

Abstract
Perforin-deficient mice serve as models for familial hemophagocytic lymphohistiocytosis (FHL), a uniformly fatal disease associated with viral infection of perforin-deficient humans. Naïve perforin-deficient (PKO) BALB/c mice survive while vaccinated PKO mice containing virus-specific memory CD8 T-cells rapidly succumb to LCMV infection. Thus, vaccination converts a non-lethal persistent infection into a fatal disease mediated by virus-specific memory CD8 T-cells. Here, we determine the extent to which vaccination-induced mortality in PKO mice following LCMV challenge is due to differences in vaccine modalities, the quantity or epitope specificity of memory CD8 T-cells. We show that LCMV-induced mortality in immune PKO mice is independent of vaccine modalities and that the starting number of memory CD8 T-cells specific to the immunodominant epitope NP118-226 dictates the magnitude of secondary CD8 T-cell expansion, the inability to regulate production of CD8 T-cell-derived IFN-γ, and mortality in the vaccinated PKO mice. Importantly, mortality is determined by the epitope specificity of memory CD8 T-cells and the associated degree of functional exhaustion and cytokine dysregulation but not the absolute magnitude of CD8 T-cell expansion. These data suggest that a deeper understanding of the parameters that influence the outcome of vaccine-induced diseases would aid rational vaccine design to minimize adverse outcomes after infection.
Introduction
Following infection or immunization, Ag-specific CD8 T-cells undergo vigorous expansion in numbers and differentiation into effector cells [1-6] that are capable of perforin-dependent cytolysis and production of cytokines such as IFN-γ and TNF [7]. Tight regulation of cytolysis and cytokine production by effector and memory CD8 T-cells is thought to minimize immunopathology [8]. CD8 T-cell responses to infection can be associated with lethal immunopathology as evidenced by uniform, perforin-dependent mortality after intracranial injection of mice with lymphocytic choriomeningitis (LCMV) [9, 10]. In addition to its cytotoxic function in the granule exocytosis effector pathway in CD8 T-cells and NK cells [11], perforin has also been shown to regulate other aspects of the Ag-specific CD8 T-cell response, including the degree of proliferative expansion in a bacterial infection [12], exhaustion in chronic viral infection [13, 14], and survival of CD8 T-cells in models of graft-versus-host disease [15]. However, the precise role of perforin in regulating these aspects of the CD8 T-cell response is still unclear. In particular, the role of perforin in regulating the secondary CD8 T-cell response to infection has not been well characterized.
Additionally, perforin-deficient (PKO) mice serve as a clinically relevant model for the human disease, Familial Hemophagocytic Lymphohistiocytosis (FHL) [16-19]. FHL is a rare, but uniformly fatal, autosomal recessive immune disorder that is characterized by massive activation of T cells and macrophages with an associated “inflammatory cytokine storm” that results in host mortality [20]. The clinical manifestation of FHL in humans are often linked to viral infections [21, 22] and the clinical severity and age of disease onset correlate with the degree to which perforin function is impaired [20, 23-25].
The number of memory CD8 T-cells generated by infection or vaccination correlates strongly with the degree of protection observed. Thus, effective vaccination strategies aim to increase the number of protective memory CD8 T-cells. Since perforin is a critical cytotoxic CD8 T-cell effector molecule, perforin deficiency results in immunocompromised state in the host. However, in some models of infection (ie, Listeria monocytogenes infection), immunity can be restored by increasing memory CD8 T-cell numbers even in the absence of perforin [26]. Thus, PKO hosts should theoretically benefit from vaccination to increase memory CD8 T-cell responses. PKO mice fail to clear primary LCMV infection [9, 11]. However, in contrast to improved immunity against LM by vaccination [27], we showed that vaccination of PKO BALB/c mice with attenuated recombinant LM expressing the dominant LCMV NP118-126 epitope resulted in massive LCMV-specific CD8 T-cell expansion, dysregulated production of CD8 T-cell-derived IFN-γ, and increased mortality following LCMV challenge [16]. Thus, while vaccination generally enhances antimicrobial immunity, it can also evoke lethal immunopathology or exacerbate the disease.
MATERIALS AND METHODS
Mice
BALB/c-PKO mice (H-2d MHC; 8-16 weeks of age) [12, 27] were maintained by brother-sister mating under specific pathogen-free conditions until initiation of experiments. Following LCMV infection PKO mice were monitored daily for weight loss. Mice that lost > 30% of their starting weight were euthanized per IACUC guidelines.
Dendritic cells, Bacteria, Virus and Immunization
Peptide-coated splenic DC were generated as described [28]. Attenuated (actA-deficient) Listeria monocytogenes strains DP-L1942 (att LM) [29], XFL303actA-(att LM-NP118) [30], and att LM-CS252 [31] are resistant to streptomycin and were used as described [16]. The Armstrong strain of LCMV was prepared as described [12]. Viral titers in homogenates of spleen were determined by plaque assay on VERO cells as described [32].
Adoptive-Transfer Experiments
Naïve female PKO mice were immunized with 1×107 cfu att LM-NP118 and the memory time point (day 100) spleen cells were analyzed for the frequency and phenotype of NP118-specific CD8 T-cells via FACS. For adoptive transfer experiment, groups of naïve PKO mice received splenocytes from memory mice containing the indicated numbers of NP118-specific memory CD8 T-cells one day before LCMV-Arm infection.
Quantification and phenotypic analysis of antigen-specific T-cells
The magnitude of the epitope-specific CD8 T-cell response was determined either by intracellular IFN-γ staining (ICS) after 5-6hr incubation in brefeldin A, in the presence or absence of 200 nM of indicated peptide or MHC class I tetramer staining as described [33]. ICS from blood was done in the presence of peptide-coated P815 cells.
Antibodies, Peptides, and MHC Class I Tetramers
We used antibodies with the indicated specificity and with appropriate combination of fluorochromes: IFN-γ (clone XMG1.2, eBioscience), CD8 (53-6.7, BD), Thy1.2 (53-2.1, BD), TNF (MP6-XT22, eBioscience), CD127 (A7R34, eBioscience), CD43 (1B11, BD), CD27 (LG.7F9, eBioscience), IL-2 (JES6-5H4, BD), CD62L (MEL-14, eBioscience), PD-1 (J43, eBioscience), KLRG-1 (2F1, eBioscience), Granzyme B (Caltag) and appropriate isotype controls. MHC class I tetramers specific for NP118 and GP283 prepared using published protocols [34, 35].
