

Abstract
Bacillus subtilis biofilm formation is tightly regulated by elaborate signaling pathways. In contrast to domesticated lab strains of B. subtilis which form smooth, essentially featureless colonies, undomesticated strains such as NCIB 3610 form architecturally complex biofilms. NCIB 3610 also contains an 80-kb plasmid absent from laboratory strains, and mutations in a plasmid-encoded homolog of a Rap protein, RapP, caused a hyperrugose biofilm phenotype. Here we explored the role of rapP phrP in biofilm formation. We found that RapP is a phosphatase that dephosphorylates the intermediate response regulator Spo0F. RapP appears to employ a catalytic glutamate to dephosphorylate the Spo0F aspartyl phosphate, and the implications of the RapP catalytic glutamate are discussed. In addition to regulating B. subtilis biofilm formation, we found that RapP regulates sporulation and genetic competence as a result of its ability to dephosphorylate Spo0F. Interestingly, while rap phr gene cassettes routinely form regulatory pairs; i.e., the mature phr gene product inhibits the activity of the rap gene product, the phrP gene product did not inhibit RapP activity in our assays. RapP activity was, however, inhibited by PhrH in vivo but not in vitro. Additional genetic analysis suggests that RapP is directly inhibited by peptide binding. We speculate that PhrH could be subject to posttranslational modification in vivo and directly inhibit RapP activity or, more likely, PhrH upregulates the expression of a peptide that, in turn, directly binds to RapP and inhibits its Spo0F phosphatase activity.
INTRODUCTION
Bacillus subtilis biofilm communities consist of subpopulations of developmentally different cell types, including motile, matrix-secreting, and sporulating cells (for a review, see reference 1). B. subtilis biofilm cells follow a defined lineage; i.e., motile cells become matrix-producing cells, which in turn become sporulating cells (2). The ratio of cell types in a B. subtilis biofilm population changes over time, and these changes are regulated in large part by the spatial arrangement of the cells in the architecturally complex biofilm. In fact, the biofilm population heterogeneity appears to hinge on the formation of an architecturally complex biofilm, and strains defective in extracellular matrix production contain very few motile cells and essentially no sporulating cells.
In contrast to wild (undomesticated) B. subtilis strains such as NCIB 3610 (3610), which form architecturally complex wrinkled biofilms on solid surfaces and thick pellicles at the air-liquid interfaces of static cultures, domesticated lab strains such as strain 168 form smooth, flat, essentially featureless colonies (3, 4). The genetic basis underlying the differences in wild and domesticated B. subtilis strain biofilm formation was previously examined (3–7). In one study, four point mutations were shown to impair biofilm formation in common B. subtilis lab strains; however, reverting these mutations to their wild-type identity resulted in strains that formed hyperwrinkled rather than wild-type biofilms (4). Importantly, the formation of biofilms with a wild-type architecture required not only the reversion of the four biofilm-impairing mutations but also the expression of the rapP phrP gene cassette.
The rapP phrP cassette is carried on an 80-kb plasmid present in wild B. subtilis strains and absent from domesticated strains. While no functional activity had been assigned to RapP, its name resulted from the similarity of the RapP amino acid sequence to that of other members of the Rap protein family (4). Rap proteins derive their name from the original members of the family, RapA and RapB, which were shown to be response regulator aspartate phosphatases (8). RapA and RapB dephosphorylate Spo0F, which is an intermediate response regulator in the B. subtilis phosphorelay pathway central to the regulation of both sporulation and biofilm formation (Fig. 1) (8, 9). In addition to RapA and RapB, many other Rap proteins, including RapE, RapH, and RapJ, function similarly to dephosphorylate Spo0F (10–12). The ultimate effect of dephosphorylating Spo0F is a decrease in the cellular concentration of phosphorylated Spo0A (Spo0A∼P). Because the expression of the B. subtilis biofilm extracellular polysaccharide (EPS) and structural matrix-associated protein TasA requires Spo0A∼P (13, 14) (Fig. 1), inhibition of the phosphorelay also inhibits biofilm development.
Fig 1.

B. subtilis sporulation, biofilm formation, and genetic competence signaling pathways. Phosphoryl groups from histidine kinases are transferred to Spo0F, Spo0B, and ultimately Spo0A. Spo0F dephosphorylation causes the direction of phosphoryl flow to reverse; i.e., phosphoryl groups are transferred from Spo0A to Spo0B and in turn to Spo0F (not shown). Some (but not all) pro-phr genes are regulated by σH. Arrows indicate the positive control, while perpendiculars indicate the negative control. Solid lines represent regulation by protein-protein interaction, and dashed lines represent regulation at the level of gene transcription. REC, receiver domain; H, histidine; D, aspartic acid; P, phosphoryl group.
More specifically, EPS is produced by the gene products of the epsA-epsO (epsA-O) operon (3, 15) and TasA is encoded in the tapA(yqxM)-sipW-tasA operon (16). The transcription factor SinR functions as the master regulator of B. subtilis biofilm formation by repressing the expression of the epsA-O and tapA(yqxM)-sipW-tasA operons (Fig. 1) (15, 17). SinR is antagonized by SinI, which binds to SinR and inhibits its interaction with target DNA promoters (18). Spo0A∼P triggers biofilm formation at least in part by driving the transcription of sinI (19).
Despite their sequence and structural similarity to the Rap proteins that dephosphorylate Spo0F, some B. subtilis Rap proteins, such as RapC, RapF, and RapG, are transcriptional antiactivator proteins that inhibit the function of their response regulator targets without dephosphorylating their receiver domains (20–23). For example, RapC and RapF bind to the DNA binding domain of the response regulator ComA (20, 23) and RapG binds to DegU (22). We previously showed that RapF functions mechanistically to sterically block the interaction of ComA with DNA promoters and disrupt the formation of active ComA dimers (21).
ComA drives the expression of the srf operon, which contains, among other genes, that for the nonribosomal peptide synthetase that produces the lipopeptide surfactin (24). Surfactin is a secreted surfactant required for B. subtilis swarming motility (5), and surfactin is also a B. subtilis paracrine signal that triggers the formation of membrane pores and potassium leakage (7). Potassium leakage activates the sporulation phosphorelay kinase KinC, resulting in Spo0A phosphorylation and biofilm matrix production (7). Thus, Rap proteins that inhibit ComA activity would be expected to inhibit both surfactin production and, in turn, biofilm formation.
As described above, rapP exists in a gene cassette with phrP, which overlaps the 3′ end of rapP. phr genes typically encode pro-Phr polypeptides, which are secreted from the cell, where they undergo proteolytic maturation (25, 26). Mature Phr pentapeptides (25, 27) or hexapeptides (28) are imported into the cell (29), where each peptide inhibits its cognate Rap protein. For example, PhrA binds to RapA and inhibits its phosphatase activity (30) and PhrF binds to RapF and disrupts the binding of RapF to ComA (23). What role, if any, PhrP plays in the regulation of B. subtilis biofilm formation was unknown.
Here we explored the function of RapP and PhrP in the regulation of biofilm formation in undomesticated B. subtilis. We discovered that RapP dephosphorylates Spo0F directly, and unlike previously characterized Rap protein Spo0F phosphatases, RapP employs a catalytic glutamate rather than glutamine to accelerate the rate of Spo0F dephosphorylation. Consistent with its role as a Spo0F phosphatase, in addition to regulating biofilm formation, RapP also regulates sporulation and the development of genetic competence. Finally, we show that the phrP gene product does not counteract RapP function, and our results suggest that a peptide not encoded within phrP directly regulates RapP-dependent dephosphorylation of Spo0F.
MATERIALS AND METHODS
Strains and growth conditions.
Constructs were first introduced into domesticated strain PY79 by natural competence and then transferred to the 3610 background by SPP1-mediated generalized phage transduction as described below.
