

Abstract
The permeability transition (PT) denotes an increase of the mitochondrial inner membrane permeability to solutes with molecular masses up to about 1500 Da. It is presumed to be mediated by opening of a channel, the permeability transition pore (PTP), whose molecular nature remains a mystery. Here I briefly review the history of the PTP, discuss existing models, and present our new results indicating that reconstituted dimers of the FOF1 ATP synthase form a channel with properties identical to those of the mitochondrial megachannel (MMC), the electrophysiological equivalent of the PTP. Open questions remain, but there is now promise that the PTP can be studied by genetic methods to solve the large number of outstanding problems.
Keywords: mitochondria, permeability transition, calcium, FOF1 ATP synthase
The mitochondrial permeability transition
The permeability transition (PT) defines an increase of mitochondrial inner membrane permeability to ions and solutes with molecular masses up to about 1500 Da leading to matrix swelling. The occurrence of swelling in isolated mitochondria, its strict dependence on matrix Ca2+, stimulation by Pi and fatty acids, and inhibition by Mg2+ and adenine nucleotides has been recognized very early (Raaflaub, 1953a,b; Brenner-Holzach and Raaflaub, 1954; Hunter and Ford, 1955; Tapley, 1956; Lehninger, 1959; Lehninger and Remmert, 1959; Wojtczak and Lehninger, 1961; Zborowski and Wojtczak, 1963; Azzi and Azzone, 1965; Azzone and Azzi, 1965; Chappell and Crofts, 1965; Crofts and Chappell, 1965); and its detrimental effects on energy conservation have been described before the emergence of chemiosmotic concepts (Mitchell, 1961, 2011). As a result, the PT has initially been considered an in vitro artifact of questionable pathophysiological relevance, although a few Authors did recognize its potential importance in pathophysiology (Rasola and Bernardi, 2007).
Pfeiffer and Coworkers suggested that the permeabilizing effect of Ca2+ had a physiological role in steroidogenesis. These Authors showed that Ca2+ induces a “transformation” of adrenal cortex mitochondria allowing diffusion of extramitochondrial pyridine nucleotides through the otherwise impermeable inner membrane, and that NADPH entering in this way supports the 11-β hydroxylation of deoxycorticosterone (Pfeiffer and Tchen, 1973, 1975; Pfeiffer et al., 1976). These findings match those of Vinogradov et al., who demonstrated the Ca2+-dependent release of matrix pyridine nucleotides in liver mitochondria (Vinogradov et al., 1972). The term “permeability transition” was introduced by Haworth and Hunter, who thoroughly characterized its basic features in heart mitochondria. These Authors provided the key insight that the PT was due to reversible opening of a proteinaceous pore in the inner mitochondrial membrane, the permeability transition pore (PTP), whose physiological role remained undefined (Hunter et al., 1976; Hunter and Haworth, 1979a,b; Haworth and Hunter, 1979).
Studies on the PT were not too popular at this time, a possible consequence of the general acceptance of the chemiosmotic hypothesis which had just been fully recognized with the award of the Nobel Prize to Peter Mitchell in 1978. As noted earlier (Bernardi, 1999) studies of mitochondrial ion transport were indeed carried out in the same laboratories involved in clarifying the mechanisms of energy conservation, and they tended to become tests of the predictions of the chemiosmotic theory. In retrospect it is not too surprising that the existence of a large pore in the inner membrane appeared to contradict the basic tenets of chemiosmosis. As a result, and with very few exceptions (see e.g., Beatrice et al., 1980; Coelho and Vercesi, 1980; Bernardi and Pietrobon, 1982; Lê-Quôc and Lê-Quôc, 1982, 1985; Siliprandi et al., 1983; Vercesi, 1984; Riley and Pfeiffer, 1985; Al Nasser and Crompton, 1986; Sokolove and Shinaberry, 1988) research in this area did not enjoy much popularity.
The discovery that in mammalian mitochondria the PT can be inhibited by submicromolar concentrations of cyclosporin (Cs) A (Fournier et al., 1987; Crompton et al., 1988; Broekemeier et al., 1989; Davidson and Halestrap, 1990) changed the field substantially. CsA inhibits the PTP after binding to matrix cyclophilin (CyP)D, a peptidyl-prolyl cis-trans isomerase (PPIase) whose enzymatic activity is blocked by CsA (Fischer et al., 1989; Takahashi et al., 1989) in the same range of concentrations inhibiting the pore (Connern and Halestrap, 1992, 1994; Nicolli et al., 1996; Woodfield et al., 1997). A second fundamental finding was the demonstration that mitochondria possess ion channels that can be studied by electrophysiology (Sorgato et al., 1987). This seminal study was soon followed by the demonstration that the inner mitochondrial membrane is endowed with a high-conductance (≈1–1.3 nS) channel, the “mitochondrial megachannel” (MMC) (Kinnally et al., 1989; Petronilli et al., 1989). The MMC is inhibited by CsA (Szabó and Zoratti, 1991) and possesses all the key regulatory features of the PTP (Bernardi et al., 1992; Szabó et al., 1992) leaving little doubt that the PTP and the MMC are two aspects of the same molecular entity (Szabó and Zoratti, 1992). Electrophysiology has greatly contributed to our understanding of the MMC–PTP, and to acceptance of the pore theory of the PT (Zoratti et al., 2005).
Involvement of the PT in cell death was hypothesized 25 years ago (Crompton and Costi, 1988). Early support was obtained in hepatocytes subjected to oxidative stress (Broekemeier et al., 1992; Imberti et al., 1992), anoxia (Pastorino et al., 1993), or treatment with ATP (Zoeteweij et al., 1993); and in cardiomyocytes (Duchen et al., 1993) and isolated hearts (Griffiths and Halestrap, 1995) exposed to ischemia followed by reperfusion. The exponential increase in experimental papers dealing with the PT as an effector mechanism of cell death, however, only followed the demonstration that in the course of apoptosis cytochrome c is released into the cytosol (Liu et al., 1996) together with apoptosis-inducing factor (Susin et al., 1996) and a set of other proteins involved in the effector phase of apoptosis (Du et al., 2000; Ekert et al., 2000; Hegde et al., 2001; Li et al., 2001).
The molecular basis of the PT is still the matter of conjectures (Siemen and Ziemer, 2013). The various models and working hypotheses proposed over the years had to cope with the lack of selectivity for the permeating species, the strict requirement for matrix Ca2+, which has recently been established also for yeast mitochondria (Yamada et al., 2009), the stimulation by oxidants, the existence of a vast number of “inducers” without common structural features, and of a more limited but still substantial number of inhibitors (Gunter and Pfeiffer, 1990).
Many suspects, no culprits
Adenine nucleotide translocator, VDAC, and contact sites
Inhibition of mitochondrial swelling by adenine nucleotides has long been appreciated (Raaflaub, 1953b; Brenner-Holzach and Raaflaub, 1954), but the idea that the adenine nucleotide translocator (ANT) is causally involved in the PT probably originates from the finding that Ca2+-dependent membrane permeabilization is affected by the ANT inhibitors atractylate and bongkrekate, atractylate being a PT inducer and bongkrekate a PT inhibitor (Hunter and Haworth, 1979a). While early studies concluded (correctly, as it turns out) that the PT is an inner membrane event, the outer membrane was called into the picture after the work of Brdiczka and coworkers on inner-outer membrane contact sites, i.e., specialized structures where the two membranes form close contacts mediated by protein–protein interactions (Kottke et al., 1988). The contact sites were proposed to facilitate channeling of adenine nucleotides to and from mitochondria, and to comprise hexokinase on the cytosolic surface and VDAC within the outer membrane, creatine kinase and nucleoside diphosphate kinase (in tissues that express these proteins), and ANT in the inner membrane (Adams et al., 1989; Bucheler et al., 1991). The connection with the PTP was made in 1996, when the same laboratory found that hexokinase-enriched fractions formed channels with the conductance expected of the PTP, and conferred permeability properties to liposomes that could be inhibited by N-methylVal-4-cyclosporin (Beutner et al., 1996). Unlike the case of MMC, however, currents were inhibited rather than induced by atractylate; the active fractions were not enriched in VDAC or ANT; and the preparation contained a very large number of proteins (Beutner et al., 1996) making assignment of the channel activity to a specific species quite problematic. The same hexokinase-enriched fractions were used to study the interactions with proteins of the Bcl-2 family (Marzo et al., 1998). The hexokinase/VDAC/ANT model of the PTP was extended to include outer membrane TSPO [formerly known as peripheral benzodiazepine (Bz) receptor] and Bcl-2 family members (Zamzami and Kroemer, 2001). No further progress was made in the purification of the component(s) mediating channel activity and membrane permeabilization, yet this model became widely accepted as the “structure” of the PTP.
What did we learn from genetic inactivation of putative PTP components
Conclusive evidence that the ANT is not essential for PTP formation comes from analysis of mitochondria lacking all ANT isoforms, which revealed that a Ca2+-dependent PT took place (Kokoszka et al., 2004). The PT could be inhibited by CsA and triggered by thiol oxidants, demonstrating that the ANT is neither the obligatory binding partner of CyPD nor the site of action of oxidants (Kokoszka et al., 2004) as suggested earlier (Costantini et al., 1998). In addition, hepatocytes prepared from control and ANT-deficient livers showed identical responses to activation of receptor-mediated apoptotic pathways initiated by TNFα and Fas (Kokoszka et al., 2004). It has been argued that a low, undetectable level of ANT expression could explain the PT observed in ANT-null mitochondria (Halestrap, 2004). I believe that this possibility is quite remote because the PTP of mitochondria lacking ANT was insensitive to opening by atractylate and to closure by ADP (Kokoszka et al., 2004). As noted elsewhere (Bernardi et al., 2006) it is very difficult to envisage how any ANT molecules present in mutant mitochondria would promote a CsA-sensitive PT and yet not respond to atractylate and ADP.
A detailed comparison of the PT in mitochondria from wild-type and Vdac1−/− mice demonstrated that VDAC1 is fully dispensable for PTP opening and regulation (Krauskopf et al., 2006). Furthermore, the PT of mitochondria prepared from Vdac3−/− and Vdac1−/−Vdac3−/− mice, or from fibroblasts lacking all three VDAC isoforms, was identical to that of strain-matched wild-type mitochondria (Baines et al., 2007). Taken together, these results clearly indicate that VDACs are not components of the PTP although, as discussed later in the review, the outer membrane appears to participate in PTP regulation (Sinha Roy et al., 2009; Šileikyte et al., 2011).
