

Abstract
The carotid body is a sensory organ for detecting arterial blood O2 levels and reflexly mediates systemic cardiac, vascular and respiratory responses to hypoxia. This article presents a brief review of the roles of gaseous messengers in the sensory transduction at the carotid body, genetic and epigenetic influences on hypoxic sensing and the role of the carotid body chemoreflex in cardiorespiratory diseases. Type I (also called glomus) cells, the site of O2 sensing in the carotid body, express haem oxygenase-2 and cystathionine-γ-lyase, the enzymes which catalyse the generation of CO and H2S, respectively. Physiological studies have shown that CO is an inhibitory gas messenger, which contributes to the low sensory activity during normoxia, whereas H2S is excitatory and mediates sensory stimulation by hypoxia. Hypoxia-evoked H2S generation in the carotid body requires the interaction of cystathionine-γ-lyase with haem oxygenase-2, which generates CO. Hypoxia-inducible factors 1 and 2 constitute important components of the genetic make-up in the carotid body, which influence hypoxic sensing by regulating the intracellular redox state via transcriptional regulation of pro- and antioxidant enzymes. Recent studies suggest that developmental programming of the carotid body response to hypoxia involves epigenetic changes, e.g. DNA methylation of genes encoding redox-regulating enzymes. Emerging evidence implicates heightened carotid body chemoreflex in the progression of autonomic morbidities associated with cardiorespiratory diseases, such as sleep-disordered breathing with apnoea, congestive heart failure and essential hypertension.
 |
Nanduri R. Prabhakar received a PhD in Physiology from Baroda, India and DSc in Biology from Ruhr-University, Germany. In 1984, he joined the Case Western Reserve University, Cleveland, OH, USA as an Assistant Professor and became Professor and Vice-Chairman of the Department of Physiology (1997–2007). In 2007, he joined the University of Chicago. He is currently Harold H. Hines Professor and Inaugural Director of the Institute for Integrative Physiology and Center for Systems Biology of O2 sensing at the University of Chicago, IL, USA. He is a leading authority on O-sensing mechanisms and physiological consequences of hypoxia and has published more than 200 papers.
Introduction
Oxygen is an essential substrate for generating ATP, which is a major source of energy in mammalian cells. Vertebrates evolved complex respiratory and cardiovascular systems to insure optimal O2 delivery to tissues to maintain energy homeostasis. All mammalian cells respond to decreased O2 availability or hypoxia, albeit to different degrees. The systemic cardiorespiratory responses to hypoxia are reflex in nature and are initiated by specialized sensory organs called ‘peripheral chemoreceptors’, which monitor changes in arterial blood O2 levels. Heymans & Heymans (1927) were some of the first to report that stimulation of breathing by hypoxia is a reflex triggered by the carotid body, and they proposed the existence of similar structures in the aortic arch (aortic bodies) (Heymans et al. 1931). A subsequent study by Comroe (1939) provided firm evidence for reflex stimulation of breathing by aortic bodies as well. For the discovery of the sensory nature of the carotid body, C. F. Heymans received the Nobel Prize in Physiology in 1938, while J. H. Comroe Jr was given an honorary doctorate from the Karolinska Institute, Stockholm for elucidating the functional role of aortic bodies. Tissues with morphology similar to carotid and aortic bodies have also been described in the thorax and abdomen, and might serve as ancillary chemoreceptors (Deane et al. 1975; Easton & Howe, 1983).
Much of the information on the mechanisms of hypoxic sensing by the peripheral chemoreceptors has come from the studies on the carotid body. Innumerable studies have investigated how the carotid body detects hypoxia. An account of these early studies can be found in previous reviews (Fidone & Gonzalez, 1986; Gonzalez et al. 1994; Prabhakar, 2000). A more comprehensive and contemporary analysis of the structure and function of the carotid body and the physiological significance of the chemoreflex is available in a recent review (Kumar & Prabhakar, 2012). The present article focuses on recent studies addressing the following factors: (i) the roles of gaseous messengers in the hypoxic sensing by the carotid body; (ii) modulation of hypoxic sensing by genetic and epigenetic factors; and (iii) the role of the carotid body chemoreflex in cardiorespiratory diseases.
Physiology of carotid body hypoxic sensing
Carotid bodies, which reside bilaterally in the bifurcation of the common carotid arteries, receive sensory innervation from a branch of the glossopharengeal nerve called the ‘carotid sinus nerve’. The sensory discharge frequency of the carotid sinus nerve is low during normoxia (arterial 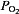 ∼100 mmHg), but increases dramatically with even a modest drop in arterial
∼100 mmHg), but increases dramatically with even a modest drop in arterial 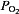 (e.g.
(e.g. 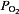 80–60 mmHg). The sensory response to low oxygen is rapid and occurs within seconds after the onset of hypoxia. The remarkable sensitivity and the speed with which it responds to hypoxia make the carotid body a unique sensory receptor for monitoring changes in the arterial blood
80–60 mmHg). The sensory response to low oxygen is rapid and occurs within seconds after the onset of hypoxia. The remarkable sensitivity and the speed with which it responds to hypoxia make the carotid body a unique sensory receptor for monitoring changes in the arterial blood 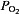 . The chemoreceptor tissue is composed of two major cell types, called type I (also called glomus) cells and type II cells. A substantial body of evidence suggests that type I cells are the initial sites of hypoxic sensing, and they work in concert with the nearby sensory nerve ending as a ‘sensory unit’ (see Kumar & Prabhakar, 2012 for references). The general consensus is that hypoxia inhibits certain K+ channels in type I cells, and the resulting depolarization leads to Ca2+-dependent release of excitatory neurotransmitter(s), which stimulates the nearby sensory nerve ending, leading to an increase in sensory discharge (Fig. 1). Despite intensive investigations, neither the mechanism(s) by which hypoxia inhibits K+ channels nor the identity of the chemical messenger(s) mediating the sensory excitation by low O2 are certain.
. The chemoreceptor tissue is composed of two major cell types, called type I (also called glomus) cells and type II cells. A substantial body of evidence suggests that type I cells are the initial sites of hypoxic sensing, and they work in concert with the nearby sensory nerve ending as a ‘sensory unit’ (see Kumar & Prabhakar, 2012 for references). The general consensus is that hypoxia inhibits certain K+ channels in type I cells, and the resulting depolarization leads to Ca2+-dependent release of excitatory neurotransmitter(s), which stimulates the nearby sensory nerve ending, leading to an increase in sensory discharge (Fig. 1). Despite intensive investigations, neither the mechanism(s) by which hypoxia inhibits K+ channels nor the identity of the chemical messenger(s) mediating the sensory excitation by low O2 are certain.
Figure 1. Schematic illustration of transduction of the hypoxic stimulus in a type I cell of the carotid body.
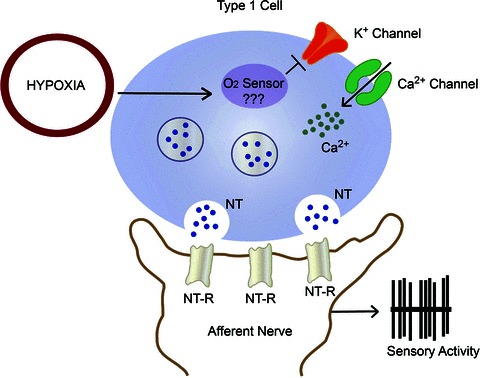
Abbreviations: NT, neurotransmitter(s); and NT-R, neurotransmitter receptor.
Role of gaseous messengers in hypoxic sensing
Nearly four decades ago, Brian Lloyd, Daniel Cunningham and their colleagues from the University of Oxford made an intriguing observation that brief inhalation of carbon monoxide inhibits ventilatory stimulation by hypoxia in conscious human subjects (Lloyd et al. 1968). Given that the affinity of haemoglobin for CO is much greater than for O2, it was proposed that the deoxy- conformation of a haem-containing protein(s) might be critical for initiating the carotid body response to hypoxia. Interestingly, CO, which was once used as an experimental tool to understand hypoxic sensing by the carotid body, is now known to be produced by the same chemoreceptor tissue. The following section provides a brief summary of our current understanding of the role of endogenous CO in the carotid body.