The bacterial strains and plasmids used in this study are listed in Table 1. Escherichia coli strain DH5α was used for the construction and maintenance of plasmids. E. coli strain DH5α was grown in LB medium (10 g/liter tryptone, 5 g/liter yeast extract, 5 g/liter NaCl). As indicated, B. subtilis strains were grown in TY medium (LB with the addition of 10 mM MgSO4 and 100 μM MnSO4), MSgg medium (3), Difco sporulation medium (DSM) (31), and competence medium (CM) (32). Solid medium contained 1.5% Bacto agar. Pellicle biofilms were grown in six-well microtiter plates in 10 ml liquid MSgg medium inoculated with 10 μl of an LB starter culture grown overnight at room temperature and were incubated for 3 days at 25°C. Colony biofilms were toothpick inoculated onto MSgg medium fortified with 1.5% Bacto agar and incubated for 3 days at 25°C. The antibiotics ampicillin (100 μg/ml), spectinomycin (100 μg/ml), chloramphenicol (5 μg/ml), erythromycin (0.5 μg/ml), lincomycin (2.5 μg/ml), kanamycin (5 μg/ml), and erythromycin (1 μg/ml) plus lincomycin (25 μg/ml) (MLS) were included as appropriate.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotypea |
|---|---|
| Strains | |
| 3610 | Undomesticated wild strain B. subtilis NCIB 3610 |
| BD4798 | IS75 PsrfA::luc |
| DS1381 | rapP::Tn10 spec |
| DS7906 | ΔrapP |
| DS7974 | ΔphrP |
| DS8796 | ΔrapPphrP |
| DS8819 | ΔrapPphrP, amyE::Phs-rapP(E49A) spec |
| DS8820 | ΔrapPphrP amyE::Phs-rapP spec |
| DS8823 | ΔrapPphrP amyE::Phs-phrP spec |
| DS8824 | ΔrapP phrP amyE::Phs-rapPphrP spec |
| IS75 | his leu-8 metB5, laboratory strain |
| PP533 | IS75 PspoIIG::luc |
| PY79 | swrAPY79 sfp0 (laboratory strain) |
| VP041 | Peps::luc 3610 |
| VP042 | Peps::luc ΔphrP |
| VP044 | Peps::luc ΔrapPphrP |
| VP049 | PrapP::luc |
| VP051 | PspoIIG::luc 3610 |
| VP055 | PspoIIG::luc ΔphrP |
| VP056 | PspoIIG::luc ΔrapPphrP |
| VP057 | PspoIIG::luc ΔrapPphrP, amy::Phs-rapP |
| VP064 | Peps::luc ΔrapPphrP, amy::Phs-rapP |
| VP065 | Peps::luc ΔrapPphrP, amy::Phs-rapP(E49A) |
| VP073 | PspoIIG::luc ΔrapPphrP, amy::Phs-rapP(E49A) |
| VP089 | PspoIIG::luc ΔrapPphrP, amy::Phs-rapPphrP |
| VP122 | ΔrapP phrP amyE::Phs-rapP(E49A L60A) spec |
| VP126 | Peps::luc ΔrapPphrP, amy::Phs-rapP(E49A, L60A) |
| VP128 | PspoIIG::luc ΔrapPphrP, amy::Phs-rapP(E49A L60A) |
| VP134 | PsrfA::luc ,3610 |
| VP136 | PsrfA::luc ΔrapPphrP |
| VP138 | PsrfA::luc ΔrapPphrP, amy::Phs-rapP |
| VP161 | PspoIIG::luc ΔrapPphrP, amy::Phs-rapP(D203A) |
| VP163 | PsrfA::luc, ΔrapPphrP, amy::Phs-rapP(D203A) |
| Plasmids | |
| pMP52 | amyE::Phs-rapP spec amp |
| pMP69 | ΩΔphrP mls amp |
| pDR111 | amyE::Phs spec amp |
| pMP87 | amyE::Phs-phrP spec amp |
| pMP80 | amyE::Phs-rapP(E49A) spec amp |
| pDG364 | amyE::cat amp |
| pMP88 | amyE::Phs-rapPphrP spec amp |
| pMP91 | ΩΔrapPphrP mls amp |
| pTBP | pTB146 with rapP cloned into SapI/XhoI sites |
| pTBP49A | pTB146 with rapP(E49A) cloned into SapI/XhoI sites |
| pMiniMAD2 | oriBsTs amp mls amp |
| pUC18Cm-Luc | Plasmid containing PstI-KpnI-RBS (spoVG)-luciferase (Photinus pyralis) (strain ED1377) cat |
Laboratory strains are specifically indicated. All other strains are derivatives of 3610. Strains BD4798 and PP533 and plasmid pUC18Cm-Luc were kind gifts from D. Dubnau, Public Health Research Institute, University of Medicine and Dentistry of New Jersey. pDR111 was a gift from David Rudner, Harvard Medical School. RBS, ribosome binding site.
SPP1 phage transduction.
As described below, SPP1 phage transduction was performed as described in reference 33, with some modifications. To 0.2 ml of dense culture grown in TY broth, serial dilutions of SPP1 phage stock were added and statically incubated for 15 min at 37°C. To each mixture, 3 ml of soft TY agar (molten TY supplemented with 0.5% agar) was added, poured atop fresh TY plates, and incubated at 30°C overnight. Top agar from the plate containing nearly confluent plaques was harvested by scraping into a 15-ml conical tube, vortexed, and centrifuged at 5,000 × g for 10 min. The supernatant was treated with 25 μg/ml (final concentration) DNase for 10 min before being passed through a 0.45-μm syringe filter and stored at 4°C. Recipient cells were grown to stationary phase in 3 ml of TY broth at 37°C. A total of 1 ml of cells was mixed with 25 μl of SPP1 donor phage stock. A total of 9 ml of TY broth was added to the mixture and allowed to stand at 37°C for 30 min. The transduction mixture was then centrifuged at 5,000 × g for 5 min, the supernatant was discarded, and the pellet was resuspended in the remaining volume. A total of 100 μl of cell suspension was then plated on LB fortified with 1.5% agar, the appropriate antibiotic, and 10 mM sodium citrate.
In-frame deletions.
To generate the ΔrapP phrP in-frame markerless deletion construct, the region upstream of rapP was amplified with the primer pair 2575/2576 and digested with EcoRI and BamHI. The region downstream of phrP was PCR amplified, respectively, with the primer pair 2581/2582 and digested with XhoI and BamHI. The two fragments were then simultaneously ligated, respectively, into the EcoRI/BamHI sites of pMiniMAD2 (34), which carries a temperature-sensitive origin of replication and an erythromycin resistance cassette to generate pMP91. Plasmid pMP91 was introduced into PY79 by transformation and maintained as a plasmid at the permissive temperature for plasmid replication (22°C) with MLS resistance as a selection. The plasmid was then transduced into 3610 at the permissive temperature. To force the integration of the plasmid into the 3610 endogenous plasmid, the strain was incubated in 3 ml LB broth with MLS selection at a temperature permissive for plasmid replication (22°C) for 14 h, diluted in fresh LB broth, and incubated for 12 h at a temperature restrictive for plasmid replication (42°C) while maintaining MLS selection. To evict the integrated plasmid, the strain was incubated in 3 ml LB broth at a temperature permissive for plasmid replication (22°C) for 14 h, diluted 30-fold in fresh LB broth, and incubated at 22°C for another 8 h. Dilution and outgrowth were repeated two more times. Cells were then serially diluted and plated on LB agar at 37°C. Individual colonies were patched onto LB plates and LB plates containing MLS to identify MLS-sensitive colonies that had evicted the plasmid. Chromosomal DNA from colonies that had excised the plasmid was purified and screened by PCR with primers 2575/2582 to determine which isolate had retained the ΔrapP phrP allele. The rapP phrP deletion was confirmed by Western blotting with anti-RapP rabbit antiserum (see Fig. S1 in the supplemental material).
To generate the ΔphrP in-frame markerless deletion construct, the region upstream of phrP was amplified with the primer pair 2579/2580 and digested with EcoRI and BamHI. The region downstream of phrP was PCR amplified, respectively, with the primer pair 2581/2582 and digested with XhoI and BamHI. The two fragments were then simultaneously ligated, respectively, into the EcoRI/BamHI sites of pMiniMAD2 to generate pMP69. Plasmid pMP69 was introduced into PY79 by transformation and maintained as a plasmid at a temperature permissive for plasmid replication (22°C) with MLS resistance as a selection. The plasmid was then transduced into 3610 at the permissive temperature. The plasmid was subsequently integrated and evicted as described above. Chromosomal DNA from colonies that had excised the plasmid was purified and screened by PCR with primers 2579/2582 to determine which isolate had retained the ΔphrP allele.
Overexpression constructs.
The rapP gene was amplified with 3610 genomic DNA as the template and the primer pair 2367/2368 and then digested with NheI and SphI. The fragment was ligated into the NheI/SphI sites of pDR111 containing a spectinomycin cassette to create pMP52.
