

Abstract
We aimed to investigate the role of insulin in the bladder and its relevance for the development of overactive bladder (OAB) in insulin-resistant obese mice. Bladders from male individuals who were involved in multiple organ donations were used. C57BL6/J mice were fed with a high-fat diet for 10 weeks to induce insulin-resistant obesity. Concentration–response curves to insulin were performed in human and mouse isolated mucosa-intact and mucosa-denuded bladders. Cystometric study was performed in terminally anaesthetized mice. Western blot was performed in bladders to detect phosphorylated endothelial NO synthase (eNOS) (Ser1177) and the phosphorylated protein kinase AKT (Ser473), as well as the unfolded protein response (UPR) markers TRIB3, CHOP and ATF4. Insulin (1–100 nm) produced concentration-dependent mouse and human bladder relaxations that were markedly reduced by mucosal removal or inhibition of the PI3K/AKT/eNOS pathway. In mouse bladders, insulin produced a 3.0-fold increase in cGMP levels (P < 0.05) that was prevented by PI3K/AKT/eNOS pathway inhibition. Phosphoinositide 3-kinase (PI3K) inhibition abolished insulin-induced phosphorylation of AKT and eNOS in bladder mucosa. Obese mice showed greater voiding frequency and non-voiding contractions, indicating overactive detrusor smooth muscle. Insulin failed to relax the bladder or to increase cGMP in the obese group. Insulin-stimulated AKT and eNOS phosphorylation in mucosa was also impaired in obese mice. The UPR markers TRIB3, CHOP and ATF4 were increased in the mucosa of obese mice. The UPR inhibitor 4-phenyl butyric acid normalized all the functional and molecular parameters in obese mice. Our data show that insulin relaxes human and mouse bladder via activation of the PI3K/AKT/eNOS pathway in the bladder mucosa. Endoplasmic reticulum stress-dependent insulin resistance in bladder contributes to OAB in obese mice.
Key points
Metabolic syndrome, obesity and insulin resistance are risk factors for overactive bladder, but little is known about the impact of insulin resistance on bladder functioning.
We aimed to investigate the insulin effects in murine and human bladder, and whether its defective action contributes to the bladder dysfunction associated with obesity.
Under physiological conditions, insulin relaxes urinary bladder through the release of nitric oxide from the mucosal layer, and this mechanism is impaired in bladders from insulin-resistant obese mice.
The defective insulin action in bladder mucosa from obese mice is due to endoplasmic reticulum stress, which remarkably contributes to the bladder overactivity in obesity conditions.
Our results enable a better understanding of the mechanism of action of insulin in the urinary bladder and how its defective action in mucosa contributes to bladder dysfunction in conditions of obesity-associated insulin resistance.
Introduction
Insulin resistance, defined as the impaired capacity of insulin to stimulate glucose sequestration, plays a major role in the pathogenesis of type 2 diabetes and is associated with several peripheral complications, such as cardiovascular disease and lower urinary tract symptoms (LUTS) (Rask-Madsen & King, 2007; Hammarsten & Peeker, 2011). Besides its well-recognized relevance for glucose homeostasis, insulin is an important mediator of vasorelaxation through a mechanism that involves endothelium-dependent nitric oxide (NO) production (Scherrer et al. 1994). Insulin-mediated NO generation by endothelial cells results from upstream activation of the RAC-α serine/threonine-protein kinase (AKT) pathway (Lee & Ragolia, 2006), which in turn directly activates endothelial NO synthase (eNOS) through serine 1177 phosphorylation (Dimmeler et al. 1999). NO mediates vasodilatation mainly by diffusion to vascular smooth muscle cells, where it activates soluble guanylyl cyclase, resulting in the formation of cyclic guanosine monophosphate (cGMP) (Zeng et al. 2000). Endothelial insulin resistance and subsequent impairment of insulin-induced vasorelaxation through stimulation of NO synthesis is proposed as an important causative mechanism of diabetes-related vascular dysfunction (Vicent et al. 2003; Contreras et al. 2010).
In parallel to vascular alterations, obesity and metabolic syndrome have been identified as common risk factors for LUTS, including overactive bladder and urinary incontinence (Rohrmann et al. 2005; Laven et al. 2008; Richter et al. 2010; Tai et al. 2010). The recent dramatic increase in the incidence of diabetes, obesity and metabolic syndrome has been accompanied by a simultaneous increase in urological complications such as diabetic bladder dysfunction (DBD) (Moul & McVary, 2010). DBD affects up to 80% of the diabetic population, impacting directly on patients’ quality of life and sexual health (Irwin et al. 2008; Coyne et al. 2012). Obesity is a well-established strong risk factor for urinary incontinence in women (Hunskaar, 2008). A strong positive correlation was recently found between the presence of metabolic syndrome and overactive bladder in female patients (Uzun & Zorba, 2012). In men, obesity/metabolic syndrome has also been associated with LUTS, including overactive bladder (Rohrmann et al. 2005), which may be secondary to the development of benign prostatic hyperplasia (Hammarsten & Högstedt, 2001; Kupelian et al. 2009; Parsons et al. 2011; Lee et al. 2012).
Experimental studies also showed that rats with insulin resistance induced by long-term fructose feeding exhibit overactive bladder with increases in non-voiding contractions (Lee et al. 2008, 2011). A few experimental studies have focused on bladder activity in high-fat-diet-induced obesity. Male Sprague–Dawley rats fed with a hyperlipidaemic diet for 24 weeks showed enhanced voiding and non-voiding bladder contractions during urodynamic evaluation, suggesting bladder overactivity (Rahman et al. 2007). Additionally, bladder overactivity associated with increased tumour necrosis factor α (TNF-α) levels and Rho-kinase activity was reported in a transgenic mouse model of type 2 diabetes (Wang et al. 2012a). However, the direct action of insulin on the urinary bladder has not previously been studied, so the relevance of impaired insulin action and potential involvement of the AKT/eNOS pathway in the bladder to obesity-related OAB remains unknown.
The unfolded protein response (UPR) is a cellular response triggered by endoplasmic reticulum stress that mediates obesity-associated insulin resistance in peripheral organs such as liver (Ozcan et al. 2004). One of the mechanisms by which UPR impairs insulin signalling relies on the upregulation of tribbles homologue 3 (TRIB3), a pseudo-kinase inhibitor of AKT (Du et al. 2003). As AKT is a pivotal step in the activation of eNOS by insulin, we focused on this pathway as a candidate to explain the alteration in bladder of obese mice. We hypothesized that insulin would play a direct action in the bladder and its defective action would impair bladder function in diabetic conditions. In this study we investigated the direct effects of insulin on human and murine bladder, as well as the molecular mechanisms driving these biological effects. We demonstrated that disruption of insulin signalling in the bladder mucosa is necessary for the manifestation of OAB in obese mice. Moreover, the impaired insulin action in the bladder mucosa and the functional alterations observed in high-fat-fed obese mice were dependent on the UPR.
Methods
Human tissue preparation
All the procedures were approved by the Ethics Committee of the State University of Campinas (UNICAMP-Campinas, Brazil; protocol number 761/2005). Bladder dome pieces, from male cadaver specimens aged between 18 and 60 years, were harvested through open surgery procedure. Before bladder removal, appropriate authorization from the patients’ families for multiple organ donations was obtained. After removal, bladder fragments were immediately placed in Krebs–Henseleit solution with the following composition (mm): 117 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3 and 11 glucose. The tissues were sectioned in longitudinal strips and mounted in organ baths, as further described. Four to six longitudinal bladder strips were obtained from each human bladder.