Carbon monoxide contributes to the low sensory activity of the carotid body during normoxia
Carbon monoxide is generated during degradation of haem by haem oxygenases (HOs), with NADPH and cytochrome P450 reductase as cofactors (Maines, 1997). Molecular O2 is essential for enzymatic generation of CO. The affinity (Km) of O2 for the haem–HO complex ranges between 30 and 80 μm (Migita et al. 1998). Two HO isoforms have been identified, namely a constitutively expressed HO-2 and an inducible HO-1 (also called heat shock protein 32 or Hsp32; Maines, 1997). Haem oxygenase-2 is expressed in type I cells of cat, rat (Prabhakar et al. 1995), mouse (Ortega-Sáenz et al. 2006) and human carotid bodies (Mkrtchian et al. 2012), but not in the nerve fibres and type II cells (Prabhakar et al. 1995). Haem oxygenase-1 expression is not evident in the carotid body in basal conditions.
The following evidence suggests that endogenous CO is a physiological inhibitor of carotid body activity. Zinc protoporphyrin-9, an inhibitor of HO, increases carotid body sensory activity in a dose-dependent manner, whereas copper protoporphyrin-9, which does not inhibit HO activity, has no influence (Prabhakar et al. 1995). Haem oxygenase-2 knockout mice exhibit elevated baseline carotid body sensory activity and an augmented sensory response to hypoxia (Prabhakar, 2012). Exogenous application of low concentrations of CO, similar to O2, inhibits the carotid body activity (Prabhakar et al. 1995). Disruption of HO-2 function in type I cells, like hypoxia, inhibits the maxi-K+ channel activity (Riesco-Fagundo et al. 2001; Williams et al. 2004) and elevates [Ca2+]i (Prabhakar, 1998). Biochemical studies have shown that carotid bodies generate substantial levels of CO during normoxia, whereas hypoxia inhibits CO generation (Prabhakar, 2012). Based on these findings, it was proposed that high levels of CO generated during normoxia keep the sensory activity low, and that hypoxia-evoked carotid body stimulation is in part due to reduced formation of CO, thereby removing its inhibitory influence on the sensory activity (Prabhakar, 1999).
Recent studies have argued against a role for endogenous CO in hypoxic sensing in the carotid body. For instance, Ortega-Sáenz et al. (2006) found that the type I cell neurosecretory response to hypoxia is unaltered in HO-2 knockout mice. This study utilized carotid body slices cultured for 20–96 h. Whether the long-term culture conditions masked the expected hypoxia-evoked increase in the secretory response in HO-2 knockout mice, however, is not clear from this study. If CO is a physiological inhibitor of the carotid body, then the hypoxic ventilatory response (HVR), a hallmark reflex triggered by the carotid body, should be augmented. Contrary to this possibility, Adachi et al. (2004) reported a blunted HVR in HO-2 knockout mice. However, in their study the authors measured the HVR for 15–20 s (see Adachi et al. 2004, p. 515), which is too short a duration to monitor breathing reliably in conscious mice. Thus, these unanticipated findings from the studies described above appear to be related to the experimental conditions employed and may therefore not provide a critical assessment of the role of CO in carotid body function.
In striking contrast to inhibition of the sensory activity by low doses of CO (Prabhakar et al. 1995), exogenous application of extremely high concentrations of CO (partial pressure of CO ∼320 mmHg) stimulate carotid body activity, depolarize type I cells and facilitate voltage-gated Ca2+ influx, and these effects are antagonized by light (Barbe et al. 2002). As early as 1928, Warburg and Negelein demonstrated an interaction of CO with the mitochondrial respiratory chain, which is also antagonized by light. Inhibitors of mitochondrial oxidative phosphorylation are known for their stimulatory effects on the carotid body (see Lahiri et al. 2006 for references). It is conceivable that, like other metabolic poisons, the effect of a high concentration of CO on the carotid body is due to its inhibition of the mitochondrial electron transport chain. Thus, the findings of Barbe et al. (2002) do not necessarily negate the proposed inhibitory role of endogenous CO in the carotid body.
Hydrogen sulfide mediates the sensory excitation by hypoxia
As early as in 1931, Heymans et al. reported that systemic administration of an H2S donor stimulates breathing, and this effect is mediated by the carotid body chemoreflex. It is now well established that H2S is produced endogenously and functions as a gaseous messenger in various physiological processes (Gadalla & Snyder, 2010; Wang, 2012). Cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) are the two major enzymes that catalyse the biosynthesis of H2S. Cystathionine β-synthase is abundant in the central nervous system, whereas CSE is preponderant in peripheral tissues. Carotid body type I cells express CBS (Li et al. 2010; Telezhkin et al. 2010; Fitzgerald et al. 2011) as well as CSE (Peng et al. 2010; Mkrtchian et al. 2012).
Cystathionine γ-lyase-deficient mice and rats treated with dl-propargylglycine, an inhibitor of CSE, exhibit absence or marked attenuation of carotid body sensory function, as well as the ventilatory response to hypoxia (Peng et al. 2010), suggesting that CSE-catalysed H2S is an excitatory gas messenger and mediates the sensory excitation by hypoxia. Mice treated with aminooxyacetic acid, a putative inhibitor of CBS, also exhibit impaired carotid body and ventilatory responses to hypoxia (Li et al. 2010). However, in addition to inhibiting CBS, aminooxyacetic acid also inhibits other pyridoxal phosphate-dependent enzymes, including CSE and 4-aminobutyrate aminotransferase (Beeler & Churchich, 1976), and disrupts mitochondrial function (Kauppinen et al. 1987). Given the broad spectrum of actions of aminooxyacetic acid, the role of CBS-catalysed H2S in the carotid body remains uncertain at present.
Further evidence supporting an excitatory role for H2S in the carotid body comes from studies with H2S donors. Exogenous application of NaHS, an H2S donor, produces robust sensory excitation of the carotid body in a concentration-dependent manner in mice and rats (Li et al. 2010; Peng et al. 2010). Like the response to hypoxia, the sensory response to NaHS is rapid in onset, occurs within seconds, and returns promptly to baseline after termination of its application (Peng et al. 2010).
How might H2S activate the carotid body? Inhibition of K+ channels and the resulting depolarization-induced voltage-gated Ca2+ influx in type I cells are critical steps for producing sensory excitation by hypoxia. Sodium hydrosulfide, like hypoxia, inhibits maxi-K+ (Li et al. 2010; Telezhkin et al. 2010) and TASK-like K+ channel (Tandem Pore K+ channel family) (Buckler, 2012) and depolarizes type I cells (Buckler, 2012). Also, NaHS leads to robust elevation of [Ca2+]i in type I cells, and this effect is absent in the absence of extracellular Ca2+ (Buckler, 2012; Makarenko et al. 2012) or if the depolarization is prevented by voltage clamping the cell at the resting membrane potential (Buckler, 2012). Furthermore, nifedipine, a blocker of L-type Ca2+ channels, prevents [Ca2+]i elevation by NaHS as well as by hypoxia (Makarenko et al. 2012). Sodium hydrosulfide increases NADH autofluorescence in type I cells, suggesting that H2S might mediate its actions in part by its effects on the mitochondrial electron transport chain (Buckler, 2012). These studies taken together demonstrate that H2S, like hypoxia, depolarizes type I cells by inhibiting certain K+ channels, facilitates voltage-gated Ca2+ influx and thus produces sensory excitation of the carotid body.
