mV*s) |
Concn (μM) |
Slope for calibration (mV · s · μM−1) | Ratio (glucose:mannose) | |||
|---|---|---|---|---|---|---|---|---|
| Glucose | Mannose | Glucose | Mannose | Glucose | Mannose | |||
| Standards | ||||||||
| Glucose | 14.31 | 4,775 | 10,000 | 0.48 | ||||
| Mannose | 15.17 | 4,234 | 10,000 | 0.42 | ||||
| Background strains | ||||||||
| MW001 S1 | 14.31 | 15.19 | 645 | 68.6 | 1,351 | 162.0 | 8.3:1 | |
| MW001 S2 | 14.33 | 15.18 | 646 | 69.3 | 1,353 | 163.7 | 8.3:1 | |
| MW001 S3 | 14.32 | 15.20 | 537 | 43 | 1,125 | 101.6 | 11.1:1 | |
| Mutants | ||||||||
| Δagl16 S1 | 14.34 | 15.18 | 97.6 | 21.4 | 204 | 50.5 | 4.0:1 | |
| Δagl16 S2 | 14.35 | 15.16 | 51.16 | 9.52 | 107 | 22.5 | 4.8:1 | |
| Δagl16 S3 | 14.33 | 15.17 | 112.31 | 23.51 | 235 | 55.5 | 4.2:1 | |
Deletion of the agl16 showed a drastic reduction of motility.
We reasoned that the reduced FlaB protein concentration in the deletion mutants might be caused by a diminished stability or impaired assembly of the archaellum by the altered N-glycan composition. To test the functionality of the archaellum in the deletion strains, a motility assay was performed as previous described (33). Aliquots of the nonmotile S. acidocaldarius ΔaapF ΔflaH deletion strain (33) and the hypermotile S. acidocaldarius ΔaapF strain (37) were used as control strains. The S. acidocaldarius background strain MW001, the Δagl3 strain, the S. acidocaldarius Δagl16 strain, and the complemented strain were dropped (or dripped) onto semisolid Gelrite plates containing either 0 mM NaCl or 300 mM NaCl and incubated for 4 and 9 days. In contrast to the hypermotile S. acidocaldarius ΔaapF strain, for which swimming could be detected after 4 days of incubation, the nonmotile S. acidocaldarius ΔaapF ΔflaH deletion strain showed no motility even after 9 days of incubation (Fig. 6). The background strain MW001 showed strong motility after 9 days of incubation, whereas the Δagl3 and Δagl16 deletion strains exhibited no and drastically reduced swimming abilities, respectively. The swimming area appeared fuzzy at the colony border for the Δagl16 strain, showing a reduced but not totally impaired swimming ability, whereas the ability to swim in the Δagl3 strain seemed totally abolished. The complementation of Δagl16 restored motility, as the colony showed a swimming radius similar to that of the background strain MW001 (Fig. 6). The incubation under elevated salt conditions (300 mM NaCl) abolished motility, as even the hypermotile S. acidocaldarius ΔaapF strain did not show any swimming behavior. However, the deletion strains showed a drastic reduction of growth at high salinities, compared to the background strain or the hyper- or nonmotile strains (Fig. 6). The ability to grow at high salinity is dependent on the glycan size. The Δagl3 deletion strain showed no obvious growth after 4 days of incubation, whereas after 9 days of incubation, only a slight increase in cell density could be detected. After 4 days of incubation, the growth of the Δagl16 deletion strain was reduced compared to the background strain MW001; however, the defect was not so striking as in the Δagl3 deletion strain. Complementation of the Δagl16 recovered the growth defect, as the complementation Δagl16 + pSVA1274 strain showed the same colony density and size as the background strain MW001.
Fig 6.