Animals
All animal procedures and the experimental protocols were according to the Ethical Principles in Animal Research adopted by the Brazilian College for Animal Experimentation (COBEA) and were approved by the institutional Committee for Ethics in Animal Research/State University of Campinas. Four-week-old male C57BL6/J mice were provided by the Central Animal House Services of the State University of Campinas. Mice were killed in a CO2 chamber and urinary bladders removed and sectioned horizontally at the level of the ureters. Bladders were immediately placed in Krebs–Henseleit solution (as described above), and sectioned in longitudinal strips and mounted in organ baths, as further described. Two longitudinal strips were obtained from each mouse bladder.
Diet-induced obesity and treatments
Mice were initially divided into two groups: lean and obese. Two animals were housed per cage on a 12 h light–dark cycle, and fed for 10 weeks with either a standard chow diet (carbohydrate: 70%; protein: 20%; fat: 10%) or a high-fat diet that induces obesity (carbohydrate: 29%; protein: 16%; fat: 55%), as previously described (Calixto et al. 2010).
Lean and obese mice were further treated orally with the endoplasmic reticulum (ER) stress inhibitor 4-phenyl butyric acid (PBA, 500 mg kg−1 twice a day, given by gavage for 4 days) or its vehicle (0.9% NaCl (w/v) and 0.15 mm NaHCO3) (Ozcan et al. 2009).
Intraperitoneal insulin tolerance test
Whole-body insulin sensitivity was assessed by an intraperitoneal insulin tolerance test performed in 6 h-fasted mice. Tail blood samples were collected before (0 min) and at 5, 10, 15, 20, 25 and 30 min after an intraperitoneal injection of 1.0 U kg−1 of regular insulin (Novolin R, NovoNordisk, Bagsvaerd, Denmark). Glucose concentrations were measured using a glucometer (ACCUCHEK Performa; Roche Diagnostics, Indianapolis, IN, USA) and the values were used to calculate the constant rate for blood glucose disappearance (Kitt), based on the linear regression of the Naperian logarithm of glucose concentrations (ln[glucose]) obtained from 0 to 30 min of the test.
In vitro functional assays
Strips of mice and human bladders were mounted in 10 ml organ baths containing Krebs–Henseleit solution (as described above), pH 7.4, at 37°C and bubbled with a gas mixture of 95% O2 and 5% CO2. Experiments were also conducted with denuded muscle strips, where ophthalmic scissors under a stereomicroscope were used to scrape off the mucosal layer before mounting in the organ bath.
Changes in isometric force were recorded using a PowerLab v.7.0 system (ADInstruments, Colorado Springs, CO, USA). The resting tension was adjusted to 0.5 g or 2.0 g for mice and human bladder, respectively, at the beginning of the experiments. The equilibration period was 60 min and the bathing medium was changed every 15 min. Relaxation responses were represented as the percentage of the amplitude of the pre-contraction with KCl (80 mm).
Relaxation responses to insulin
Cumulative concentration–response curves to insulin (10−9 to 10−7 m) in KCl (80 mm) pre-contracted mouse and human mucosa-intact or mucosa-denuded bladder strips were performed. Relaxation responses to insulin were also performed in intact preparations in the presence of the NO synthase (NOS) inhibitor l-NAME (100 μm), PI3K inhibitors wortmannin (100 nm) and LY294002 (10 μm) or with AKT inhibitor (5 μm).
Western blotting
Mouse bladder tissues were homogenized in a SDS lysis buffer with a Polytron PTA 20S generator (model PT 10/35; Brinkmann Instruments, Inc., Westbury, NY, USA) operated at maximum speed for 30 s and centrifuged (12,000 g, 4°C, 20 min) to remove insoluble material. For the measurement of the phosphorylated proteins, bladders were stimulated with insulin (100 nm) for 20 min in oxygenated Krebs solution prior to extraction. Protein concentrations of the supernatants were determined by the Bradford assay, and an equal amount of protein from each sample (50 μg) was treated with Laemmli buffer containing 100 mm dithiothreitol. Samples were heated in a boiling water bath for 15 min and resolved by SDS-PAGE. Electrotransfer of proteins to nitrocellulose membrane was performed for 60 min at 15 V (constant) in a semi-dry device (Bio-Rad, Hercules, CA, USA). Non-specific protein binding to nitrocellulose was reduced by pre-incubating the membrane overnight at 4°C in blocking buffer (0.5% non-fat dried milk, 10 mm Tris, 100 mm NaCl and 0.02% Tween 20). Detection using specific antibodies, HRP-conjugated secondary antibodies, and luminol solution was performed, as described previously (Anhêet al. 2007). Anti-eNOS (ab5589) was obtained from AbCam Technology (Cambridge, UK) and anti-eNOS phospho-Ser1177 (07-428) from Millipore (Billerica, MA, USA). Anti-AKT 1/2/3 (SC8312), anti-AKT 1/2/3 phospho-Ser473 (SC7985-R), anti-TRIB3 (SC34215), anti-CHOP (SCSC575) and anti-ATF4 (SC200) were from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). Densitometry was performed using the Scion Image software (Scion Corporation, Frederick, MD, USA) and results were normalized to the density of the β-actin, except for levels of phosphorylated AKT that were normalized to total AKT (to test whether changes in AKT phosphorylation occur independently of changes in AKT content).
Determination of cGMP levels
Mouse bladders were equilibrated for 30 min in warmed and oxygenated Krebs solution. Tissues were stimulated for 20 min with insulin (100 nm) in the absence and in the presence of the inhibitors of the PI3K/AKT/eNOS pathway, l-NAME (100 μm), wortmannin (100 nm), LY294002 (10 μm) or AKT inhibitor (5 μm), and then stimulated for 20 min with insulin (100 nm). After this interval, tissues were immediately frozen in liquid nitrogen. Frozen tissues were pulverized, homogenized in trichloroacetic acid (TCA, 5% w/v), centrifuged for 10 min at 4°C at 1500 g and the supernatant was collected. The pellet was dried and weighed. TCA was extracted from the supernatant with three washes of water-saturated ether. Preparation of tracer, samples, standards and incubation with antibody were performed according to the manufacturer's protocol (Cayman Chemical Cyclic GMP EIA kit, Ann Arbor, MI, USA). The assays were performed in duplicates, and the pellet weight was used to normalize the data as picomoles per milligram of tissue.
Cystometry
Mice were anaesthetized with an intraperitoneal injection of urethane (1.8 g kg−1). A 1 cm abdominal incision was made, the bladder was exposed and a butterfly cannula (25 G) was inserted into the bladder dome. The cannula was connected to a three-way tap, one port of which was connected to a pressure transducer and the other to the infusion pump through a catheter (PE50). Before starting the cystometry, the bladder was emptied via the third port. Continuous cystometry was carried out by infusing saline into the bladder at a rate of 0.6 ml h−1. The following parameters were assessed: threshold pressure (TP; the intravesical pressure immediately before micturition); post-void pressure (PVP; the intravesical pressure immediately after micturition); peak pressure (PP; the peak pressure reached during micturition); capacity (CP; the volume of saline needed to induce the first micturition); compliance (CO; the ratio of CP to TP, the volume required for a unit rise of pressure measured during filling); frequency of voiding contractions and frequency of non-voiding contractions (NVCs). NVCs were defined as spontaneous bladder contractions greater than 4 mmHg from the baseline pressure that did not result in a void. Bladders from mice used in the cystometry were not used in the other experiments.