The generation of H2S in the carotid body is regulated by O2. Levels of H2S are low during normoxia, and hypoxia increases its levels in a stimulus-dependent manner in the carotid body (Peng et al. 2010). A similar increase in H2S generation was also seen in isolated type I cells challenged with hypoxia (Makarenko et al. 2012). Hypoxia-evoked H2S generation was absent in CSE knockout mice and in rats treated with the CSE inhibitor dl-propargylglycine (Peng et al. 2010). These observations, taken together with the finding that disruption of H2S generation prevents the carotid body and type I cell responses to hypoxia (Li et al. 2010; Peng et al. 2010; Makarenko et al. 2012), led to the suggestion that H2S is a physiological mediator of the carotid body response to hypoxia. However, as the concentrations of H2S required for eliciting type I cell and ventilatory responses are much greater than those produced endogenously, Buckler (2012) and Haouze et al. (2011) argued against a physiological role for H2S in the carotid body. The development of sensitive methods for simultaneous measurements of H2S in the carotid body along with the sensory discharge is needed in order firmly to establish the physiological role of H2S in the carotid body. Notwithstanding this limitation, current evidence from studies involving pharmacological and genetic disruption of endogenous H2S generation do provide a rather compelling case for a physiological role of H2S in mediating the carotid body response to hypoxia. In this context, it is noteworthy that H2S is an ancient gaseous messenger system that is well conserved across phyla, as evidenced by its participation in hypoxic responses of trout gill chemoreceptors (Olson et al. 2008) and Caenorhabditis elegans (Ma et al. 2012).
Mechanism(s) underlying increased H2S generation by hypoxia in the carotid body: a role for CO
How does hypoxia increase H2S generation? The findings that CO levels are relatively high and H2S levels low during normoxia prompted Peng et al. (2010) to examine whether CO keeps the generation of H2S suppressed during normoxia. Indeed, reducing CO levels by inhibition of HO markedly increased H2S generation during normoxia. Conversely, a CO donor inhibited the hypoxia-evoked H2S generation in the carotid body in a concentration-dependent manner. Given that the effects of the HO inhibitor were absent in CSE knockout mice, it was proposed that CO reduces H2S generation by inhibiting CSE enzyme activity (Peng et al. 2010). Based on these findings, it was suggested that low sensory discharge during normoxia is due to an inhibitory influence of CO on CSE, resulting in low H2S generation, whereas reduced CO generation during hypoxia lifts the inhibition on CSE, leading to elevated H2S levels and increased sensory discharge (Fig. 2). Further studies, however, are needed to delineate the mechanisms by which CO inhibits CSE activity during normoxia. In addition, as proposed by Dombkowski et al. (2006), inhibition of H2S oxidation might also contribute to its elevated levels during hypoxia, a possibility that remains to be investigated.
Figure 2. Schematic illustration of interactions between haem oxygenase-2 (HO-2)-generated carbon monoxide and cystathionine-γ-lyase (CSE)-regulated hydrogen sulfide generation and their effects on carotid body sensory activity during normoxia and hypoxia.
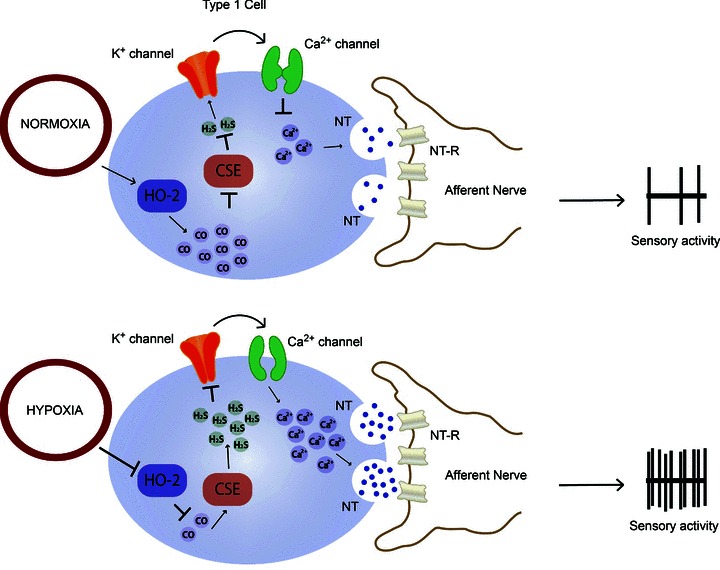
Abbreviations: NT, neurotransmitter(s); and NT-R, neurotransmitter receptor.
Is H2S an ‘O2 sensor’ or ‘mediator’ of the carotid body response to hypoxia?
Hydrogen sulfide has been proposed as an ‘O2 sensor’ in trout gill chemoreceptors (Olson et al. 2008). Can H2S be regarded as an ‘O2 sensor’ in the carotid body? Although H2S is critical for sensory excitation by hypoxia, the available evidence suggests that its generation in the carotid body is regulated by CO. Thus, interactions between the HO-2–CO and CSE–H2S systems preclude a role for H2S as an ‘O2 sensor’ in the carotid body. Rather, it would be more appropriate to regard H2S as an important ‘mediator’ of the sensory excitation during hypoxia. It was proposed that interacting proteins working in concert as a ‘chemosome’ account for the curvilinear response of the carotid body to hypoxia, providing a fail-proof redundancy of chemoreceptor response to low O2 (Prabhakar, 2006). It is likely that the enzymes generating CO and H2S, in concert with K+ channels in type I cells constitute important components of the ‘chemosome’. Recent studies suggest that AMP kinase signalling also plays a role in hypoxic sensing by the carotid body (Evans et al. 2012). It would be interesting to examine the potential cross-talk between the HO-2–CO and CSE–H2S systems with AMP kinase signalling in the carotid body in future studies.
Genetic influence on carotid body hypoxic sensing
John Weil and his co-workers from the University of Colorado were some of the first to recognize variations in the HVR in human subjects, with familial clusters and in mono- and dizygotic twins (see Weil, 2003 for references). As HVR is a hallmark reflex initiated by the carotid body, these variations were attributed to the influence of genetic factors on intrinsic hypoxic sensitivity of the chemoreceptors (Weil, 2003). Studies on spontaneously hypertensive rats and F-344 rats, which come from two genetic backgrounds, showed that the carotid body response to hypoxia was markedly augmented in spontaneously hypertensive rats, whereas it was severely impaired in F-344 rats (Weil et al. 1998), suggesting that genetic factors do influence hypoxic sensing by the carotid body. However, neither the identity of the genetic mechanism(s) nor the genes influencing the intrinsic hypoxic sensitivity of the carotid body have been explored further.
Regulation of carotid body hypoxic sensing by transcriptional activators: role of hypoxia-inducible factors (HIFs)
Hypoxia-inducible factors 1 and 2 are the well-studied members of the HIF family of transcriptional activators, which are essential for maintaining O2 homeostasis (Semenza, 2012). Hypoxia-inducible factors 1 and 2 are heterodimeric proteins, which consist of a constitutively expressed HIF-1β subunit and O2-regulated HIF-1α or HIF-2α subunits, respectively. Type I cells express both HIF-1α and HIF-2α, and the relative abundance of the latter is higher than the former (Tian et al. 1998; Roux et al. 2005).
Given that homozygous deficiency of either HIF-1α or HIF-2α is embryonically lethal (Iyer et al. 1998; Scortegagna et al. 2003), the roles of HIFs in the carotid body response to hypoxia were examined in mice with partial deficiencies in either HIF-1α (Hif-1a+/−) or HIF-2α (Hif-2a+/−). Carotid body responses to hypoxia were severely impaired in Hif-1a+/− mice (Kline et al. 2002; Peng et al. 2006), whereas responses to cyanide, a potent chemoreceptor stimulant, or to CO2, another physiological stimulus, were unaffected. In striking contrast, Hif-2a+/− mice exhibit selective heightening of the carotid body response to hypoxia (Peng et al. 2011). The enhanced hypoxic sensitivity of the carotid body in Hif-2a+/− mice was reflected in exaggerated chemoreflex function manifested by irregular breathing, with apnoea, hypertension and elevated plasma catecholamines. These observations demonstrate that carotid bodies from Hif-1a+/− mice are hyposensitive to hypoxia, whereas carotid bodies from Hif-2a+/− mice are hypersensitive.