TSPO is an 18 kDa highly hydrophobic protein, involved in cholesterol and protophorphyrin transport, which is located in the outer mitochondrial membrane (Batarseh and Papadopoulos, 2010). It was initially identified as a binding site for Bz in tissues that lack GABA receptors (Anholt et al., 1985, 1986). TSPO binds with nM affinity a variety of ligands, notably the Bz Ro5-4864 and the isoquinoline carboxamide PK11195 (Verma et al., 1987) as well as protoporphyrin IX, a potent inducer of the PTP (Pastorino et al., 1994). Treatment with TSPO ligands does affect the channel activity of the MMC–PTP in the proper concentration range (Kinnally et al., 1993) but TSPO is not the only mitochondrial protein binding these compounds, which have been shown to interact with, and inhibit, the FOF1 ATP synthase (Cleary et al., 2007). The latter finding acquires a particular meaning in the light of our recent demonstration that FOF1 ATP synthase dimers form channels indistinguishable from the PTP that can be activated by Bz-423 (Giorgio et al., 2013). Perhaps not surprisingly then, the PTP of mitochondria from mice where expression of TSPO had been conditionally inactivated could still be sensitized to Ca2+ by TSPO ligands, and had general features indistinguishable from those of mitochondria isolated from mice with wild-type genotype (Šileikytė, Forte et al., unpublished observations).
In summary, what we learned from gene inactivation studies is that none of the channel-forming proteins suggested to take part in PTP formation is necessary for its occurrence.
Alternative models for PTP formation
The Halestrap laboratory has proposed a new model for the PTP that envisions the formation of a pore by the Pi carrier (Leung et al., 2008). PTP formation would be favored by Ca2+ and CyPD binding, and the open probability would be increased by formation of a heterodimer with the ANT (Leung and Halestrap, 2008). This model still needs to be tested by reconstitution and by genetic means, but it should be noted that patch-clamp experiments with the functionally active mitochondrial Pi carrier revealed an anion-selective channel function with a mean conductance as low as 40 pS (the typical conductance of the PTP is 1.0−1.3 nS, and the channel is not ion-selective). Furthermore, the current was decreased to 25 pS by both Ca2+ (which activates the PTP) and Mg2+ (which inhibits it). Finally, and at striking variance with the pore, channel activity was inhibited by Pi and unaffected by ADP (Herick et al., 1997). These features make the Pi carrier a very unlikely candidate as a PTP component.
A radically different model of the PTP has been proposed in which the pore does not form from a specific protein but rather from aggregation of misfolded integral membrane proteins damaged by oxidant and other stresses (Kowaltowski et al., 2001; He and Lemasters, 2002). In the hypothesis of He and Lemasters conductance through misfolded protein clusters would be normally blocked by chaperone-like proteins (including CyPD) and modulated by Ca2+ and CsA. Opening of “unregulated” pores would occur when protein clusters exceed available chaperones (He and Lemasters, 2002). This model fails to account for the absolute Ca2+ requirement of PTP, and its regulation by voltage (Bernardi, 1992), matrix pH (Nicolli et al., 1993), and adenine nucleotides, all effects that are not easy to reconcile with a permeability pathway created by a heterogeneous set of denatured proteins.
It should be mentioned that Ca2+-dependent, CsA-insensitive PT-like channel activities have been described that are formed or activated by 3-hydroxybutyrate/polyphosphate (Pavlov et al., 2005) and by fatty acids (Mironova et al., 2001; Sultan and Sokolove, 2001). The latter observation links to early work demonstrating that fatty acids are potent inducers of mitochondrial swelling (Lehninger and Remmert, 1959; Wojtczak and Lehninger, 1961), which was later recognized to be due to PTP opening (Scorrano et al., 2001; Bernardi et al., 2002).
Cyclophilin D and FoF1 ATP synthase
CyPs are ubiquitous, conserved proteins possessing PPIase activity (Fischer et al., 1989; Takahashi et al., 1989) sharing a common domain of about 109 amino acids, the CyP-like domain (Wang and Heitman, 2005). In man 16 unique CyPs have been found, the prototype being cytosolic CyPA (Wang and Heitman, 2005). After binding CsA the PPIase activity is inhibited (Borel et al., 1977) and the CsA/CyPA complex binds to and inhibits the cytosolic phosphatase calcineurin (Liu et al., 1991), causing immunosuppression (Clipstone and Crabtree, 1992; Walsh et al., 1992).
CyPD is the mitochondrial CyP isoform in mammals (Connern and Halestrap, 1992; Nicolli et al., 1996; Woodfield et al., 1997), see Giorgio et al. (2010) for a review. Much on its role in PTP regulation has been learned by genetic ablation of the Ppif gene (which encodes for CyPD). CyPD is the mitochondrial receptor for CsA and modulates the PTP but is not a structural pore component (Baines et al., 2005; Basso et al., 2005; Nakagawa et al., 2005; Schinzel et al., 2005). As discussed elsewhere (Bernardi et al., 2006), the effect of CsA on the PTP is best described as “desensitization” in the sense that the PTP becomes more resistant to opening by Ca2+ and Pi. It should be stressed that pore opening readily takes place for Ca2+-Pi loads that are about twice those required in wild-type mitochondria. This indicates that Ppif−/− (CyPD-null) mitochondria are not null for the PTP. This consideration is important for the interpretation of results obtained with CsA because (similarly to the absence of CyPD) CsA can desensitize but not block the PTP; thus, lack of sensitivity to CsA does not necessarily mean that the PTP is not involved in the event being studied. Furthermore, expression of CyPD is modulated, e.g., by muscle denervation (Csukly et al., 2006), and only CyPD-expressing mitochondria respond to CsA (Li et al., 2012). No endogenous ligands of CyPD mimicking the effects of CsA are known, but other PTP (de)sensitizers might act by favoring CyPD association or dissociation, as documented for acidic pH and increasing ionic strength (Nicolli et al., 1996). Furthermore, CyPD phosphorylation (Rasola et al., 2010), acetylation (Shulga and Pastorino, 2010), and nitrosylation (Kohr et al., 2011; Nguyen et al., 2011) affect the propensity of the PTP to open (Rasola and Bernardi, 2011). Not surprisingly CyPD may interact with many proteins including Hsp90 and its related molecule TRAP-1 (Kang et al., 2007); Bcl-2 (Eliseev et al., 2009); ERK-2/GSK-3 (Rasola et al., 2010); possibly p53 (Vaseva et al., 2012), but see (Karch and Molkentin, 2012); and the FOF1 ATP synthase (Giorgio et al., 2009). The latter interaction turned out to be the key for identification of the PTP.
The FOF1 ATP synthase (or complex V) is the rotary enzyme that synthesizes the vast majority of ATP in respiring cells (Rees et al., 2009). It is formed by the catalytic F1, the membrane-bound proton-translocating FO, and the lateral stalk linking F1 and FO (Strauss et al., 2008; Thomas et al., 2008; Rees et al., 2009; Baker et al., 2012; Davies et al., 2012) (Figure 1). The lateral stalk acts as a stator preventing the α3β3 subcomplex of F1 from rotating with subunits γ, δ, and ε and the ring of FO-subunits c (Rees et al., 2009). The FOF1 ATP synthase complex associates to form dimers, which are considered to be the “building blocks” of long rows of oligomers that promote cristae formation and may optimize the enzyme's catalytic performance (Campanella et al., 2008; Strauss et al., 2008; Thomas et al., 2008; Rees et al., 2009; Baker et al., 2012; Davies et al., 2012). The angle formed by the dimers seems to display a large variability, and dimers are believed to represent the physiological unit of complex V in the inner membrane (Thomas et al., 2008).
Figure 1.
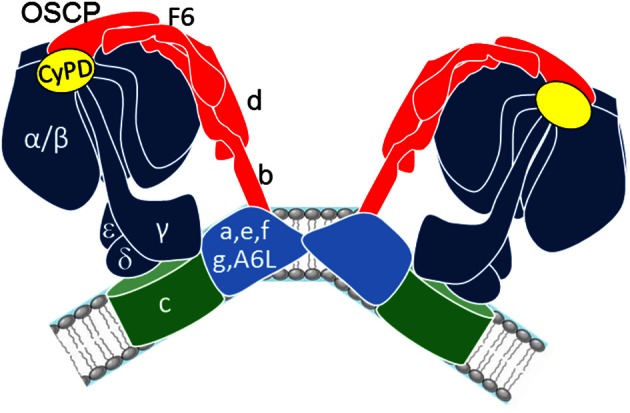
Schematic representation of FOF1 ATP synthase dimers. F1 (dark blue), FO (green and light blue), and stalk subunits (red) are illustrated based on recent structural studies (Strauss et al., 2008; Baker et al., 2012; Davies et al., 2012).
We found that CyPD binds the lateral stalk of complex V in an apparent ratio of 1:1:1:1 with the OSCP, b and d subunits (Giorgio et al., 2009). CyPD binding requires Pi and results in partial inhibition of ATP synthase activity, while CsA displaces CyPD resulting in enzyme reactivation (Giorgio et al., 2009). The stimulatory effect of CsA on enzyme catalysis is lost in Ppif−/− mitochondria, proving that it is mediated by CyPD, while assembly of the ATP synthase is not affected by CyPD ablation (Giorgio et al., 2009). In spite of the striking analogies with PTP regulation (Pi dependence, CsA sensitivity) whether the interactions of CyPD with complex V are relevant for the PT remained unknown until very recently.
Dimers of FoF1 ATP synthase form channels indistinguishable from the MMC–PTP
We have defined the binding site of CyPD on ATP synthase to the OSCP subunit, and shown that the interaction is electrostatic in nature. Modeling of surface potentials and isopotential curves suggested that CyPD interacts with OSCP in a region overlapping with helices 3 and 4, and this turned out to be a key insight (Giorgio et al., 2013). Indeed, this is also the binding site of Bz-423, a well-characterized inhibitor of the FOF1 ATP synthase (Johnson et al., 2005; Stelzer et al., 2010). If the CyPD interactions are important for the PT, it was logical to expect that Bz-423 should act as a PTP inducer, and this was the case. Bz-423 sensitized the PTP to Ca2+, and the inducing effect was blunted by Pi at concentrations that increase CyPD binding to OSCP (Giorgio et al., 2013). Consistent with an involvement of complex V in PTP formation, decreased levels of OSCP did affect the Ca2+ dependence of the pore. Indeed, mitochondria took up Ca2+ normally when ATP was used as the energy source, but the threshold Ca2+ load required for PTP opening was halved, which is the first molecular evidence that OSCP affects the probability of pore opening. Consistent with a key role of the ATP synthase, the Ca2+ affinity of the PTP was also affected by enzyme catalysis in the sense that ATP-hydrolyzing mitochondria required twice the Ca2+ load of ATP-synthesizing organelles, an effect as large as that of CsA (Giorgio et al., 2013).
Direct evidence that the PTP forms from the FOF1 ATP synthase was obtained by incorporating dimers purified by blue native electrophoresis into azolectin bilayers, and by measuring the passage of currents after application of a voltage difference. Addition of Bz-423 in the presence of Ca2+ triggered opening of high-conductance channels that were blocked by Mg2+/ADP and by the ATP synthase inhibitor AMP-PNP (γ-imino ATP, a non hydrolyzable ATP analog). Monomers of ATP synthase were devoid of channel activity in spite of the same overall subunit composition as the dimers (Tomasetig et al., 2002; Giorgio et al., 2013). The characteristics of the reconstituted pore closely matched those of MMC–PTP (Petronilli et al., 1989; Szabó and Zoratti, 1991; Szabó et al., 1992). Maximal chord conductance was between 1.0 and 1.3 nS in symmetrical KCl 150 mM, and various subconductance states were observed. Like the MMC of Ppif−/− mitochondria (De Marchi et al., 2006), and in keeping with the lack of CyPD in the preparations, the channel was insensitive to CsA. Ca2+ and Pi, which sensitize the PTP even in the absence of CyPD, sufficed to induce PTP currents when dimers were prepared in the presence of 10 mM Pi. Channel openings could not be elicited by atractyloside and, once elicited by Bz-423, were still observed in the presence of bongkrekic acid. These findings are consistent with the absence of ANT in the preparations, which also totally lacked VDAC (Giorgio et al., 2013).