The phrP gene was amplified with 3610 genomic DNA as the template and the primer pair 2732/2733 and then digested with NheI and SphI. The fragment was ligated into the NheI/SphI sites of pDR111 containing a spectinomycin cassette to create pMP87.
The rapP phrP cassette was amplified with 3610 genomic DNA as the template and the primer pair 2367/2733 and then digested with NheI and SphI. The fragment was ligated into the NheI/SphI sites of pDR111 containing a spectinomycin cassette to create pMP88.
Site-directed mutagenesis of pMP52 to change the codon encoding Glu49 to a codon encoding alanine was conducted with primer pair 2658/2659 and the QuikChange II kit (Stratagene) to create pMP80. A rapP (E49A, L60A) mutation in pMP81 was obtained with a common reverse primer (REV AMP) in combination with the mutagenic primer and the ChangeIT mutagenesis kit (USB) (see Table S1 in the supplemental material). DNA sequencing confirmed that the rapP plasmids were free of mutations other than those introduced by site-directed mutagenesis. pMP80-derived plasmids were then transformed into IS75 or PY79, which, by double-crossover recombination at the amyE locus, yields strains expressing the entire wild-type or mutant rapP locus under the control of the isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible hyperspank promoter (Phs), followed by transduction into ΔrapP phrP mutant DS8796 by SPP1 phage transduction.
Determination of transformation frequency.
To determine transformation frequency, cultures were grown to competence for 4.5 h in CM at 37°C, transformed with pDG364 containing a chloramphenicol marker (35), and grown for an additional 1.5 h at 37°C. The cultures were then plated on LB plates containing chloramphenicol to determine the number of transformants and on LB plates to determine total viability. Transformation frequency was calculated by dividing the number of transformants by the number of viable colonies per ml.
Construction of luciferase promoter fusion strains.
We PCR amplified 1,000-bp epsA and 732-bp rapP promoter fragments without the native ribosome binding site from the B. subtilis chromosome with primers PepsA_1kb_F and PepsA_Luc_Inf_R for Peps and PrapP_1kb_F and PrapP_1kb_R for PrapP (see Table S1 in the supplemental material). The gel-purified PCR product was then cloned into the PstI and KpnI sites of pUC18Cm-Luc by Infusion cloning (Clontech). The resulting plasmid, pCU18Cm-promoter::luc, which cannot replicate autonomously in B. subtilis, was used to transform B. subtilis IS75, where it was integrated by a single crossover. This event reconstructs the “normal” regulatory region in front of the fusion and a complete copy of the gene of interest downstream of the fusion. Peps::luc, PsrfA::luc, and PspoIIG::luc fusions in B. subtilis IS75 were then transduced into B. subtilis 3610 or ΔrapP phrP mutant DS8796 by SPP1 phage transduction. The chromosomal DNA was isolated from chloramphenicol-resistant colonies; screened by PCR amplification with primers luc2 and PepsA_1kb_F or PrapP_1kb_F for Peps or PrapP, respectively; and then sequenced with primer Seq_from_luc.
Luciferase assay.
For the detection of luciferase activity, strains were first grown in LB medium to an optical density at 600 nm (OD600) of 2. The cells were then centrifuged and resuspended in fresh DSM or MSgg medium, adjusting all of the cultures to an OD600 of 2. These precultures were then diluted 20-fold in fresh DSM, CM, or MSgg medium, and 200 μl was distributed into each of two wells in a 96-well black plate (Corning). Then, 10 μl of luciferin was added to each well to reach a final concentration of 1.5 mg/ml (4.7 mM). The cultures were incubated at 37°C with agitation in a Perkin-Elmer Envision 2104 multilabel reader equipped with an enhanced-sensitivity photomultiplier for luminometry. The temperature of the clear plastic lid was maintained at 38°C to avoid condensation. The relative luminescence units and OD600 were measured at 5-min intervals. Each curve is representative of at least three independent experiments performed in duplicate.
Antibody production and immunoblotting.
Anti-RapP antiserum was recovered from rabbits injected with purified RapP protein (Lampire Biological Laboratories). B. subtilis whole-cell lysates were subjected to SDS-PAGE and blotted onto polyvinylidene difluoride membrane. Immunostaining was performed with anti-RapP rabbit antiserum, followed by horseradish peroxidase-conjugated anti-rabbit IgG antibody. Protein was detected by ECL chemiluminescence (Pharmacia).
Protein expression constructs, protein purification, and phosphatase assay.
rapP was amplified from B. subtilis 3610 DNA with Phusion High-Fidelity DNA polymerase and the primer pair Sumo_RapP_Inf_Fwd and Sumo_RapP_Inf_Rev (see Table S1 in the supplemental material). The PCR product was cloned into the SapI and XhoI sites of pTB146 (36) by the In-Fusion method (Clontech) to give pTBP. A rapP (E49A) mutation in pTBP was obtained with a common reverse primer (REV AMP) in combination with the mutagenic primer and the ChangeIT mutagenesis kit (USB) (Table 1). His-Sumo-RapP was overexpressed in E. coli strain BL21(DE3) by first growing the cells at 37°C in LB medium containing 100 μM ampicillin to an OD600 of 0.6 and then inducing expression with 0.1 mM IPTG for 16 h at 16°C. All subsequent purification steps were carried out at 4°C. The cells were collected by centrifugation and lysed in buffer A (20 mM Tris-HCl [pH 8.0], 250 mM NaCl, 50 mM KCl, 10 mM MgCl2, 10 mM β-mercaptoethanol, 10% glycerol) supplemented with 1 μM pepstatin, 1 μM leupeptin, 20 μg/ml DNase, and 1 mM phenylmethanesulfonyl fluoride. The lysate supernatant was applied to Ni-nitrilotriacetic acid (NTA) agarose (Qiagen) equilibrated in buffer A. The resin was then washed and resuspended in buffer A, Sumo protease was added at 4.2 μg/ml Ni-NTA resin, and the mixture was incubated at 25°C for 2 h. RapP contained no heterologous residues following the removal of the N-terminal His-Sumo fusion. RapP was eluted with buffer A and diluted 3-fold with buffer B (20 mM Tris-HCl [pH 8.0], 10 mM MgCl2, 5 mM dithiothreitol [DTT], 10% glycerol), passed through a 0.22-μm filter, and loaded onto an anion-exchange column (Source 15Q; GE Healthcare) equilibrated in buffer B containing 50 mM KCl. RapP was then eluted in a 50 to 1,000 mM KCl gradient of buffer B. Fractions containing RapP were pooled, concentrated by ultrafiltration through a 30-kDa filter, and further purified by gel filtration with a Superdex 200 (GE Healthcare) 16/70 column equilibrated in buffer C (20 mM Tris-HCl [pH 8.0], 150 mM KCl, 5 mM MgCl2, 5 mM DTT). RapP was concentrated to 913.95 μM and stored at −80°C.
KinA and Spo0F purification, Spo0F labeling, and in vitro phosphatase assays were performed as described previously (12), where 6.5 μM RapP or mutant forms thereof were used instead of RapH.
Peptide synthesis.
Synthetic oligopeptides PhrA (NH2-ARNQT-COOH), PhrC (NH2-ERGMT-COOH), PhrE (NH2-SRNVT-COOH), PhrF (NH2-QRGMI-COOH), PhrH (NH2-TDRNTT-COOH), PhrI (NH2-DRVGA-COOH), PhrP-5mer (NH2-DRAAT-COOH), and PhrP-6mer (NH2-ADRAAT-COOH) at 95% purity were purchased from LifeTein (South Plainfield, NJ). The lyophilized oligopeptides were reconstituted as 10 mM stocks in H2O for use in in vitro phosphatase assays, or the oligopeptides were reconstituted as 10 mM stocks in MSgg medium, DSM, or CM for use in in vivo PspoIIG-luciferase reporter assays. Aliquots of the synthetic oligopeptides were stored at −20°C.
RESULTS
Biofilm phenotypes of rapP and phrP mutants.
B. subtilis strain 3610 forms architecturally complex biofilms on agar medium and thick pellicles at the air-liquid interface of static cultures (Fig. 2A) (3). Transposon screening for hyperrugose colony architecture was conducted, and in one isolate a transposon was found to be inserted in the plasmid-carried rapP gene (Fig. 2A) (4). To validate the transposon-derived phenotype, two in-frame markerless deletion mutations were generated in strain 3610, one that deleted the rapP gene, and one that simultaneously deleted rapP and the downstream phrP gene. The ΔrapP and ΔrapP phrP mutants showed the same hyperrugose phenotype as the rapP transposon insertion mutant (Fig. 2A). In contrast, an in-frame markerless deletion of just the phrP open reading frame resulted in biofilm formation similar to that of wild-type 3610 (Fig. 2A). We conclude that the expression of RapP is necessary to inhibit biofilm formation.
