

Abstract
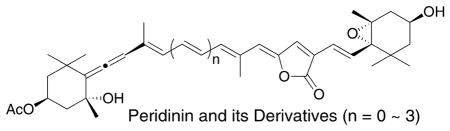
Peridinin, a nor-carotenoid, exhibits an exceptionally high energy transfer efficiency to chlorophyll a in photosynthesis in the sea. This efficiency would be related to the unique structure of peridinin. To answer the question of why peridinin possesses the irregular C37 skeleton, we have achieved the synthesis of three peridinin derivatives. Their ultrafast time-resolved optical absorption and Stark spectra measurements have shown the presence of the characteristic intramolecular charge transfer state and the featured electrostatic properties of peridinin.
The unique C37-skeletal nor-carotenoid peridinin (1) was isolated from planktonic algae dinoflagellates associated with the marine red tide.1,2 Peridinin is a representative auxiliary light-harvesting pigment for photosynthesis in the sea.3 In vivo, it is found that a supermolecular structure is composed of two chlorophyll (Chl) a and eight peridinins embedded in a protein complex denoted PCP.4 In this complex, exceptionally high (>95%) energy transfer efficiency from peridinin to Chl a has been reported.3,5 It has been postulated that the presence of an intramolecular charge transfer (ICT) state, which is formed in carbonyl-containing carotenoids, is associated with the exceptionally high energy transfer.6 In addition, an atypical and striking solvent effect on the lifetime of the lowest excited singlet state of these carbonyl-containing carotenoids has been reported.6a Although the precise nature of the ICT state is gradually beginning to unfold, there are no studies on the relationship between the structural features of peridinin and its propensity for carrying out high efficiency energy transfer in the PCP complex. In addition, Stark spectroscopy of peridinin has been performed to determine the change in electrostatic properties produced upon excitation.7
We have already established a highly efficient method for the synthesis of peridinin,8 which would be versatile and effective for providing various kinds of peridinin analogues, and have started an investigation to answer the question of why peridinin possesses an irregular C37 carbon skeleton along with allene and γ-ylidenbutenolide groups.
In a previous paper, we reported the synthesis of three peridinin derivatives as a series of allene-modified compounds and their Stark spectra. The Stark spectra showed that the allene group of peridinin effectively contributes to the generation of the large dipole moment in the molecule.9 We then turned our attention to the C37 carbon skeleton of peridinin, which makes it a “nor-carotenoid” and unlike the typical C40 system present in most carotenoids. We designed three peridinin derivatives as a series of different π-electron chain length compounds (Figure 1). We now describe the synthesis of these peridinin derivatives and the results from ultrafast time-resolved optical absorption and the Stark spectra carried out on the synthesized compounds in comparison with peridinin. The spectroscopic data point to the characteristic ICT of peridinin and bring into focus an answer to the question of why nature chose the irregular C37 carbon skeleton of peridinin.
Figure 1.

Conjugated polyene-modified peridinin derivatives.
On the basis of the stereocontrolled synthesis of peridinin by the modified Julia coupling of 8 with 9,8 we synthesized C33, C35, and C39 peridinin (Figure 2). The synthesis of C33 peridinin 2 would be realized by the Pd-catalyzed one-pot ylidenbutenolide formation between the new iodide 5 and the reported alkyne 6.8 Meanwhile, the modified Julia coupling between C15-allenic segment 710,11 and C20-ylidenbutenolide segment 8 would produce C35 peridinin 3. Furthermore, applying the same method to the coupling between C17-allenic segment 9 and C22-ylidenbutenolide segment 1010 may produce the desired C39 peridinin 4, if the coupling product would be enough stable to be handled.
Figure 2.

Synthetic strategy.