Bz-423 was characterized as an apoptosis-inducing agent acting through mitochondria (Blatt et al., 2002) and OSCP was identified as its target through the unbiased screening of a human phage display library (Johnson et al., 2005). The selectivity of action of Bz-423 on OSCP, its ability to trigger channel activity of ATP synthase dimers, and the lack of activity of monomer preparations strongly argues against the possibility that the currents observed by Giorgio et al. (2013) are due to unidentified contaminating proteins. The PTP-inducing effect of Bz-423 and CyPD (which both act through OSCP on top of the lateral stalk in the matrix) must be indirect, as it affects the permeability properties of the inner membrane. We assume that the PTP forms at the membrane interface between two adjacent FO sectors, which could also accommodate the effects of fatty acids (Lehninger and Remmert, 1959; Wojtczak and Lehninger, 1961; Bernardi et al., 2002). Matrix Ca2+ has an essential permissive role in PTP formation, and we have suggested that accessibility of Ca2+ to the metal binding sites of the catalytic F1 sector is influenced by OSCP. This subunit could indeed affect the affinity of the metal binding sites of ATP synthase and thus determine the ease with which matrix Ca2+ can replace Mg2+ causing PTP opening. Our working hypothesis is that OSCP as such is a “negative” modulator, whose effect can be counteracted by binding of the “positive” effector CyPD (which indeed increases the apparent Ca2+ affinity of the PTP). Removal of OSCP, or CyPD binding to OSCP, would induce similar conformational effects leading to increased probability of PTP opening, a working hypothesis that awaits experimental verification.
A cartoon with the hypothetical events leading to PTP formation from complex V dimers is presented in Figure 2. In the “coupled” condition the dimers bind Mg2+-ADP/ATP, and the concentration of free Ca2+ (and Pi, not shown in the Figure) is not high enough for the PT to occur (panel A). Binding of CyPD, which is favored by Pi (Giorgio et al., 2009), would cause a conformational change affecting accessibility of the metal binding sites. This in turn would decrease the matrix Ca2+ load necessary to trigger the PT (panels B,D). The transition would essentially depend on replacement of Mg2+ with Ca2+, which in the absence of CyPD binding would require higher matrix Ca2+ loads (panel C). Accessibility of the metal binding site (and probability of occurrence of the PT at a given Ca2+ load) would be increased by thiol oxidation and counteracted by thiol reduction. Ion and solute permeation would then occur at the interface between the c rings (panels C,D). The PT can be fully reverted by Ca2+ chelation with EGTA (Petronilli et al., 1994), which would restore Mg2+ binding and allow the dimer to recover the coupled structure.
Figure 2.

Hypothetical transition of FOF1 ATP synthase dimers to form the PTP. ATP synthase dimers (A) can undergo PTP formation when Ca2+ rather than Mg2+ is bound, possibly at the catalytic sites, in a reversible process favored by thiol oxidation (C). Binding of CyPD, which is favored by Pi (B) would increase the accessibility of the metal binding sites, allowing PTP formation at lower Ca2+ concentrations (as depicted here by a smaller face type) (D). Adenine nucleotides counteract PTP formation in synergy with Mg2+. Red arrows denote the hypothetical pathway for solute diffusion between two FO subunits.
A mystery solved, more mysteries to solve
Role of outer membrane
It can no longer be questioned that the PT is an inner membrane event since (1) complex V dimers suffice to induce MMC activity and (2) Ca2+-dependent PT occurs in mitoplasts, i.e., mitochondria stripped of the outer membrane (Šileikyte et al., 2011). These findings should not be taken to imply that the outer membrane does not play a role in PTP modulation, which is in fact supported by a large number of studies. An early indication was provided by Lê-Quôc and Lê-Quôc, who showed that induction of the PTP by substituted maleimides requires the outer membrane (Lê-Quôc and Lê-Quôc, 1985). We have fully confirmed this “sensitizing” role of the outer membrane by an independent approach, i.e., irradiation of mitochondria with visible light after treatment with hematoporphyrin, a strategy that allows production of singlet oxygen leading to PTP inactivation or reactivation depending on the light dose (Ricchelli et al., 2011). Low light doses inactivate the PTP through degradation of hystidyl residues that prevent cysteine oxidation on the matrix side (Salet et al., 1997), while higher light doses activate the PTP through cysteine oxidation at the outer membrane (Moreno et al., 2001; Petronilli et al., 2009). Mitoplasts are completely resistant to PTP induction by high light doses (Šileikyte et al., 2011) proving that the outer membrane can favor PTP opening through a still unidentified mechanism (Ricchelli et al., 2011).
Bcl-2 family proteins, which are generally believed to locate to the outer membrane, affect the PTP (Forte and Bernardi, 2006). According to several studies Bcl-2 increases resistance to the PT (Susin et al., 1996; Decaudin et al., 1997) while Bax and/or Bad activated by tBid favor PTP opening (Pastorino et al., 1998, 1999; Scorrano et al., 2002; Sinha Roy et al., 2009). Although CsA did not prevent cytochrome c release in one study where tBid was used, it is not known whether the cells expressed CyPD and were therefore sensitive to CsA inhibition (Garcia-Perez et al., 2012), an issue that may explain discrepancies with earlier studies (Scorrano et al., 2002; Tafani et al., 2002). Occurrence of a PT is instead opposed by outer membrane binding of hexokinase II (Pastorino et al., 2002, 2005; Chiara et al., 2008), which counteracts the inducing effects of Bax (Pastorino et al., 2002). It must be mentioned that Bcl-2 family proteins also interact with other targets. Indeed, tBid initiates a CsA-sensitive process of cristae remodeling that increases cytochrome c availability in the intermembrane space, which is consistent with a PT-dependent facilitation of cytochrome c release even when the outer membrane is intact (Scorrano et al., 2002, 2003); and Bax inhibits the mitochondrial inner membrane potassium channel Kv1.3 (Szabó et al., 2008), a proapoptotic effect that critically depends on Bax residue K128 and is abrogated by a K128E mutation, while an E158K mutation in antiapoptotic Bcl-xL (position 158 of Bcl-xL corresponds to position 128 of Bax) turns Bcl-xL into a proapoptotic protein (Szabó et al., 2011).
The detailed mechanisms through which the outer membrane affects formation of the PTP remain to be addressed. I would like to stress that the vast majority of complex V is located in long rows of oligomers deep inside mitochondrial cristae (Strauss et al., 2008; Davies et al., 2011, 2012) where direct physical interaction with the outer membrane is obviously not possible. Thus, either the PTP forms in the small population of dimers facing the intermembrane space, where direct contact with the outer membrane can occur; or the effect is exerted by controlling the diffusion of PTP-regulating metabolites and ions including Ca2+ itself (Madesh and Hajnoczky, 2001; Rapizzi et al., 2002) in a process that would be greatly favored by cristae remodeling (Scorrano et al., 2002).
The PTP as a calcium release channel
We have recently reviewed the hypothesis (Bernardi and Petronilli, 1996; Ichas et al., 1997) that the PTP may open transiently (Hüser et al., 1998; Hüser and Blatter, 1999; Petronilli et al., 1999, 2001) to operate as a mitochondrial Ca2+ release channel under physiological conditions (Bernardi and von Stockum, 2012). The basis for this hypothesis goes back to 1992, when Altschuld et al. reported that CsA significantly increases net Ca2+ uptake and decreases Ca2+ efflux in isolated cardiomyocytes, as measured by radiolabeled 45Ca2+, through a demonstrably mitochondrial effect that has no impact on cell morphology or viability (Altschuld et al., 1992). Three recent publications based on Ppif−/− cells and mice support this hypothesis (Elrod et al., 2010; Barsukova et al., 2011; Parone et al., 2013).
An age-related phenotype was discovered in the hearts of Ppif−/− mice, which dysplayed decreased contractile reserve and increased shortening and relaxation times, with longer decay of cytosolic Ca2+ transients (Elrod et al., 2010). Direct measurement of total mitochondrial Ca2+ content of Ppif−/− hearts showed a 2.6-fold increase, which was matched by larger Ca2+ transients in mitochondria of myocytes treated with CsA. Under continuous pacing PTP desensitization with CsA decreased the rise time in Ca2+ accumulation and prolonged the recovery time after pacing, findings that are entirely consistent with the PTP acting as a Ca2+ release channel to prevent Ca2+ overload (Elrod et al., 2010). Consistent with this idea, cytosolic [Ca2+] increases induced by either ATP or depolarizing concentrations of KCl gave comparable transient increases of mitochondrial [Ca2+] in adult cortical neurons from wild type and Ppif−/− mice; while application of the two stimuli together resulted in much higher levels of mitochondrial [Ca2+] in the Ppif−/− neurons, suggesting that the threshold for PTP activation had been reached in the wild type but not in the CyPD-null mitochondria in situ. These data indicate that the regulatory role of CyPD (and thus PTP opening) in Ca2+ homeostasis may be essential only for relatively large mitochondrial Ca2+ loads (Barsukova et al., 2011). Mice expressing amyotrophic lateral sclerosis-linked mutants of superoxide dismutase (SOD) 1 develop a motor neuron disease with many pathological hallmarks seen in patients (Gurney et al., 1994). Spinal cord mitochondria from these mice display decreased Ca2+ retention capacity long before onset of motor weakness and neuronal death (Damiano et al., 2006), and this was corrected by genetic elimination of the Ppif−/− gene (Parone et al., 2013). Improved mitochondrial Ca2+ buffering was matched by improved mitochondrial ATP synthesis and reduced swelling, attenuation of glial activation, reduction of misfolded SOD1 aggregates in the spinal cord, and significant suppression of motor neuron death throughout disease, although survival was unfortunately not improved (Parone et al., 2013).
Transient PTP openings appear also to be at the basis of “superoxide flashes” observed with mitochondrially targeted, circularly permuted yellow fluorescent protein (mt-cpYFP) in a variety of cell types (Wang et al., 2008, 2013; Fang et al., 2011). There is an ongoing debate on whether the flashes are in fact partially or totally due to changes of matrix pH (Schwarzlander et al., 2011, 2012), although it is fair to say that the measured matrix alkalinization appears to be way too small to account for the fluorescence changes of mt-cpYFP (Wei-Lapierre et al., 2013).
The recent finding that Drosophila mitochondria possess a PTP-related but CsA-insensitive Ca2+ release channel selective for Ca2+ and H+ (von Stockum et al., 2011) provides a nice model system, and perhaps the missing link between the PTP of yeast and mammals (Azzolin et al., 2010). I am confident that it will be now possible to address the potential role of the PTP as a Ca2+ release channel through inhibition of the “pore function” of ATP synthase dimers through appropriate drugs and through genetic manipulation.