Fig 2.
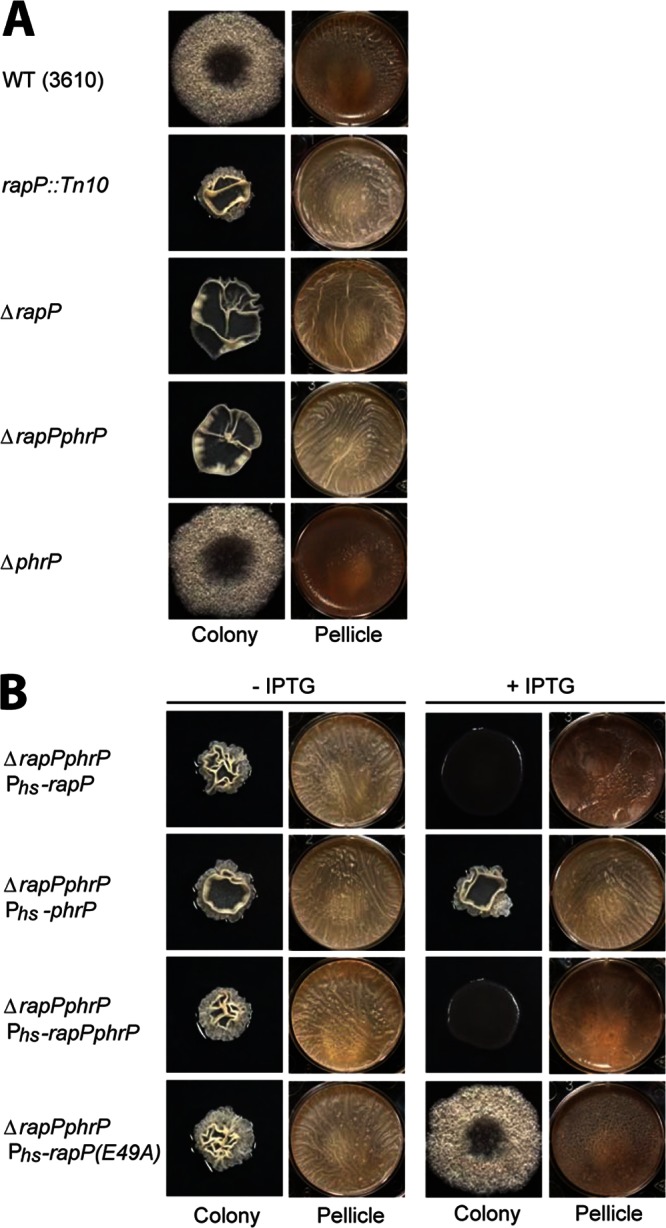
Colony and pellicle phenotypes of rapP and phrP mutant strains. (A and B) Colonies were grown on MSgg agar medium for 3 days at 25°C. Pellicles were grown in six-well microtiter plates for 3 days at 25°C. The strains are the wild type (WT; 3610) and rapP::Tn10 (DS1381), ΔrapP (DS7906), ΔrapP phrP (DS8796), ΔphrP (DS7974), ΔrapP phrP Phs-rapP (DS8820), ΔrapP phrP Phs-phrP (DS8823), ΔrapP phrP Phs-rapP phrP (DS8824), and ΔrapP phrP Phs-rapP(E49A) (DS8819) mutants. (B) Overexpression of rapP and rapP phrP was controlled by Phs with 1 mM IPTG.
To further explore the role of RapP and PhrP in B. subtilis biofilm formation, we overexpressed rapP, phrP, or rapP phrP under IPTG-inducible control in strains with the endogenous rapP phrP locus deleted. As expected, in the absence of IPTG, all of the rapP phrP deletion strains formed hyperwrinkled biofilms (Fig. 2B). Furthermore, overexpression of phrP alone in the rapP ΔphrP background resulted in biofilm formation identical to that of the noninduced control. However, overexpression of rapP or rapP phrP resulted in the inhibition of biofilms, as indicated by the growth of completely smooth, featureless colonies and thin pellicles (Fig. 2). We conclude that the expression of RapP is sufficient to inhibit biofilm formation.
RapP regulates eps expression at the level of Spo0A.
One way in which RapP might repress B. subtilis biofilm formation is by inhibiting the expression of genes required for matrix biosynthesis. To monitor matrix gene expression, we placed firefly luciferase under the control of the Peps promoter that drives the expression of the eps operon. Consistent with the idea that RapP inhibits eps expression, deletion of the rapP phrP cassette caused an increase in Peps expression in comparison to that of the wild-type control strain (Fig. 3A). Furthermore, overexpression of rapP phrP under the control of IPTG-inducible Phs in the rapP phrP deletion strain inhibited Peps expression (Fig. 3A). Thus, RapP inhibits the expression of the eps operon.
Fig 3.

Luciferase reporter assays. (A) Peps and (B) PspoIIG expression was measured with luciferase as a reporter. Overexpression of rapP and rapP phrP was controlled by Phs with 100 μM IPTG. The luciferase strains are derivatives of the corresponding strains used in Fig. 2. Each curve is representative of at least three independent experiments performed in duplicate. RLU, relative luminescence units.
Spo0A∼P derepresses eps operon expression. More specifically, Spo0A∼P drives the expression of SinI, which antagonizes the master repressor of the eps operon, SinR (15, 17–19, 37). Furthermore, Spo0A∼P directly inhibits the expression of abrB, whose gene product represses eps operon transcription (38, 39). One way in which RapP might repress the expression of eps is by inhibiting the phosphorylation of Spo0A. If RapP were to act at the level of Spo0A∼P, one would expect that RapP would repress a gene directly activated by Spo0A∼P. To determine the effect of RapP on Spo0A-dependent genes, we placed firefly luciferase under the control of the PspoIIG promoter, which drives the expression of spoIIG, a late-stage sporulation gene whose expression is directly regulated by Spo0A∼P (40). Consistent with the idea that RapP regulates phosphorelay signal transduction, overexpression of rapP or rapP phrP in the rapP phrP deletion strain completely repressed PspoIIG activity (Fig. 3B). In addition, PspoIIG expression was induced slightly earlier in the rapP phrP deletion strain than in the wild-type control, which is expected if RapP dephosphorylates Spo0F and in turn lowers the cellular level of Spo0A∼P. We conclude that RapP regulates multiple genes under Spo0A control and likely exerts its regulatory effects at or upstream of Spo0A activation.
RapP is a Spo0F phosphatase.
Many (but not all) Rap proteins dephosphorylate Spo0F. To determine if RapP dephosphorylates Spo0F, we phosphorylated Spo0F in vitro with purified KinA and then measured the ability of RapP to dephosphorylate Spo0F∼P (Fig. 4A). Indeed, RapP greatly accelerated Spo0F∼P dephosphorylation compared to the control reaction mixture containing no RapP. In fact, after 5 min, 90% of the Spo0F∼P remained in the control reaction mixture containing no RapP and only 21% of the Spo0F∼P remained in the reaction mixture containing RapP. Therefore, we conclude that RapP is a Spo0F phosphatase.
Fig 4.
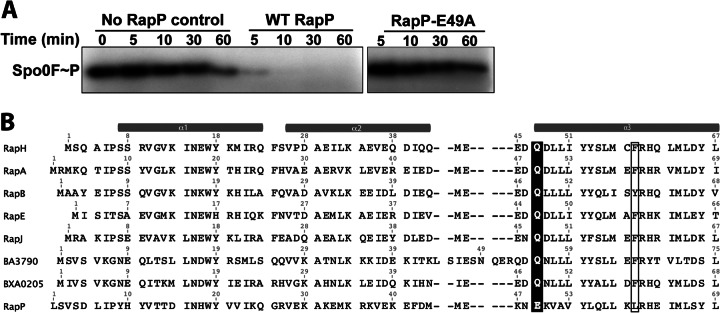
Mechanism of RapP phosphatase activity. (A) RapP dephosphorylates Spo0F∼P in vitro, and RapP-E49A is catalytically inactive. The gel is representative of phosphatase assays repeated three times. WT, wild type. (B) The amino acid sequences of Bacillus Rap proteins previously demonstrated to dephosphorylate Spo0F in vitro were aligned with RapP and the Blosum62 matrix in Geneious Pro (Biomatters Ltd.). For simplicity, only the amino acids corresponding to RapP residues 1 to 69 are shown. Catalytic residues (Gln or Glu) are shaded. Residues corresponding to RapH Phe58 and RapP Lue60 are boxed.