Drugs
Urethane, LY294002, wortmannin, l-Nω-nitro-l-arginine and 4-phenyl butyric acid were obtained from Sigma (St Louis, MO, USA). Regular insulin (Novolin R) was purchased from Novo Nordisk (Bagsvaerd, Denmark). The AKT inhibitor was purchased from Merck Millipore (Darmstadt, Germany).
Statistical analysis
Data are expressed as mean ± SEM of n experiments. The program Instat (GraphPad Software) was used for statistical analysis. We have used one- or two-way analysis of variances (ANOVA) followed by a Tukey post test. Student's t test was also used when appropriate. P < 0.05 was accepted as significant.
Results
Insulin relaxes human bladder via PI3K/AKT/eNOS pathway
In order to evaluate whether insulin exerts a relaxant effect on human bladder we performed concentration–response curves in pre-contracted detrusor strips extracted from cadavers. Insulin produced concentration-dependent bladder relaxation reaching the largest relaxant response at 100 nm (32.6 ± 2.9%; Fig. 1A and B). Insulin-induced relaxations were markedly reduced in mucosa-denuded bladder strips (n= 4, P < 0.05; Fig. 1C). In addition, inhibition of the PI3K/AKT/eNOS axis with the NO synthase (NOS) inhibitor l-NAME (100 μm), the PI3K inhibitors wortmannin and LY294002 (100 nm and 10 μm, respectively) or with an AKT inhibitor (5 μm) in intact preparations markedly reduced the bladder relaxations (n= 4–5, P < 0.05; Fig. 1D–G).
Figure 1. Insulin evoked concentration-dependent relaxation in bladder strips from human patients.

A, concentration–response curve to insulin (1–100 nm) in carbachol-pre-contracted bladder. B, representative traces of the relaxant effect of insulin in the bladder. C, concentration–response curves to insulin in mucosa-intact and mucosa-denuded detrusor smooth muscle (DSM). D–G, concentration–response curves to insulin in the presence of the NOS inhibitor l-NAME (D), PI3K inhibitors wortmannin (E) and LY294002 (F), and AKT inhibitor (G). Results are expressed as mean ± SEM from 4–5 patients. #P < 0.05 in comparison with mucosa-intact preparations. *P < 0.05 in comparison with untreated bladder strips.
Insulin-resistant obese mice exhibit impaired insulin-stimulated relaxation
High-fat-diet-fed obese mice showed increased body weight, epididymal fat mass and fasting glucose (P < 0.001) compared to their lean counterparts (Supplemental Table 1; Supplemental material is available online only). The insulin tolerance test revealed that high-fat-diet-fed obese mice exhibit whole body insulin resistance as demonstrated by reduced Kitt levels in comparison with lean mice (1.50 ± 0.65% min−1 and 4.70 ± 0.88% min−1, respectively, P < 0.05).
Insulin produced concentration-dependent relaxations in the bladder from both lean and obese mice. However, relaxation in response to insulin was significantly reduced in obese mice in comparison to lean mice (17.1 ± 3.9% and 35.5 ± 3.3% relaxation to 100 nm, respectively, n= 11–13, P < 0.01; Fig. 2A and B). We found that the insulin-induced relaxation was significantly reduced in mucosa-denuded bladder strips from lean mice (n= 5, P < 0.05; Fig. 2C and D). On the other hand, mucosal removal caused no further impairment of relaxation in bladder from obese mice (Fig. 2C and D). In intact mucosal strips, pre-incubation with the NOS inhibitor l-NAME significantly reduced the relaxation responses in bladder from lean mice (n= 5, P < 0.05; Fig. 2E). Pre-incubation with PI3K inhibitors wortmannin or LY294002 also prevented the relaxation responses to insulin in lean mice (n= 4–6, P < 0.05; Fig. 2F and G). In vitro pre-incubation with the AKT inhibitor (30 min) also significantly inhibited insulin-induced bladder relaxation in lean mice (n= 5, P < 0.05; Fig. 2H). Pre incubation with l-NAME, wortmannin, LY294002 or the AKT inhibitor did not significantly affect bladder relaxations in obese mice (n= 4–7; Fig. 2E–H).
Figure 2. Obese mice exhibit insulin resistance in the bladder as demonstrated by impaired insulin-induced bladder relaxation.
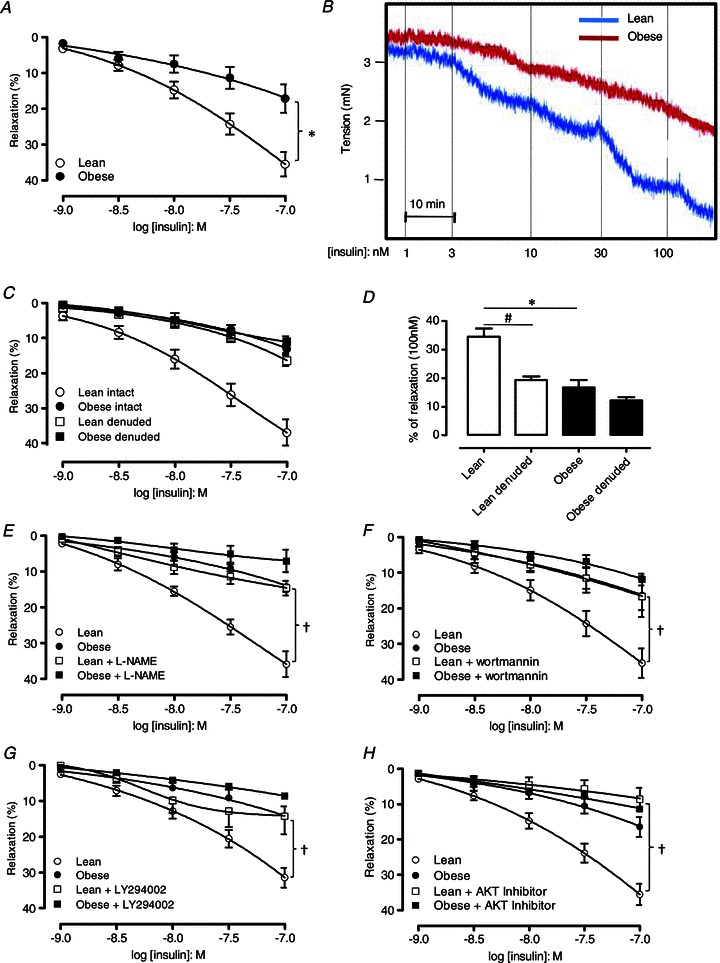
A, concentration–response curves to insulin in bladder strips from lean and obese mice. B, representative trace of an insulin-induced relaxation in the bladder strips from lean and obese mice. C, concentration–response curves to insulin in mucosa-intact and mucosa-denuded bladders of lean and obese mice. D, bar graph expressing relaxation response to insulin (100 nm) in intact and denuded bladders of lean and obese mice. Relaxant–response curves to insulin in the presence of NOS inhibitor l-NAME (E), PI3K inhibitors wortmannin (F) and LY294002 (G) and AKT inhibitor 124005 (H). Results are expressed as mean ± SEM from at least 4 animals. *P < 0.05 vs. lean group. #P < 0.05 vs. intact detrusor. †P < 0.05 vs. untreated bladder preparations.