Effects on the motility from S. acidocaldarius by interfering with the N-glycan assembly. Equivalent amounts of cells (5 to 6 μl) from S. acidocaldarius ΔaapF ΔflaH (lane 1), S. acidocaldarius ΔaapF (lane 2), the background strain S. acidocaldarius MW001(lane 3), S. acidocaldarius Δagl3 (lane 4), S. acidocaldarius Δagl16, and S. acidocaldarius Δagl16 complemented with pSVA1274 (lane 6), were dripped onto a 0.15% Gelrite plate and incubated for 4 and 9 days at 75°C. The panels on the right show the enlarged colony border and the motility zone (taken from the left panel: 0 mM NaCl, 9-day incubation), as well as the N-glycan composition of MW001 and the Δagl16 and Δagl3 mutants. The ability to swim corresponds to the N-glycan size.
The cells of the agl3 and agl16 deletion mutants appeared nonarchaeallated.
To confirm that the impaired or reduced motility of the agl mutants was caused by unstable archaella due to the N-glycan defects, the presence of cell appendages in each mutant was analyzed by transmission electron microscopy (TEM). In contrast to the MW001 background strain, which exhibits Aap pili with a diameter of 8 to 10 nm and archaella with a diameter of 10 to 14 nm (Fig. 7A), no archaella could be detected in either agl deletion mutant (Fig. 7B and C). The analyses were repeated with cell cultures that had undergone tryptone starvation to induce archaellum production, as previously described (33). Even under inducing conditions, archaella could not be detected in either mutant by TEM. This confirmed that the archaellum is unstable or cannot be assembled on the cell surface of the agl deletion mutants. Although the agl16 deletion mutant showed little motility on semisolid Gelrite plates (Fig. 6), this analysis underlines the importance of the terminal hexose for stable archaellum assembly. However, in contrast to the archaella, the pilins were present in each of the mutants. Also, the Aap pili are predicted to be N-glycosylated but with a lower ratio than the archaellins; moreover, the pili seem to be much more stable even under harsh detergent treatment (A. Henche and S.-V. Albers, personal communication). The EM images gave further indication that the S-layer stability or assembly is affected in these agl mutants. In contrast to that of the MW001 background strain, the S-layer of the mutants showed defects causing the cytoplasmic membrane to bulge out (Fig. 7). This instable S-layer might not affect the growth under normal growth condition but is more obvious at elevated salt concentrations, in which the stability of the S-layer is more affected.
Fig 7.

TEM analysis of negatively stained cells of MW001 (A), the Δagl3 mutant (B), and the Δagl16 mutant (C) showed the lack of archaella on each of the agl deletion mutants. The MW001 background strain exhibits Aap pili (8 to 10 nm; white arrow) as well as the archaella (10 to 14 nm, black arrow); in the agl deletion mutants, only the Aap pili could be detected. Bars, 200 nm (A) and 100 nm (B and C).
The alteration of the N-glycan resulted in growth retardation upon salt stress.
As shown in the motility assay defects in the N-glycan biosynthesis have strong effects on the growth under elevated salinities. Previously we reported the significant reduction of the growth rate for the Δagl3 strain under salt stress, in which the biosynthesis of the sulfoquinovose incorporated in the N-glycan was interrupted (14). The S-layer as the sole surface envelope in S. acidocaldarius has to stabilize the cell surface and protect it from external conditions. As defects in the biogenesis of these glycoproteins presumably have an effect on the interaction and the stability of the protective S-layer, we measured the growth rate for the background and the Δagl16 strains. Under standard growth conditions (Brock medium, 0.1% NZ-amine and 0.1% dextrin, 0 mM NaCl, 79°C and pH 3), no significant changes in the doubling times of the mutant (Td = 8.59 ± 0.23 h) compared to the background strain MW001 (Td = 8.72 ± 0.26 h) were observed (Fig. 8). The effect of the reduced glycan structure became evident when the strains were grown at higher salinities. At 200 mM NaCl, the doubling time of the Δagl16 strain was increased (Td = 14.82 ± 1.22 h), whereas the doubling time of the background strain (Td = 8.62 ± 0.55 h) was not altered compared to that under standard growth conditions. At 300 mM NaCl, the growth of the background strain was only slightly affected (Td = 13.01 ± 0.37 h), while the deletion mutant was profoundly affected. The doubling time of the mutant increased to twice (Td = 24.57 ± 1.75 h) the doubling time of the background strain. In comparison to the Δagl3 strain, for which the doubling time was 63.9 ± 2 h (14) at this salt concentration, the effect of one missing sugar in the S-layer N-glycan had a much lower impact than the deletion of three sugars from the glycan tree. However, at 400 mM NaCl, only the background strain, MW001, was capable of sustaining growth after a long lag phase (Td = 34.84 h ± 6.48 h), whereas the mutant showed no obvious growth.