Mechanism of RapP phosphatase activity.
A catalytic glutamine is conserved in every Rap protein previously demonstrated to dephosphorylate Spo0F (Fig. 4B), and we previously showed that this residue is inserted into the Spo0F active site to catalyze the hydrolysis reaction (12). RapP, however, contains a glutamate rather than a glutamine at corresponding residue 49 (Fig. 4B). To determine whether RapP Glu49 is functionally important, we purified RapP-E49A and measured its ability to dephosphorylate Spo0F∼P. While Spo0F∼P was rapidly dephosphorylated in the presence of wild-type RapP, the reaction mixture containing RapP-E49A appeared identical to the control reaction mixture containing no RapP (Fig. 4A). Therefore, Glu49 is required for RapP phosphatase activity, and we infer that that Glu49 is inserted into the Spo0F active site. Consistent with a catalytic glutamate, we note that in previous studies of RapH, the replacement of Gln47 with glutamate retained wild-type phosphatase activity (12).
Furthermore, in these previous studies, we also showed that the RapH-Q47N mutant is catalytically dead; i.e., it cannot dephosphorylate Spo0F (12). However, we also found that RapH-Q47N could bind to Spo0F and disrupt phosphotransfer both from KinA to Spo0F and from Spo0F to Spo0B in vitro and in vivo. Thus, the RapH-Q47N mutant is catalytically dead but still sterically interferes with the phosphorelay. Consistent with the fact that RapP Glu49 is the catalytic residue, overexpression of RapP-E49A in the rapP phrP deletion strain resulted in biofilm formation essentially identical to that displayed by wild-type 3610, which expresses RapP at modest levels under the control of its endogenous promoter (Fig. 2A and B; see Fig. S1 in the supplemental material). The biofilm phenotype of the strain overexpressing RapP-E49A is expected if the mutant is catalytically dead but still capable of binding to Spo0F and sterically disrupting the phosphorelay (12).
In previous studies, we also showed that an additional mutation at the RapH-Spo0F interface, e.g., RapH-F58A, caused the RapH-Q47N mutant to lose its ability to sterically inhibit phosphotransfer. Sequence analysis (Fig. 4B) and comparative modeling (data not shown) suggested that RapP L60 is structurally equivalent to RapH F58, but we were unable to detect a drastic difference in biofilm architecture when we compared strains overexpressing RapP-E49A (Fig. 2B) or RapP-E49A,L60A (data not shown). We did, however, detect significant differences in RapP-E49A and RapP-E49A,L60A activity in comparison to overexpressed wild-type RapP with the luciferase reporter assays (Fig. 3A and B), which can provide a more sensitive readout of Rap activity than gross biofilm architecture. More specifically, the RapP-E49A,L60A double mutant displayed a greater loss of function than the RapP-E49A single mutant in the Peps (Fig. 3A) and PspoIIG (Fig. 3B) luciferase reporter assays. Thus, we conclude that the overexpression of RapP-E49A results in biofilm formation similar to that observed with wild-type 3610 expressing RapP from its endogenous promoter because, in all likelihood and similar to RapH-Q47N, the catalytically dead RapP mutant can sterically block phosphotransfer from KinA to Spo0F and from Spo0F to Spo0B.
RapP regulates the expression of srfA.
As discussed above, surfactin plays an important role in B. subtilis biofilm formation, and we investigated the possibility that RapP regulates the expression of the srfA operon. Deletion of rapP phrP resulted in increased PsrfA expression in vivo; i.e., PsrfA was expressed at an earlier time point and the maximum level of PsrfA activity was higher at all time points (Fig. 5A). Consistent with these results, overexpression of RapP inhibited PsrfA activity throughout the time course of the experiment (Fig. 5A).
Fig 5.

RapP inhibits genetic competence. (A) RapP inhibits PsrfA-luc expression in vivo. The overexpression of rapP and rapP phrP was controlled by Phs with 100 μM IPTG. The strains used are derivatives of the corresponding strains used in Fig. 2. Each curve is representative of at least three independent experiments performed in duplicate. RLU, relative luminescence units. (B) Strain 3610 is poorly naturally competent in comparison to strain PY79. rapP overexpression lowers the transformation frequency in strain PY79. The overexpression of rapP was controlled by Phs with IPTG at the indicated concentrations. WT, wild type.
Embedded within the srfA operon is comS, which encodes a small protein whose expression results in the accumulation of the master regulator of competence gene expression, ComK (41). If RapP inhibits the expression of PsrfA, then it was logical to expect RapP to also inhibit genetic competence. Indeed, while the deletion of rapP phrP had little or no effect on transformation frequency by natural competence in strain 3610, overexpression of RapP significantly reduced the transformation frequency in both undomesticated strain 3610 and laboratory strain PY79 (Fig. 5B; see Fig. S2 in the supplemental material). It is worth noting that in comparison to the lab strain, the undomesticated strain is poorly naturally competent (42). We conclude that RapP inhibits srfA operon expression, which would, in turn, affect biofilm formation and the development of genetic competence.
What is the mechanism of RapP-mediated srfA regulation? One Rap protein, RapH, both dephosphorylates Spo0F and binds to the ComA DNA binding domain, directly inhibiting its interaction with DNA and allosterically inhibiting ComA dimerization (11, 21). Therefore, we explored the possibility that RapP might function to both dephosphorylate Spo0F and bind ComA, inhibiting ComA-driven srfA expression. Preliminary studies comparing the amino acid sequence of RapP with that of the ComA-binding protein RapF suggested that RapP would not form a stable complex with the ComA DNA binding domain (ComAC). More specifically, we previously determined the X-ray crystal structure of RapF in complex with ComAC and identified six RapF residues critical to the RapF-ComAC interaction (21, 23); RapP conserves only one of these six residues. Consistent with the prediction that RapP would not form a stable complex with ComAC, RapP does not interact with ComAC, as determined by native PAGE analysis, RapP does not dephosphorylate ComA∼P in vitro, and RapP does not interact with full-length ComA, the ComA DNA binding domain, or the ComA REC domain, as determined by bacterial two-hybrid analysis (data not shown). On the basis of the above results, we conclude that RapP is not a ComA antiactivator protein.
It is possible that RapP-mediated dephosphorylation of Spo0F and the resulting decrease in Spo0A∼P are sufficient to decrease PsrfA expression and competence development. More specifically, Spo0A∼P inhibits the expression of AbrB, which represses the expression of σH (39, 43) (Fig. 1). σH drives the expression of numerous genes encoding Phr peptides, including, among others, PhrC, PhrF, and PhrK (44, 45). PhrC, PhrF, and PhrK inhibit the ComA antiactivators RapC, RapF, and RapK, respectively (27, 29), resulting in srfA operon upregulation (29). Thus, RapP inhibition of the phosphorelay could be responsible for the RapP-dependent downregulation of PsrfA expression, but the possibility remains that RapP could regulate a target other than Spo0F or ComA, resulting in PsrfA downregulation.
The phrP gene product does not affect RapP regulation of Peps, PspoIIG, or PsrfA expression.
Genes that encode Rap proteins often have small open reading frames immediately downstream that encode Rap antagonist peptides called Phr peptides. Downstream of rapP is the open reading frame phrP (4). As described above, an in-frame markerless deletion of the phrP open reading frame resulted in biofilm formation similar to that of wild-type 3610 (Fig. 2A). Furthermore, the ΔrapP and ΔrapP phrP mutant strains, as well as the ΔrapP phrP mutant strain overexpressing phrP, displayed identical hyperrugose phenotypes (Fig. 2A and B). These findings led us to question whether phrP has an effect on the RapP regulation of Peps, PspoIIG, and PsrfA expression. On the basis of the extensive in vivo results (Fig. 3A and B and 5A), we conclude that phrP has no effect on the expression of Peps, PspoIIG, or PsrfA, respectively.