Statistical analysis
Significant differences between two groups were evaluated using a two tailed Student t test.
Results
Mortality in vaccinated PKO mice following LCMV-Arm challenge is independent of immunization modalities
Several experimental animal models demonstrated that vaccination to increase pathogen-specific memory CD8 T-cells can provide enhanced resistance against pathogen challenge in immunocompromised hosts. For example, PKO mice and IFN-γ- and TNF-deficient mice vaccinated with attenuated LM were better protected against virulent LM challenge in a CD8 T-cell dependent manner [27, 36-38]. However, robust memory CD8 T-cell recall responses to pathogen challenge could also lead to severe immunopathology and mortality. C57BL/6 mice vaccinated with recombinant Vaccinia virus expressing LCMV proteins succumbed to fatal meningitis after intracranial infection with a normally non-lethal dose of LCMV [39]. Similarly, we showed that BALB/c-PKO mice that were vaccinated with attenuated LM expressing the dominant LCMV epitope (NP118-126; H-2Ld restricted) succumbed to LCMV infection despite massive expansion of CD8 T-cells [16]. In contrast, PKO mice immunized with control attenuated LM survived the LCMV infection [16]. In this case, of the presence of that NP118-specific memory CD8 T cells in PKO hosts converts a non-lethal viral infection into devastating disease. However, it is unclear whether the vaccine-induced mortality in PKO mice is a unique consequence of Listeria-based vaccination.
To test the hypothesis that NP118-specific memory CD8 T-cells are the sole determinant of LCMV-induced mortality regardless of vaccine delivery systems, we immunized PKO with either attenuated LM expressing the H-2Ld restricted dominant epitope, NP118 (att LM-NP118) or with NP118-peptide coated dendritic cells (DC-NP118) (Figure 1A). Both immunization protocols generated NP118-specific memory CD8 T-cells with similar frequency, phenotype (CD127hi, KLRG-1lo, CD27hi, CD43lo) and functionality (IFN-γ, TNF and Granzyme B expression) (Figure 1B-D).
Figure 1. Mortality in vaccinated PKO mice following LCMV-Arm challenge is independent of immunization modalities.

A) Naïve BALB/c-PKO mice (15 /group) were immunized with either att LM-NP118 (~106 cfu) or DC-coated with NP118 peptide or PBS (non-immunized group). On day 102 p.i. all groups of mice were infected with LCMV-Arm (~5×105 pfu/mouse; i.p.). B) Representative dot plots and histograms showing the frequency and phenotype of NP118-specific memory CD8 T-cells as detected by peptide stimulated intracellular IFN-γ staining (ICS) at day 102 p.i. Frequency C) and total number D) of NP118-specific CD8 T-cells in the spleen as measured by Ld/NP118 tetramers at memory time point and 5 days following LCMV challenge (day 102+5). E) Survival after LCMV infection (9 /group). F) Frequency and phenotype of NP118-specific secondary effector CD8 T-cells as detected by ICS at day 5 post LCMV challenge. The numbers represent percentage of IFN-γ (B and F) or Ld/NP118 tetramer positive cells (C) of all splenic CD8 T-cells. The numbers inside the histograms represent the percentage of NP118-specific CD8 T-cells positive for the indicated marker. Shaded histograms represent isotype-control staining. Data are representative of at least 2 independent experiments. N.D. = not done.
Mice from both vaccinated groups and non-immunized controls were then challenged with LCMV-Arm. Consistent with our previously results [16], the NP118-specific CD8 T-cells in the att LM-NP118-vaccinated PKO mice underwent massive expansion, constituting ~75% of all CD8 T-cells in the spleen (~ 6-7×107 per spleen), at day 5 after LCMV challenge (Figure 1C-D). 100% of these mice succumbed to the infection based on morbidity criteria by day 11 post LCMV challenge (Figure 1E). In sharp contrast, non-immunized PKO mice exhibited relatively modest expansion of NP118-specific CD8 T-cells at day 5 post-LCMV infection and none of these mice succumbed (Figure 1C-E). Interestingly, massive expansion of that NP118-specific CD8 T cells was also observed in DC-NP118-vaccinated mice and all of those mice succumbed to LCMV infection (Figure 1C-E). Finally, the NP118-specific secondary effector CD8 T-cells at day 5 post LCMV challenge exhibited similar phenotypes in the two vaccinated groups (Figure 1F). These results suggest that mortality in vaccinated PKO mice following LCMV-Arm challenge is independent of immunization modalities.
The number of NP118-specific memory CD8 T-cells at the time of LCMV infection dictates secondary expansion and mortality in vaccinated PKO mice
Current literature suggests that the magnitude of CD8 T-cell expansion after primary infection is related to the number of precursors recruited into the response [40, 41]. However, it remains unclear whether the number of LCMV-specific memory CD8 T-cells at the time of LCMV infection determines the magnitude of secondary expansion and subsequent mortality in PKO mice. To address this question, we generated different levels of memory CD8 T-cells either by varying the dose of att LM-NP118 used for immunization or by adoptive transfer of different numbers NP118-specific memory CD8 T-cells into naïve PKO mice.
Naïve PKO mice were immunized with 5×106 cfu (high dose) or 5×102 cfu (low dose) of att LM-NP118. In order to control the extent of inflammation elicited by two different doses of infection used, mice that received a low dose of att LM-NP118 were co-infected with 5×106 cfu of the Att LM strain that does not express the NP118 epitope (Figure 2A). Approximately 4-fold fewer NP118-specific memory CD8 T-cells (detected in PBL) were present in ‘low dose’ compared to ‘high dose’ immunized groups of mice (Figure 2B). At day 70 p.i. mice from both experimental groups and an additional control (non-immunized) group were challenged with LCMV-Arm. Despite having 4-fold difference in starting memory numbers, similar numbers of secondary effector NP118-specific CD8 T-cells (>107 cells/spleen) were detected in both groups of vaccinated PKO mice at days 5 and 7 post LCMV challenge (Figure 2C). Importantly, all vaccinated mice rapidly lost weight and succumbed to LCMV infection while non-vaccinated mice exhibited less weight loss and survived (Figure 2D-E).