Impaired activation of PI3K/AKT/eNOS by insulin in mucosal layer correlates with bladder dysfunction in obese mice
As shown in Fig. 3A, insulin stimulation produced a ∼3-fold increase in cGMP production in mucosa-intact bladder strips from lean (P < 0.01) but not from obese mice. Moreover, insulin-stimulated cGMP production was prevented by pre-incubating with the inhibitors of the PI3K/AKT/eNOS pathway, l-NAME, wortmannin, LY294002 or the AKT inhibitor (Fig. 3A). To assess the molecular mechanism involved in the impaired insulin-induced relaxant effect on the bladder we quantified AKT phosphorylation at Ser473 (pAKT) and eNOS phosphorylation at Ser1177 (p-eNOS) after stimulation with insulin. Insulin stimulation produced a ∼3-fold increase in pAKT (P < 0.01; Fig. 3B) and ∼2-fold increase in p-eNOS (P < 0.01; Fig. 3C) in bladder isolated from lean mice, while no increase was observed in obese animals. eNOS phosphorylation was normalized to the expression of β-actin because eNOS levels were significantly reduced in bladders from obese mice (n= 5–6; Supplemental Fig. 1).
Figure 3. Impairment of insulin-mediated activation of PI3K/AKT/eNOS signalling pathway in the bladder.

A, cGMP levels in basal and insulin-stimulated bladder tissue from lean and obese mice in the absence and in the presence of different inhibitors of the PI3K/AKT/eNOS pathway. Protein expression of pAKT (Ser473) (B) and p-eNOS (Ser1177) (C) in bladder tissues from lean and obese mice. Results are expressed as mean ± SEM from 4–6 animals. *P < 0.01 vs. absence of insulin; **P < 0.001 vs. lean groups.
Given that the relaxant effect of insulin was dependent on the presence of an intact mucosa, we next moved on to determine the precise bladder territory in which obesity impaired insulin signal transmission. Therefore, insulin stimulation of AKT/eNOS was evaluated in isolated mucosa and in mucosa-denuded muscle strips. We found that insulin increased AKT and eNOS phosphorylation in the isolated mucosa from lean mice while no modulation was found in the obese group (n= 4–9; Fig. 4A and B). When mucosa-denuded bladders were used, we observed an upregulation of AKT phosphorylation after insulin stimulation in lean but not in obese mice (n= 4–6; Fig. 4C). Insulin stimulation did not modify eNOS phosphorylation in mucosa-denuded bladders from lean or obese mice (n= 4, Fig. 4D).
Figure 4. Insulin resistance in bladder mucosa of obese mice evidenced by the impaired insulin/PI3K-mediated phosphorylation of AKT and eNOS.

Protein levels of phosphorylated AKT (A) and eNOS (B) in the mucosal layer of lean (open bars) and obese mice (filled bars) before and after stimulation with insulin (100 nm). Protein levels of phosphorylated AKT (C) and eNOS (D) in the denuded bladders of lean and obese mice before and after stimulation with insulin. Protein levels of phosphorylated AKT (E) and eNOS (F) before and after stimulation with insulin in the absence or in the presence of pre-incubated PI3K inhibitor wortmannin. Results are expressed as mean ± SEM from at least 4 animals. *P < 0.01 vs. absence of insulin; **P < 0.01 vs. lean groups.
Insulin-induced AKT and eNOS phosphorylation in the isolated mucosal layer from lean mice was completely abolished by pre-incubation with the PI3K inhibitor wortmannin (n= 4, 20 min; Fig. 4E and F). Altogether, these data suggest that insulin promotes its relaxant effects in the bladder through mucosal activation of the PI3K/AKT/eNOS pathway, which in turn releases NO that diffuses to the detrusor smooth muscle, triggering relaxation. This mechanism is impaired in bladder from obese mice due to urothelial insulin resistance.
Increased expression of TRIB3, ATF4 and CHOP in mucosal layer from obese mice
The endoplasmic reticulum stress-activated unfolded protein response (UPR) triggers several signalling pathways that lead to tribble 3 (TRIB3) expression and are of putative relevance for insulin resistance. TRIB3 is a pseudo-kinase inhibitor of AKT that is directly involved in the pathogenesis of insulin resistance in several tissues with metabolic relevance (Ti et al. 2011; Wang et al. 2012b). Thus, we next determined whether changes in TRIB3 content correlated with insulin resistance in the mucosal layer from lean and obese mice. TRIB3 levels were enhanced in mucosa from obese mice when compared to mucosa from lean mice (n= 4–5, P < 0.05; Fig. 5A and B). TRIB3 expression is induced by the UPR through a mechanism dependent on C/EBP homologous protein (CHOP) and activating transcription factor 4 (ATF4) (Ohoka et al. 2005). Accordingly, both CHOP and ATF4 are also increased in the mucosal layer of obese mice compared to mucosa from lean mice (n= 4–5, P < 0.05; Fig. 5A, C and D).
Figure 5. Obese mice exhibit enhanced TRIB3, ATF4 and CHOP protein expression in bladder mucosa.
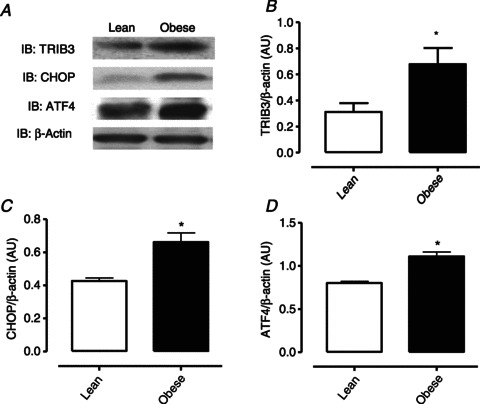
A, representative images of western blotting determination for TRIB3, CHOP, ATF4 and β-actin. Protein values for TRIB3 (B), CHOP (C) and ATF4 (D) in the mucosal layer from obese and lean mice. Results are expressed as mean ± SEM from 4–5 animals. *P < 0.05 vs. lean groups.
UPR suppression by PBA treatment restores insulin sensitivity and bladder function
To assess whether UPR is involved in the mucosal insulin resistance and in the bladder dysfunction in obese mice, animals were treated with oral phenyl butyric acid (PBA). PBA is a chemical chaperone that inhibits ER stress activation of the UPR and restores glucose homeostasis in obese mice (Ozcan et al. 2004). Obese mice treated with PBA showed an improvement in whole body insulin sensitivity when compared to vehicle-treated obese mice as demonstrated by Kitt values (1.75 ± 0.29 and 0.7 ± 0.09% min−1, respectively, n= 5; P < 0.05; Fig. 6A). PBA completely restored insulin-stimulated AKT phosphorylation (n= 4, P < 0.05; Fig. 6B) and significantly enhanced insulin-stimulated eNOS phosphorylation in mucosa from obese mice (n= 4–6, P < 0.05; Fig. 6C). After PBA treatment, mucosal expression of TRIB3, ATF4 and CHOP in obese mice was reduced to values similar to that found in lean mice (P < 0.05; Fig. 6D–G). In vitro bladder strip relaxation to insulin (100 nm) was also normalized by PBA treatment compared with strips from untreated obese mice (39.1 ± 3.34% and 6.1 ± 1.8%, respectively, n= 4–5; Fig. 7A). In lean mice, treatment with PBA did not significantly affect insulin-induced relaxations (100 nm: 39.4 ± 7.22 and 39.81 ± 7.39%, n= 4; Fig. 7A). Figure 7B shows that insulin produced a significant (P < 0.05) increase in cGMP levels in obese mice treated with PBA while no modulation in cGMP production was found in obese mice treated with vehicle. The insulin-dependent increase in cGMP levels was reduced in lean mice treated with PBA compared with vehicle group, but it remained greater than basal cGMP production (Fig. 7B, P < 0.05). Treatment with PBA also improved cystometric parameters, such as frequency of non-voiding contractions, voiding frequency and the post-void pressure, indicating an improvement of bladder function and voiding efficiency in obese mice resulting from UPR inhibition (Fig. 7C–H). Bladder compliance, capacity, threshold pressure and peak pressure did not change among the studied groups (Supplemental Table 1, n= 5).