Complex I
The PTP has the remarkable feature of being modulated by electron flow through complex I (Fontaine et al., 1998a), which may in part be linked to the fact that NADH oxidation favors PTP opening (Costantini et al., 1996). A specific role of complex I, however, is suggested by the striking discovery that in mammalian mitochondria rotenone can be an effective inhibitor of the pore (Chauvin et al., 2001; Li et al., 2012). Inhibition of the PT by rotenone is maximal in mitochondria from tissues that express low levels of CyPD, where CsA has little inhibitory effect; while inhibition by CsA is maximal in mitochondria from tissues with high levels of expression of CyPD, where rotenone does not affect the PTP. Finally, tissues with mitochondria expressing intermediate levels of CyPD are sensitive to both rotenone and CsA, with additive effects of the two inhibitors (Li et al., 2012). Intriguingly, the inhibitory effect of rotenone is potentiated by Pi, which is reminiscent of the Pi requirement for inhibition by CsA (Basso et al., 2008). The concentrations of rotenone required for PT inhibition matched quite precisely those required to inhibit respiration, and a similar PT-inhibiting effect was seen with metformin, which partially inhibits complex I as well (El Mir et al., 2000; Guigas et al., 2004). Since rotenone and metformin do not share common structural features, it appears likely that modulation of the PTP by inhibition of complex I is indirect, and possibly mediated by inhibition of complex I-derived reactive oxygen species (Batandier et al., 2004). An indirect effect is also suggested by the observations (1) that a Ca2+-dependent (albeit CsA-insensitive) PT can occur in mitochondria from yeast strains that do not possess an energy-conserving complex I (Jung et al., 1997; Yamada et al., 2009; Uribe-Carvajal et al., 2011); (2) that complex I purified by blue native electrophoresis did not form Ca2+-activated channels in lipid bilayers under conditions that instead promote MMC opening after incorporation of complex V (Giorgio et al., 2013); and (3) that complex I does not colocalize with ATP synthase oligomers (Davies et al., 2011).
Quinones
The regulatory effects of quinones on the PTP has been discovered in our laboratory in 1998 (Fontaine et al., 1998a,b) and thoroughly characterized by Fontaine and Coworkers (Walter et al., 2000, 2002; Devun et al., 2010). Ub0 (i.e., ubiquinone without a side chain) is the most potent inhibitor of the PTP; the dose-dependence displays a bell-shaped curve, however, a feature that is also typical of other inhibitory quinones like decylubiquinone (Fontaine et al., 1998a,b; Fontaine and Bernardi, 1999; Walter et al., 2000). PTP-inducing quinones include idebenone (Walter et al., 2000) while idebenol (the reduced form) is no longer effective, suggesting that the effect on the PTP is related to oxidation-reduction events (Giorgio et al., 2012). The structure–function relationship of quinones for PTP inhibition/activation has not been solved, and further complexity arises from the fact that the same quinone can have opposite effects depending on the cell type (Devun et al., 2010). It appears possible that, like substituted maleimides, Ub0 affects PTP formation in part at least through its ability to form adducts with SH groups, which is not shared by short-chain quinones. ROS-forming ability of the latter does not always correlate with their PTP-inducing features (Devun et al., 2010) and the general mechanism of action of quinones on the PT remains to be solved.
Conclusions and perspectives
The findings that (1) dimers of the ATP synthase treated with Ca2+ generate currents indistinguishable from the MMC–PTP; (2) channel openings are favored by Bz-423 and thiol oxidants; (3) channel openings are inhibited by adenine nucleotides and Mg2+; and (4) monomers lack any channel activity, all represent strong indications that the PTP forms from a specific, Ca2+-dependent conformation of the dimers. The FOF1 ATP synthase readily accommodates key pathophysiological effectors of the PT since Ca2+, Mg2+, adenine nucleotides, and Pi bind the catalytic core at F1; and the membrane potential and matrix pH, which are key PTP modulators (Bernardi et al., 2006), are also key regulators of the ATP synthase. On the other hand, several regulators of the PTP (specifically, rotenone and quinones) are not easily accounted for by the dimer hypothesis, and their mechanism of action will need further studies. However, I believe that our findings provide a novel and stimulating working hypothesis that should help address outstanding issues including species-specific features of the PTP of rats (Ricchelli et al., 2005; Zulian et al., 2007), yeast (Jung et al., 1997; Manon et al., 1998; Yamada et al., 2009; Uribe-Carvajal et al., 2011), and Drosophila melanogaster (von Stockum et al., 2011); and the mystery of Artemia franciscana, an anoxia and salt-tolerant brine shrimp whose mitochondria are refractory to PTP opening (Menze et al., 2005, 2010). Together our results suggest a dual function for complex V, ATP synthesis and PTP formation. The key enzyme of life appears therefore to be also the molecular switch that signals the presence of fully depolarized, dysfunctional mitochondria to promote cell death (Rasola and Bernardi, 2011) and/or mitophagy (Youle and Narendra, 2011).
Conflict of interest statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to thank Mike Forte, Valentina Giorgio, Giovanna Lippe, Valeria Petronilli, and Andrea Rasola for helpful comments on the manuscript. Research in our laboratory is supported by AIRC (IG 13392), MIUR (FIRB RBAP11S8C3 and PRIN 20107Z8XBW), NIH-PHS (R01GM069883 and R03DA033978-01), Telethon (GGP 11082 and GPP10005A), and Università di Padova (Progetti Strategici di Ateneo).
References
- Adams V., Bosch W., Schlegel J., Wallimann T., Brdiczka D. (1989). Further characterization of contact sites from mitochondria of different tissues: topology of peripheral kinases. Biochim. Biophys. Acta 981, 213–225 [DOI] [PubMed] [Google Scholar]
- Al Nasser I., Crompton M. (1986). The reversible Ca2+-induced permeabilization of rat liver mitochondria. Biochem. J. 239, 19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschuld R. A., Hohl C. M., Castillo L. C., Garleb A. A., Starling R. C., Brierley G. P. (1992). Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. Am. J. Physiol. 262, H1699–H1704 [DOI] [PubMed] [Google Scholar]
- Anholt R. R., De Souza E. B., Oster-Granite M. L., Snyder S. H. (1985). Peripheral-type benzodiazepine receptors: autoradiographic localization in whole-body sections of neonatal rats. J. Pharmacol. Exp. Ther. 233, 517–526 [PubMed] [Google Scholar]
- Anholt R. R., Pedersen P. L., De Souza E. B., Snyder S. H. (1986). The peripheral-type benzodiazepine receptor. Localization to the mitochondrial outer membrane. J. Biol. Chem. 261, 576–583 [PubMed] [Google Scholar]
- Azzi A., Azzone G. F. (1965). Swelling and shrinkage phenomena in liver mitochondria. I. Large amplitude swelling induced by inorganic phosphate and by ATP. Biochim. Biophys. Acta 105, 253–264 [DOI] [PubMed] [Google Scholar]
- Azzolin L., von Stockum S., Basso E., Petronilli V., Forte M. A., Bernardi P. (2010). The mitochondrial permeability transition from yeast to mammals. FEBS Lett. 584, 2504–2509 10.1016/j.febslet.2010.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzone G. F., Azzi A. (1965). Volume changes in liver mitochondria. Proc. Natl. Acad. Sci. U.S.A. 53, 1084–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines C. P., Kaiser R. A., Purcell N. H., Blair N. S., Osinska H., Hambleton M. A., et al. (2005). Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662 10.1038/nature03434 [DOI] [PubMed] [Google Scholar]
- Baines C. P., Kaiser R. A., Sheiko T., Craigen W. J., Molkentin J. D. (2007). Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 9, 550–555 10.1038/ncb1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker L. A., Watt I. N., Runswick M. J., Walker J. E., Rubinstein J. L. (2012). Arrangement of subunits in intact mammalian mitochondrial ATP synthase determined by cryo-EM. Proc. Natl. Acad. Sci. U.S.A. 109, 11675–11680 10.1073/pnas.1204935109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsukova A., Komarov A., Hajnoczky G., Bernardi P., Bourdette D., Forte M. (2011). Activation of the mitochondrial permeability transition pore modulates Ca2+ responses to physiological stimuli in adult neurons. Eur. J. Neurosci. 33, 831–842 10.1111/j.1460-9568.2010.07576.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso E., Fante L., Fowlkes J., Petronilli V., Forte M. A., Bernardi P. (2005). Properties of the permeability transition pore in mitochondria devoid of Cyclophilin, D. J. Biol. Chem. 280, 18558–18561 10.1074/jbc.C500089200 [DOI] [PubMed] [Google Scholar]
- Basso E., Petronilli V., Forte M. A., Bernardi P. (2008). Phosphate is essential for inhibition of the mitochondrial permeability transition pore by cyclosporin A and by cyclophilin D ablation. J. Biol. Chem. 283, 26307–26311 10.1074/jbc.C800132200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batandier C., Leverve X., Fontaine E. (2004). Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex, I. J. Biol. Chem. 279, 17197–17204 10.1074/jbc.M310329200 [DOI] [PubMed] [Google Scholar]
- Batarseh A., Papadopoulos V. (2010). Regulation of translocator protein 18kDa (TSPO) expression in health and disease states. Mol. Cell. Endocrinol. 327, 1–12 10.1016/j.mce.2010.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatrice M. C., Palmer J. W., Pfeiffer D. R. (1980). The relationship between mitochondrial membrane permeability, membrane potential, and the retention of Ca2+ by mitochondria. J. Biol. Chem. 255, 8663–8671 [PubMed] [Google Scholar]
- Bernardi P. (1992). Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J. Biol. Chem. 267, 8834–8839 [PubMed] [Google Scholar]
- Bernardi P. (1999). Mitochondrial transport of cations: channels, exchangers and permeability transition. Physiol. Rev. 79, 1127–1155 [DOI] [PubMed] [Google Scholar]
- Bernardi P., Krauskopf A., Basso E., Petronilli V., Blachly-Dyson E., Di Lisa F., et al. (2006). The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 273, 2077–2099 10.1111/j.1742-4658.2006.05213.x [DOI] [PubMed] [Google Scholar]
- Bernardi P., Penzo D., Wojtczak L. (2002). Mitochondrial energy dissipation by fatty acids. Mechanisms and implications for cell death. Vitam. Horm. 65, 97–126 [DOI] [PubMed] [Google Scholar]