Figure 2. Low frequency of NP118-specific memory CD8 T-cells is sufficient to cause mortality in vaccinated PKO mice following LCMV challenge.

A) Naïve PKO mice (12 /group) were immunized with either ~5×106 or ~5×102 cfu att LM-NP118 or PBS (non-immunized group) and infected with LCMV-Arm (day 70 p.i.). Frequency B) and total number C) of NP118-specific CD8 T-cells in PBL as measured by ICS at day 70 p.i. (mean + SD). D) NP118-specific secondary effector CD8 T-cell responses in the spleen were detected by Ld/NP118 tetramers staining (mean ± SD, n=3 at each time point) and enumerated at day 5 and 7 following LCMV infection. Morbidity measured as weight loss E) and survival F) of PKO mice after LCMV infection (at least 6 /group). Data are representative of at least 2 independent experiments. LOD = limit of detection.
In order to further decrease the number of memory CD8 T-cells we performed adoptive transfer of different numbers of NP118-specific memory CD8 T-cells (ranging from 8×102 cells to 8×105 cells per mouse) into naïve PKO hosts. The NP118-specific population of memory CD8 T-cells transferred exhibited a late memory phenotype (CD127hi, CD62Lhi, KLRG-1lo, CD27hi) and function (IL-2 and TNF cytokine production upon re-stimulation with NP118 peptide) (Figure 3A). All recipient mice and a group that did not receive memory CD8 T-cells were challenged with LCMV-Arm. Mice receiving 8×105 and 8×104 NP118-specific memory CD8 T-cells rapidly lost weight and succumbed following LCMV infection (Figure 3B-C). Interestingly, mice receiving 8×103 NP118-specific memory CD8 T-cells lost weight during the first week after LCMV infection but recovered without any mortality. On the other hand, mice receiving 8×102 NP118-specific memory CD8 T-cells exhibited only slight weight loss and did not succumb, similar to control mice that did not receive any memory CD8 T-cells (Figure 3B-C). Consistent with their poor outcome, mice receiving either 8×105 or 8×104 NP118-specific memory CD8 T-cells had high numbers (>107 cells/spleen) at 5 days post LCMV infection. Importantly, a substantial fraction of NP118-specific secondary effector CD8 T-cells in the groups receiving the highest numbers of memory CD8 T-cells produced IFN-γ directly ex-vivo even in the absence of exogenous peptide stimulation (Figure 3D). Together, these results suggested that secondary CD8 T-cell expansion and mortality in PKO mice are dictated by the starting number of NP118-specific memory CD8 T-cells at the time of LCMV challenge.
Figure 3. Starting number of NP118-specific memory CD8 T-cells at the time of LCMV infection dictate secondary expansion and mortality in vaccinated PKO mice.

Five groups of naïve PKO mice (10 /group) received splenocytes from previously immunized donor PKO memory mice containing 8×105 or 8×104 or 8×103 or 8×102 or 0 NP118-specific memory CD8 T-cells one day before LCMV-Arm challenge .A) Phenotype of NP118-specific memory CD8 T-cells prior to adoptive transfer as detected by direct ex-vivo Ld/NP118 tetramer staining without antigen stimulation (top row) or intracellular IFN-γ after in vitro antigen stimulation (bottom row). Survival B) and morbidity measured as weight loss C) of the recipient PKO mice after LCMV-Arm infection (7 /group). D) Number of IFN-γ+ CD8+ T cells (mean + SD, n=3) in the spleen of recipient PKO mice in the absence (left panel) or presence (right panel) of NP118 peptide stimulation at day 5 post LCMV infection. Data are representative of at least 2 independent experiments.
Epitope specificity dictates vaccination-induced mortality in PKO mice following LCMV challenge
Naïve PKO mice survive LCMV-Arm infection by exhausting their NP118-specific CD8 T-cells [16]. Furthermore, more than 98% of the CD8 T-cell response to LCMV infection in BALB/c mice is directed at the dominant NP118 epitope, with subdominant responses directed to GP283 and GP96 epitopes [42, 43]. Previously work showed that vaccination to generate wild-type memory CD8 T-cells against subdominant epitopes may be effective at protecting from both LCMV and LM infection [44, 45].However, it remains unknown whether memory CD8 T-cells specific for subdominant LCMV epitopes will also lead to vaccine-induced mortality in perforin-deficient hosts.
To address this issue, we immunized naïve PKO mice with 5×105 DC coated with either the dominant NP118 or subdominant GP283 LCMV epitopes, while mice in the control group received DC coated with a P. berghei CS252 epitope. At day 102 after immunization all groups of mice were challenged with LCMV-Arm and the kinetics of the CD8 T-cell response and morbidity/mortality were analyzed (Figure 4A). Interestingly, the majority of mice vaccinated with the subdominant GP283 epitope survived the LCMV infection as did the control mice vaccinated with the control P. berghei CS252 epitope. As previously observed, the majority of mice vaccinated with the dominant NP118 epitope succumbed to the LCMV infection (Figure 4B (Figure 1E). Importantly, the NP118- and the GP283-specific memory CD8 T-cells exhibited similar memory phenotype and function (CD127hi, KLRG-1lo, CD27hi, CD43lo and high frequencies of these cells produce IL-2 and TNF upon specific peptide restimulation) at the time of LCMV infection (Figure 4C) suggesting the difference in outcome was not an issue of memory quality. However, a statistically significant difference (p=0.03) in total number of NP118- and GP283-specific memory CD8 T-cells in the spleen of vaccinated PKO mice prior to LCMV challenge was observed (Figure 4D).
Figure 4. Epitope specificity dictates vaccination-induced mortality in PKO mice following LCMV infection, independent of magnitude of memory CD8 T-cell expansion.