First, the synthesis of C33 peridinin 2 is described (Scheme 1). Aldehyde 11, which was obtained by MnO2 oxidation followed by acetylation of the corresponding known C15-allenic triol10–12 prepared from (−)-actinol, was transformed into the desired iodide 5 by the Wittig reaction with triphenylphosphonium ethylide and NIS in 78% yield. Unfortunately, the major product was the 11Z-isomer. The desired 11E-isomer 5 isolated easily isomerized into a 1/3 mixture of 11E/11Z at room temperature, and hence we used the mixture as the allenic half-segment. The crucial one-pot ylidenbutenolide formation from 5 and segment 6 (Figure 2) was explored as the key step in the synthesis of C33 peridinin 2. Thus, a mixture of 5 and 6 was stirred in the presence of catalytic amounts of Pd(PPh3)4 and cuprous iodide in triethylamine at 45 °C for 10 min. After the complete consumption of 6 was ascertained by TLC, formic acid was added, and then the mixture was stirred at 45 °C to produce C33 peridinins in 35% yield as a mixture of stereoisomers. The undesired 11Z-isomer 5 resulted in the undesired isomer of compound 2. The resulting mixture was then allowed to isomerize in benzene at room temperature under fluorescent light in an argon atmosphere.9 As shown in Figure 3, after 2 days, we observed that the initially generated major peak (peak 2) transformed into another major peak (peak 1) in the HPLC. In addition, peak 3 became larger in the equilibrium state. We isolated all peaks by mobile-phase HPLC and elucidated their structures by NMR (400 MHz). Thus, we understood that peak 1 was fortunately (11E,11′Z)-all-trans C33 peridinin 2, peak 2 was (11Z,11′Z)-isomer 2′, and peak 3 was (11E,11′E)-isomer 2″, respectively. Interestingly, (11E,11′E)-isomer 2″ became the second largest isomer in the equilibrium state.
Scheme 1.
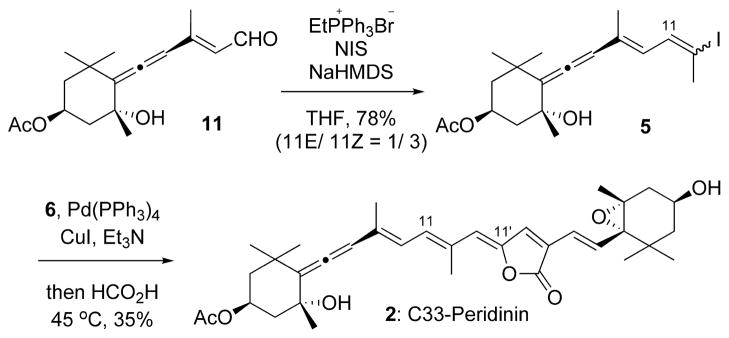
Synthesis of C33 Peridinin 2
Figure 3.

Isomerization and structures of C33 peridinin.
Our next interest is the synthesis of C35 pedirinin 3 by the modified Julia olefination13 of C15 allenic segment 7 with C20 ylidenbutenolide segment 88 (Figure 2 and Scheme 2). Sulfone 7 was obtained from 11 by a sequence of reduction, Mitsunobu reaction, and then oxidation of the resulting sulfide in exellent yield. The reaction of an anion derived from 7 with 8 at −78 °C proceeded smoothly within 5 min in the dark to produce C35 peridinin 3 in 39% yield as a mixture of stereoisomers. Similar to the previous results of the modified Julia olefination,8,9,14 the Z-isomer was predominant in a ratio of 15:85, which was changed to 84: 16 of the 11E- to 11Z-isomers by the isomerization under the usual conditions in an equilibrium state after 2 days. Thus, we obtained C35 pedirinin 3 in our hands.
Scheme 2.