- Bernardi P., Petronilli V. (1996). The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J. Bioenerg. Biomembr. 28, 131–138 [DOI] [PubMed] [Google Scholar]
- Bernardi P., Pietrobon D. (1982). On the nature of Pi-induced, Mg2+-prevented Ca2+ release in rat liver mitochondria. FEBS Lett. 139, 9–12 10.1016/0014-5793(82)80475-4 [DOI] [PubMed] [Google Scholar]
- Bernardi P., Vassanelli S., Veronese P., Colonna R., Szabo I., Zoratti M. (1992). Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J. Biol. Chem. 267, 2934–2939 [PubMed] [Google Scholar]
- Bernardi P., von Stockum S. (2012). The permeability transition pore as a Ca2+ release channel: new answers to an old question. Cell Calcium 52, 22–27 10.1016/j.ceca.2012.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner G., Rück A., Riede B., Welte W., Brdiczka D. (1996). Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Lett. 396, 189–195 10.1016/0014-5793(96)01092-7 [DOI] [PubMed] [Google Scholar]
- Blatt N. B., Bednarski J. J., Warner R. E., Leonetti F., Johnson K. M., Boitano A., et al. (2002). Benzodiazepine-induced superoxide signals B cell apoptosis: mechanistic insight and potential therapeutic utility. J. Clin. Invest. 110, 1123–1132 10.1172/JCI16029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borel J. F., Feurer C., Magnee C., Stahelin H. (1977). Effects of the new anti-lymphocytic peptide cyclosporin A in animals. Immunology 32, 1017–1025 [PMC free article] [PubMed] [Google Scholar]
- Brenner-Holzach O., Raaflaub J. (1954). Die korrelation zwischen der schwellung isolierter mitochondrien und dem abbau intramitochondrialen adenosinnucleotide (ATP, ADP, AMP, CoA). Helv. Physiol. Pharmacol. Acta 12, 242–252 [PubMed] [Google Scholar]
- Broekemeier K. M., Carpenter Deyo L., Reed D. J., Pfeiffer D. R. (1992). Cyclosporin A protects hepatocytes subjected to high Ca2+ and oxidative stress. FEBS Lett. 304, 192–194 [DOI] [PubMed] [Google Scholar]
- Broekemeier K. M., Dempsey M. E., Pfeiffer D. R. (1989). Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J. Biol. Chem. 264, 7826–7830 [PubMed] [Google Scholar]
- Bucheler K., Adams V., Brdiczka D. (1991). Localization of the ATP/ADP translocator in the inner membrane and regulation of contact sites between mitochondrial envelope membranes by ADP. A study on freeze-fractured isolated liver mitochondria. Biochim. Biophys. Acta 1056, 233–242 [DOI] [PubMed] [Google Scholar]
- Campanella M., Casswell E., Chong S., Farah Z., Wieckowski M. R., Abramov A. Y., et al. (2008). Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 8, 13–25 10.1016/j.cmet.2008.06.001 [DOI] [PubMed] [Google Scholar]
- Chappell J. B., Crofts A. R. (1965). Calcium ion accumulation and volume changes of isolated liver mitochondria. Calcium ion-induced swelling. Biochem. J. 95, 378–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvin C., De Oliveira F., Ronot X., Mousseau M., Leverve X., Fontaine E. (2001). Rotenone inhibits the mitochondrial permeability transition-induced cell death in U937 and KB cells. J. Biol. Chem. 276, 41394–41398 10.1074/jbc.M106417200 [DOI] [PubMed] [Google Scholar]
- Chiara F., Castellaro D., Marin O., Petronilli V., Brusilow W. S., Juhaszova M., et al. (2008). Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS ONE 3:e1852 10.1371/journal.pone.0001852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary J., Johnson K. M., Opipari A. W., Jr., Glick G. D. (2007). Inhibition of the mitochondrial F1F0-ATPase by ligands of the peripheral benzodiazepine receptor. Bioorg. Med. Chem. Lett. 17, 1667–1670 10.1016/j.bmcl.2006.12.102 [DOI] [PubMed] [Google Scholar]
- Clipstone N. A., Crabtree G. R. (1992). Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature 357, 695–697 10.1038/357695a0 [DOI] [PubMed] [Google Scholar]
- Coelho J. L., Vercesi A. E. (1980). Retention of Ca2+ by rat liver and rat heart mitochondria: effect of phosphate, Mg2+, and NAD(P) redox state. Arch. Biochem. Biophys. 204, 141–147 10.1016/0003-9861(80)90016-8 [DOI] [PubMed] [Google Scholar]
- Connern C. P., Halestrap A. P. (1992). Purification and N-terminal sequencing of peptidyl-prolyl cis-trans-isomerase from rat liver mitochondrial matrix reveals the existence of a distinct mitochondrial cyclophilin. Biochem. J. 284, 381–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connern C. P., Halestrap A. P. (1994). Recruitment of mitochondrial cyclophilin to the mitochondrial inner membrane under conditions of oxidative stress that enhance the opening of a calcium-sensitive non-specific channel. Biochem. J. 302, 321–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini P., Chernyak B. V., Petronilli V., Bernardi P. (1996). Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J. Biol. Chem. 271, 6746–6751 10.1074/jbc.271.12.6746 [DOI] [PubMed] [Google Scholar]
- Costantini P., Colonna R., Bernardi P. (1998). Induction of the mitochondrial permeability transition by N-ethylmaleimide depends on secondary oxidation of critical thiol groups. Potentiation by copper-ortho-phenanthroline without dimerization of the adenine nucleotide translocase. Biochim. Biophys. Acta 1365, 385–392 10.1016/S0005-2728(98)00090-5 [DOI] [PubMed] [Google Scholar]
- Crofts A. R., Chappell J. B. (1965). Calcium ion accumulation and volume changes of isolated rat liver mitochondria. Reversal of calcium ion-induced swelling. Biochem. J. 95, 387–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M., Costi A. (1988). Kinetic evidence for a heart mitochondrial pore activated by Ca2+, inorganic phosphate and oxidative stress. A potential mechanism for mitochondrial dysfunction during cellular Ca2+ overload. Eur. J. Biochem. 178, 489–501 10.1111/j.1432-1033.1988.tb14475.x [DOI] [PubMed] [Google Scholar]
- Crompton M., Ellinger H., Costi A. (1988). Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 255, 357–360 [PMC free article] [PubMed] [Google Scholar]
- Csukly K., Ascah A., Matas J., Gardiner P. F., Fontaine E., Burelle Y. (2006). Muscle denervation promotes opening of the permeability transition pore and increases the expression of cyclophilin D. J. Physiol. 574, 319–327 10.1113/jphysiol.2006.109702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiano M., Starkov A. A., Petri S., Kipiani K., Kiaei M., Mattiazzi M., et al. (2006). Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J. Neurochem. 96, 1349–1361 10.1111/j.1471-4159.2006.03619.x [DOI] [PubMed] [Google Scholar]
- Davidson A. M., Halestrap A. P. (1990). Partial inhibition by cyclosporin A of the swelling of liver mitochondria in vivo and in vitro induced by sub-micromolar [Ca2+], but not by butyrate. Evidence for two distinct swelling mechanisms. Biochem. J. 268, 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies K. M., Anselmi C., Wittig I., Faraldo-Gomez J. D., Kühlbrandt W. (2012). Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc. Natl. Acad. Sci. U.S.A. 109, 13602–13607 10.1073/pnas.1204593109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies K. M., Strauss M., Daum B., Kief J. H., Osiewacz H. D., Rycovska A., et al. (2011). Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc. Natl. Acad. Sci. U.S.A. 108, 14121–14126 10.1073/pnas.1103621108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaudin D., Geley S., Hirsch T., Castedo M., Marchetti P., Macho A., et al. (1997). Bcl-2 and Bcl-XL antagonize the mitochondrial dysfunction preceding nuclear apoptosis induced by chemotherapeutic agents. Cancer Res. 57, 62–67 [PubMed] [Google Scholar]
- De Marchi U., Basso E., Szabó I., Zoratti M. (2006). Electrophysiological characterization of the Cyclophilin D-deleted mitochondrial permeability transition pore. Mol. Membr. Biol. 23, 521–530 10.1080/09687860600907644 [DOI] [PubMed] [Google Scholar]
- Devun F., Walter L., Belliere J., Cottet-Rousselle C., Leverve X., Fontaine E. (2010). Ubiquinone analogs: a mitochondrial permeability transition pore-dependent pathway to selective cell death. PLoS ONE 5:e11792 10.1371/journal.pone.0011792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C., Fang M., Li Y., Li L., Wang X. (2000). Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102, 33–42 10.1016/S0092-8674(00)00008-8 [DOI] [PubMed] [Google Scholar]
- Duchen M. R., McGuinness O., Brown L. A., Crompton M. (1993). On the involvement of a cyclosporin A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovasc. Res. 27, 1790–1794 10.1093/cvr/27.10.1790 [DOI] [PubMed] [Google Scholar]
- Ekert P. G., Silke J., Connolly L. M., Reid G. E., Moritz R. L., Vaux D. L. (2000). Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102, 43–53 10.1016/S0092-8674(00)00009-X [DOI] [PubMed] [Google Scholar]
- Eliseev R. A., Malecki J., Lester T., Zhang Y., Humphrey J., Gunter T. E. (2009). Cyclophilin D interacts with Bcl2 and exerts an anti-apoptotic effect. J. Biol. Chem. 284, 9692–9699 10.1074/jbc.M808750200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Mir M. Y., Nogueira V., Fontaine E., Averet N., Rigoulet M., Leverve X. (2000). Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 275, 223–228 10.1074/jbc.275.1.223 [DOI] [PubMed] [Google Scholar]
- Elrod J. W., Wong R., Mishra S., Vagnozzi R. J., Sakthievel B., Goonasekera S. A., et al. (2010). Cyclophilin D controls mitochondrial pore-dependent Ca2+ exchange, metabolic flexibility, and propensity for heart failure in mice. J. Clin. Invest. 120, 3680–3687 10.1172/JCI43171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H., Chen M., Ding Y., Shang W., Xu J., Zhang X., et al. (2011). Imaging superoxide flash and metabolism-coupled mitochondrial permeability transition in living animals. Cell Res. 21, 1295–1304 10.1038/cr.2011.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G., Wittmann-Liebold B., Lang K., Kiefhaber T., Schmid F. X. (1989). Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 337, 476–478 10.1038/337476a0 [DOI] [PubMed] [Google Scholar]
- Fontaine E., Bernardi P. (1999). Progress on the mitochondrial permeability transition pore: regulation by complex I and ubiquinone analogs. J. Bioenerg. Biomembr. 31, 335–345 [DOI] [PubMed] [Google Scholar]
- Fontaine E., Eriksson O., Ichas F., Bernardi P. (1998a). Regulation of the permeability transition pore in skeletal muscle mitochondria. Modulation by electron flow through the respiratory chain complex I. J. Biol. Chem. 273, 12662–12668 10.1074/jbc.273.20.12662 [DOI] [PubMed] [Google Scholar]