Figure 6. Oral treatment with 4-phenyl butyric acid (PBA) restored insulin resistance and UPR in bladder mucosa of obese mice.

A, the constant rate for blood glucose disappearance (Kitt), based on the linear regression of the Naperian logarithm of glucose concentrations, obtained from indicated time points. B, insulin-stimulated AKT phosphorylation (Ser473) in mucosa of obese and lean mice treated with PBA or vehicle. C, expression of insulin-stimulated eNOS phosphorylation (Ser1177) in mucosa of obese and lean mice treated with PBA (500 mg kg−1, twice a day for 4 days by gavage) or its vehicle (0.9% NaCl and 0.15 mm NaHCO3). D, representative images of western blot determination of TRIB3, CHOP, ATF4 and β-actin levels in mucosa of obese and lean mice treated with PBA or vehicle. Expression of TRIB3 (E), CHOP (F) and ATF4 (G) in mucosa from obese and lean mice treated with PBA or vehicle. Results are expressed as mean ± SEM from 4–6 animals. *P < 0.05 vs. lean groups; **P < 0.05 compared with obese + Vehicle groups; #P < 0.05 vs. absence of insulin; †P < 0.05 vs. vehicle-treated mice.
Figure 7. The ER stress inhibitor 4-phenyl butyric acid (PBA) improved bladder function as evidenced by the restoration of the relaxant response and void pattern in obese group.

A, concentration–response curves to insulin in bladder strips from lean and obese mice treated with PBA (500 mg kg−1, twice a day for 4 days by gavage) or its vehicle (0.9% NaCl and 0.15 mm NaHCO3). B, insulin-induced cGMP production in the bladders from lean and obese mice treated with PBA or vehicle. Cystometric parameters were accessed in obese and lean mice treated with PBA or vehicle: void frequency (C), frequency of non-void contractions (D) and post-void pressure (E). Representative traces of cystometry in lean (F), obese (G) and PBA-treated obese mice (H). Results are expressed as mean ± SEM from 3–5 animals. *P < 0.05 vs. vehicle-treated obese mice; **P < 0.05 vs. absence of insulin; #P < 0.05 compared with respective group in the absence of insulin; †P < 0.05 vs. lean groups.
Discussion
In this study we provide the first demonstration of insulin-induced relaxation of human bladder smooth muscle. We also demonstrate that relaxation triggered by insulin is highly dependent on activation of the PI3K/AKT/eNOS signalling pathway in the mucosal layer, which in turn releases NO to relax the detrusor smooth muscle. Moreover, this relaxant response to insulin is impaired in OAB from obese mice due to UPR-mediated insulin resistance in the mucosal layer.
In addition to its classical metabolic actions, insulin displays a potent vasorelaxant ability that is dependent on its action on the endothelial layer. The mechanism underlying insulin-induced vasorelaxation relies on the release of NO via activation of the PI3K/AKT/eNOS pathway (Kim et al. 2006). Endothelial insulin resistance has been described as mediating cardiovascular and haemodynamic alteration found in rodent models of obesity thus implicating insulin resistance in pathologies beyond the immediate disruption of glucose homeostasis (Zecchin et al. 2003, 2007). The urothelium and vascular endothelium share many anatomical and functional features. They both overlie a smooth muscle layer, and function both as a barrier and as a highly active and specialized sensory structure. The urothelium and endothelium can recognize distinct mechanical, chemical and thermal stimuli and transduce information both to sensory afferents and the smooth muscle by releasing an array of chemical mediators (Birder, 2005; Gevaert et al. 2007; Mochizuki et al. 2009; Zagorodnyuk et al. 2009). Here, we show that insulin causes a concentration-dependent relaxation in smooth muscle from human and murine bladders. Relaxations in both were abrogated by pre-incubation with inhibitors of the PI3K/AKT/eNOS signalling cascade, such as l-NAME, wortmannin and AKT inhibitor. Thus, the present study supports a previously undescribed role for insulin in the bladder in which it causes detrusor smooth muscle relaxation by means of eNOS activation that is analogous to that already described for insulin in the vasculature (Lee & Ragolia, 2006).
Given this newly described physiological action of insulin, we next hypothesized that insulin resistance taking place in bladder could contribute to DBD found in insulin-resistant patients. In line with the progressive increase in the incidence of insulin resistance and diabetes, epidemiological studies have reported a concomitant increase in the occurrence of resultant deleterious complications, such as DBD. Although several experimental studies and clinical trials have pointed to a correlation between type 2 diabetes/metabolic syndrome and bladder dysfunction (Rohrmann et al. 2005; Laven et al. 2008; Richter et al. 2010; Tai et al. 2010), no mechanistic explanation for the impact of insulin resistance on bladder functioning has been proposed. In order to study the pathophysiology of insulin resistance in the bladder we first assessed the ability of insulin to stimulate relaxation in bladders from insulin-resistant mice.
We used mice made insulin resistant by high-fat diet feeding. After 10 weeks feeding with a high-fat diet, mice exhibited increased body weight and epididymal fat mass, along with alterations in metabolic parameters such as fasting blood glucose and whole body insulin resistance. We have previously shown that these high-fat-fed animals also exhibit impaired glucose tolerance and an increase in low-density lipoprotein (LDL) levels (Toque et al. 2011). It has been reported that high-fat-fed obese rats exhibit impairment in the insulin and acetylcholine-stimulated PI3K/AKT/eNOS pathway in the aorta, leading to a decreased vasodilatation (Zecchin et al. 2007). The components of the PI3K/AKT/eNOS pathway have also been identified in murine bladder, where bladder stretch and release of Ca2+ from sarcoplasmic reticulum is reported to stimulate NO production (Wei et al. 2008). In line with this, we showed that bladders removed from obese insulin-resistant mice show reduced relaxation to insulin in vitro in comparison to lean mice. To confirm the molecular basis of the insulin action in this tissue, we measured AKT (Ser473) and eNOS (Ser1177) phosphorylation and cGMP production resultant from insulin stimulation. We found that eNOS expression and insulin-induced AKT/eNOS phosphorylation and cGMP production were impaired in bladder from obese mice, thus providing a molecular explanation for the inefficient relaxation to insulin observed in bladders from insulin-resistant mice. In further accordance with our data, it was previously reported that eNOS expression is regulated by insulin (Kuboki et al. 2000).