A) Naïve PKO mice (9 /group) were immunized with either DC-NP118 or DC-GP283 or DC-CS252 (106 cells/mouse) and infected (day 102 p.i.) with LCMV-Arm (5×105 pfu). B) Survival of vaccinated PKO mice after LCMV infection (n=6 mice/group). C) Representative dot plots and histograms showing the frequency and phenotype of NP118- and GP283-specific memory CD8 T-cells as detected by ICS prior to LCMV infection. D) Total number (mean + SD, n=3) of NP118-specific (black bar) or GP283-specific (white bar) CD8 T-cells in the spleen as measured by tetramers staining at days 102 and 102+5. (E and F) In a subsequent experiment, naïve PKO mice were immunized with either ~1×105 DC-NP118 or ~1.6×106 DC-GP283 or 1.6×106 DC-CS252 per mouse (n=10 mice/group) and infected (day 124 p.i.) with LCMV-Arm (5×105 pfu). E) Total number (mean + SD, n=3) of NP118- or GP283-specific CD8 T-cells in the spleen as measured by tetramers staining at memory time point (day 124) and 5 and 7 days following LCMV challenge. F) Survival of vaccinated PKO mice after LCMV infection (7 /group).
To determine if the difference in the starting number of memory CD8 T-cells of different Ag-specificity controls the difference in susceptibility to the LCMV challenge, groups of naïve PKO mice were immunized with different numbers of peptide-coated DCs to equalize the number of memory CD8 T-cells At day 124 following DC immunization, the frequency of GP283-specific memory CD8 T-cells was approximately equal to that of NP118-specific memory CD8 T-cells (Figure 4E). More importantly, the magnitude of expansion was also similar between GP283- and NP118-specific CD8 T-cells at days 5 and 7 after LCMV infection (Figure 4E). However, we observed 100% mortality in DC-NP118 vaccinated mice but 0% mortality in DC-GP283- or DC-CS252 – vaccinated groups of mice (Figure 4F). Thus, PKO mice containing memory CD8 T-cells against a dominant epitope, but not a subdominant epitope, are predisposed to LCMV-induced mortality, under conditions where the starting number and magnitude of expansion of memory CD8 T-cells are similar. These results suggested that the epitope specificity dictates vaccination-induced mortality in BALB/c-PKO mice following LCMV challenge.
Since vaccination of naïve PKO with the subdominant epitope did not result in mortality following LCMV challenge, we also sought to determine whether these vaccinated mice showed enhanced resistance against LCMV infection. Similar to the DC-NP118-vaccinated PKO mice, the DC-GP283-vaccinated mice had significantly reduced viral load at day 5 post LCMV infection compared to the non-immunized mice. However, the viral load reduction was not sustained by day 7 post LCMV (Figure 5). Thus, although CD8 T-cell-mediated LCMV-induced mortality can be avoided by vaccination of PKO mice with the subdominant instead of the dominant epitope, this immunization did not provide sustained virus control
Figure 5. Robust expansion of both NP118- and GP283-specific memory CD8 T-cells correlates with early viral control but these mice failed to clear the virus after LCMV infection.
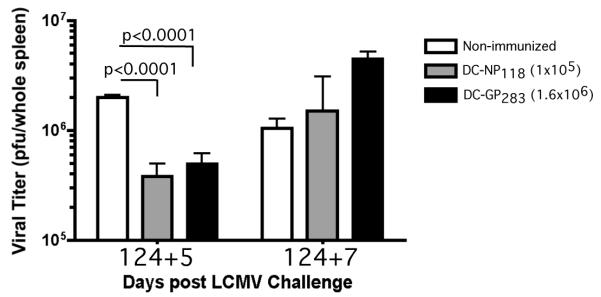
Spleens of mice as in Figure 5E were assessed for viral load (pfu/spleen; mean + SD, n=3) at days 5 and 7 following LCMV-Arm infection. Data are representative of at least 2 independent experiments.
Degree of cytokine dysregulation and functional exhaustion correlates with survival of PKO mice vaccinated with subdominant GP283-epitope
In general, CD8 T-cells exhibit tight regulation of cytokine production and do not produce IFN-γ directly ex vivo unless they receive Ag-stimulation. Following LCMV infection, the mortality in vaccinated PKO mice containing NP118-specific memory CD8 T-cells is associated with massive secondary expansion of cells exhibiting high levels of IFN-γ directly ex vivo without further Ag stimulation. The inability to regulate cytokine production is likely a major contributor to the mortality in PKO mice since treatment with neutralizing anti-IFN-γ antibodies prevents mortality in vaccinated BALB/c-PKO as well as in naïve C57BL/6-PKO mice after LCMV infection [16, 18]. The discrepancy in survival in mice containing NP118- vs GP273-specific memory CD8 T cells could be explained by the extent to which Ag-specific CD8 T-cells can regulate cytokine production. To test this notion, we examined the IFN-γ production and the phenotype of CD8 T-cells post LCMV challenge in vaccinated as well as in control mice. Five and 7 days after LCMV infection, a substantial percentage of total splenic CD8 T-cells exhibited IFN-γ production in the absence of exogenous peptide stimulation (no peptide) in the DC-NP118-vaccinated mice (Figure 6A, middle row) while there was little difference in the DC-GP283-vaccinated or non-vaccinated mice (Figure 6A, top and bottom rows). This resulted in significantly (p =0.0017) higher number of total splenic CD8 T-cells (~ 10-fold) producing IFN-γ directly ex vivo at day 5 post LCMV in DC-NP118-vaccinated mice (Figure 6B). In addition, stimulation of splenic CD8 T-cells isolated from DC-NP118-vaccinated mice at 5 and 7 days post LCMV infection with GP283 peptide did not increase the frequency of IFN-γ-producing cells over the baseline (no peptide), suggesting that most of these IFN-γ-producing CD8 T-cells are NP118-specific (Figure 6A, middle row). Finally, the GP283-specific secondary effector CD8 T-cells from DC-GP283-vaccinated mice had lower expression of Programmed Death 1 receptor (PD-1) and higher fraction of these cells producing TNF when compared to NP118-specific CD8 T-cells from DC-NP118-vaccinated mice (Figure 6C). While PD-1 is upregulated in effector cells, sustained expression requires continued antigen-stimulation [46, 47]. This phenotype suggested a lesser degree of functional exhaustion in the GP283-specific CD8 T-cells since increased PD-1 expression and loss of TNF production have been shown to correspond to exhaustion of antigen-specific CD8 T-cell in chronic viral infection model [46, 47]. These results demonstrated that CD8 T-cell epitope specificity impacts both, functional exhaustion and the ability to tightly regulate CD8 T-cell-derived cytokine secretion, rather than the absolute number or magnitude of CD8 T-cell expansion.