How might HIFs contribute to hypoxic sensing by the carotid body? The HIFs regulate a variety of gene products associated with the maintenance of O2 homeostasis (Prabhakar & Semenza, 2012). Hypoxia-inducible factor-regulated gene products may affect carotid body function either by affecting its morphology and/or independent of structural changes. The defective hypoxic sensing in Hif-1a+/− mice and the exaggerated hypoxic response in Hif-2a+/− mice appear not to be secondary to changes in carotid body morphology (Kline et al. 2002; Peng et al. 2011). Although HIFs regulate a variety of gene products, including some ion channels (see Prabhakar & Semenza, 2012 for references), recent studies have shown that HIF-1 and HIF-2 regulate the expression of gene products with opposing functions that regulate the redox state. For instance, HIF-1 regulates the expression of pro-oxidant enzymes, including NADPH oxidases (Diebold et al. 2010; Yuan et al. 2011), whereas HIF-2 regulates the expression of antioxidant enzymes (Scortegagna et al. 2003; Nanduri et al. 2009). Given that hypoxia alters the redox equilibrium in cells towards a more reduced state, it is likely that the contrasting effects of carotid body responses to hypoxia in HIF-1a+/− versus HIF-2a+/− mice might be due to differential expression of pro- and antioxidant enzymes and the ensuing changes in the redox state. Indeed, carotid bodies from Hif-2a+/− mice showed reduced levels of mRNAs encoding antioxidant enzymes and oxidative stress, and antioxidant treatment prevented the augmented hypoxic sensitivity of the carotid body and corrected the oxidative stress in these mice (Peng et al. 2011). Given that HIF-1 regulates the transcription of pro-oxidant enzymes (Diebold et al. 2010; Yuan et al. 2011), it is likely that reduced pro-oxidant enzyme expression and the resulting reduction in the intracellular redox state might contribute to the impaired hypoxic sensitivity in Hif-1a+/− mice, a possibility that remains to be established. Taken together, these studies suggest that a subset of HIF-regulated genes associated with the maintenance of the intracellular redox state might account in part for genetic variability in the carotid body response to hypoxia. The effects of partial deficiencies of HIF-α isoforms on the carotid body activity and the downstream redox-regulating enzyme genes are summarized in Fig. 3. Further studies are needed to determine whether HIFs also regulate the enzymes generating CO and H2S as well as ion channels and to elucidate the role of other transcriptional activators that may also contribute to genetic variability of the carotid body response to hypoxia.
Figure 3.
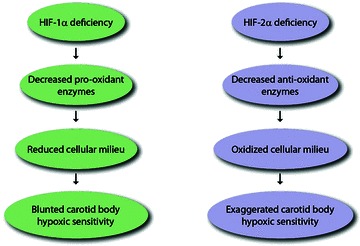
Molecular mechanisms underlying the carotid body responses to hypoxia in mice with partial deficiencies of hypoxia-inducible factors HIF-1α and HIF-2α
Epigenetic regulation of hypoxic sensing
Systemic or environmental perturbations in O2 levels in the early neonatal period influence the carotid body response to hypoxia in adulthood. For instance, adult rats treated with perinatal intermittent hypoxia (IH), simulating the apnoea of prematurity, exhibit carotid body hypersensitivity to hypoxia (Peng et al. 2004; Pawar et al. 2008, 2009). The mechanisms underlying the long-term consequences of O2 perturbations in the early developmental period are not known.
Emerging evidence suggests that aberrant epigenetic regulation is an important mechanism mediating the effects of environmental perturbations in the neonatal period in adulthood (Feinberg, 2007). Epigenetic mechanisms are heritable modifications of DNA that do not involve changes in the DNA primary sequence (Barker et al. 1993; Gluckman et al. 2005). Silencing RNAs, DNA methylation and histone modifications are three well-studied epigenetic mechanisms, which either individually or collectively determine whether a gene is activated or silenced (Feinberg, 2007). Of the three mechanisms, DNA methylation has been implicated in mediating the gene expression associated with developmental programming (Waterland and Jirtle, 2003). The DNA methylation is catalysed by DNA methyltransferases or Dnmts and occurs at the 5′ position of cytosine of CpG dinucleotide clusters in the gene promoter called ‘CpG’ islands (Gardiner-Garden & Frommer, 1987; Feinberg, 2007). In general, DNA hypermethylation leads to repression of gene transcription, whereas hypomethylation causes transcriptional activation.
A recent study examined the role of DNA methylation in mediating the long-term effect of neonatal IH on carotid body responses to hypoxia in adult rats (Nanduri et al. 2012). Adult rats exposed to neonatal IH exhibit augmented hypoxic sensitivity of the carotid body, respiratory abnormalities manifested by a greater number of spontaneous apnoeas, hypertension and elevated plasma catecholamines. The mRNA levels of Dnmt1 and Dnmt3b mRNA and the corresponding protein levels were elevated and global DNA methylation was increased in the carotid bodies from adult rats exposed to neonatal IH. Genes encoding antioxidant enzymes were downregulated, and there was increased oxidative stress in the carotid bodies. Given that DNA hypermethylation suppresses gene expression, whether increased DNA methylation contributes to the reduced antioxidant gene expression was examined. A detailed analysis of the promoter region of the superoxide dismutase 2 gene (Sod2) revealed marked hypermethylation of a single CpG dinucleotide close to the transcription start site in adult rats exposed to neonatal IH. Remarkably, neonatal treatment with decitabine, an inhibitor of DNA methylation, eliminated DNA hypermethylation of the single CpG dinucleotide in the Sod2 gene, corrected the oxidative stress, prevented the enhanced hypoxic sensitivity of the carotid body, and normalized the cardiorespiratory functions in adult rats exposed to neonatal IH (Nanduri et al. 2012). These findings implicate a hitherto uncharacterized role for epigenetic mechanisms mediating the effects of perturbations in O2 levels in the neonatal period on the carotid body response to hypoxia in adulthood (Fig. 4). These studies are only the beginning in this area, and further investigations are certainly needed to delineate the role of other epigenetic mechanisms, including the role of histone modifications and/or silencing RNAs, and to identify the target genes associated with developmental programming of the carotid body response to hypoxia.
Figure 4.
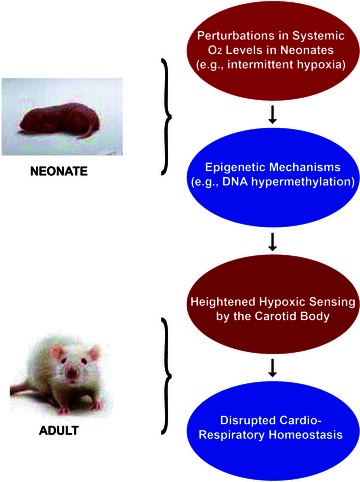
Schematic illustration of epigenetic mechanisms (e.g. DNA hypermethylation) contributing to augmented carotid body sensitivity to hypoxia in adult rats exposed to neonatal intermittent hypoxia and its impact on cardiorespiratory homeostasis
Carotid body chemoreflex in cardiorespiratory diseases
The importance of the carotid body chemoreflex in maintaining cardiorespiratory homeostasis during a variety of physiological conditions is well documented. For instance, the chemoreflex is critical for ventilatory adaptations to high altitude, as well as for cardiorespiratory responses to exercise (see Kumar & Prabhakar, 2012). Emerging evidence suggests that the carotid body chemoreflex plays a more prominent role than hitherto appreciated in the progression of autonomic morbidities associated with cardiorespiratory diseases. For example, heightened chemoreflex function has been implicated in autonomic dysfunction caused by sleep-disordered breathing with apnoea (Kara et al. 2003; Prabhakar et al. 2007), congestive heart failure (Schultz & Li, 2007) and neurogenic hypertension (Abdala et al. 2012). Owing to space constraints, the following section will focus on studies assessing the effects of chronic intermittent hypoxia, a hallmark of sleep-disordered breathing with apnoea, on carotid body and chemoreflex function.