- Fontaine E., Ichas F., Bernardi P. (1998b). A ubiquinone-binding site regulates the mitochondrial permeability transition pore. J. Biol. Chem. 273, 25734–25740 10.1074/jbc.273.40.25734 [DOI] [PubMed] [Google Scholar]
- Forte M., Bernardi P. (2006). The permeability transition and BCL-2 family proteins in apoptosis: co-conspirators or independent agents? Cell Death Differ. 13, 1287–1290 10.1038/sj.cdd.4401957 [DOI] [PubMed] [Google Scholar]
- Fournier N., Ducet G., Crevat A. (1987). Action of cyclosporine on mitochondrial calcium fluxes. J. Bioenerg. Biomembr. 19, 297–303 [DOI] [PubMed] [Google Scholar]
- Garcia-Perez C., Roy S. S., Naghdi S., Lin X., Davies E., Hajnoczky G. (2012). Bid-induced mitochondrial membrane permeabilization waves propagated by local reactive oxygen species (ROS) signaling. Proc. Natl. Acad. Sci. U.S.A. 109, 4497–4502 10.1073/pnas.1118244109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio V., Bisetto E., Soriano M. E., Dabbeni-Sala F., Basso E., Petronilli V., et al. (2009). Cyclophilin D modulates mitochondrial FOF1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 284, 33982–33988 10.1074/jbc.M109.020115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio V., Petronilli V., Ghelli A., Carelli V., Rugolo M., Lenaz G., et al. (2012). The effects of idebenone on mitochondrial bioenergetics. Biochim. Biophys. Acta 1817, 363–369 10.1016/j.bbabio.2011.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio V., Soriano M. E., Basso E., Bisetto E., Lippe G., Forte M. A., et al. (2010). Cyclophilin D in mitochondrial pathophysiology. Biochim. Biophys. Acta 1797, 1113–1118 10.1016/j.bbabio.2009.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio V., von Stockum S., Antoniel M., Fabbro A., Fogolari F., Forte M., et al. (2013). Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. U.S.A. 110, 5887–5892 10.1073/pnas.1217823110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths E. J., Halestrap A. P. (1995). Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 307, 93–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigas B., Detaille D., Chauvin C., Batandier C., De Oliveira F., Fontaine E., et al. (2004). Metformin inhibits mitochondrial permeability transition and cell death: a pharmacological in vitro study. Biochem. J. 382, 877–884 10.1042/BJ20040885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter T. E., Pfeiffer D. R. (1990). Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 258, C755–C786 [DOI] [PubMed] [Google Scholar]
- Gurney M. E., Pu H., Chiu A. Y., Dal Canto M. C., Polchow C. Y., Alexander D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264, 1772–1775 10.1126/science.8209258 [DOI] [PubMed] [Google Scholar]
- Halestrap A. P. (2004). Mitochondrial permeability: dual role for the ADP/ATP translocator? Nature 430, 1 p following 983. [DOI] [PubMed] [Google Scholar]
- Haworth R. A., Hunter D. R. (1979). The Ca2+-induced membrane transition of rat liver mitochondria. II. Nature of the Ca2+ trigger site. Arch. Biochem. Biophys. 195, 460–467 [DOI] [PubMed] [Google Scholar]
- He L., Lemasters J. J. (2002). Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett. 512, 1–7 [DOI] [PubMed] [Google Scholar]
- Hegde R., Srinivasula S. M., Zhang Z., Wassell R., Mukattash R., Cilenti L., et al. (2001). Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 277, 432–438 10.1074/jbc.M109721200 [DOI] [PubMed] [Google Scholar]
- Herick K., Krämer R., Lühring H. (1997). Patch clamp investigation into the phosphate carrier from Saccharomyces cerevisiae mitochondria. Biochim. Biophys. Acta 1321, 207–220 10.1016/S0005-2728(97)00050-9 [DOI] [PubMed] [Google Scholar]
- Hunter D. R., Haworth R. A. (1979a). The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch. Biochem. Biophys. 195, 453–459 [DOI] [PubMed] [Google Scholar]
- Hunter D. R., Haworth R. A. (1979b). The Ca2+-induced membrane transition in mitochondria. III. Transitional Ca2+ release. Arch. Biochem. Biophys. 195, 468–477 [DOI] [PubMed] [Google Scholar]
- Hunter D. R., Haworth R. A., Southard J. H. (1976). Relationship between configuration, function, and permeability in calcium-treated mitochondria. J. Biol. Chem. 251, 5069–5077 [PubMed] [Google Scholar]
- Hunter F. E., Jr., Ford L. (1955). Inactivation of oxidative and phosphorylative systems in mitochondria by preincubation with phosphate and other ions. J. Biol. Chem. 216, 357–369 [PubMed] [Google Scholar]
- Hüser J., Blatter L. A. (1999). Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem. J. 343, 311–317 [PMC free article] [PubMed] [Google Scholar]
- Hüser J., Rechenmacher C. E., Blatter L. A. (1998). Imaging the permeability pore transition in single mitochondria. Biophys. J. 74, 2129–2137 10.1016/S0006-3495(98)77920-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichas F., Jouaville L. S., Mazat J. P. (1997). Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 89, 1145–1153 10.1016/S0092-8674(00)80301-3 [DOI] [PubMed] [Google Scholar]
- Imberti R., Nieminen A. L., Herman B., Lemasters J. J. (1992). Synergism of cyclosporin A and phospholipase inhibitors in protection against lethal injury to rat hepatocytes from oxidant chemicals. Res. Commun. Chem. Pathol. Pharmacol. 78, 27–38 [PubMed] [Google Scholar]
- Johnson K. M., Chen X., Boitano A., Swenson L., Opipari A. W., Jr., Glick G. D. (2005). Identification and validation of the mitochondrial F1F0-ATPase as the molecular target of the immunomodulatory benzodiazepine Bz-423. Chem. Biol. 12, 485–496 10.1016/j.chembiol.2005.02.012 [DOI] [PubMed] [Google Scholar]
- Jung D. W., Bradshaw P. C., Pfeiffer D. R. (1997). Properties of a cyclosporin-insensitive permeability transition pore in yeast mitochondria. J. Biol. Chem. 272, 21104–21112 10.1074/jbc.272.34.21104 [DOI] [PubMed] [Google Scholar]
- Kang B. H., Plescia J., Dohi T., Rosa J., Doxsey S. J., Altieri D. C. (2007). Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 131, 257–270 10.1016/j.cell.2007.08.028 [DOI] [PubMed] [Google Scholar]
- Karch J., Molkentin J. D. (2012). Is p53 the long-sought molecular trigger for cyclophilin D-regulated mitochondrial permeability transition pore formation and necrosis? Circ. Res. 111, 1258–1260 10.1161/CIRCRESAHA.112.280990 [DOI] [PubMed] [Google Scholar]
- Kinnally K. W., Campo M. L., Tedeschi H. (1989). Mitochondrial channel activity studied by patch-clamping mitoplasts. J. Bioenerg. Biomembr. 21, 497–506 [DOI] [PubMed] [Google Scholar]
- Kinnally K. W., Zorov D. B., Antonenko Y. N., Snyder S. H., McEnery M. W., Tedeschi H. (1993). Mitochondrial benzodiazepine receptor linked to inner membrane ion channels by nanomolar actions of ligands. Proc. Natl. Acad. Sci. U.S.A. 90, 1374–1378 10.1073/pnas.90.4.1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohr M. J., Aponte A. M., Sun J., Wang G., Murphy E., Gucek M., et al. (2011). Characterization of potential S-nitrosylation sites in the myocardium. Am. J. Physiol. Heart Circ. Physiol. 300, H1327–H1335 10.1152/ajpheart.00997.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokoszka J. E., Waymire K. G., Levy S. E., Sligh J. E., Cai J., Jones D. P., et al. (2004). The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465 10.1038/nature02229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottke M., Adam V., Riesinger I., Bremm G., Bosch W., Brdiczka D., et al. (1988). Mitochondrial boundary membrane contact sites in brain: points of hexokinase and creatine kinase location, and control of Ca2+ transport. Biochim. Biophys. Acta 935, 87–102 10.1016/0005-2728(88)90111-9 [DOI] [PubMed] [Google Scholar]
- Kowaltowski A. J., Castilho R. F., Vercesi A. E. (2001). Mitochondrial permeability transition and oxidative stress. FEBS Lett. 495, 12–15 [DOI] [PubMed] [Google Scholar]
- Krauskopf A., Eriksson O., Craigen W. J., Forte M. A., Bernardi P. (2006). Properties of the permeability transition in VDAC1(-/-) mitochondria. Biochim. Biophys. Acta 1757, 590–595 10.1016/j.bbabio.2006.02.007 [DOI] [PubMed] [Google Scholar]
- Lehninger A. L. (1959). Reversal of various types of mitochondrial swelling by adenosine triphosphate. J. Biol. Chem. 234, 2465–2471 [PubMed] [Google Scholar]
- Lehninger A. L., Remmert L. F. (1959). An endogenous uncoupling and swelling agent in liver mitochondria and its enzymic function. J. Biol. Chem. 234, 2459–2464 [PubMed] [Google Scholar]
- Lê-Quôc K., Lê-Quôc D. (1982). Control of the mitochondrial inner membrane permeability by sulfhydryl groups. Arch. Biochem. Biophys. 216, 639–651 [DOI] [PubMed] [Google Scholar]
- Lê-Quôc K., Lê-Quôc D. (1985). Crucial role of sulfhydryl groups in the mitochondrial inner membrane structure. J. Biol. Chem. 260, 7422–7428 [PubMed] [Google Scholar]
- Leung A. W., Halestrap A. P. (2008). Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 1777, 946–952 10.1016/j.bbabio.2008.03.009 [DOI] [PubMed] [Google Scholar]
- Leung A. W., Varanyuwatana P., Halestrap A. P. (2008). The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem. 283, 26312–26323 10.1074/jbc.M805235200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Chauvin C., De P. D., De O. F., Gharib A., Vial G., et al. (2012). Inhibition of complex I regulates the mitochondrial permeability transition through a phosphate-sensitive inhibitory site masked by cyclophilin D. Biochim. Biophys. Acta 1817, 1628–1634 10.1016/j.bbabio.2012.05.011 [DOI] [PubMed] [Google Scholar]
- Li L. Y., Luo X., Wang X. (2001). Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412, 95–99 10.1038/35083620 [DOI] [PubMed] [Google Scholar]
- Liu J., Farmer J. D. J., Lane W. S., Friedman J., Weissman I., Schreiber S. L. (1991). Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66, 807–815 10.1016/0092-8674(91)90124-H [DOI] [PubMed] [Google Scholar]
- Liu X., Kim C. N., Yang J., Jemmerson R., Wang X. (1996). Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157 10.1016/S0092-8674(00)80085-9 [DOI] [PubMed] [Google Scholar]
- Madesh M., Hajnoczky G. (2001). VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol. 155, 1003–1015 10.1083/jcb.200105057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manon S., Roucou X., Guerin M., Rigoulet M., Guerin B. (1998). Characterization of the yeast mitochondria unselective channel: a counterpart to the mammalian permeability transition pore? J. Bioenerg. Biomembr. 30, 419–429 [DOI] [PubMed] [Google Scholar]