In order to investigate whether insulin relaxation is mediated indirectly via the mucosa rather than through direct action on the detrusor smooth muscle, we examined AKT/eNOS activation by insulin in the isolated mucosa and detrusor smooth muscle. We found that AKT phosphorylation increased in response to insulin in both mucosa and detrusor smooth muscle, whilst an increase in eNOS phosphorylation was detected exclusively in mucosa. Moreover, insulin-mediated bladder AKT/eNOS activation was dependent on PI3K activation and was abrogated in mucosa isolated from bladders removed from obese mice. Removal of the mucosa substantially inhibited detrusor relaxation to insulin in the lean group, further supporting an indirect mechanism of relaxation, rather than a direct effect of insulin on detrusor. Altogether, these results suggest that insulin mediates bladder relaxation through activation of the mucosal PI3K/AKT/eNOS pathway, which in turns releases NO that diffuses to the detrusor smooth muscle resulting in cGMP production and bladder relaxation. Studies using bladders from eNOS knockout mice, which would be expected to show impaired relaxation in response to insulin, would provide further support for these proposed mechanisms. Bladder mucosa is composed of transitional epithelium (urothelium), basement membrane and lamina propria. Findings from several studies suggest that urothelium acts both as a sensor (detecting mechanical and chemical stimuli) and transducer (by releasing transmitter substances) (Birder 2006). As the urothelium has the capacity to release NO and communicate with neighbouring cells, we believe that the insulin-induced production of NO takes place via PI3K/AKT/eNOS activation in the urothelial layer. Interestingly, an increase in phosphorylation of AKT (but not eNOS) was also detected in response to insulin treatment of mucosa-denuded bladders from lean mice, suggesting the presence of AKT pathways independent of eNOS. These may play a role in processes such as insulin-dependent glucose uptake by muscle cells. Of interest, a previous study has shown that stretching of bladder smooth muscle strips increases the phosphorylation of AKT (Wei et al. 2008).
The last part of our study focused on the molecular mechanisms underlying insulin resistance in the mucosal layer of obese mice. Endoplasmic reticulum stress is currently considered a crucial event that drives insulin resistance in peripheral tissues (Ozcan et al. 2004; Raciti et al. 2010). ER stress occurs due to an imbalance of luminal ER homeostasis (e.g. increasing protein loading to the ER) thus triggering the unfolded protein response (UPR) (Ron & Walter, 2007). Among the myriad of pathways that are sequentially activated by UPR, those leading to tribble 3 (TRIB3) expression are of putative relevance for insulin resistance (Oskolkova et al. 2008; Bromati et al. 2011). TRIB3 is a pseudo-kinase that inhibits AKT and abrogates insulin signalling leading to glucose intolerance (Du et al. 2003). UPR stimulates TRIB3 expression through the stimulation of heterodimerization between the transcription factors ATF4 and CHOP (Ohoka et al. 2005).
Humans with a gain-of-function mutation in TRIB3 show increased insulin resistance and diabetes-associated complications (Gong et al. 2009; Oberkofler et al. 2010). In line with this, we show that bladder mucosa isolated from high-fat-diet-fed obese mice expresses greater levels of TRIB3 as well as CHOP and ATF4. In accordance with our data, recent studies report an increase in the TRIB3 expression concomitant with impaired pAKT levels in rodent models of type 2 diabetes. In these studies, TRIB3 gene silencing ameliorated diabetic complications such as cardiomyopathy and atherosclerosis, as well as normalizing insulin sensitivity and pAKT levels (Ti et al. 2011; Wang et al. 2012b).
To further test the relationship between UPR and mucosal insulin resistance we treated obese mice with PBA. PBA is classified as a chemical chaperone able to block UPR and restore insulin sensitivity (Ozcan et al. 2006). Oral treatment with PBA was effective in reducing TRIB3, CHOP and ATF4 levels in the bladder mucosa of obese mice. In accordance with previous studies (Ti et al. 2011), we also demonstrated that TRIB3 suppression resulted in improvement of the whole body insulin sensitivity and fully restored the insulin-stimulated mucosal AKT phosphorylation. Insulin-induced eNOS phosphorylation, although not restored to the levels observed in lean mice, was higher in obese mice treated with PBA than in obese mice that received vehicle. Thus UPR inhibition was reflected in a marked improvement in bladder function, as obese animals treated with PBA had higher insulin-stimulated bladder relaxation and cGMP production.
According to the International Continence Society definition, OAB is clinically characterized by the presence of urinary urgency, accompanied by frequency and nocturia, with or without urgency urinary incontinence (Abrams et al. 2002). Cystometrograms from patients with OAB usually show evidence of detrusor overactivity that is characterized by an increase in NVCs, voiding frequency and increased PVP, which indicates impairment in the efficiency of bladder emptying. A recent study showed that mice with type 2 diabetes presented OAB characterized by an increase in NVCs and decreased voided volume along with an enhancement in the number of voids in a period of 3 h (Wang et al. 2012a). We also observed cystometric alterations in insulin-resistant mice (e.g. increases in NVCs, voiding frequency and PVP) that parallel those seen in human OAB. These alterations were normalized by oral treatment with PBA, suggesting that UPR-dependent mucosal insulin resistance importantly contributed to diabetic bladder dysfunction (DBD) in obese animals, and that smooth muscle tonus regulation by insulin is important for the maintenance of normal bladder function.
In summary, our study provides the first demonstration that insulin causes bladder relaxation through activation of the mucosal PI3K/AKT/eNOS pathway, releasing NO from this layer to detrusor smooth muscle. Insulin-resistant obese mice exhibited UPR-dependent impairment of insulin action in the mucosal layer that was associated with DBD and a reduction in bladder relaxant response to insulin (Fig. 8). These findings highlight a new mechanism for the regulation of detrusor function that influences the pathogenesis of DBD. In addition it opens new perspectives to study the integrative role of metabolic and motor disorders induced by diabetes in specific tissues.
Figure 8. Diagram of the proposed pathway.
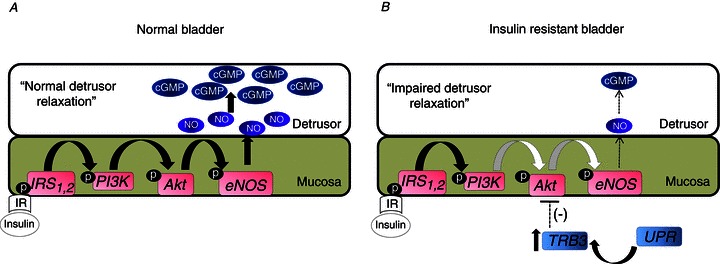
A, insulin binds to insulin receptor (IR) in the mucosal layer triggering the canonical signalling pathway of insulin that in turns activates the PI3K/AKT/eNOS signalling pathway promoting nitric oxide (NO) release from mucosa to relax the detrusor smooth muscle via cGMP production. B, insulin-resistant bladder exhibits ER stress characterized by increased TRIB3 levels that causes AKT inactivation in bladder mucosa. The downstream signalling pathway to AKT is impaired in mucosa from obese mice resulting in reduced NO release and cGMP production and reduced detrusor relaxation. IRS, insulin receptor substrate.
Acknowledgments
L.O.L. and E.A. thank Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) for financial support.
Glossary
- ATF4
activating transcription factor 4
- AKT
RAC-α serine/threonine-protein kinase
- cGMP
cyclic guanosine monophosphate
- CHOP
C/EBP homologous protein
- DBD
diabetic bladder dysfunction
- eNOS
endothelial NO synthase
- ER
endoplasmic reticulum
- LUTS
lower urinary tract symptoms
- NO
nitric oxide
- NVCs
non-voiding contractions
- OAB
overactive bladder
- PBA
4-phenyl butyric acid
- PI3K
phosphoinositide 3-kinase
- PVP
post-void pressure
- TRIB3
(Drosophila) tribbles homologue 3
- UPR
unfolded protein response
Author contributions
L.O.L., G.D.N., A.D.G., G.F.A. and E.A. conceived and designed the experiments. The experiments were carried out by L.O.L., C.S., F.R.B. and F.Z.M. in the laboratories of G.D.N., G.F.A. and E.A. The data were collected, analysed and interpreted by L.O.L., A.D.G., C.L.D., G.F.A. and E.A. The manuscript was written by L.O.L., A.D.G., G.F.A. and E.A. All authors approved the final version of the manuscript.