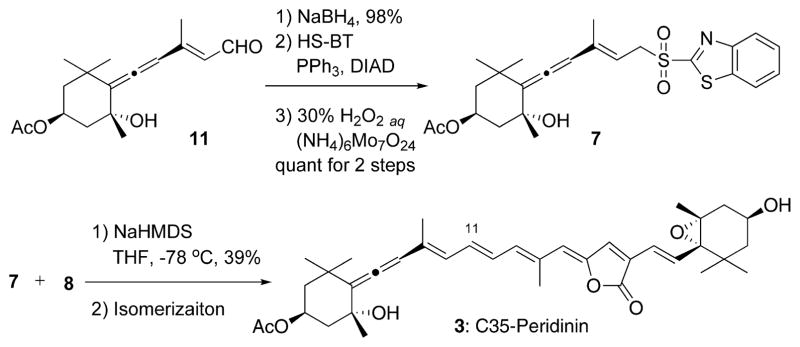
Synthesis of C35 Peridinin 3
Our next interest was the synthesis of C39 pedirinin 4. If this longer conjugating polyene derivative would stably exist, the very mild modified Julia olefination between 9 and 10 would yield the desired coupling product, which would be transformed into all-trans 4 by isomerization according to the same synthetic strategy (Scheme 3). The stereocontrolled preparation of C22 ylidenbutenolide segment 10 from alkyne 6 was fortunately successful by the one-pot procedure; a mixture of 6 and vinyl iodide 12, which was prepared from corresponding ester,8 was stirred at 45 °C for 10 min to produce the desired ylidenbutenolide 13 in 40% yield as a 13′E/13′Z mixture (10:1). The reaction with the corresponding hydroxy derivative of 12 did not give the desired result because of its instability. The anion derived from 98 (Figure 2), which was the allenic half-segment of the established peridinin synthesis, was stirred with 10 under the same condition. Fortunately, the reaction was over within 5 min in the dark to produce the coupling products as a mixture of the stereoisomers in almost 35% amount, in which the 13Z-isomer was estimated to be 48% of the mixture by HPLC analysis (13E-isomer was 19%). Isomerization to the desired 4 was again attempted by the same method. After 2 days, most of the 13Z-isomer of 4 changed to the all-trans C39 peridinin 4 (57% based on HPLC analysis) in an equilibrium state. We then isolated both compounds and elucidated their structures by NMR (750 MHz). The synthesized C39 peridinin gradually decomposed within 1 month under an argon gas atmosphere at around −20 °C. This instability was in contrast to the case of peridinin, which could be stored without any remarkable decomposition under the same conditions.
Scheme 3.
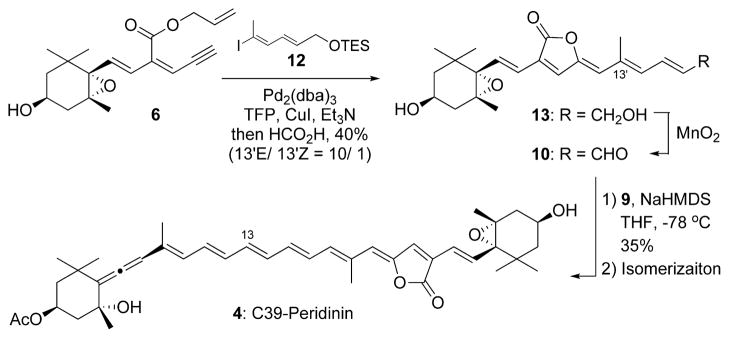
Synthesis of C39 Peridinin 4
Ultrafast time-resolved optical absorption experiments with peridinin (1) and the C33, C35, and C39 derivatives 2–4 were performed. The lifetime of the lowest excited singlet state of peridinin (1) and many other carbonyl-containing carotenoids have been reported to depend on the polarity of the solvent.6b,c This effect is attributed to the presence of an intramolecular charge transfer (ICT) state in the manifold of the excited states for these molecules. The lifetime of the lowest excited singlet state of the four compounds was then measured, and the results are listed in Table 1.15 The data listed in Table 1 show that the lifetime is shorter in a polar solvent, methanol, and is longer in a nonpolar solvent, n-hexane. The lifetime of the lowest excited singlet state of C33 peridinin 2 is the one most strongly dependent on the solvent polarity. In fact, this is the strongest solvent dependence on the lifetime of the carotenoid excited state so far yet reported. Moreover, the most striking observation in the data is that the lifetime of the ICT state converges to a value of 10 ± 1 ps in the polar solvent, methanol, for all the peridinin analogues regardless of the extent of π-electron conjugation. These results strongly support the idea that the S1 and ICT states act as independent states, either uncoupled or due to different minima on the same potential energy surface.15
Table 1.