Fig 8.
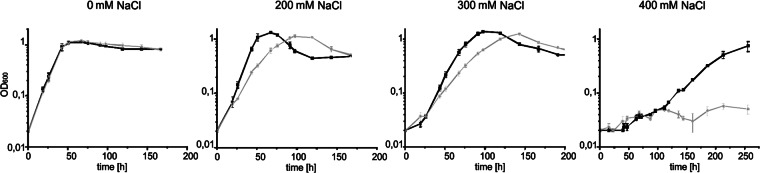
Response to salt stress in the Δagl16 strain. S. acidocaldarius MW001 (black line) or Δagl16 (gray line) were grown in Brock medium at different salt concentrations (0 to 400 mM NaCl). Growth was measured as the optical density at 600 nm (OD600). Shown are values obtained from an experiment done in triplicate; repeats generated a similar result.
DISCUSSION
Various surface-exposed proteins from S. acidocaldarius have been shown to be modified by the attachment of glycan, e.g., S-layer protein (7), cytochrome b558/566 (23, 25), archaellins (14), or sugar-binding proteins (24). Most of them are shown to be modified via the N-glycosylation process. Recently, we identified the first enzyme participating in the N-glycosylation process in S. acidocaldarius. Agl3, a UDP-sulfoquinovose synthase, creates the nucleotide-activated sugar precursor UDP-sulfoquinovose, which is assembled to form a branch of the N-glycan (14). However, additional enzymes, in particular glycosyltransferases (GTases), necessary for the assembly of the full-length N-linked glycan are still unidentified in S. acidocaldarius (38). Unlike the agl gene cluster within Haloferax volcanii, in which the oligosaccharyltransferase AglB and other enzymes involved in the N-glycosylation process are found (39), genes coding for predicted GTases are scattered all over the S. acidocaldarius genome and not located near the aglB gene (Table 3). This scattered distribution of GTase seems common within thermophilic organisms (19), making the identification of N-GTases in thermophilic Archaea more difficult. Various deletion studies of genes coding for predicted GTases within the genome of S. acidocaldarius so far have identified only one GTase involved in the biosynthesis of the N-glycan of the S-layer glycoprotein. In the present study, we report this first identified glycosyltransferase involved in the N-glycosylation in a crenarchaeon. The markerless in-frame deletion of agl16 in S. acidocaldarius resulted in a reduced molecular size of the large S-layer protein SlaA and the archaellin FlaB. The slight reduction of the molecular size of these proteins implied that one or more sugar residues are absent in the N-glycan structure. Nano-LC-MS/MS analysis confirmed the presence of a pentasaccharide lacking one terminal hexose residue. HPLC analyses of the sugar composition of isolated S-layer proteins from the Δagl16 and the background strains revealed a reduced Glc concentration in the agl16 mutant, which indicate that Agl16 is important for the transfers of the terminal glucose residue onto the premature N-glycan. The recently developed expression system in S. acidocaldarius (28) enables us to express and purify Agl16 homologously. The characterization of Agl16 will give us new insight in the glycosyltransferase reaction, e.g., which donor and acceptor are used. For instance Agl16 might use soluble nucleotide-activated (UPD or GDP) glucose precursors or dolichyl phosphate-linked glucose residues as donors. Additionally, an activity assay of Agl16 would also reveal the preferred acceptor, whether it is dolichyl phosphate or directly the premature N-glycan tree linked to dolichyl phosphate. In Haloferax volcanii, the last assembly step of the N-glycan is mediated by AglS, which transfers the terminal mannose of the pentasaccharide (40). In contrast to the soluble Agl16, AglS is a membrane-bound GTase, which transfers the last mannose residue from a dolichyl phosphate-activated mannose onto the N-glycan tree. This assembly step is similar to the last eukaryal N-glycan biosynthesis step, in which the membrane-bound GTase uses dolichyl phosphate-linked mannose or glucose residues as the sugar donor (41, 42). All other soluble eukaryal GTases use nucleotide-activated sugar residues in the first N-glycan biosynthesis steps. Therefore, it is most likely that Agl16 also uses a nucleotide-activated sugar precursor as the sugar donor.