Supplementary material
Supplemental Table 1
Supplemental Fig. 1
References
- Abrams P, Cardozo L, Fall M, Griffiths D, Rosier P, Ulmsten U, van Kerrebroeck P, Victor A, Wein A. The standardisation of terminology of lower urinary tract function: report from the Standardisation Sub-committee of the International Continence Society. Neurourol Urodyn. 2002;21:167–178. doi: 10.1002/nau.10052. [DOI] [PubMed] [Google Scholar]
- Anhê GF, Hirabara SM, Turrer TC, Caperuto LC, Anhê FF, Ribeiro LM, Marçal AC, Carvalho CR, Curi R, Machado UF, Bordin S. Postpartum glycemic homeostasis in early lactating rats is accompanied by transient and specific increase of soleus insulin response through IRS2/AKT pathway. Am J Physiol Regul Integr Comp Physiol. 2007;292:R2225–R2233. doi: 10.1152/ajpregu.00902.2006. [DOI] [PubMed] [Google Scholar]
- Birder LA. More than just a barrier: urothelium as a drug target for urinary bladder pain. Am J Physiol Renal Physiol. 2005;289:F489–E495. doi: 10.1152/ajprenal.00467.2004. [DOI] [PubMed] [Google Scholar]
- Birder LA. Urinary bladder urothelium: Molecular sensors of chemical/thermal/mechanical stimuli. Vasc Pharmacol. 2006;45:221–226. doi: 10.1016/j.vph.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Bromati CR, Lellis-Santos C, Yamanaka TS, Nogueira TC, Leonelli M, Caperuto LC, Gorjão R, Leite AR, Anhê GF, Bordin S. UPR induces transient burst of apoptosis in islets of early lactating rats through reduced AKT phosphorylation via ATF4/CHOP stimulation of TRB3 expression. Am J Physiol Regul Integr Comp Physiol. 2011;300:R92–R100. doi: 10.1152/ajpregu.00169.2010. [DOI] [PubMed] [Google Scholar]
- Calixto MC, Lintomen L, Schenka A, Saad MJ, Zanesco A, Antunes E. Obesity enhances eosinophilic inflammation in a murine model of allergic asthma. Br J Pharmacol. 2010;159:617–625. doi: 10.1111/j.1476-5381.2009.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras C, Sánchez A, Martínez P, Raposo R, Climent B, García-Sacristán A, Benedito S, Prieto D. Insulin resistance in penile arteries from a rat model of metabolic syndrome. Br J Pharmacol. 2010;161:350–364. doi: 10.1111/j.1476-5381.2010.00825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne KS, Sexton CC, Thompson CL, Clemens JQ, Chen CI, Bavendam T, Dmochowski R. Impact of overactive bladder on work productivity. Urology. 2012;80:97–103. doi: 10.1016/j.urology.2012.03.039. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–1577. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- Gevaert T, Vriens J, Segal A, Everaerts W, Roskams T, Talavera K, Owsianik G, Liedtke W, Daelemans D, Dewachter I, Van Leuven F, Voets T, De Ridder D, Nilius B. Deletion of the transient receptor potential cation channel TRPV4 impairs murine bladder voiding. J Clin Invest. 2007;117:3453–3462. doi: 10.1172/JCI31766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong HP, Wang ZH, Jiang H, Fang NN, Li JS, Shang YY, Zhang Y, Zhong M, Zhang W. TRIB3 functional Q84R polymorphism is a risk factor for metabolic syndrome and carotid atherosclerosis. Diabetes Care. 2009;32:1311–1313. doi: 10.2337/dc09-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarsten J, Högstedt B. Hyperinsulinaemia as a risk factor for developing benign prostatic hyperplasia. Eur Urol. 2001;39:151–158. doi: 10.1159/000052430. [DOI] [PubMed] [Google Scholar]
- Hammarsten J, Peeker R. Urological aspects of the metabolic syndrome. Nat Rev Urol. 2011;8:483–494. doi: 10.1038/nrurol.2011.112. [DOI] [PubMed] [Google Scholar]
- Hunskaar S. A systematic review of overweight and obesity as risk factors and targets for clinical intervention for urinary incontinence in women. Neurourol Urodyn. 2008;27:749–757. doi: 10.1002/nau.20635. [DOI] [PubMed] [Google Scholar]
- Irwin DE, Milsom I, Reilly K, Hunskaar S, Kopp Z, Herschorn S, Coyne KS, Kelleher CJ, Artibani W, Abrams P. Overactive bladder is associated with erectile dysfunction and reduced sexual quality of life in men. J Sex Med. 2008;5:2904–2910. doi: 10.1111/j.1743-6109.2008.01000.x. [DOI] [PubMed] [Google Scholar]
- Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- Kuboki K, Jiang ZY, Takahara N, Ha SW, Igarashi M, Yamauchi T, Feener EP, Herbert TP, Rhodes CJ, King GL. Regulation of endothelial constitutive nitric oxide synthase gene expression in endothelial cells and in vivo: a specific vascular action of insulin. Circulation. 2000;101:676–681. doi: 10.1161/01.cir.101.6.676. [DOI] [PubMed] [Google Scholar]
- Kupelian V, McVary KT, Kaplan SA, Hall SA, Link CL, Aiyer LP, Mollon P, Tamimi N, Rosen RC, McKinlay JB. Association of lower urinary tract symptoms and the metabolic syndrome: results from the Boston Area Community Health Survey. J Urol. 2009;182:616–624. doi: 10.1016/j.juro.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laven BA, Orsini N, Andersson SO, Johansson JE, Gerber GS, Wolk A. Birth weight, abdominal obesity and the risk of lower urinary tract symptoms in a population based study of Swedish men. J Urol. 2008;179:1891–1895. doi: 10.1016/j.juro.2008.01.029. [DOI] [PubMed] [Google Scholar]
- Lee JH, Ragolia L. AKT phosphorylation is essential for insulin-induced relaxation of rat vascular smooth muscle cells. Am J Physiol Cell Physiol. 2006;291:C1355–C1365. doi: 10.1152/ajpcell.00125.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WC, Chien CT, Yu HJ, Lee SW. Bladder dysfunction in rats with metabolic syndrome induced by long-term fructose feeding. J Urol. 2008;179:2470–2476. doi: 10.1016/j.juro.2008.01.086. [DOI] [PubMed] [Google Scholar]
- Lee WC, Chuang YC, Chiang PH, Chien CT, Yu HJ, Wu CC. Pathophysiological studies of overactive bladder and bladder motor dysfunction in a rat model of metabolic syndrome. J Urol. 2011;186:318–325. doi: 10.1016/j.juro.2011.03.037. [DOI] [PubMed] [Google Scholar]
- Lee RK, Chung D, Chughtai B, Te AE, Kaplan SA. Central obesity as measured by waist circumference is predictive of severity of lower urinary tract symptoms. BJU Int. 2012;110:540–545. doi: 10.1111/j.1464-410X.2011.10819.x. [DOI] [PubMed] [Google Scholar]
- Mochizuki T, Sokabe T, Araki I, Fujishita K, Shibasaki K, Uchida K, Naruse K, Koizumi S, Takeda M, Tominaga M. The TRPV4 cation channel mediates stretch evoked Ca2+ influx and ATP release in primary urothelial cell cultures. J Biol Chem. 2009;284:21257–21264. doi: 10.1074/jbc.M109.020206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moul S, McVary KT. Lower urinary tract symptoms, obesity and the metabolic syndrome. Curr Opin Urol. 2010;20:7–12. doi: 10.1097/MOU.0b013e3283336f3f. [DOI] [PubMed] [Google Scholar]