Carotid body function in patients with recurrent apnoea
Sleep-disordered breathing with obstructive sleep apnoea is a major clinical disorder that is reaching epidemic proportions (Nieto et al. 2000; Shahar et al. 2001). In severely affected patients, the frequency of apnoea may exceed 60 episodes h−1, and arterial blood O2 saturation can be reduced to as low as 50%. Patients with recurrent apnoea exhibit elevated sympathetic nerve activity, plasma and urinary catecholamines, and are prone to develop hypertension. It has been proposed that carotid bodies constitute the ‘frontline’ defense system for detecting periodic hypoxaemia associated with apnoea and that the chemoreflex contributes to elevated sympathetic activity and blood pressure (Cistulli & Sullivan, 1994). Patients with recurrent apnoea exhibit an augmented hypoxic ventilatory response (Narkiewicz et al. 1999; Kara et al. 2003), a reflex initiated by the carotid body. Obstructive sleep apnoea patients exhibit a more pronounced decrease in sympathetic activity in response to brief hyperoxia (Dejour's test; an indirect measure of peripheral chemoreceptor sensitivity) than control subjects (Tafil-Klawe et al. 1991; Kara et al. 2003), suggesting augmented chemoreflex function. Remarkably, carotid body-resected subjects with sleep apnoea do not develop hypertension (see discussion by Somers & Abboud, 1993). These studies suggest heightened carotid body chemoreflex function in patients with recurrent apnoea.
Effects of chronic intermittent hypoxia on carotid body response to hypoxia
Recurrent apnoea produces chronic IH and hypercapnia. A major advance in the field is the demonstration that exposure of experimental animals to chronic IH alone is sufficient to cause autonomic morbidities similar to those described in recurrent apnoea patients (Fletcher, 2001; Peng et al. 2006). Rats exposed to chronic IH also develop hypertension similar to that seen in patients with recurrent apnoea, and bilateral sectioning of the carotid sinus nerves prevents this response (Lesske et al. 1997). Direct recording of carotid body sensory activity has shown selective enhancement of the hypoxic response in animals exposed to chronic IH (Peng et al. 2003, 2006; Peng & Prabhakar, 2004; Rey et al. 2004, 2006). Furthermore, repetitive hypoxia produces a long-lasting increase in baseline sensory activity in rodents exposed to chronic IH, and this phenomenon was termed ‘sensory long-term facilitation’ or sensory LTF (Peng et al. 2003, 2006). Chronic IH had no significant effect on carotid body morphology (Peng et al. 2003). In striking contrast, the carotid body response to hypoxia was normal in rats exposed to chronic continuous hypoxia (Peng et al. 2003), suggesting that the effects are unique to IH. The absence of a heightened carotid body response to hypoxia with chronic continuous hypoxia might explain why high-altitude residents, who are adapted to a low-O2 environment, do not exhibit autonomic dysfunction, as opposed to patients with recurrent apnoea, who experience chronic IH.
Reactive oxygen species (ROS) mediate the effects of chronic IH on the carotid body
The following findings suggest that ROS signalling mediates the effects of chronic IH on the carotid body. First, ROS levels were elevated in carotid bodies from chronic IH-treated rodents (Peng et al. 2003, 2006; Pawar et al. 2009). Second, antioxidant treatment prevented the chronic IH-evoked augmented hypoxic sensitivity and sensory LTF (Peng et al. 2003; Pawar et al. 2009). Available evidence suggests that increased transcription and activation of the pro-oxidant enzyme, NADPH oxidase-2, and decreased transcription and activity of the antioxidant enzymes (e.g. superoxide dismutase 2) contribute to the increase in ROS levels induced by IH (Peng et al. 2009; Nanduri et al. 2009). In addition, IH increases ROS in the mitochondrial compartment by inhibiting complex I of the mitochondrial electron transport chain (Peng et al. 2003). A recent study by Khan et al. (2011) showed that ROS generated by NADPH oxidase-2 inhibit the mitochondrial complex I in IH-exposed cells, involving S-glutathionylation of the complex I subunits. The selective contribution of ROS generated in the mitochondrial compartment to the altered carotid body function, however, remains to be investigated.
Analysis of the molecular mechanisms revealed that activation of HIF-1 mediates the increase in NADPH oxidase-2 transcription induced by IH (Peng et al. 2006; Yuan et al. 2011). On the contrary, IH decreases HIF-2 activity, which in turn leads to insufficient transcription of superoxide dismutase 2 (Nanduri et al. 2009). Thus, the imbalance between HIF-1 and HIF-2 and resulting changes in pro- and antioxidant enzyme expressions contribute to IH-induced oxidative stress in the carotid body. How might ROS mediate the effects of chronic IH on the carotid body? Available evidence suggests that ROS-dependent recruitment of endothelin-1 and 5-HT signalling contribute to the chronic IH-evoked augmented hypoxic response and sensory LTF, respectively (Pawar et al. 2009; Peng et al. 2009). However, the effects of chronic IH on recently identified HO-2–CO and CSE–H2S signalling in the carotid body remain to be investigated.
Heightened carotid body chemoreflex mediates autonomic dysfunction caused by chronic IH
What might be the functional significance of chronic IH-induced changes in carotid body function? It was proposed that during each episode of apnoea the increased carotid body sensitivity to hypoxia can lead to a greater magnitude of hyperventilation, thus driving the respiratory controller below the apnoeic threshold for CO2, leading to a greater number of apnoeas (Prabhakar, 2001). In other words, the heightened hypoxic sensitivity of the carotid body might act as a ‘positive feedback’, thereby exacerbating the occurrence of apnoeas (Fig. 5A). Supporting such a possibility is the finding that chronic IH-exposed rats with intact carotid bodies exhibit a greater incidence of spontaneous apnoeas, and this effect was absent in carotid body-sectioned rats exposed to chronic IH (Fig. 5B). Patients with sleep-disordered breathing exhibit elevated sympathetic nerve activity during the day time, wherein apnoeas are absent (Kara et al. 2003). Given that the carotid body chemoreflex stimulates sympathetic nerve activity, it was proposed that the sensory LTF contributes to the day-time increase in sympathetic nerve activity in patients with recurrent apnoea (Prabhakar, 2001; Prabhakar et al. 2012; Fig. 5C). Indeed, rats exposed to chronic IH showed elevated sympathetic nerve activity, and this effect was absent following chronic bilateral sectioning of the carotid sinus nerves (Fig. 5D). These findings suggest that the heightened carotid body chemoreflex contributes to autonomic dysfunction in patients with sleep-disordered breathing with apnoea.
Figure 5. Contribution of carotid body chemoreflex to cardiorespiratory responses to chronic intermittent hypoxia (CIH).
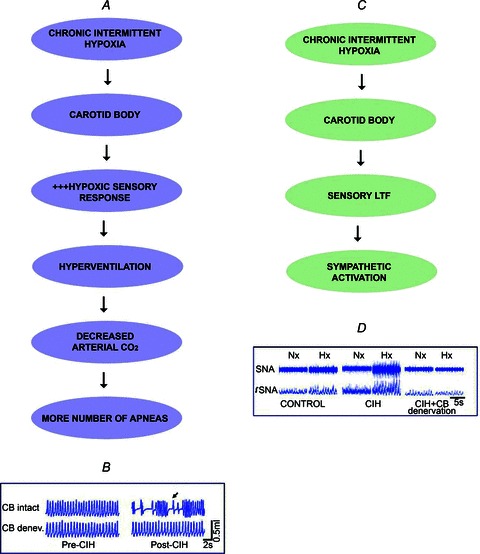
A, the heightened carotid body response to hypoxia induced by CIH leads to a greater number of spontaneous apnoeas. B, example of breathing responses in awake rats before and after exposure to CIH with intact and denervated carotid bodies (CB). Arrow represents irregular breathing with apnoea. C, CIH-induced sensory long-term facilitation (LTF) of the carotid body reflexly stimulates sympathetic activity. D, examples of splanchnic nerve activity (SNA) in normoxia (Nx) and hypoxia (Hx, 12% O2) in anaesthetized, mechanically ventilated rats exposed to normoxia (control) or CIH with intact carotid bodies and following carotid body denervation. Note the enhanced SNA response to Hx in the CIH-exposed rat and the absence of this response in the CB-denervated rat exposed to CIH.