Figure 6. GP283-specific CD8 T-cells exhibited less cytokine dysregulation and functional exhaustion following LCMV-Arm challenge.

Naïve PKO mice were immunized with either ~1×105 DC-NP118 or ~1.6×106 DC-GP283. These mice and non-immunized controls were infected with LCMV-Arm on day 117 post immunization. A) Representative dot plots showing gating strategy and IFN-γ production by the splenic CD8 T-cells in the absence or presence of NP118 or GP283 peptide stimulation at day 5 and 7 post LCMV infection. B) Number of IFN-γ+ CD8 T-cells (mean + SD, n=3) in the spleen in the absence of peptide stimulation at day 5 and 7 post LCMV infection. C) Representative histograms and D) cumulative data showing PD-1 or TNF expression by the NP118- and GP283-specific CD8 T-cells in the vaccinated PKO mice at days 5 and 7 following LCMV infection. The numbers inside the histograms represent the percentage of antigen-specific CD8 T-cells positive for the indicated markers.
DISCUSSION
Memory CD8 T-cells provide enhanced resistance to re-infection by the same pathogen. Moreover, the number of memory CD8 T-cells correlates strongly with the level of protection in experimental models of infection [1, 3]. The ultimate goal of any vaccine regimen is to induce protective immunity against the targeted pathogens. Thus, vaccines to promote cellular immunity should logically focus on achieving a sufficiently high number of memory cells for protection. However, strong CD8 T-cell recall responses have also been demonstrated to cause undesired and sometimes lethal immunopathology in certain circumstances [9, 10, 16, 39]. Therefore, rational vaccine design needs to take into account the delicate balance between robust immunity and lethal CD8 T-cell mediated immunopathology.
Following LCMV-Arm infection, wild-type mice mount vigorous antiviral CD8 T-cell responses and clear the virus in a perforin-dependent manner [48]. PKO mice fail to clear LCMV-Arm and develop chronic infections [14]. Moreover, the requirement for perforin-mediated cytolysis in resistance to primary infection with LCMV is well documented [49] and PKO mice are models for FHL [16-19], a uniformly fatal disease associated with viral infection in human with mutations in perforin gene [20, 23-25, 50]. Thus, perforin deficiency represents an immunocompromised state in which defective antiviral CD8 T-cell response results in the establishment of chronic infection [16]. Previous work in our laboratory demonstrated that vaccination to generate memory CD8 T-cells can overcome perforin deficiency and provide enhanced resistance against intracellular infection with Listeria monocytogenes [27, 38]. In contrast, vaccination of BALB/c-PKO mice results in accelerated mortality following LCMV infection [16]. In this case, vaccination of PKO hosts converts a non-lethal persistent infection into a rapidly fatal disease mediated by CD8 T-cells.
To understand why vaccination leads to mortality in the absence of perforin, we analyzed multiple parameters that could potentially contribute to the drastic, and ultimately fatal response observed. We have shown that vaccination-induced mortality is mediated by massive expansion of NP118-specific memory CD8 T-cells and the associated aberrant cytokine production in PKO mice. Different vaccine strategies did not alter the outcome as long as the number of NP118-specific memory CD8 T-cells exceeds a certain threshold number.
In our adoptive transfer experiments (Figure 3), we observed that the majority of PKO mice succumbed to LCMV infection if they received at least 8×104 NP118-specific CD8 T-cells. Assuming 10% “take” of the transferred number, this result indicated that as few as 8,000 NP118-specific CD8 T-cells in the spleen at the time of LCMV infection would be sufficient to cause mortality in these PKO mice. Although we did not observe any mortality in mice that received 8×103 NP118-specific memory CD8 T-cells (i.e. 800 memory cells in the spleen, assuming 10% take), we documented severe morbidity as significant weight loss in these mice following LCMV infection (Figure 3C). Thus, even a small number of NP118-specific memory CD8 T-cells is sufficient to cause immunopathology after LCMV infection of PKO mice. These results could impact vaccine design strategies by challenging the notion that “more is always better” in the level of memory T cells generated after vaccination. In addition, these data also support the notion that the secondary CD8 T-cell response exhibits elements of “programming” [51] since the NP118-specific CD8 T-cell expansion after LCMV infection is proportional to the initial memory levels in PKO mice, suggesting all recruited cells underwent a similar number of divisions (Figure 3D).
We observed minor differences in the phenotype of Ag-specific CD8 T-cells between DC- and att LM-primed PKO mice at memory time points. For example, the frequency of KLRG-1-expressing memory CD8 T-cells is higher in LM infected compared to DC primed mice. The extent to which such phenotypic differences influence the ability of memory cells to respond to LCMV infection may be minimal, since we observed the same massive expansion of that NP118-specific memory cells in both groups. In addition, recent data suggested that KLRG-1 was dispensable for normal CD8 T-cell differentiation and function after viral infections [52].
Tight regulation of cytolysis and cytokine production by effector and memory CD8 T-cells in the presence of antigen has been proposed as a likely mechanism to minimize immunopathology [8, 53]. IFN-γ production by wild-type NP118-specific CD8 T-cells from LCMV infected mice is not detected in direct ex vivo assays at any time post infection without addition of antigen [54, 55]. In addition, IFN-γ production by these cells is rapidly extinguished by removal of antigen [54, 55]. Thus, it is likely that failure to clear LCMV in vaccinated PKO mice causes chronic stimulation of the massively expanded NP118-specific CD8 T-cell population, resulting in dysregulated production of cytokines and mortality. Interestingly, we observed significant reduction of LCMV viral titer in the spleen of NP118-vaccinated PKO mice at day 5 post LCMV infection compared to control mice (Figure 5). We would have predicted that lower viral titer would correspond with lower systemic cytokine levels. However, in this case, lower viral titer may be the result of increased systemic cytokine (i.e. cytokine storm) that potentially interferes with viral replication. The inability to clear the virus leads to rebound of LCMV titer in these vaccinated PKO mice suggesting that despite enormous number of Ag-specific CD8 T-cells perforin-mediated cytolysis is absolutely required to control LCMV infection and provide sterilizing immunity. Thus, the early substantial reduction in viral titers is still associated with mortality in these PKO mice. In addition, this result also suggested that cytokine dysregulation is a property inherent to PKO-derived memory CD8 T-cell response as has been suggested from in vitro studies [56].