Lowest Excited Singlet State Lifetime of Peridinin and Derivatives
| solvent | lifetime (ps)
|
|||
|---|---|---|---|---|
| C33-peridinin | C35-peridinin | peridinin | C39-peridinin | |
| n-hexane | 4200 ± 200 | 1000 ± 100 | 186 ± 4 | 41 ± 1 |
| methanol | 11 ± 3 | 9 ± 1 | 10 ± 1 | 9 ± 1 |
| τ(n-hex)/τ(meth) | 382 | 111 | 19 | 5 |
In addition, the Stark spectra of peridinin (1) and C35 (3) and C39 (4) derivatives were recorded in methyl methacrylate polymer at 77 K.16 The Stark spectra can determine the change in electrostatic properties and estimate the change in the static dipole moment (|Δμ|) between the ground state and the excited state. The |Δμ| values are determined to the corresponding charge transfer (CT) absorption band. As a result, peridinin (1) showed the largest |Δμ| value among all of them (Table 2). The |Δμ| value of peridinin agrees with that reported by van Grondelle et al.7 Although peridinin possesses fewer conjugated double bonds and shows a λmax rather shorter than that of C39 peridinin 4, the |Δμ| value of peridinin (1) is the largest among the three compounds. Thus, the C37 skeleton of peridinin (1) would contribute to the large dipole moment of the molecule in the exited state to facilitate energy transfer. This would be at least a partial answer to the question of why peridinin (1) possesses an irregular C37 skeleton.
Table 2.
λmax and Stark Spectral Data of Peridinin and Derivatives
|
λmax (nm)
|
|Δμ| (×10−29 C·m) | ||
|---|---|---|---|
| hexane | methanol | ||
| C33-peridinin 2 | 416.0 | 429.5 | |
| C35-peridinin 3 | 436.5 | 455.0 | 4.25 |
| peridinin 1 | 454.0 | 472.0 | 5.42 |
| C39-peridinin 4 | 469.0 | 488.5 | 5.29 |
In summary, we have achieved the synthesis of three different peridinin derivatives as a series modifying the length of the π-electron conjugated chain. The data of ultrafast time-resolved optical absorption spectra of these derivatives including peridinin (1) strongly supported the notion that the S1 and ICT states behave independently. In addition, the Stark spectra of peridinin and its derivatives show that the |Δμ| value of peridinin was largest among the C35 and C39 peridinin. Thus, peridinin (1) represents a molecule having a remarkablely large static dipole moment that would facilitate energy transfer suggesting a rationale for why peridinin possesses the irregular C37 skeleton.
Supplementary Material
Acknowledgments
We thank Dr. Thomas Netscher of DSM Nutritional Products, Ltd. for the donation of (−)-actinol. We also would like to thank Nirmalya Chatterjee for his help with the ultrafast experiments. This work was supported by a Grant-in-Aid for Science Research on Priority Areas 16073222 from the Ministry of Education, Culture, Sports, Science and Technology, and Matching Fund Subsidy for a Private University, Japan. This work was also supported in the lab of H.A.F. by a grant from the National Institutes of Health (GM-30353) and by the University of Connecticut Research Foundation. H.H. thanks Nissan Science Foundation and HFSP for financial support.