Concluding remarks
Vertebrates are endowed with a variety of sensory receptors for detecting diverse sensory modalities, including mechanical, thermal, visual, olfactory and taste sensations. Two principal categories of sensory transduction machineries have been indentified. One involves ion channels, which mediate the mechanical and thermal sensations, and the other is G-protein-coupled receptors, which contribute to the transduction of visual, olfactory and taste sensations (Julius & Nathans, 2012). Although ion channels were implicated in carotid body hypoxic sensing, emerging evidence suggests a biochemical mechanism associated with O2-dependent enzymatic generation of gas messengers (e.g. CO and H2S) as an important initial step in the transduction cascade of the hypoxic stimulus in the carotid body. Thus, the transduction machinery in the carotid body appears to be distinctly different from other known sensory receptors. Evidence is emerging that genetic and epigenetic mechanisms profoundly impact the carotid body response to hypoxia by altering the intracellular redox state. Elucidation of the mechanisms by which redox state affects the transduction machinery of the carotid body is an important area for future research.
Emerging evidence suggests that either hypo- or hypersensitivity of the carotid body to hypoxia has adverse physiological consequences. For instance, people with blunted HVR, a hallmark reflex response of the carotid body, exhibit impaired ventilatory adaptations to high altitude, manifested by pulmonary oedema (Hohenhaus et al. 1995). On the contrary, hypersensitivity to hypoxia contributes to the autonomic dysfunction associated with highly prevalent cardiorespiratory diseases, such as sleep-disordered breathing with apnoea, congestive heart failure and neurogenic hypertension. Future development of new therapeutic interventions targeting the carotid body to correct either the hypo- or hypersensitivity to hypoxia is of greater translational significance than hitherto appreciated.
Acknowledgments
This article is based on 2012 J.H. Comroe Distinguished Lecture of the American Physiological Society. It is a great privilege to acknowledge the collaborations of Professors Solomon H. Snyder and Gregg L. Semenza of the Johns Hopkins School of Medicine. The studies presented in this article would have not been possible without the participation of my colleagues Professor G. K. Kumar, Professor J. Nanduri and Drs Y-J. Peng and G. Yuan. I am greatly indebted to Ms Shanna L. Williams for preparing the figures. The research in my laboratory is supported by grants from National Institutes of Health, Heart, Lung and Blood Institute, R01-HL76537, P01-HL90554 and R01-HL86493.
Glossary
- CBS
cystathionine β-synthase
- CSE
cystathionine-γ-lyase
- HIF
hypoxia-inducible factor
- HO
haem oxygenase
- HVR
hypoxic ventilatory response
- IH
intermittent hypoxia
- LTF
long-term facilitation
- ROS
reactive oxygen species
References
- Abdala AP, McBryde FD, Marina N, Hendy EB, Engelman ZJ, Fudim M, Sobotka PA, Gourine AV, Paton JF. Hypertension is critically dependent on the carotid body input in the spontaneously hypertensive rat. J Physiol. 2012;590:4269–4277. doi: 10.1113/jphysiol.2012.237800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi T, Ishikawa K, Hida W, Matsumoto H, Masuda T, Date F, Ogawa K, Takeda K, Furuyama K, Zhang Y, Kitamuro T, Ogawa H, Maruyama Y, Shibahara S. Hypoxemia and blunted hypoxic ventilatory responses in mice lacking heme oxygenase-2. Biochem Biophys Res Commun. 2004;320:514–522. doi: 10.1016/j.bbrc.2004.05.195. [DOI] [PubMed] [Google Scholar]
- Barbé C, Al-Hashem F, Conway AF, Dubuis E, Vandier C, Kumar P. A possible dual site of action for carbon monoxide-mediated chemoexcitation in the rat carotid body. J Physiol. 2002;543:933–945. doi: 10.1113/jphysiol.2001.015750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, Osmond C, Simmonds SJ, Wield GA. The relation of small head circumference and thinness at birth to death from cardiovascular disease in adult life. BMJ. 1993;306:422–426. doi: 10.1136/bmj.306.6875.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler T, Churchich JE. Reactivity of the phosphopyridoxal groups of cystathionase. J Biol Chem. 1976;251:5267–5271. [PubMed] [Google Scholar]
- Buckler KJ. Effects of exogenous hydrogen sulphide on calcium signalling, background (TASK) K channel activity and mitochondrial function in chemoreceptor cells. Pflugers Arch. 2012;463:743–754. doi: 10.1007/s00424-012-1089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cistulli P, Sullivan C. Pathophysiology of sleep apnea. In: Saunders N, Sullivan C, editors. Sleep and Breathing. New York: Marcel Dekker; 1994. pp. 405–448. [Google Scholar]
- Comroe JH. The location and function of the chemoreceptors of the aorta. Am J Physiol. 1939;127:176–191. [Google Scholar]
- Deane BM, Howe A, Morgan M. Abdominal vagal paraganglia: distribution and comparison with carotid body, in the rat. Acta Anat (Basel) 1975;93:19–28. doi: 10.1159/000144493. [DOI] [PubMed] [Google Scholar]
- Diebold I, Petry A, Djordjevic T, BelAiba RS, Fineman J, Black S, Schreiber C, Fratz S, Hess J, Kietzmann T, Görlach A. Reciprocal regulation of Rac1 and PAK-1 by HIF-1α: a positive-feedback loop promoting pulmonary vascular remodeling. Antioxid Redox Signal. 2010;13:399–412. doi: 10.1089/ars.2009.3013. [DOI] [PubMed] [Google Scholar]
- Dombkowski RA, Doellman MM, Head SK, Olson KR. Hydrogen sulfide mediates hypoxia-induced relaxation of trout urinary bladder smooth muscle. J Exp Biol. 2006;209:3234–3240. doi: 10.1242/jeb.02376. [DOI] [PubMed] [Google Scholar]
- Easton J, Howe A. The distribution of thoracic glomus tissue (aortic bodies) in the rat. Cell Tissue Res. 1983;232:349–356. doi: 10.1007/BF00213792. [DOI] [PubMed] [Google Scholar]
- Evans AM, Peers C, Wyatt CN, Kumar P, Hardie DG. Ion channel regulation by the LKB1-AMPK signalling pathway: the key to carotid body activation by hypoxia and metabolic homeostasis at the whole body level. Adv Exp Med Biol. 2012;758:81–90. doi: 10.1007/978-94-007-4584-1_11. [DOI] [PubMed] [Google Scholar]
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- Fidone SJ, Gonzalez C. Handbook of Physiology, section 3, The Respiratory System, vol. II, Control of Breathing. Bethesda, MD, USA: American Physiological Society; 1986. Initiation and control of chemoreceptor activity in the carotid body; pp. 247–312. [Google Scholar]
- Fitzgerald RS, Shirahata M, Chang I, Kostuk E, Kiihl S. The impact of hydrogen sulfide (H2S) on neurotransmitter release from the cat carotid body. Respir Physiol Neurobiol. 2011;176:80–89. doi: 10.1016/j.resp.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher EC. Physiological consequences of intermittent hypoxia: systemic blood pressure. J Appl Physiol. 2001;90:1600–1605. doi: 10.1152/jappl.2001.90.4.1600. [DOI] [PubMed] [Google Scholar]