The deletion of agl16 in S. acidocaldarius causes significant effects on the phenotype, as the presence of only the partial N-glycan structure on the S-layer protein and the archaellins resulted in a reduced growth rate at elevated salt concentration as well as impaired function of the archaellum. A similar effect of a truncated N-glycan was also observed in other Archaea. The observation that underglycosylation affects archaellum filament assembly was first made in Methanococcus maripaludis, where deletion of MMP0350, encoding a putative acetyltransferase, was proposed to be responsible for the acetylation of the first two N-glycan sugar residues (43). Also, in Methanococcus voltae, deletion of aglA, involved in the final biosynthetic step of the trisaccharide, had a similar effect on the stability of the archaella, as the ΔaglA strain exhibited only few and shortened archaella (22). The finding that archaellins need to be modified with at least a subset of the N-glycan sugar to assemble and function has recently also been shown in H. volcanii (17). Defects in the N-glycosylation introduced by agl gene deletions as well as the replacement of the asparagines within the N-glycosylation sites in the archaellins resulted in the loss of motility and archaellum assembly in H. volcanii (17). However, in contrast to H. volcanii and M. voltae in which a trisaccharide is the minimal N-glycan size to allow motility (17, 22, 44), S. acidocaldarius displays a strong dependency on the fully assembled tribranched hexasaccharide, as even one missing sugar residue in the N-glycan almost completely abolished motility. A correlation of salinity and growth was also shown in H. volcanii, in which the deletion of aglD resulted in a reduction of the growth rate in media with high salinities (3.7 M or 4.8 M NaCl), whereas under standard growth conditions (1.7 M NaCl), no change in growth was observed (10).
Beside agl16, the genes saci0802 and saci0806, lying upstream of agl16, are predicted to be involved in the N-glycan biosynthesis step. Mutants with deletions of these genes will help determine whether these gene products, as it is proposed in Fig. 1A, activate the last hexose residue before it is transferred by Agl16. Additionally, analyses of gene expression might give insight into a coordinated regulation of these first genes involved in the N-glycosylation process.
In conclusion, this study identified the first crenarchaeal GTase, Agl16, catalyzing the last biosynthesis step of the N-glycan in S. acidocaldarius. The deletion of agl16 demonstrated the importance of the fully assembled N-linked hexasaccharide for the growth and proper assembly and function of the archaellum of S. acidocaldarius. The comparison to the Δagl16 mutant and the previous reported Δagl3 mutant demonstrated that motility is strongly dependent on N-glycan size.
This study has now extended the field of N-glycosylation research to crenarchaeotes and contributes to the increasing data available on archaeal N-glycosylation and enzymes responsible for biosynthesis and assembly of N-glycans. Further studies will be undertaken to uncover the complete N-glycosylation pathway in S. acidocaldarius.
ACKNOWLEDGMENTS
E.P., P.H., and A.D. were supported by the Biotechnology and Biological Sciences Research Council (BBF0083091, BBC5196701) and the UK Research Councils' Basic Technology Initiative Translational Grant (EP/G037604/1). B.H.M. was partially supported by the Collaborative Research Centre 987 by the DFG, C.D. was supported by the International Max-Planck Research School, and S.-V.A. was supported by intramural funds of the Max Planck Society.