- Marzo I., Brenner C., Zamzami N., Susin S. A., Beutner G., Brdiczka D., et al. (1998). The permeability transition pore complex: a target for apoptosis regulation by caspases and bcl-2-related proteins. J. Exp. Med. 187, 1261–1271 10.1084/jem.187.8.1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menze M. A., Fortner G., Nag S., Hand S. C. (2010). Mechanisms of apoptosis in Crustacea: what conditions induce versus suppress cell death? Apoptosis 15, 293–312 10.1007/s10495-009-0443-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menze M. A., Hutchinson K., Laborde S. M., Hand S. C. (2005). Mitochondrial permeability transition in the crustacean Artemia franciscana: absence of a calcium-regulated pore in the face of profound calcium storage. Am. J. Physiol. 289, R68–R76 10.1152/ajpregu.00844.2004 [DOI] [PubMed] [Google Scholar]
- Mironova G. D., Gateau-Roesch O., Levrat C., Gritsenko E., Pavlov E., Lazareva A. V., et al. (2001). Palmitic and stearic acids bind Ca2+ with high affinity and form nonspecific channels in black-lipid membranes. Possible relation to Ca2+-activated mitochondrial pores. J. Bioenerg. Biomembr. 33, 319–331 [DOI] [PubMed] [Google Scholar]
- Mitchell P. (1961). Coupling of phosphorylation to electron and hydrogen transfer by a chemio-osmotic type of mechanism. Nature 191, 144–148 [DOI] [PubMed] [Google Scholar]
- Mitchell P. (2011). Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. 1966. Biochim. Biophys. Acta 1807, 1507–1538 10.1016/j.bbabio.2011.09.018 [DOI] [PubMed] [Google Scholar]
- Moreno G., Poussin K., Ricchelli F., Salet C. (2001). The effects of singlet oxygen produced by photodynamic action on the mitochondrial permeability transition differ in accordance with the localization of the sensitizer. Arch. Biochem. Biophys. 386, 243–250 10.1006/abbi.2000.2200 [DOI] [PubMed] [Google Scholar]
- Nakagawa T., Shimizu S., Watanabe T., Yamaguchi O., Otsu K., Yamagata H., et al. (2005). Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434, 652–658 10.1038/nature03317 [DOI] [PubMed] [Google Scholar]
- Nguyen T. T., Stevens M. V., Kohr M., Steenbergen C., Sack M. N., Murphy E. (2011). Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J. Biol. Chem. 286, 40184–40192 10.1074/jbc.M111.243469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolli A., Basso E., Petronilli V., Wenger R. M., Bernardi P. (1996). Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, a cyclosporin A-sensitive channel. J. Biol. Chem. 271, 2185–2192 10.1074/jbc.271.4.2185 [DOI] [PubMed] [Google Scholar]
- Nicolli A., Petronilli V., Bernardi P. (1993). Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by matrix pH. Evidence that the pore open-closed probability is regulated by reversible histidine protonation. Biochemistry 32, 4461–4465 [DOI] [PubMed] [Google Scholar]
- Parone P. A., Da C. S., Han J. S., McAlonis-Downes M., Vetto A. P., Lee S. K., et al. (2013). Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J. Neurosci. 33, 4657–4671 10.1523/JNEUROSCI.1119-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino J. G., Chen S. T., Tafani M., Snyder J. W., Farber J. L. (1998). The overexpression of Bax produces cell death upon induction of the mitochondrial permeability transition. J. Biol. Chem. 273, 7770–7775 10.1074/jbc.273.13.7770 [DOI] [PubMed] [Google Scholar]
- Pastorino J. G., Hoek J. B., Shulga N. (2005). Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 65, 10545–10554 10.1158/0008-5472.CAN-05-1925 [DOI] [PubMed] [Google Scholar]
- Pastorino J. G., Shulga N., Hoek J. B. (2002). Mitochondrial binding of hexokinase II inhibits bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 277, 7610–7618 10.1074/jbc.M109950200 [DOI] [PubMed] [Google Scholar]
- Pastorino J. G., Simbula G., Gilfor E., Hoek J. B., Farber J. L. (1994). Protoporphyrin, IX, an endogenous ligand of the peripheral benzodiazepine receptor, potentiates induction of the mitochondrial permeability transition and the killing of cultured hepatocytes by rotenone. J. Biol. Chem. 269, 31041–31046 [PubMed] [Google Scholar]
- Pastorino J. G., Snyder J. W., Serroni A., Hoek J. B., Farber J. L. (1993). Cyclosporin and carnitine prevent the anoxic death of cultured hepatocytes by inhibiting the mitochondrial permeability transition. J. Biol. Chem. 268, 13791–13798 [PubMed] [Google Scholar]
- Pastorino J. G., Tafani M., Rothman R. J., Marcineviciute A., Hoek J. B., Farber J. L. (1999). Functional consequences of the sustained or transient activation by Bax of the mitochondrial permeability transition pore. J. Biol. Chem. 274, 31734–31739 10.1074/jbc.274.44.31734 [DOI] [PubMed] [Google Scholar]
- Pavlov E., Zakharian E., Bladen C., Diao C. T., Grimbly C., Reusch R. N., et al. (2005). A large, voltage-dependent channel, isolated from mitochondria by water-free chloroform extraction. Biophys. J. 88, 2614–2625 10.1529/biophysj.104.057281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronilli V., Miotto G., Canton M., Brini M., Colonna R., Bernardi P., et al. (1999). Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys. J. 76, 725–734 10.1016/S0006-3495(99)77239-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronilli V., Nicolli A., Costantini P., Colonna R., Bernardi P. (1994). Regulation of the permeability transition pore, a voltage-dependent mitochondrial channel inhibited by cyclosporin A. Biochim. Biophys. Acta 1187, 255–259 [DOI] [PubMed] [Google Scholar]
- Petronilli V., Penzo D., Scorrano L., Bernardi P., Di Lisa F. (2001). The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem. 276, 12030–12034 10.1074/jbc.M010604200 [DOI] [PubMed] [Google Scholar]
- Petronilli V., Šileikyte J., Zulian A., Dabbeni-Sala F., Jori G., Gobbo S., et al. (2009). Switch from inhibition to activation of the mitochondrial permeability transition during hematoporphyrin-mediated photooxidative stress. Unmasking pore-regulating external thiols. Biochim. Biophys. Acta 1787, 897–904 10.1016/j.bbabio.2009.03.014 [DOI] [PubMed] [Google Scholar]
- Petronilli V., Szabó I., Zoratti M. (1989). The inner mitochondrial membrane contains ion-conducting channels similar to those found in bacteria. FEBS Lett. 259, 137–143 10.1016/0014-5793(89)81513-3 [DOI] [PubMed] [Google Scholar]
- Pfeiffer D. R., Kuo T. H., Tchen T. T. (1976). Some effects of Ca2+, Mg2+, and Mn2+ on the ultrastructure, light-scattering properties, and malic enzyme activity of adrenal cortex mitochondria. Arch. Biochem. Biophys. 176, 556–563 [DOI] [PubMed] [Google Scholar]
- Pfeiffer D. R., Tchen T. T. (1973). The role of Ca2+ in control of malic enzyme activity in bovine adrenal cortex mitochondria. Biochem. Biophys. Res. Commun. 50, 807–813 10.1016/0006-291X(73)91316-8 [DOI] [PubMed] [Google Scholar]
- Pfeiffer D. R., Tchen T. T. (1975). The activation of adrenal cortex mitochondrial malic enzyme by Ca2+ and Mg2+. Biochemistry 14, 89–96 [DOI] [PubMed] [Google Scholar]
- Raaflaub J. (1953a). Die schwellung isolierter leberzell mitochondrien und ihre physikalisch beeinfluβarkeit. Helv. Physiol. Pharmacol. Acta 11, 142–156 [PubMed] [Google Scholar]
- Raaflaub J. (1953b). Über den wirkungsmechanismus von adenosintriphosphat (ATP) als cofaktor isolierter mitochondrien. Helv. Physiol. Pharmacol. Acta 11, 157–165 [PubMed] [Google Scholar]
- Rapizzi E., Pinton P., Szabadkai G., Wieckowski M. R., Vandecasteele G., Baird G., et al. (2002). Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 159, 613–624 10.1083/jcb.200205091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasola A., Bernardi P. (2007). The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 12, 815–833 10.1007/s10495-007-0723-y [DOI] [PubMed] [Google Scholar]
- Rasola A., Bernardi P. (2011). Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 50, 222–233 10.1016/j.ceca.2011.04.007 [DOI] [PubMed] [Google Scholar]
- Rasola A., Sciacovelli M., Chiara F., Pantic B., Brusilow W. S., Bernardi P. (2010). Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc. Natl. Acad. Sci. U.S.A. 107, 726–731 10.1073/pnas.0912742107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees D. M., Leslie A. G., Walker J. E. (2009). The structure of the membrane extrinsic region of bovine ATP synthase. Proc. Natl. Acad. Sci. U.S.A. 106, 21597–21601 10.1073/pnas.0910365106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricchelli F., Dabbeni-Sala F., Petronilli V., Bernardi P., Hopkins B., Bova S. (2005). Species-specific modulation of the mitochondrial permeability transition by norbormide. Biochim. Biophys. Acta 1708, 178–186 10.1016/j.bbabio.2005.03.002 [DOI] [PubMed] [Google Scholar]
- Ricchelli F., Šileikyte J., Bernardi P. (2011). Shedding light on the mitochondrial permeability transition. Biochim. Biophys. Acta 1807, 482–490 10.1016/j.bbabio.2011.02.012 [DOI] [PubMed] [Google Scholar]
- Riley W. W., Jr., Pfeiffer D. R. (1985). Relationships between Ca2+ release, Ca2+ cycling, and Ca2+- mediated permeability changes in mitochondria. J. Biol. Chem. 260, 12416–12425 [PubMed] [Google Scholar]
- Salet C., Moreno G., Ricchelli F., Bernardi P. (1997). Singlet oxygen produced by photodynamic action causes inactivation of the mitochondrial permeability transition pore. J. Biol. Chem. 272, 21938–21943 10.1074/jbc.272.35.21938 [DOI] [PubMed] [Google Scholar]
- Schinzel A. C., Takeuchi O., Huang Z., Fisher J. K., Zhou Z., Rubens J., et al. (2005). Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. U.S.A. 102, 12005–12010 10.1073/pnas.0505294102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzlander M., Logan D. C., Fricker M. D., Sweetlove L. J. (2011). The circularly permuted yellow fluorescent protein cpYFP that has been used as a superoxide probe is highly responsive to pH but not superoxide in mitochondria: implications for the existence of superoxide ‘flashes’. Biochem. J. 437, 381–387 10.1042/BJ20110883 [DOI] [PubMed] [Google Scholar]
- Schwarzlander M., Murphy M. P., Duchen M. R., Logan D. C., Fricker M. D., Halestrap A. P., et al. (2012). Mitochondrial ‘flashes’: a radical concept repHined. Trends Cell Biol. 22, 503–508 10.1016/j.tcb.2012.07.007 [DOI] [PubMed] [Google Scholar]
- Scorrano L., Ashiya M., Buttle K., Weiler S., Oakes S. A., Mannella C. A., et al. (2002). A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2, 55–67 10.1016/S1534-5807(01)00116-2 [DOI] [PubMed] [Google Scholar]
- Scorrano L., Oakes S. A., Opferman J. T., Cheng E. H., Sorcinelli M. D., Pozzan T., et al. (2003). BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300, 135 10.1126/science.1081208 [DOI] [PubMed] [Google Scholar]