- Gadalla MM, Snyder SH. Hydrogen sulfide as a gasotransmitter. J Neurochem. 2010;113:14–26. doi: 10.1111/j.1471-4159.2010.06580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Cutfield W, Hofman P, Hanson MA. The fetal, neonatal, and infant environments—the long-term consequences for disease risk. Early Hum Dev. 2005;81:51–59. doi: 10.1016/j.earlhumdev.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiol Rev. 1994;74:829–898. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- Haouzi P, Bell H, Van de Louw A. Hypoxia-induced arterial chemoreceptor stimulation and hydrogen sulfide: too much or too little. Respir Physiol Neurobiol. 2011;179:97–102. doi: 10.1016/j.resp.2011.09.009. [DOI] [PubMed] [Google Scholar]
- Heymans C, Bouckaert JJ, Dautrebande L. Sinus carotidien et reflexes respiratoires: sensibilite des sinus carotidiens aux substances chimiques. Action stimulante respiratoire reflexe du sulfure de sodium, du cyanure de potassium, de la nicotine et de la lobeline. Arch Int Pharmacodyn Ther. 1931;40:54–91. [Google Scholar]
- Heymans J, Heymans C. Sur les modifications directes et sur la regulation reflexede l’activitie du centre respiratory de la tete isolee du chien. Arch Int Pharmacodyn Ther. 1927;33:273–372. [Google Scholar]
- Hohenhaus E, Paul A, McCullough RE, Kücherer H, Bärtsch P. Ventilatory and pulmonary vascular response to hypoxia and susceptibility to high altitude pulmonary oedema. Eur Respir J. 1995;8:1825–1833. doi: 10.1183/09031936.95.08111825. [DOI] [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julius D, Nathans J. Signaling by sensory receptors. Cold Spring Harb Perspect Biol. 2012;4:a005991. doi: 10.1101/cshperspect.a005991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kara T, Narkiewicz K, Somers VK. Chemoreflexes – physiology and clinical implications. Acta Physiol Scand. 2003;177:377–384. doi: 10.1046/j.1365-201X.2003.01083.x. [DOI] [PubMed] [Google Scholar]
- Kauppinen RA, Sihra TS, Nicholls DG. Aminooxyacetic acid inhibits the malate-aspartate shuttle in isolated nerve terminals and prevents the mitochondria from utilizing glycolytic substrates. Biochim Biophys Acta. 1987;930:173–178. doi: 10.1016/0167-4889(87)90029-2. [DOI] [PubMed] [Google Scholar]
- Khan SA, Nanduri J, Yuan G, Kinsman B, Kumar GK, Joseph J, Kalyanaraman B, Prabhakar NR. NADPH oxidase 2 mediates intermittent hypoxia-induced mitochondrial complex I inhibition: relevance to blood pressure changes in rats. Antioxid Redox Signal. 2011;14:533–542. doi: 10.1089/ars.2010.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Peng YJ, Manalo DJ, Semenza GL, Prabhakar NR. Defective carotid body function and impaired ventilatory responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. Proc Natl Acad Sci U S A. 2002;99:821–826. doi: 10.1073/pnas.022634199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Prabhakar NR. Peripheral chemoreceptors: function and plasticity of the carotid body. Compr Physiol. 2012;2:141–219. doi: 10.1002/cphy.c100069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S, Roy A, Baby SM, Hoshi T, Semenza GL, Prabhakar NR. Oxygen sensing in the body. Prog Biophys Mol Biol. 2006;91:249–286. doi: 10.1016/j.pbiomolbio.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Lesske J, Fletcher EC, Bao G, Unger T. Hypertension caused by chronic intermittent hypoxia – influence of chemoreceptors and sympathetic nervous system. J Hypertens. 1997;15:1593–1603. doi: 10.1097/00004872-199715120-00060. [DOI] [PubMed] [Google Scholar]
- Li Q, Sun B, Wang X, Jin Z, Zhou Y, Dong L, Jiang LH, Rong W. A crucial role for hydrogen sulfide in oxygen sensing via modulating large conductance calcium-activated potassium channels. Antioxid Redox Signal. 2010;12:1179–1189. doi: 10.1089/ars.2009.2926. [DOI] [PubMed] [Google Scholar]
- Lloyd BB, Cunningham DJC, Goode RC. Depression of hypoxic hyperventilation in man by sudden inspiration of carbon monoxide. In: Torrance RW, editor. Arterial Chemoreceptors. Oxford: Blackwell; 1968. pp. 145–150. [Google Scholar]
- Ma DK, Vozdek R, Bhatla N, Horvitz HR. CYSL-1 interacts with the O2-sensing hydroxylase EGL-9 to promote H2S-modulated hypoxia-induced behavioral plasticity in C. elegans. Neuron. 2012;73:925–940. doi: 10.1016/j.neuron.2011.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- Makarenko VV, Nanduri J, Raghuraman G, Fox AP, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. Endogenous H2S is required for hypoxic sensing by carotid body glomus cells. Am J Physiol Cell Physiol. 2012;303:C916–C923. doi: 10.1152/ajpcell.00100.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migita CT, Matera KM, Ikeda-Saito M, Olson JS, Fujii H, Yoshimura T, Zhou H, Yoshida T. The oxygen and carbon monoxide reactions of heme oxygenase. J Biol Chem. 1998;273:945–949. doi: 10.1074/jbc.273.2.945. [DOI] [PubMed] [Google Scholar]
- Mkrtchian S, Kåhlin J, Ebberyd A, Gonzalez C, Sanchez D, Balbir A, Kostuk EW, Shirahata M, Fagerlund MJ, Eriksson LI. The human carotid body transcriptome with focus on oxygen sensing and inflammation – a comparative analysis. J Physiol. 2012;590:3807–3819. doi: 10.1113/jphysiol.2012.231084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanduri J, Makarenko V, Reddy VD, Yuan G, Pawar A, Wang N, Khan SA, Zhang X, Kinsman B, Peng YJ, Kumar GK, Fox AP, Godley LA, Semenza GL, Prabhakar NR. Epigenetic regulation of hypoxic sensing disrupts cardiorespiratory homeostasis. Proc Natl Acad Sci U S A. 2012;109:2515–2520. doi: 10.1073/pnas.1120600109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanduri J, Wang N, Yuan G, Khan SA, Souvannakitti D, Peng YJ, Kumar GK, Garcia JA, Prabhakar NR. Intermittent hypoxia degrades HIF-2α via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc Natl Acad Sci U S A. 2009;106:1199–1204. doi: 10.1073/pnas.0811018106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJ, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation. 1999;99:1183–1189. doi: 10.1161/01.cir.99.9.1183. [DOI] [PubMed] [Google Scholar]
- Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D’Agostino RB, Newman AB, Lebowitz MD, Pickering TG. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA. 2000;283:1829–1836. doi: 10.1001/jama.283.14.1829. [DOI] [PubMed] [Google Scholar]
- Olson KR, Healy MJ, Qin Z, Skovgaard N, Vulesevic B, Duff DW, Whitfield NL, Yang G, Wang R, Perry SF. Hydrogen sulfide as an oxygen sensor in trout gill chemoreceptors. Am J Physiol Regul Integr Comp Physiol. 2008;295:R669–R680. doi: 10.1152/ajpregu.00807.2007. [DOI] [PubMed] [Google Scholar]