Footnotes
Supporting Information Available: Experimental details and characterization data of 2–5 and 13. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Strain HH, WA, Svec K, Aitzetmuller MC, Grandolfo JJ, Katz H, Kjosen S, Norgard S, Liaaen-Jensen FT, Haxo P, Wegfahrt H, Rapoport J Am Chem Soc. 1971;93:1823. [Google Scholar]; (b) Johansen JE, Borch G, Liaaen-Jensen S. Photochemistry. 1980;19:441. [Google Scholar]
- 2.Strain HH, Svec WA, Wegfahrt P, Rapoport H, Haxo FT, Norgard S, Kjosen H, Liaaen-Jensen S. Acta Chem Scand Ser B. 1976;30:109. [Google Scholar]
- 3.Song PS, Koka P, Prezelin BB, Haxo FT. Biochemistry. 1976;15:4422. doi: 10.1021/bi00665a012. [DOI] [PubMed] [Google Scholar]
- 4.Hofmann E, Wrench P, Diedrichs K, Sharples FP, Hiller RG, Welte W, Diedrichs K. Science. 1996;272:1788. doi: 10.1126/science.272.5269.1788. [DOI] [PubMed] [Google Scholar]
- 5.Siefermann-Harms D. Photochem Photobiol. 1982;35:719. doi: 10.1111/j.1751-1097.1985.tb01646.x. [DOI] [PubMed] [Google Scholar]
- 6.(a) Bautista JA, Connors RE, Raju BB, Hiller RG, Sharples FP, Gosztola D, Wasielewski MR, Frank HA. J Phys Chem B. 1999;103:8751. [Google Scholar]; (b) Frank HA, Bautista JA, Josue J, Pendon Z, Hiller RG, Sharples FP, Gosztola D, Wasielewski MR. J Phys Chem B. 2000;104:4569. [Google Scholar]; (c) Zigmantas D, Hiller RG, Sharples FP, Frank HA, Sundstrom V, Polivka T. Phys Chem Chem Phys. 2004;6:3009. [Google Scholar]
- 7.Premvardhan L, Papagiannakis E, Hiller RG, van Grondelle R. J Phys Chem B. 2005;109:15589. doi: 10.1021/jp052027g. [DOI] [PubMed] [Google Scholar]
- 8.(a) Furuichi N, Hara H, Osaki T, Mori H, Katsumura S. Angew Chem, Int Ed. 2002;41:1023. doi: 10.1002/1521-3773(20020315)41:6<1023::aid-anie1023>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]; (b) Furuichi N, Hara H, Osaki T, Nakano M, Mori H, Katsumura S. J Org Chem. 2004;69:7949. doi: 10.1021/jo048852v. [DOI] [PubMed] [Google Scholar]
- 9.Kajikawa T, Aoki K, Singh RS, Iwashita T, Kusumoto T, Frank HA, Hashimoto H, Katsumura S. Org Biomol Chem. 2009;7:3723. doi: 10.1039/b907456b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olpp T, Brückner R. Angew Chem, Int Ed. 2006;45:4023. doi: 10.1002/anie.200600502. [DOI] [PubMed] [Google Scholar]
- 11.Vaz B, Dominguez M, Alvarez R, de Lera AR. Chem Eur J. 2007;13:1273. doi: 10.1002/chem.200600959. [DOI] [PubMed] [Google Scholar]
- 12.Nakano M, Furuichi N, Mori H, Katsumura S. Tetrahedron Lett. 2001;42:7307. [Google Scholar]
- 13.(a) Baudin JB, Hareau G, Julia SA, Ruel O. Tetrahedron Lett. 1991;32:1175. [Google Scholar]; (b) Baudin JB, Hareau G, Julia SA, Lorne R, Ruel O. Bull Soc Chim Fr. 1993;130:856. [Google Scholar]; (c) Blakemore PR, Cole WJ, Kocienski PJ, Morley A. Synlett. 1998:26. [Google Scholar]
- 14.(a) Brückner R, Song A. Synlett. 2005;2:289. [Google Scholar]; (b) Vaz B, Alvarez R, Souto JA, de Lera AR. Synlett. 2005;2:294. [Google Scholar]
- 15.The detailed results and analysis of the ultrafast time-resolved optical absorption experiments about C35-peridinin have been reported: Chatterjee N, Niedzwiedzki DM, Kajikawa T, Hasegawa S, Katsumura S, Frank HA. Chem Phys Lett. 2008;463:219. doi: 10.1016/j.cplett.2008.08.056. Further detailed results and analysis about other derivatives will be reported in a paper in the field of physical chemistry.
- 16.Detailed results and analysis of the Stark spectra will be reported in a paper in the field of physics. For the experimental procedure, see: Yanagi K, Kobayashi T, Hashimoto H. Phys Rev B. 2003;67:115122.Yanagi K, Shimizu M, Hashimoto H. J Phys Chem B. 2005;109:992. doi: 10.1021/jp046929d.Nakagawa K, Suzuki S, Fujii R, Gardiner AT, Cogdell RJ, Nango M, Hashimoto H. J Phys Chem B. 2008;112:9467. doi: 10.1021/jp801773j.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.