Footnotes
Published ahead of print 8 March 2013
REFERENCES
- 1. Albers SV, Meyer BH. 2011. The archaeal cell envelope. Nat. Rev. Microbiol. 9:414–426 [DOI] [PubMed] [Google Scholar]
- 2. Zeitler R, Hochmuth E, Deutzmann R, Sumper M. 1998. Exchange of Ser-4 for Val, Leu or Asn in the sequon Asn-Ala-Ser does not prevent N-glycosylation of the cell surface glycoprotein from Halobacterium halobium. Glycobiology 8:1157–1164 [DOI] [PubMed] [Google Scholar]
- 3. Voisin S, Houliston RS, Kelly J, Brisson JR, Watson D, Bardy SL, Jarrell KF, Logan SM. 2005. Identification and characterization of the unique N-linked glycan common to the flagellins and S-layer glycoprotein of Methanococcus voltae. J. Biol. Chem. 280:16586–16593 [DOI] [PubMed] [Google Scholar]
- 4. Sumper M, Berg E, Mengele R, Strobel I. 1990. Primary structure and glycosylation of the S-layer protein of Haloferax volcanii. J. Bacteriol. 172:7111–7118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Paul G, Lottspeich F, Wieland F. 1986. Asparaginyl-N-acetylgalactosamine. Linkage unit of halobacterial glycosaminoglycan. J. Biol. Chem. 261:1020–1024 [PubMed] [Google Scholar]
- 6. Mescher MF, Strominger JL. 1976. Purification and characterization of a prokaryotic glycoprotein from cell envelope of Halobacterium salinarium. J. Biol. Chem. 251:2005–2014 [PubMed] [Google Scholar]
- 7. Peyfoon E, Meyer B, Hitchen PG, Panico M, Morris HR, Haslam SM, Albers SV, Dell A. 2010. The S-layer glycoprotein of the crenarchaeote Sulfolobus acidocaldarius is glycosylated at multiple sites with chitobiose-linked N-glycans. Archaea 2010:754101 doi:10.1155/2010/754101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kessel M, Volker S, Santarius U, Huber R, Baumeister W. 1990. Three dimensional reconstruction of the surface protein of the extremely thermophilic archaebacterium Archaeoglobus fulgidus. Syst. Appl. Microbiol. 13:207–213 [Google Scholar]
- 9. Kessel M, Wildhaber I, Cohen S, Baumeister W. 1988. Three dimensional structure of the regular surface glycoprotein layer of Halobacterium volcanii from the Dead Sea. EMBO J. 7:1549–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abu-Qarn M, Yurist-Doutsch S, Giordano A, Trauner A, Morris HR, Hitchen P, Medalia O, Dell A, Eichler J. 2007. Haloferax volcanii AglB and AglD are involved in N-glycosylation of the S-layer glycoprotein and proper assembly of the surface layer. J. Mol. Biol. 374:1224–1236 [DOI] [PubMed] [Google Scholar]
- 11. Ng SY, Wu J, Nair DB, Logan SM, Robotham A, Tessier L, Kelly JF, Uchida K, Aizawa S, Jarrell KF. 2011. Genetic and mass spectrometry analyses of the unusual type IV-like pili of the archaeon Methanococcus maripaludis. J. Bacteriol. 193:804–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Elferink MG, Albers SV, Konings WN, Driessen AJ. 2001. Sugar transport in Sulfolobus solfataricus is mediated by two families of binding protein-dependent ABC transporters. Mol. Microbiol. 39:1494–1503 [DOI] [PubMed] [Google Scholar]
- 13. Ng SY, Chaban B, Jarrell KF. 2006. Archaeal flagella, bacterial flagella and type IV pili: a comparison of genes and posttranslational modifications. J. Mol. Microbiol. Biotechnol. 11:167–191 [DOI] [PubMed] [Google Scholar]