The above results showed that the phrP gene product does not significantly affect RapP function under any of our in vivo assay conditions (Fig. 2, 3A and B, and 5A). In line with these in vivo studies, the synthetic penta- and hexapeptides DRAAT and ADRAAT derived from the C-terminal sequence of the theoretical pro-PhrP polypeptide did not inhibit the RapP dephosphorylation of Spo0F in vitro (see Fig. S3 in the supplemental material).
Inhibition of RapP activity.
While the above results demonstrate that RapP is not inhibited by a phrP gene product, we speculated that RapP is perhaps inhibited by a Phr peptide encoded by a gene other than phrP. There is precedent for cross regulation, as RapB is regulated by PhrC, which is encoded in an operon with RapC (27). To determine if RapP is inhibited by a peptide encoded by a gene other than phrP, we evaluated the abilities of a number of known Phr peptides to inhibit RapP-mediated inhibition of PspoIIG expression (Fig. 6A). Strikingly, when RapP was overexpressed, added synthetic PhrH pentapeptide (DRNTT) or hexapeptide (TDRNTT) upregulated the expression of PspoIIG-luc. We previously described why we believe the hexapeptide may be the biologically important mature PhrH peptide (28). In contrast, other Phr peptides, including PhrA, PhrC, PhrE, PhrF, and PhrI, as well as the pro-PhrP-derived peptides DRAAT and ADRAAT, had essentially no effect on PspoIIG-luc expression. Furthermore, added synthetic PhrH upregulated the expression of PsrfA-luc (Fig. 6B). However, neither pentapeptide (DRNTT) nor hexapeptide (TDRNTT) PhrH inhibited the RapP-mediated dephosphorylation of Spo0F in vitro (data not shown). Finally, we note that PhrH inhibition of RapP was observed in DSM (PspoIIG) (Fig. 6A) and CM (PsrfA) (Fig. 6B) but not in MSgg medium (Peps) (data not shown).
Fig 6.

PhrH inhibits RapP activity in vivo. (A) Synthetic Phr peptides were evaluated for the ability to counteract RapP-mediated inhibition of PspoIIG-luc expression. (B) PhrH counteracts the RapP-mediated inhibition of PsrfA-luc expression. PhrH does not counteract the RapP-D203A-mediated inhibition of PspoIIG-luc (C) and PsrfA-luc (D) expression. The strain background is 3610, ΔrapP phrP, and Phs-rapP in panels C and D. The overexpression of rapP was controlled by Phs with 100 μM IPTG (A and C) or 50 μM IPTG (B and D). The final concentration of added Phr peptide was 1 mM. Each curve is representative of at least three independent experiments performed in duplicate. RLU, relative luminescence units.
Interestingly, we found that mutant RapP containing an alanine substituted for aspartate in position 203 (RapP-D203A) inhibited PspoIIG-luc and PsrfA-luc expression in vivo but that RapP-D203A inhibition of PspoIIG-luc and PsrfA-luc expression was not counteracted by PhrH (Fig. 6C and D). Substitution mutations at positions corresponding to RapP Asp203 were previously shown to render Rap proteins resistant to the inhibitory effects of their cognate Phr peptides (8, 20, 27, 46). Moreover, we recently showed that B. subtilis Rap proteins have aspartate or, less commonly, glutamate at the position corresponding to RapP Asp203, and these conserved acidic residues form salt bridges with conserved basic residues in their cognate Phr peptides (47). On the basis of the above results and as discussed below, we propose that RapP Asp203 makes critical contacts with a regulatory peptide.
DISCUSSION
How Rap proteins and their regulatory Phr peptides control B. subtilis biofilm formation was unknown. The studies presented here focused on the rapP phrP operon, which was previously shown to be required for the formation of B. subtilis biofilms with wild-type architecture (4). While the signals that regulate the expression of RapP are unknown, our results show that RapP modulates the flow of phosphoryl groups along the phosphorelay, regulating biofilm formation, sporulation, and genetic competence.
We previously determined the X-ray crystal structure of the Spo0F phosphatase RapH complexed with Spo0F and found that Rap protein residues that lie in the RapH-Spo0F interface are highly conserved among the Rap proteins known to dephosphorylate Spo0F (12). One of these conserved RapH residues, Gln47, is inserted into the Spo0F active site to facilitate nucleophilic attack on the aspartyl phosphate phosphorus atom (12). Our in vivo results suggested that RapP might be a Spo0F phosphatase; however, we did not find significant similarity of the RapP amino acid sequence to the sequences conserved in Rap proteins known to dephosphorylate Spo0F. More specifically, RapP contains glutamate at position 49, which corresponds to RapH catalytic residue Gln47 (Fig. 4B), and very few of the other Rap-Spo0F interface residues are conserved in RapP (see Table S2 in the supplemental material). This led us to question whether RapP was indeed capable of dephosphorylating Spo0F. Nonetheless, our previous mutagenesis studies substituting the Rap protein catalytic glutamine for glutamate showed that glutamate could support Spo0F dephosphorylation (12). This led us to hypothesize that RapP could dephosphorylate Spo0F, which indeed turned out to be true, as RapP dephosphorylated Spo0F in vitro. Moreover, our computational modeling of the RapP-Spo0F structure (not shown) suggested that significantly divergent amino acids in RapP might undergo relatively subtle compensatory rearrangements to bind Spo0F. It also suggested how the Rap-Spo0F interface could adjust to accommodate structurally dissimilar residues in positions that are relatively well conserved. However, since alanine substitution of the predicted RapP catalytic residue, Glu49, resulted in complete loss of RapP phosphatase activity in vitro, we propose that the structure of the RapP-Spo0F active site is essentially identical to that of RapH-Spo0F. Previous biochemical and genetic studies of the Spo0A phosphatase Spo0E also suggest that an aspartate residue in Spo0E likely plays a similar role coordinating water for nucleophilic attack (48). In contrast, mutation of the catalytic residue Gln404 to glutamate in the histidine kinase NarX was not sufficient to support its phosphatase activity (49). We conclude that an acidic side chain O− group in response regulator aspartate phosphatases can, at least in some cases, coordinate the attacking water molecule, considering that (i) RapH-Q47E is catalytically active (12), (ii) Spo0E likely employs a catalytic aspartate (48), and (iii) RapP naturally contains glutamate in a position we previously identified as the catalytic position (12)
As described below, Spo0F dephosphorylation is likely responsible for the RapP-mediated effects on the expression of (i) eps, which is important for biofilm formation; (ii) srfA, an operon that contains genes important for biofilm formation and genetic competence development; and (iii) spoIIG, a late-stage sporulation gene. The level of RapP in the cell might be an important factor in determining the degree to which RapP affects Spo0F dephosphorylation and, in turn, biofilm formation, genetic competence development, and sporulation. Consistent with this idea, we found that the promoter driving the expression of the rapP phrP operon is differentially regulated in MSgg medium, DSM, and CM (see Fig. S5 in the supplemental material).
RapP-mediated dephosphorylation of Spo0F would drain phosphoryl groups from Spo0A, resulting in the decreased expression of eps, srfA, and spoIIG. Linkages between Spo0A phosphorylation and the expression of eps, srfA, and spoIIG are well established. More specifically, Spo0A∼P upregulates the expression of the antirepressor SinI (18). SinI antagonizes the repressor SinR; therefore, RapP-dependent dephosphorylation of Spo0F would decrease Spo0A∼P levels, resulting in the repression of the epsA-epsO (epsA-O) and tapA(yqxM)-sipW-tasA biofilm operons (50, 51) (Fig. 1). Similarly, a decrease in the cellular concentration of phosphorylated Spo0A results in the increased expression of abrB (52), and the transcriptional repressor AbrB, in turn, downregulates the expression of the srfA (43, 53), epsA-O, and tapA(yqxM)-sipW-tasA operons (14) (Fig. 1). Finally, RapP-dependent dephosphorylation of Spo0F and the subsequent decrease in Spo0A∼P would result in the decreased expression of spoIIG because its expression is directly driven by Spo0A∼P (40) (Fig. 1).
It is also worthwhile to note that the pattern of PsrfA expression is markedly different in undomesticated strain 3610 (Fig. 5A) and laboratory strain IS75 (21) (see Fig. S4 in the supplemental material). In the undomesticated strain, the maximum srfA transcription level is reached at 2 h and srfA is expressed only transiently; i.e., the srfA expression level increases over a 2-h period and then rapidly returns to the baseline over the next 2 h (Fig. 5A). In the laboratory strain, while the maximum transcription level is reached at 2 h, the level is higher than it is in 3610, and the level remains close to the maximum for ∼8 h before declining rapidly (21) (see Fig. S4 in the supplemental material). The pattern of srfA expression in the rapP phrP 3610 deletion strain (Fig. 5A) suggests that RapP is important for the transient expression of srfA in the undomesticated strain.