Naïve BALB/c-PKO mice (H-2d) survive LCMV Arm infection by exhausting their NP118-specific CD8 T-cells [16]. In contrast, a substantial number of, although not all, naïve PKO mice on the B6 background (H-2b) succumb to LCMV Arm infection, apparently as a consequence of failure to exhaust their antigen-specific CD8 T-cells [13, 14]. The differences in the complexity of the CD8 T-cell response or the influence of background genes (ex. extent of IFN-γ production) may account for the results. Using LCMV infection of naïve C57BL-6-PKO mice Lykens et. al. recently showed that heightened antigenic stimulation is responsible for exaggerated T cell activation [57]. They suggested that perforin-dependent cytotoxicity, in addition to promoting viral clearance, regulates T cell activation by modulating Ag presentation [57]. Despite the differences in susceptibility of naïve BALB/c and C57BL/6 PKO mice to LCMV infection, we also observed massive CD8 T-cell expansion and accelerated LCMV induced mortality in GP33 vaccinated compared to naïve C57BL/6-PKO mice (data not shown). Thus, the vaccine-induced sensitization to mortality associated with PKO memory CD8 T-cells after LCMV infection is not restricted to BALB/c background. In addition, functional exhaustion of antigen-specific CD8 T-cells is not always associated with chronic infection [58, 59]. Chronic infection may be pathogen or host specific and it does not necessarily lead to Ag-specific CD8 T-cell exhaustion in all the cases. Although we observed lesser degree of “exhaustion” as characterized by TNF and PD-1 expression in GP283-specific CD8 T-cells compared to NP118-specific CD8 T cells, viral control was not achieved in the absence of perforin in both cases (Fig. 5). In the absence of perforin, the phenotype of GP283-specific CD8 T-cells appeared “less exhausted” at the time we analyzed them could reflect the extent that these cells can regulate cytokine production. In addition, it remained to be elucidated whether encounter with antigen is similar between the NP118- and GP283-specific memory CD8 T-cells in the PKO mice, not just initially, but throughout the infection course.
Previous studies using different models of infection showed that protective immunity mediated by pathogen-specific CD8 T-cells did not correlate with immunodominance hierarchies after infection [44, 45]. Based on the results with PKO mice vaccinated with dominant NP118 epitope, we expected that massive antigen-specific memory CD8 T-cell expansion contributed to the LCMV induced mortality independent of epitope specificity. Interestingly, PKO mice vaccinated with subdominant GP283 epitope survived the LCMV infection even though they contained similar starting memory CD8 T-cell numbers and underwent similar expansion in numbers as NP118-specfic CD8 T-cells. These results suggested that epitope specificity dictates the LCMV induced mortality in vaccinated PKO mice. Furthermore, we also observed less cytokine dysregulation, in particular IFN-γ, by GP283-specific CD8 T-cells following LCMV infection. It is unclear which specific parameter(s) influence the cytokine profile of these GP283-specific CD8 T-cells and subsequent vaccine-induced mortality in PKO mice. Whether it is the duration or the amount of functional antigen display of the subdominant epitope, and/or the intrinsic properties of GP283-specific CD8 T-cells remains to be elucidated. Importantly, GP283-vaccinated PKO mice survive the LCMV infection but viral titers in these mice were only transiently reduced suggesting that sterilizing CD8 T-cell-mediated immunity was not achieved. Therefore, our results suggested that vaccination of perforin-deficient hosts (and perhaps FHL patients) against either dominant or subdominant epitopes may not be beneficial but rather could potentially cause harmful outcome for the hosts. In addition, exhaustion of immunodominant NP118-specfic memory CD8 T-cells following primary LCMV infection of BALB/c PKO mice is thought to limit cytokine dysregulation and establish chronic infection. Whether secondary GP283-specific memory CD8 T-cells following LCMV challenge will also undergo exhaustion after massive primary response and the impact on the chronic infection by LCMV remain to be elucidated.
ACKNOWLEDGEMENTS
We sincerely thank all members of the Harty laboratory for helpful discussion.
Footnotes
Supported by NIH grants AI46653, AI150073 and AI42767.
References
- 1.Badovinac VP, Harty JT. Programming, demarcating, and manipulating CD8+ T-cell memory. Immunol Rev. 2006;211:67–80. doi: 10.1111/j.0105-2896.2006.00384.x. [DOI] [PubMed] [Google Scholar]
- 2.Harty JT, Badovinac VP. Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol. 2008;8:107–119. doi: 10.1038/nri2251. [DOI] [PubMed] [Google Scholar]
- 3.Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2:251–262. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 4.Sallusto F, Lanzavecchia A, Araki K, Ahmed R. From vaccines to memory and back. Immunity. 2010;33:451–463. doi: 10.1016/j.immuni.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78:5535–5545. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. 2007;25:171–192. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- 7.Harty JT, Badovinac VP. Influence of effector molecules on the CD8(+) T cell response to infection. Curr Opin Immunol. 2002;14:360–365. doi: 10.1016/s0952-7915(02)00333-3. [DOI] [PubMed] [Google Scholar]
- 8.Slifka MK, Whitton JL. Antigen-specific regulation of T cell-mediated cytokine production. Immunity. 2000;12:451–457. doi: 10.1016/s1074-7613(00)80197-1. [DOI] [PubMed] [Google Scholar]