- 14. Meyer BH, Zolghadr B, Peyfoon E, Pabst M, Panico M, Morris HR, Haslam SM, Messner P, Schaffer C, Dell A, Albers S. 2011. Sulfoquinovose synthase—an important enzyme in the N-glycosylation pathway of Sulfolobus acidocaldarius. Mol. Microbiol. 82:1150–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jarrell KF, Albers SV. 2012. The archaellum: an old motility structure with a new name. Trends Microbiol. 20:307–312 [DOI] [PubMed] [Google Scholar]
- 16. Lu D, Yang C, Liu Z. 2012. How hydrophobicity and the glycosylation site of glycans affect protein folding and stability: a molecular dynamics simulation. J. Phys. Chem. B 116:390–400 [DOI] [PubMed] [Google Scholar]
- 17. Tripepi M, You J, Temel S, Onder O, Brisson D, Pohlschroder M. 2012. N-glycosylation of Haloferax volcanii flagellins requires known Agl proteins and is essential for biosynthesis of stable flagella. J. Bacteriol. 194:4876–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nothaft H, Szymanski CM. 2010. Protein glycosylation in bacteria: sweeter than ever. Nat. Rev. Microbiol. 8:765–778 [DOI] [PubMed] [Google Scholar]
- 19. Magidovich H, Eichler J. 2009. Glycosyltransferases and oligosaccharyltransferases in Archaea: putative components of the N-glycosylation pathway in the third domain of life. FEMS Microbiol. Lett. 300:122–130 [DOI] [PubMed] [Google Scholar]
- 20. Maita N, Nyirenda J, Igura M, Kamishikiryo J, Kohda D. 2010. Comparative structural biology of eubacterial and archaeal oligosaccharyltransferases. J. Biol. Chem. 285:4941–4950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Calo D, Kaminski L, Eichler J. 2010. Protein glycosylation in Archaea: Sweet and extreme. Glycobiology 20:1065–1076 [DOI] [PubMed] [Google Scholar]
- 22. Chaban B, Voisin S, Kelly J, Logan SM, Jarrell KF. 2006. Identification of genes involved in the biosynthesis and attachment of Methanococcus voltae N-linked glycans: insight into N-linked glycosylation pathways in Archaea. Mol. Microbiol. 61:259–268 [DOI] [PubMed] [Google Scholar]
- 23. Hettmann T, Schmidt CL, Anemuller S, Zahringer U, Moll H, Petersen A, Schafer G. 1998. Cytochrome b558/566 from the archaeon Sulfolobus acidocaldarius. J. Biol. Chem. 273:12032–12040 [DOI] [PubMed] [Google Scholar]
- 24. Albers SV, Elferink MG, Charlebois RL, Sensen CW, Driessen AJ, Konings WN. 1999. Glucose transport in the extremely thermoacidophilic Sulfolobus solfataricus involves a high-affinity membrane-integrated binding protein. J. Bacteriol. 181:4285–4291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zahringer U, Moll H, Hettmann T, Knirel VA, Schafer G. 2000. Cytochrome b558/566 from the archaeon Sulfolobus acidocaldarius has a unique Asn-linked highly branched hexasaccharide chain containing 6-sulfoquinovose. Eur. J. Biochem. 267:4144–4149 [DOI] [PubMed] [Google Scholar]
- 26. Heinz E. (ed). 1993. Recent investigations on the biosynthesis of the plant sulfolipid. SPB Academic Publishers, The Hague, Netherlands [Google Scholar]
- 27. Benning C. 1998. Biosynthesis and function of the sulfolipid sulfoquinovosyl diacylglycerol. Annu. Rev. Plant Physiol. Plant Mol. Biol. 49:53–75 [DOI] [PubMed] [Google Scholar]
- 28. Wagner M, van Wolferen M, Wagner A, Lassak K, Meyer BH, Reimann J, Albers SV. 2012. Versatile genetic tool Box for the crenarchaeote Sulfolobus acidocaldarius. Front. Microbiol. 3:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brock TD, Brock KM, Belly RT, Weiss RL. 1972. Sulfolobus: a new genus of sulfur-oxidizing bacteria living at low pH and high temperature. Arch. Microbiol. 84:54–68 [DOI] [PubMed] [Google Scholar]