Finally, while the deletion or overexpression of rapP had profound effects on biofilm architecture, the presence or absence of phrP had no effect (Fig. 2). Synthetic penta- or hexapeptides derived from the theoretical pro-PhrP C terminus similarly had no effect on the RapP-dependent dephosphorylation of Spo0F in vitro (see Fig. S3 in the supplemental material), and phrP had no effect on the RapP regulation of eps, spoIIG, and srfA expression (Fig. 3 and 5A). Interestingly, however, PhrH counteracted the RapP-dependent inhibition of PspoIIG and PsrfA expression in DSM and CM, respectively (Fig. 6A and B) but PhrH did not counteract the RapP-dependent inhibition of Peps in MSgg medium (data not shown). Synthetic PhrH peptide did not inhibit the RapP-mediated dephosphorylation of Spo0F in vitro, but a RapP mutation that disrupts Phr peptide binding in other Rap proteins rendered RapP resistant to the effects of PhrH in vivo (Fig. 6C and D). Thus, we hypothesize that RapP may be inhibited directly by a regulatory peptide. The identity of this peptide is unknown, but we speculate that PhrH could be subject to posttranslational modification in vivo and directly inhibit RapP activity or, more likely, PhrH could upregulate the expression of a peptide that, in turn, directly binds to RapP, inhibiting its Spo0F phosphatase activity. While ongoing studies will reveal how PhrH functions mechanistically to counteract the RapP-dependent inhibition of target gene expression in DSM and CM but not in MSgg medium, we speculate that MSgg medium might not support PhrH modification or the PhrH-induced expression of the RapP inhibitory peptide.
Concluding remarks.
In addition to RapP, two other plasmid-encoded Rap proteins, Bacillus anthracis BXA0205 and B. subtilis Rap60, were previously shown to regulate the phosphorelay (54, 55). Whether these Rap proteins regulate biofilm formation was not directly examined; however, biochemical and genetic results showed that the Rap protein BXA0205 dephosphorylates Spo0F (54), and on the basis of genetic results, it was proposed that Rap60 dephosphorylates a component of the sporulation phosphorelay (55). It is therefore likely that RapP, BXA0205, and Rap60 function similarly, but their effects on biofilm formation may not be identical because of possible differences in the timing of Rap or cognate Phr peptide expression, the catalytic efficiency of the Rap proteins, or the inhibitory constants of the Phr peptides.
Finally, B. subtilis is commonly used commercially to produce secreted enzymes such as the protease AprE, and Rap-Phr systems have been the targets of studies designed to improve AprE expression, which is under AbrB control (56). In light of the fact that RapP functions to inhibit the phosphorelay, we note that the presence of rapP or the 80-kb rapP-containing plasmid could be counterproductive to the overexpression of AprE or other proteins whose expression is similarly regulated by the phosphorelay. Conversely, protocols designed to overproduce proteins whose expression is inhibited by Spo0A∼P may, in fact, benefit from the presence of rapP.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge David Dubnau and Atul Khataokar for critical review of the manuscript, Marta Perego for the ComP overexpression construct pMALc2-ComP, Eugenie Dubnau and Miguel Dias for technical advice, and Lauren McEllen and Timothy Berghold for technical assistance.
This work was supported by Indiana University Genetics, Molecular and Cellular Sciences Training Grant T32-GM007757 to M.A.K. and National Institutes of Health (NIH) grants R01 GM093030 to D.B.K. and R01 AI081736 to M.B.N.
Footnotes
Published ahead of print 22 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02030-12.
REFERENCES
- 1. López D, Kolter R. 2010. Extracellular signals that define distinct and coexisting cell fates in Bacillus subtilis. FEMS Microbiol. Rev. 34:134–149 [DOI] [PubMed] [Google Scholar]
- 2. Vlamakis H, Aguilar C, Losick R, Kolter R. 2008. Control of cell fate by the formation of an architecturally complex bacterial community. Genes Dev. 22:945–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Branda SS, Gonzalez-Pastor JE, Ben-Yehuda S, Losick R, Kolter R. 2001. Fruiting body formation by Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 98:11621–11626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McLoon AL, Guttenplan SB, Kearns DB, Kolter R, Losick R. 2011. Tracing the domestication of a biofilm-forming bacterium. J. Bacteriol. 193:2027–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kearns DB, Losick R. 2003. Swarming motility in undomesticated Bacillus subtilis. Mol. Microbiol. 49:581–590 [DOI] [PubMed] [Google Scholar]
- 6. Nakano MM, Corbell N, Besson J, Zuber P. 1992. Isolation and characterization of sfp: a gene that functions in the production of the lipopeptide biosurfactant, surfactin, in Bacillus subtilis. Mol. Gen. Genet. 232:313–321 [DOI] [PubMed] [Google Scholar]
- 7. López D, Fischbach MA, Chu F, Losick R, Kolter R. 2009. Structurally diverse natural products that cause potassium leakage trigger multicellularity in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 106:280–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Perego M, Hanstein C, Welsh KM, Djavakhishvili T, Glaser P, Hoch JA. 1994. Multiple protein-aspartate phosphatases provide a mechanism for the integration of diverse signals in the control of development in B. subtilis. Cell 79:1047–1055 [DOI] [PubMed] [Google Scholar]
- 9. Burbulys D, Trach KA, Hoch JA. 1991. Initiation of sporulation in B. subtilis is controlled by a multicomponent phosphorelay. Cell 64:545–552 [DOI] [PubMed] [Google Scholar]
- 10. Jiang M, Grau R, Perego M. 2000. Differential processing of propeptide inhibitors of Rap phosphatases in Bacillus subtilis. J. Bacteriol. 182:303–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smits WK, Bongiorni C, Veening JW, Hamoen LW, Kuipers OP, Perego M. 2007. Temporal separation of distinct differentiation pathways by a dual specificity Rap-Phr system in Bacillus subtilis. Mol. Microbiol. 65:103–120 [DOI] [PubMed] [Google Scholar]
- 12. Parashar V, Mirouze N, Dubnau DA, Neiditch MB. 2011. Structural basis of response regulator dephosphorylation by Rap phosphatases. PLoS Biol. 9:e1000589 doi:10.1371/journal.pbio.1000589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Branda SS, Gonzalez-Pastor JE, Dervyn E, Ehrlich SD, Losick R, Kolter R. 2004. Genes involved in formation of structured multicellular communities by Bacillus subtilis. J. Bacteriol. 186:3970–3979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hamon MA, Lazazzera BA. 2001. The sporulation transcription factor Spo0A is required for biofilm development in Bacillus subtilis. Mol. Microbiol. 42:1199–1209 [DOI] [PubMed] [Google Scholar]
- 15. Kearns DB, Chu F, Branda SS, Kolter R, Losick R. 2005. A master regulator for biofilm formation by Bacillus subtilis. Mol. Microbiol. 55:739–749 [DOI] [PubMed] [Google Scholar]
- 16. Branda SS, Chu F, Kearns DB, Losick R, Kolter R. 2006. A major protein component of the Bacillus subtilis biofilm matrix. Mol. Microbiol. 59:1229–1238 [DOI] [PubMed] [Google Scholar]
- 17. Chu F, Kearns DB, Branda SS, Kolter R, Losick R. 2006. Targets of the master regulator of biofilm formation in Bacillus subtilis. Mol. Microbiol. 59:1216–1228 [DOI] [PubMed] [Google Scholar]
- 18. Bai U, Mandic-Mulec I, Smith I. 1993. SinI modulates the activity of SinR, a developmental switch protein of Bacillus subtilis, by protein-protein interaction. Genes Dev. 7:139–148 [DOI] [PubMed] [Google Scholar]
- 19. Shafikhani SH, Mandic-Mulec I, Strauch MA, Smith I, Leighton T. 2002. Postexponential regulation of sin operon expression in Bacillus subtilis. J. Bacteriol. 184:564–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Core L, Perego M. 2003. TPR-mediated interaction of RapC with ComA inhibits response regulator-DNA binding for competence development in Bacillus subtilis. Mol. Microbiol. 49:1509–1522 [DOI] [PubMed] [Google Scholar]