- 9.Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- 10.Walsh CM, Matloubian M, Liu CC, Ueda R, Kurahara CG, Christensen JL, Huang MT, Young JD, Ahmed R, Clark WR. Immune function in mice lacking the perforin gene. Proc Natl Acad Sci U S A. 1994;91:10854–10858. doi: 10.1073/pnas.91.23.10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kagi D, Vignaux F, Ledermann B, Burki K, Depraetere V, Nagata S, Hengartner H, Golstein P. Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science. 1994;265:528–530. doi: 10.1126/science.7518614. [DOI] [PubMed] [Google Scholar]
- 12.Badovinac VP, Tvinnereim AR, Harty JT. Regulation of antigen-specific CD8+ T cell homeostasis by perforin and interferon-gamma. Science. 2000;290:1354–1358. doi: 10.1126/science.290.5495.1354. [DOI] [PubMed] [Google Scholar]
- 13.Gallimore A, Glithero A, Godkin A, Tissot AC, Pluckthun A, Elliott T, Hengartner H, Zinkernagel R. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med. 1998;187:1383–1393. doi: 10.1084/jem.187.9.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matloubian M, Suresh M, Glass A, Galvan M, Chow K, Whitmire JK, Walsh CM, Clark WR, Ahmed R. A role for perforin in downregulating T-cell responses during chronic viral infection. J Virol. 1999;73:2527–2536. doi: 10.1128/jvi.73.3.2527-2536.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spaner D, Raju K, Rabinovich B, Miller RG. A role for perforin in activation-induced T cell death in vivo: increased expansion of allogeneic perforin-deficient T cells in SCID mice. J Immunol. 1999;162:1192–1199. [PubMed] [Google Scholar]
- 16.Badovinac VP, Hamilton SE, Harty JT. Viral infection results in massive CD8+ T cell expansion and mortality in vaccinated perforin-deficient mice. Immunity. 2003;18:463–474. doi: 10.1016/s1074-7613(03)00079-7. [DOI] [PubMed] [Google Scholar]
- 17.Fischer A, Latour S, de Saint Basile G. Genetic defects affecting lymphocyte cytotoxicity. Curr Opin Immunol. 2007;19:348–353. doi: 10.1016/j.coi.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 18.Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104:735–743. doi: 10.1182/blood-2003-10-3413. [DOI] [PubMed] [Google Scholar]
- 19.Menasche G, Feldmann J, Fischer A, de Saint Basile G. Primary hemophagocytic syndromes point to a direct link between lymphocyte cytotoxicity and homeostasis. Immunol Rev. 2005;203:165–179. doi: 10.1111/j.0105-2896.2005.00224.x. [DOI] [PubMed] [Google Scholar]
- 20.Gholam C, Grigoriadou S, Gilmour KC, Gaspar HB. Familial haemophagocytic lymphohistiocytosis: advances in the genetic basis, diagnosis and management. Clin Exp Immunol. 2011;163:271–283. doi: 10.1111/j.1365-2249.2010.04302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henter JI, Ehrnst A, Andersson J, Elinder G. Familial hemophagocytic lymphohistiocytosis and viral infections. Acta Paediatr. 1993;82:369–372. doi: 10.1111/j.1651-2227.1993.tb12699.x. [DOI] [PubMed] [Google Scholar]
- 22.Katano H, Cohen JI. Perforin and lymphohistiocytic proliferative disorders. Br J Haematol. 2005;128:739–750. doi: 10.1111/j.1365-2141.2004.05305.x. [DOI] [PubMed] [Google Scholar]
- 23.Clementi R, Emmi L, Maccario R, Liotta F, Moretta L, Danesino C, Arico M. Adult onset and atypical presentation of hemophagocytic lymphohistiocytosis in siblings carrying PRF1 mutations. Blood. 2002;100:2266–2267. doi: 10.1182/blood-2002-04-1030. [DOI] [PubMed] [Google Scholar]
- 24.Molleran Lee S, Villanueva J, Sumegi J, Zhang K, Kogawa K, Davis J, Filipovich AH. Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J Med Genet. 2004;41:137–144. doi: 10.1136/jmg.2003.011528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ueda I, Kurokawa Y, Koike K, Ito S, Sakata A, Matsumora T, Fukushima T, Morimoto A, Ishii E, Imashuku S. Late-onset cases of familial hemophagocytic lymphohistiocytosis with missense perforin gene mutations. Am J Hematol. 2007;82:427–432. doi: 10.1002/ajh.20878. [DOI] [PubMed] [Google Scholar]
- 26.Messingham KA, Badovinac VP, Harty JT. Deficient anti-listerial immunity in the absence of perforin can be restored by increasing memory CD8+ T cell numbers. J Immunol. 2003;171:4254–4262. doi: 10.4049/jimmunol.171.8.4254. [DOI] [PubMed] [Google Scholar]
- 27.White DW, MacNeil A, Busch DH, Pilip IM, Pamer EG, Harty JT. Perforin-deficient CD8+ T cells: in vivo priming and antigen-specific immunity against Listeria monocytogenes. J Immunol. 1999;162:980–988. [PubMed] [Google Scholar]
- 28.Pham NL, Badovinac VP, Harty JT. A default pathway of memory CD8 T cell differentiation after dendritic cell immunization is deflected by encounter with inflammatory cytokines during antigen-driven proliferation. J Immunol. 2009;183:2337–2348. doi: 10.4049/jimmunol.0901203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brundage RA, Smith GA, Camilli A, Theriot JA, Portnoy DA. Expression and phosphorylation of the Listeria monocytogenes ActA protein in mammalian cells. Proc Natl Acad Sci U S A. 1993;90:11890–11894. doi: 10.1073/pnas.90.24.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tvinnereim AR, Hamilton SE, Harty JT. CD8(+)-T-cell response to secreted and nonsecreted antigens delivered by recombinant Listeria monocytogenes during secondary infection. Infect Immun. 2002;70:153–162. doi: 10.1128/IAI.70.1.153-162.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt NW, Podyminogin RL, Butler NS, Badovinac VP, Tucker BJ, Bahjat KS, Lauer P, Reyes-Sandoval A, Hutchings CL, Moore AC, Gilbert SC, Hill AV, Bartholomay LC, Harty JT. Memory CD8 T cell responses exceeding a large but definable threshold provide long-term immunity to malaria. Proc Natl Acad Sci U S A. 2008;105:14017–14022. doi: 10.1073/pnas.0805452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen H, Miller JF, Fan X, Kolwyck D, Ahmed R, Harty JT. Compartmentalization of bacterial antigens: differential effects on priming of CD8 T cells and protective immunity. Cell. 1998;92:535–545. doi: 10.1016/s0092-8674(00)80946-0. [DOI] [PubMed] [Google Scholar]