- Scorrano L., Penzo D., Petronilli V., Pagano F., Bernardi P. (2001). Arachidonic acid causes cell death through the mitochondrial permeability transition. Implications for tumor necrosis factor-α apoptotic signaling. J. Biol. Chem. 276, 12035–12040 10.1074/jbc.M010603200 [DOI] [PubMed] [Google Scholar]
- Shulga N., Pastorino J. G. (2010). Ethanol sensitizes mitochondria to the permeability transition by inhibiting deacetylation of cyclophilin-D mediated by sirtuin-3. J. Cell Sci. 123, 4117–4127 10.1242/jcs.073502 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Siemen D., Ziemer M. (2013). What is the nature of the mitochondrial permeability transition pore and what is it not? IUBMB Life 65, 255–262 10.1002/iub.1130 [DOI] [PubMed] [Google Scholar]
- Šileikyte J., Petronilli V., Zulian A., Dabbeni-Sala F., Tognon G., Nikolov P., et al. (2011). Regulation of the inner membrane mitochondrial permeability transition by the outer membrane translocator protein (peripheral benzodiazepine receptor). J. Biol. Chem. 286, 1046–1053 10.1074/jbc.M110.172486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliprandi D., Siliprandi N., Toninello A. (1983). On the relationship between calcium and phosphate transport, transmembrane potential and acetoacetate-induced oxidation of pyridine nucleotides in rat liver mitochondria. Eur. J. Biochem. 130, 173–175 10.1111/j.1432-1033.1983.tb07133.x [DOI] [PubMed] [Google Scholar]
- Sinha Roy S., Madesh M., Davies E., Antonsson B., Danial N., Hajnoczky G. (2009). Bad targets the permeability transition pore independent of Bax or Bak to switch between Ca2+-dependent cell survival and death. Mol. Cell 33, 377–388 10.1016/j.molcel.2009.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolove P. M., Shinaberry R. G. (1988). Na+-independent release of Ca2+ from rat heart mitochondria. Induction by adriamycin aglycone. Biochem. Pharmacol. 37, 803–812 [DOI] [PubMed] [Google Scholar]
- Sorgato M. C., Keller B. U., Stühmer W. (1987). Patch-clamping of the inner mitochondrial membrane reveals a voltage-dependent ion channel. Nature 330, 498–500 10.1038/330498a0 [DOI] [PubMed] [Google Scholar]
- Stelzer A. C., Frazee R. W., Van Huis C., Cleary J., Opipari A. W., Jr., Glick G. D., et al. (2010). NMR studies of an immunomodulatory benzodiazepine binding to its molecular target on the mitochondrial F1F0-ATPase. Biopolymers 93, 85–92 10.1002/bip.21306 [DOI] [PubMed] [Google Scholar]
- Strauss M., Hofhaus G., Schroder R. R., Kühlbrandt W. (2008). Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 27, 1154–1160 10.1038/emboj.2008.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan A., Sokolove P. M. (2001). Free fatty acid effects on mitochondrial permeability: an overview. Arch. Biochem. Biophys. 386, 52–61 10.1006/abbi.2000.2195 [DOI] [PubMed] [Google Scholar]
- Susin S. A., Zamzami N., Castedo M., Hirsch T., Marchetti P., Macho A., et al. (1996). Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J. Exp. Med. 184, 1331–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó I., Bernardi P., Zoratti M. (1992). Modulation of the mitochondrial megachannel by divalent cations and protons. J. Biol. Chem. 267, 2940–2946 [PubMed] [Google Scholar]
- Szabó I., Bock J., Grassme H., Soddemann M., Wilker B., Lang F., et al. (2008). Mitochondrial potassium channel Kv1.3 mediates Bax-induced apoptosis in lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 105, 14861–14866 10.1073/pnas.0804236105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó I., Soddemann M., Leanza L., Zoratti M., Gulbins E. (2011). Single-point mutations of a lysine residue change function of Bax and Bcl-xL expressed in Bax- and Bak-less mouse embryonic fibroblasts: novel insights into the molecular mechanisms of Bax-induced apoptosis. Cell Death Differ. 18, 427–438 10.1038/cdd.2010.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó I., Zoratti M. (1991). The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J. Biol. Chem. 266, 3376–3379 [PubMed] [Google Scholar]
- Szabó I., Zoratti M. (1992). The mitochondrial megachannel is the permeability transition pore. J. Bioenerg. Biomembr. 24, 111–117 [DOI] [PubMed] [Google Scholar]
- Tafani M., Karpinich N. O., Hurster K. A., Pastorino J. G., Schneider T., Russo M. A., et al. (2002). Cytochrome c release upon Fas receptor activation depends on translocation of full-length bid and the induction of the mitochondrial permeability transition. J. Biol. Chem. 277, 10073–10082 10.1074/jbc.M111350200 [DOI] [PubMed] [Google Scholar]
- Takahashi N., Hayano T., Suzuki M. (1989). Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature 337, 473–475 10.1038/337473a0 [DOI] [PubMed] [Google Scholar]
- Tapley D. F. (1956). The effect of thyroxine and other substances on the swelling of isolated rat liver mitochondria. J. Biol. Chem. 222, 325–339 [PubMed] [Google Scholar]
- Thomas D., Bron P., Weimann T., Dautant A., Giraud M. F., Paumard P., et al. (2008). Supramolecular organization of the yeast F1Fo-ATP synthase. Biol. Cell 100, 591–601 10.1042/BC20080022 [DOI] [PubMed] [Google Scholar]
- Tomasetig L., Di Pancrazio F., Harris D. A., Mavelli I., Lippe G. (2002). Dimerization of F0F1ATP synthase from bovine heart is independent from the binding of the inhibitor protein IF1. Biochim. Biophys. Acta 1556, 133–141 10.1016/S0005-2728(02)00344-4 [DOI] [PubMed] [Google Scholar]
- Uribe-Carvajal S., Luévano-Martìnez L. A., Guerrero-Castillo S., Cabrera-Orefice A., Corona-de-la-Peña N. A., Gutiérrez-Aguilar M. (2011). Mitochondrial unselective channels throughout the eukaryotic domain. Mitochondrion 11, 382–390 10.1016/j.mito.2011.02.004 [DOI] [PubMed] [Google Scholar]
- Vaseva A. V., Marchenko N. D., Ji K., Tsirka S. E., Holzmann S., Moll U. M. (2012). p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149, 1536–1548 10.1016/j.cell.2012.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercesi A. E. (1984). Dissociation of NAD(P)+-stimulated mitochondrial Ca2+ efflux from swelling and membrane damage. Arch. Biochem. Biophys. 232, 86–91 10.1016/0003-9861(84)90523-X [DOI] [PubMed] [Google Scholar]
- Verma A., Nye J. S., Snyder S. H. (1987). Porphyrins are endogenous ligands for the mitochondrial (peripheral-type) benzodiazepine receptor. Proc. Natl. Acad. Sci. U.S.A. 84, 2256–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov A., Scarpa A., Chance B. (1972). Calcium and pyridine nucleotide interaction in mitochondrial membranes. Arch. Biochem. Biophys. 152, 646–654 10.1016/0003-9861(72)90261-5 [DOI] [PubMed] [Google Scholar]
- von Stockum S., Basso E., Petronilli V., Sabatelli P., Forte M. A., Bernardi P. (2011). Properties of Ca2+ transport in mitochondria of Drosophila melanogaster. J. Biol. Chem. 286, 41163–41170 10.1074/jbc.M111.268375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. T., Zydowsky L. D., McKeon F. D. (1992). Cyclosporin, A, the cyclophilin class of peptidylprolyl isomerases, and blockade of T cell signal transduction. J. Biol. Chem. 267, 13115–13118 [PubMed] [Google Scholar]
- Walter L., Miyoshi H., Leverve X., Bernardi P., Fontaine E. (2002). Regulation of the mitochondrial permeability transition pore by ubiquinone analogs. A progress report. Free Radic. Res. 36, 405–412 [DOI] [PubMed] [Google Scholar]
- Walter L., Nogueira V., Leverve X., Bernardi P., Fontaine E. (2000). Three classes of ubiquinone analogs regulate the mitochondrial permeability transition pore through a common site. J. Biol. Chem. 275, 29521–29527 10.1074/jbc.M004128200 [DOI] [PubMed] [Google Scholar]
- Wang J. Q., Chen Q., Wang X., Wang Q. C., Wang Y., Cheng H. P., et al. (2013). Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in Huntington disease. J. Biol. Chem. 288, 3070–3084 10.1074/jbc.M112.407726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P., Heitman J. (2005). The cyclophilins. Genome Biol. 6, 226.1–226.6 10.1186/gb-2005-6-7-226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Fang H., Groom L., Cheng A., Zhang W., Liu J., et al. (2008). Superoxide flashes in single mitochondria. Cell 134, 279–290 10.1016/j.cell.2008.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei-Lapierre L., Gong G., Gerstner B. J., Ducreux S., Yule D. I., Pouvreau S., et al. (2013). Respective contribution of mitochondrial superoxide and pH to Mt-cpYFP flash activity. J. Biol. Chem. 288, 10567–10577 10.1074/jbc.M113.455709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtczak L., Lehninger A. L. (1961). Formation and disappearance of an endogenous uncoupling factor during swelling and contraction of mitochondria. Biochim. Biophys. Acta 51, 442–456 [DOI] [PubMed] [Google Scholar]
- Woodfield K. Y., Price N. T., Halestrap A. P. (1997). cDNA cloning of rat mitochondrial cyclophilin. Biochim. Biophys. Acta 1351, 27–30 10.1016/S0167-4781(97)00017-1 [DOI] [PubMed] [Google Scholar]
- Yamada A., Yamamoto T., Yoshimura Y., Gouda S., Kawashima S., Yamazaki N., et al. (2009). Ca2+-induced permeability transition can be observed even in yeast mitochondria under optimized experimental conditions. Biochim. Biophys. Acta 1787, 1486–1491 10.1016/j.bbabio.2009.07.001 [DOI] [PubMed] [Google Scholar]
- Youle R. J., Narendra D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell. Biol. 12, 9–14 10.1038/nrm3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami N., Kroemer G. (2001). The mitochondrion in apoptosis: how Pandora's box opens. Nat. Rev. Mol. Cell. Biol. 2, 67–71 10.1038/35048073 [DOI] [PubMed] [Google Scholar]
- Zborowski J., Wojtczak L. (1963). Induction of swelling of liver mitochondria by fatty acids of various chain length. Biochim. Biophys. Acta 70, 596–598 [DOI] [PubMed] [Google Scholar]
- Zoeteweij J. P., van de Water B., de Bont H. J., Mulder G. J., Nagelkerke J. F. (1993). Calcium-induced cytotoxicity in hepatocytes after exposure to extracellular ATP is dependent on inorganic phosphate. Effects on mitochondrial calcium. J. Biol. Chem. 268, 3384–3388 [PubMed] [Google Scholar]
- Zoratti M., Szabó I., De Marchi U. (2005). Mitochondrial permeability transitions: how many doors to the house? Biochim. Biophys. Acta 1706, 40–52 10.1016/j.bbabio.2004.10.006 [DOI] [PubMed] [Google Scholar]
- Zulian A., Petronilli V., Bova S., Dabbeni-Sala F., Cargnelli G., Cavalli M., et al. (2007). Assessing the molecular basis for rat-selective induction of the mitochondrial permeability transition by norbormide. Biochim. Biophys. Acta 1767, 980–988 10.1016/j.bbabio.2007.04.002 [DOI] [PubMed] [Google Scholar]