- 21. Baker MD, Neiditch MB. 2011. Structural basis of response regulator inhibition by a bacterial anti-activator protein. PLoS Biol. 9:e1001226 doi:10.1371/journal.pbio.1001226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ogura M, Shimane K, Asai K, Ogasawara N, Tanaka T. 2003. Binding of response regulator DegU to the aprE promoter is inhibited by RapG, which is counteracted by extracellular PhrG in Bacillus subtilis. Mol. Microbiol. 49:1685–1697 [DOI] [PubMed] [Google Scholar]
- 23. Bongiorni C, Ishikawa S, Stephenson S, Ogasawara N, Perego M. 2005. Synergistic regulation of competence development in Bacillus subtilis by two Rap-Phr systems. J. Bacteriol. 187:4353–4361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nakano MM, Marahiel MA, Zuber P. 1988. Identification of a genetic locus required for biosynthesis of the lipopeptide antibiotic surfactin in Bacillus subtilis. J. Bacteriol. 170:5662–5668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Solomon JM, Lazazzera BA, Grossman AD. 1996. Purification and characterization of an extracellular peptide factor that affects two different developmental pathways in Bacillus subtilis. Genes Dev. 10:2014–2024 [DOI] [PubMed] [Google Scholar]
- 26. Lanigan-Gerdes S, Dooley AN, Faull KF, Lazazzera BA. 2007. Identification of subtilisin, Epr and Vpr as enzymes that produce CSF, an extracellular signalling peptide of Bacillus subtilis. Mol. Microbiol. 65:1321–1333 [DOI] [PubMed] [Google Scholar]
- 27. Perego M. 1997. A peptide export-import control circuit modulating bacterial development regulates protein phosphatases of the phosphorelay. Proc. Natl. Acad. Sci. U. S. A. 94:8612–8617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mirouze N, Parashar V, Baker MD, Dubnau DA, Neiditch MB. 2011. An atypical Phr peptide regulates the developmental switch protein RapH. J. Bacteriol. 193:6197–6206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lazazzera BA, Solomon JM, Grossman AD. 1997. An exported peptide functions intracellularly to contribute to cell density signaling in B. subtilis. Cell 89:917–925 [DOI] [PubMed] [Google Scholar]
- 30. Perego M, Hoch JA. 1996. Cell-cell communication regulates the effects of protein aspartate phosphatases on the phosphorelay controlling development in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 93:1549–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schaeffer P, Millet J, Aubert JP. 1965. Catabolic repression of bacterial sporulation. Proc. Natl. Acad. Sci. U. S. A. 54:704–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Albano M, Hahn J, Dubnau D. 1987. Expression of competence genes in Bacillus subtilis. J. Bacteriol. 169:3110–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yasbin RE, Young FE. 1974. Transduction in Bacillus subtilis by bacteriophage SPP1. J. Virol. 14:1343–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Patrick JE, Kearns DB. 2008. MinJ (YvjD) is a topological determinant of cell division in Bacillus subtilis. Mol. Microbiol. 70:1166–1179 [DOI] [PubMed] [Google Scholar]
- 35. Guérout-Fleury AM, Frandsen N, Stragier P. 1996. Plasmids for ectopic integration in Bacillus subtilis. Gene 180:57–61 [DOI] [PubMed] [Google Scholar]
- 36. Bendezú FO, Hale CA, Bernhardt TG, de Boer PA. 2009. RodZ (YfgA) is required for proper assembly of the MreB actin cytoskeleton and cell shape in E. coli. EMBO J. 28:193–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chai Y, Norman T, Kolter R, Losick R. 2011. Evidence that metabolism and chromosome copy number control mutually exclusive cell fates in Bacillus subtilis. EMBO J. 30:1402–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hamon MA, Stanley NR, Britton RA, Grossman AD, Lazazzera BA. 2004. Identification of AbrB-regulated genes involved in biofilm formation by Bacillus subtilis. Mol. Microbiol. 52:847–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Strauch M, Webb V, Spiegelman G, Hoch JA. 1990. The SpoOA protein of Bacillus subtilis is a repressor of the abrB gene. Proc. Natl. Acad. Sci. U. S. A. 87:1801–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Satola S, Kirchman PA, Moran CP., Jr 1991. Spo0A binds to a promoter used by sigma A RNA polymerase during sporulation in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 88:4533–4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Turgay K, Hamoen LW, Venema G, Dubnau D. 1997. Biochemical characterization of a molecular switch involving the heat shock protein ClpC, which controls the activity of ComK, the competence transcription factor of Bacillus subtilis. Genes Dev. 11:119–128 [DOI] [PubMed] [Google Scholar]
- 42. Nijland R, Burgess JG, Errington J, Veening JW. 2010. Transformation of environmental Bacillus subtilis isolates by transiently inducing genetic competence. PLoS One 5:e9724 doi:10.1371/journal.pone.0009724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weir J, Predich M, Dubnau E, Nair G, Smith I. 1991. Regulation of spo0H, a gene coding for the Bacillus subtilis sigma H factor. J. Bacteriol. 173:521–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carter HL, III, Tatti KM, Moran CP., Jr 1990. Cloning of a promoter used by sigma H RNA polymerase in Bacillus subtilis. Gene 96:101–105 [DOI] [PubMed] [Google Scholar]
- 45. McQuade RS, Comella N, Grossman AD. 2001. Control of a family of phosphatase regulatory genes (phr) by the alternate sigma factor sigma-H of Bacillus subtilis. J. Bacteriol. 183:4905–4909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Diaz AR, Core LJ, Jiang M, Morelli M, Chiang CH, Szurmant H, Perego M. 2012. Bacillus subtilis RapA phosphatase domain interaction with its substrate, phosphorylated Spo0F, and its inhibitor, the PhrA peptide. J. Bacteriol. 194:1378–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Parashar V, Jefferey PD, Neiditch MB. 2013. Conformational change-induced repeat domain expansion regulates Rap phosphatase quorum-sensing signal receptors. PLoS Biol. 11:e1001512 doi:10.1371/journal.pbio.1001512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Diaz AR, Stephenson S, Green JM, Levdikov VM, Wilkinson AJ, Perego M. 2008. Functional role for a conserved aspartate in the Spo0E signature motif involved in the dephosphorylation of the Bacillus subtilis sporulation regulator Spo0A. J. Biol. Chem. 283:2962–2972 [DOI] [PubMed] [Google Scholar]
- 49. Huynh TN, Noriega CE, Stewart V. 2010. Conserved mechanism for sensor phosphatase control of two-component signaling revealed in the nitrate sensor NarX. Proc. Natl. Acad. Sci. U. S. A. 107:21140–21145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fujita M, Gonzalez-Pastor JE, Losick R. 2005. High- and low-threshold genes in the Spo0A regulon of Bacillus subtilis. J. Bacteriol. 187:1357–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Molle V, Fujita M, Jensen ST, Eichenberger P, Gonzalez-Pastor JE, Liu JS, Losick R. 2003. The Spo0A regulon of Bacillus subtilis. Mol. Microbiol. 50:1683–1701 [DOI] [PubMed] [Google Scholar]
- 52. Perego M, Spiegelman GB, Hoch JA. 1988. Structure of the gene for the transition state regulator, abrB: regulator synthesis is controlled by the spo0A sporulation gene in Bacillus subtilis. Mol. Microbiol. 2:689–699 [DOI] [PubMed] [Google Scholar]
- 53. Lazazzera BA, Kurtser IG, McQuade RS, Grossman AD. 1999. An autoregulatory circuit affecting peptide signaling in Bacillus subtilis. J. Bacteriol. 181:5193–5200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bongiorni C, Stoessel R, Shoemaker D, Perego M. 2006. Rap phosphatase of virulence plasmid pXO1 inhibits Bacillus anthracis sporulation. J. Bacteriol. 188:487–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Koetje EJ, Hajdo-Milasinovic A, Kiewiet R, Bron S, Tjalsma H. 2003. A plasmid-borne Rap-Phr system of Bacillus subtilis can mediate cell-density controlled production of extracellular proteases. Microbiology 149:19–28 [DOI] [PubMed] [Google Scholar]
- 56. Tjalsma H, Koetje EJ, Kiewiet R, Kuipers OP, Kolkman M, van der Laan J, Daskin R, Ferrari E, Bron S. 2004. Engineering of quorum-sensing systems for improved production of alkaline protease by Bacillus subtilis. J. Appl. Microbiol. 96:569–578 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.