- 33.Badovinac VP, Harty JT. Intracellular staining for TNF and IFN-gamma detects different frequencies of antigen-specific CD8(+) T cells. J Immunol Methods. 2000;238:107–117. doi: 10.1016/s0022-1759(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 34.Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. [PubMed] [Google Scholar]
- 35.Busch DH, Pilip IM, Vijh S, Pamer EG. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity. 1998;8:353–362. doi: 10.1016/s1074-7613(00)80540-3. [DOI] [PubMed] [Google Scholar]
- 36.Harty JT, Bevan MJ. Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity. 1995;3:109–117. doi: 10.1016/1074-7613(95)90163-9. [DOI] [PubMed] [Google Scholar]
- 37.White DW, Badovinac VP, Fan X, Harty JT. Adaptive immunity against Listeria monocytogenes in the absence of type I tumor necrosis factor receptor p55. Infect Immun. 2000;68:4470–4476. doi: 10.1128/iai.68.8.4470-4476.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.White DW, Harty JT. Perforin-deficient CD8+ T cells provide immunity to Listeria monocytogenes by a mechanism that is independent of CD95 and IFN-gamma but requires TNF-alpha. J Immunol. 1998;160:898–905. [PubMed] [Google Scholar]
- 39.Oehen S, Ohashi PS, Aichele P, Burki K, Hengartner H, Zinkernagel RM. Vaccination or tolerance to prevent diabetes. Eur J Immunol. 1992;22:3149–3153. doi: 10.1002/eji.1830221218. [DOI] [PubMed] [Google Scholar]
- 40.Badovinac VP, Haring JS, Harty JT. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T cell response to infection. Immunity. 2007;26:827–841. doi: 10.1016/j.immuni.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaech SM, Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol. 2001;2:415–422. doi: 10.1038/87720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Most RG, Concepcion RJ, Oseroff C, Alexander J, Southwood S, Sidney J, Chesnut RW, Ahmed R, Sette A. Uncovering subdominant cytotoxic T-lymphocyte responses in lymphocytic choriomeningitis virus-infected BALB/c mice. J Virol. 1997;71:5110–5114. doi: 10.1128/jvi.71.7.5110-5114.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weidt G, Utermohlen O, Heukeshoven J, Lehmann-Grube F, Deppert W. Relationship among immunodominance of single CD8+ T cell epitopes, virus load, and kinetics of primary antiviral CTL response. J Immunol. 1998;160:2923–2931. [PubMed] [Google Scholar]
- 44.Gallimore A, Dumrese T, Hengartner H, Zinkernagel RM, Rammensee HG. Protective immunity does not correlate with the hierarchy of virus-specific cytotoxic T cell responses to naturally processed peptides. J Exp Med. 1998;187:1647–1657. doi: 10.1084/jem.187.10.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skoberne M, Holtappels R, Hof H, Geginat G. Dynamic antigen presentation patterns of Listeria monocytogenes-derived CD8 T cell epitopes in vivo. J Immunol. 2001;167:2209–2218. doi: 10.4049/jimmunol.167.4.2209. [DOI] [PubMed] [Google Scholar]
- 46.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 47.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, Ahmed R. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 49.Kagi D, Ledermann B, Burki K, Zinkernagel RM, Hengartner H. Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Annu Rev Immunol. 1996;14:207–232. doi: 10.1146/annurev.immunol.14.1.207. [DOI] [PubMed] [Google Scholar]
- 50.Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, Henter JI, Bennett M, Fischer A, de Saint Basile G, Kumar V. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286:1957–1959. doi: 10.1126/science.286.5446.1957. [DOI] [PubMed] [Google Scholar]
- 51.Badovinac VP, Porter BB, Harty JT. Programmed contraction of CD8(+) T cells after infection. Nat Immunol. 2002;3:619–626. doi: 10.1038/ni804. [DOI] [PubMed] [Google Scholar]
- 52.Grundemann C, Schwartzkopff S, Koschella M, Schweier O, Peters C, Voehringer D, Pircher H. The NK receptor KLRG1 is dispensable for virus-induced NK and CD8+ T-cell differentiation and function in vivo. Eur J Immunol. 2010;40:1303–1314. doi: 10.1002/eji.200939771. [DOI] [PubMed] [Google Scholar]
- 53.Slifka MK, Whitton JL. Clinical implications of dysregulated cytokine production. J Mol Med (Berl) 2000;78:74–80. doi: 10.1007/s001090000086. [DOI] [PubMed] [Google Scholar]
- 54.Badovinac VP, Corbin GA, Harty JT. Cutting edge: OFF cycling of TNF production by antigen-specific CD8+ T cells is antigen independent. J Immunol. 2000;165:5387–5391. doi: 10.4049/jimmunol.165.10.5387. [DOI] [PubMed] [Google Scholar]
- 55.Slifka MK, Rodriguez F, Whitton JL. Rapid on/off cycling of cytokine production by virus-specific CD8+ T cells. Nature. 1999;401:76–79. doi: 10.1038/43454. [DOI] [PubMed] [Google Scholar]
- 56.Sad S, Kagi D, Mosmann TR. Perforin and Fas killing by CD8+ T cells limits their cytokine synthesis and proliferation. J Exp Med. 1996;184:1543–1547. doi: 10.1084/jem.184.4.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lykens JE, Terrell CE, Zoller EE, Risma K, Jordan MB. Perforin is a critical physiologic regulator of T-cell activation. Blood. 2011;118:618–626. doi: 10.1182/blood-2010-12-324533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chentoufi AA, Kritzer E, Tran MV, Dasgupta G, Lim CH, Yu DC, Afifi RE, Jiang X, Carpenter D, Osorio N, Hsiang C, Nesburn AB, Wechsler SL, BenMohamed L. The herpes simplex virus 1 latency-associated transcript promotes functional exhaustion of virus-specific CD8+ T cells in latently infected trigeminal ganglia: a novel immune evasion mechanism. J Virol. 2011;85:9127–9138. doi: 10.1128/JVI.00587-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zelinskyy G, Myers L, Dietze KK, Gibbert K, Roggendorf M, Liu J, Lu M, Kraft AR, Teichgraber V, Hasenkrug KJ, Dittmer U. Virus-specific CD8+ T cells upregulate programmed death-1 expression during acute friend retrovirus infection but are highly cytotoxic and control virus replication. J Immunol. 2011;187:3730–3737. doi: 10.4049/jimmunol.1101612. [DOI] [PMC free article] [PubMed] [Google Scholar]