- Oberkofler H, Pfeifenberger A, Soyal S, Felder T, Hahne P, Miller K, Krempler F, Patsch W. Aberrant hepatic TRIB3 gene expression in insulin-resistant obese humans. Diabetologia. 2010;53:1971–1975. doi: 10.1007/s00125-010-1772-2. [DOI] [PubMed] [Google Scholar]
- Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskolkova OV, Afonyushkin T, Leitner A, von Schlieffen E, Gargalovic PS, Lusis AJ, Binder BR, Bochkov VN. ATF4-dependent transcription is a key mechanism in VEGF up-regulation by oxidized phospholipids: critical role of oxidized sn-2 residues in activation of unfolded protein response. Blood. 2008;112:330–339. doi: 10.1182/blood-2007-09-112870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG, Jr, Ozcan U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9:35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Görgün CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JK, Messer K, White M, Barrett-Connor E, Bauer DC, Marshall LM. Obesity increases and physical activity decreases lower urinary tract symptom risk in older men: the Osteoporotic Fractures in Men study. Eur Urol. 2011;60:1173–1180. doi: 10.1016/j.eururo.2011.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raciti GA, Iadicicco C, Ulianich L, Vind BF, Gaster M, Andreozzi F, Longo M, Teperino R, Ungaro P, Di Jeso B, Formisano P, Beguinot F, Miele C. Glucosamine-induced endoplasmic reticulum stress affects GLUT4 expression via activating transcription factor 6 in rat and human skeletal muscle cells. Diabetologia. 2010;53:955–965. doi: 10.1007/s00125-010-1676-1. [DOI] [PubMed] [Google Scholar]
- Rahman NU, Phonsombat S, Bochinski D, Carrion RE, Nunes L, Lue TF. An animal model to study lower urinary tract and erectile dysfunction: the hyperlipidaemic rat. BJU Int. 2007;100:658–663. doi: 10.1111/j.1464-410X.2007.07069.x. [DOI] [PubMed] [Google Scholar]
- Rask-Madsen C, King GL. Mechanisms of disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab. 2007;3:46–56. doi: 10.1038/ncpendmet0366. [DOI] [PubMed] [Google Scholar]
- Richter HE, Kenton K, Huang L, Nygaard I, Kraus S, Whitcomb E, Chai TC, Lemack G, Sirls L, Dandreo KJ, Stoddard A. The impact of obesity on urinary incontinence symptoms, severity, urodynamic characteristics and quality of life. J Urol. 2010;183:622–628. doi: 10.1016/j.juro.2009.09.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrmann S, Smit E, Giovannucci E, Platz EA. Association between markers of the metabolic syndrome and lower urinary tract symptoms in the Third National Health and Nutritional Examination Survey (NHANES III) Int J Obes (Lond) 2005;29:310–316. doi: 10.1038/sj.ijo.0802881. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Scherrer U, Randin D, Vollenweider P, Vollenweider L, Nicod P. Nitric oxide release accounts for insulin's vascular effects in humans. J Clin Invest. 1994;94:2511–2515. doi: 10.1172/JCI117621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai HC, Chung SD, Ho CH, Tai TY, Yang WS, Tseng CH, Wu HP, Yu HJ. Metabolic syndrome components worsen lower urinary tract symptoms in women with type 2 diabetes. J Clin Endocrinol Metab. 2010;95:1143–1150. doi: 10.1210/jc.2009-1492. [DOI] [PubMed] [Google Scholar]
- Ti Y, Xie GL, Wang ZH, Bi XL, Ding WY, Wang J, Jiang GH, Bu PL, Zhang Y, Zhong M, Zhang W. TRB3 gene silencing alleviates diabetic cardiomyopathy in a type 2 diabetic rat model. Diabetes. 2011;60:2963–2974. doi: 10.2337/db11-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toque HA, da Silva FH, Calixto MC, Lintomen L, Schenka AA, Saad MJ, Zanesco A, Antunes E. High-fat diet associated with obesity induces impairment of mouse corpus cavernosum responses. BJU Int. 2011;107:1628–1634. doi: 10.1111/j.1464-410X.2010.09704.x. [DOI] [PubMed] [Google Scholar]
- Uzun H, Zorba OÜ. Metabolic syndrome in female patients with overactive bladder. Urology. 2012;79:72–75. doi: 10.1016/j.urology.2011.08.050. [DOI] [PubMed] [Google Scholar]
- Vicent D, Ilany J, Kondo T. The role of endothelial insulin signaling in the regulation of vascular tone and insulin resistance. J Clin Invest. 2003;111:1373–1380. doi: 10.1172/JCI15211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Cheng Z, Cristofaro V, Li J, Xiao X, Gomez P, Ge R, Gong E, Strle K, Sullivan MP, Adam RM, White MF, Olumi AF. Inhibition of TNF-α improves the bladder dysfunction that is associated with type 2 diabetes. Diabetes. 2012a;61:2134–2145. doi: 10.2337/db11-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZH, Shang YY, Zhang S, Zhong M, Wang XP, Deng JT, Pan J, Zhang Y, Zhang W. Silence of TRIB3 suppresses atherosclerosis and stabilizes plaques in diabetic ApoE–/–/LDL receptor–/– mice. Diabetes. 2012b;61:463–473. doi: 10.2337/db11-0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei B, Chen Z, Zhang X, Feldman M, Dong XZ, Doran R, Zhao BL, Yin WX, Kotlikoff MI, Ji G. Nitric oxide mediates stretch-induced Ca2+ release via activation of phosphatidylinositol 3-kinase-Akt pathway in smooth muscle. PLoS One. 2008;3:e2526. doi: 10.1371/journal.pone.0002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagorodnyuk VP, Brookes SJ, Spencer NJ, Gregory S. Mechanotransduction and chemosensitivity of two major classes of bladder afferents with endings in the vicinity to the urothelium. J Physiol. 2009;587:3523–3538. doi: 10.1113/jphysiol.2009.172577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecchin HG, Bezerra RM, Carvalheira JB, Carvalho-Filho MA, Metze K, Franchini KG, Saad MJ. Insulin signalling pathways in aorta and muscle from two animal models of insulin resistance–the obese middle-aged and the spontaneously hypertensive rats. Diabetologia. 2003;46:479–491. doi: 10.1007/s00125-003-1073-0. [DOI] [PubMed] [Google Scholar]
- Zecchin HG, Priviero FB, Souza CT, Zecchin KG, Prada PO, Carvalheira JB, Velloso LA, Antunes E, Saad MJ. Defective insulin and acetylcholine induction of endothelial cell-nitric oxide synthase through insulin receptor substrate/Akt signaling pathway in aorta of obese rats. Diabetes. 2007;56:1014–1024. doi: 10.2337/db05-1147. [DOI] [PubMed] [Google Scholar]
- Zeng G, Nystrom FH, Ravichandran LV, Cong LN, Kirby M, Mostowski H, Quon MJ. Roles for insulin receptor, PI3-kinase, and Akt in insulin-signaling pathways related to production of nitric oxide in human vascular endothelial cells. Circulation. 2000;101:1539–1545. doi: 10.1161/01.cir.101.13.1539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.