- Ortega-Sáenz P, Pascual A, Gómez-Díaz R, López-Barneo J. Acute oxygen sensing in heme oxygenase-2 null mice. J Gen Physiol. 2006;128:405–411. doi: 10.1085/jgp.200609591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawar A, Nanduri J, Yuan G, Khan SA, Wang N, Kumar GK, Prabhakar NR. Reactive oxygen species-dependent endothelin signaling is required for augmented hypoxic sensory response of the neonatal carotid body by intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol. 2009;296:R735–R742. doi: 10.1152/ajpregu.90490.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawar A, Peng YJ, Jacono FJ, Prabhakar NR. Comparative analysis of neonatal and adult rat carotid body responses to chronic intermittent hypoxia. J Appl Physiol. 2008;104:1287–1294. doi: 10.1152/japplphysiol.00644.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Khan SA, Yuan G, Wang N, Kinsman B, Vaddi DR, Kumar GK, Garcia JA, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 2α (HIF-2α) heterozygous-null mice exhibit exaggerated carotid body sensitivity to hypoxia, breathing instability, and hypertension. Proc Natl Acad Sci U S A. 2011;108:3065–3070. doi: 10.1073/pnas.1100064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Raghuraman G, Souvannakitti D, Gadalla MM, Kumar GK, Snyder SH, Prabhakar NR. H2S mediates O2 sensing in the carotid body. Proc Natl Acad Sci U S A. 2010;107:10719–10724. doi: 10.1073/pnas.1005866107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Yuan G, Wang N, Deneris E, Pendyala S, Natarajan V, Kumar GK, Prabhakar NR. NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J Neurosci. 2009;29:4903–4910. doi: 10.1523/JNEUROSCI.4768-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci U S A. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol. 2004;96:1236–1242. doi: 10.1152/japplphysiol.00820.2003. discussion 1196. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Rennison J, Prabhakar NR. Intermittent hypoxia augments carotid body and ventilatory response to hypoxia in neonatal rat pups. J Appl Physiol. 2004;97:2020–2025. doi: 10.1152/japplphysiol.00876.2003. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Yuan G, Ramakrishnan D, Sharma SD, Bosch-Marce M, Kumar GK, Semenza GL, Prabhakar NR. Heterozygous HIF-1α deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol. 2006;577:705–716. doi: 10.1113/jphysiol.2006.114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR. Endogenous carbon monoxide in control of respiration. Respir Physiol. 1998;114:57–64. doi: 10.1016/s0034-5687(98)00072-3. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. NO and CO as second messengers in oxygen sensing in the carotid body. Respir Physiol. 1999;115:161–168. doi: 10.1016/s0034-5687(99)00019-5. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. Oxygen sensing by the carotid body chemoreceptors. J Appl Physiol. 2000;88:2287–2295. doi: 10.1152/jappl.2000.88.6.2287. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. Oxygen sensing during intermittent hypoxia: cellular and molecular mechanisms. J Appl Physiol. 2001;90:1986–1994. doi: 10.1152/jappl.2001.90.5.1986. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. O2 sensing at the mammalian carotid body: why multiple O2 sensors and multiple transmitters. Exp Physiol. 2006;91:17–23. doi: 10.1113/expphysiol.2005.031922. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. Carbon monoxide (CO) and hydrogen sulfide (H2S) in hypoxic sensing by the carotid body. Respir Physiol Neurobiol. 2012;184:165–169. doi: 10.1016/j.resp.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Dinerman JL, Agani FH, Snyder SH. Carbon monoxide: a role in carotid body chemoreception. Proc Natl Acad Sci U S A. 1995;92:1994–1997. doi: 10.1073/pnas.92.6.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Kumar GK, Nanduri J, Semenza GL. ROS signaling in systemic and cellular responses to chronic intermittent hypoxia. Antioxid Redox Signal. 2007;9:1397–1403. doi: 10.1089/ars.2007.1732. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Kumar GK, Peng YJ. Sympatho-adrenal activation by chronic intermittent hypoxia. J Appl Physiol. 2012;113:1304–1310. doi: 10.1152/japplphysiol.00444.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Semenza GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev. 2012;92:967–1003. doi: 10.1152/physrev.00030.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey S, Del Rio R, Alcayaga J, Iturriaga R. Chronic intermittent hypoxia enhances cat chemosensory and ventilatory responses to hypoxia. J Physiol. 2004;560:577–586. doi: 10.1113/jphysiol.2004.072033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey S, Del Rio R, Iturriaga R. Contribution of endothelin-1 to the enhanced carotid body chemosensory responses induced by chronic intermittent hypoxia. Brain Res. 2006;1086:152–159. doi: 10.1016/j.brainres.2006.02.082. [DOI] [PubMed] [Google Scholar]
- Riesco-Fagundo AM, Pérez-García MT, González C, López-López JR. O2 modulates large-conductance Ca2+-dependent K+ channels of rat chemoreceptor cells by a membrane-restricted and CO-sensitive mechanism. Circ Res. 2001;89:430–436. doi: 10.1161/hh1701.095632. [DOI] [PubMed] [Google Scholar]
- Roux JC, Brismar H, Aperia A, Lagercrantz H. Developmental changes in HIF transcription factor in carotid body: relevance for O2 sensing by chemoreceptors. Pediatr Res. 2005;58:53–57. doi: 10.1203/01.PDR.0000163390.78239.EA. [DOI] [PubMed] [Google Scholar]
- Schultz HD, Li YL. Carotid body function in heart failure. Respir Physiol Neurobiol. 2007;157:171–185. doi: 10.1016/j.resp.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scortegagna M, Ding K, Oktay Y, Gaur A, Thurmond F, Yan LJ, Marck BT, Matsumoto AM, Shelton JM, Richardson JA, Bennett MJ, Garcia JA. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat Genet. 2003;35:331–340. doi: 10.1038/ng1266. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahar E, Whitney CW, Redline S, Lee ET, Newman AB, Javier Nieto F, O’Connor GT, Boland LL, Schwartz JE, Samet JM. Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am J Respir Crit Care Med. 2001;163:19–25. doi: 10.1164/ajrccm.163.1.2001008. [DOI] [PubMed] [Google Scholar]
- Somers VK, Abboud FM. Chemoreflexes–responses, interactions and implications for sleep apnea. Sleep. 1993;16:S30–S33. discussion S33–S34. [PubMed] [Google Scholar]
- Tafil-Klawe M, Thiele AE, Raschke F, Mayer J, Peter JH, von Wichert W. Peripheral chemoreceptor reflex in obstructive sleep apnea patients; a relationship between ventilatory response to hypoxia and nocturnal bradycardia during apnea events. Pneumologie. 1991;45(Suppl 1):309–311. [PubMed] [Google Scholar]
- Telezhkin V, Brazier SP, Cayzac SH, Wilkinson WJ, Riccardi D, Kemp PJ. Mechanism of inhibition by hydrogen sulfide of native and recombinant BKCa channels. Respir Physiol Neurobiol. 2010;172:169–178. doi: 10.1016/j.resp.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Tian H, Hammer RE, Matsumoto AM, Russell DW, McKnight SL. The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev. 1998;12:3320–3324. doi: 10.1101/gad.12.21.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- Warburg O, Negelin E. Über die photochemische Dissoziation bei intermittierender Beilichtung und das absolute Absorptionsspektrum des Atmungs ferments. Biochem. Z. 1928;202:202–228. [Google Scholar]
- Waterland RA, Jirtle RL. Transposable elements: targets for epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil JV. Variation in human ventilatory control—genetic influence on the hypoxic ventilatory response. Respir Physiol Neurobiol. 2003;135:239–246. doi: 10.1016/s1569-9048(03)00048-x. [DOI] [PubMed] [Google Scholar]
- Weil JV, Stevens T, Pickett CK, Tatsumi K, Dickinson MG, Jacoby CR, Rodman DM. Strain-associated differences in hypoxic chemosensitivity of the carotid body in rats. Am J Physiol Lung Cell Mol Physiol. 1998;274:L767–L774. doi: 10.1152/ajplung.1998.274.5.L767. [DOI] [PubMed] [Google Scholar]
- Williams SE, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, Peers C, Kemp PJ. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science. 2004;306:2093–2097. doi: 10.1126/science.1105010. [DOI] [PubMed] [Google Scholar]
- Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol. 2011;226:2925–2933. doi: 10.1002/jcp.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]