- 30. Zolghadr B, Weber S, Szabo Z, Driessen AJ, Albers SV. 2007. Identification of a system required for the functional surface localization of sugar binding proteins with class III signal peptides in Sulfolobus solfataricus. Mol. Microbiol. 64:795–806 [DOI] [PubMed] [Google Scholar]
- 31. Wagner M, Berkner S, Ajon M, Driessen AJ, Lipps G, Albers SV. 2009. Expanding and understanding the genetic toolbox of the hyperthermophilic genus Sulfolobus. Biochem. Soc. Trans. 37:97–101 [DOI] [PubMed] [Google Scholar]
- 32. Kurosawa N, Grogan DW. 2005. Homologous recombination of exogenous DNA with the Sulfolobus acidocaldarius genome: properties and uses. FEMS Microbiol. Lett. 253:141–149 [DOI] [PubMed] [Google Scholar]
- 33. Lassak K, Neiner T, Ghosh A, Klingl A, Wirth R, Albers SV. 2012. Molecular analysis of the crenarchaeal flagellum. Mol. Microbiol. 83:110–124 [DOI] [PubMed] [Google Scholar]
- 34. Berkner S, Wlodkowski A, Albers SV, Lipps G. 2010. Inducible and constitutive promoters for genetic systems in Sulfolobus acidocaldarius. Extremophiles 14:249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu D, Haase AM, Lindqvist L, Lindberg AA, Reeves PR. 1993. Glycosyl transferases of O-antigen biosynthesis in Salmonella enterica: identification and characterization of transferase genes of groups B, C2, and E1. J. Bacteriol. 175:3408–3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Drummelsmith J, Whitfield C. 1999. Gene products required for surface expression of the capsular form of the group 1 K antigen in Escherichia coli (O9a:K30). Mol. Microbiol. 31:1321–1332 [DOI] [PubMed] [Google Scholar]
- 37. Henche AL, Koerdt A, Ghosh A, Albers SV. 2012. Influence of cell surface structures on crenarchaeal biofilm formation using a thermostable green fluorescent protein. Environ. Microbiol. 14:779–793 [DOI] [PubMed] [Google Scholar]
- 38. Meyer BH, Albers SV. 2013. Hot and sweet: protein glycosylation in Crenarchaeota. Biochem. Soc. Trans. 41:384–392 [DOI] [PubMed] [Google Scholar]
- 39. Yurist-Doutsch S, Eichler J. 2009. Manual annotation, transcriptional analysis, and protein expression studies reveal novel genes in the agl cluster responsible for N-glycosylation in the halophilic archaeon Haloferax volcanii. J. Bacteriol. 191:3068–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cohen-Rosenzweig C, Yurist-Doutsch S, Eichler J. 2012. AglS, a novel component of the Haloferax volcanii N-glycosylation pathway, is a dolichol phosphate-mannose mannosyltransferase. J. Bacteriol. 194:6909–6916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burda P, Aebi M. 1999. The dolichol pathway of N-linked glycosylation. Biochim. Biophys. Acta 1426:239–257 [DOI] [PubMed] [Google Scholar]
- 42. Weerapana E, Imperiali B. 2006. Asparagine-linked protein glycosylation: from eukaryotic to prokaryotic systems. Glycobiology 16:91R–101R [DOI] [PubMed] [Google Scholar]
- 43. VanDyke DJ, Wu J, Logan SM, Kelly JF, Mizuno S, Aizawa S, Jarrell KF. 2009. Identification of genes involved in the assembly and attachment of a novel flagellin N-linked tetrasaccharide important for motility in the archaeon Methanococcus maripaludis. Mol. Microbiol. 72:633–644 [DOI] [PubMed] [Google Scholar]
- 44. Chaban B, Logan SM, Kelly JF, Jarrell KF. 2009. AglC and AglK are involved in biosynthesis and attachment of diacetylated glucuronic acid to the N-glycan in Methanococcus voltae. J. Bacteriol. 191:187–195 [DOI] [PMC free article] [PubMed] [Google Scholar]