

Abstract
The heptapeptide angiotensin-(1–7) is a biologically active metabolite of angiotensin II, the predominant peptide of the renin–angiotensin system. Recently, we have shown that the receptor Mas is associated with angiotensin-(1–7)-induced signalling and mediates, at least in part, the vasodilatory properties of angiotensin-(1–7). However, it remained controversial whether an additional receptor could account for angiotensin-(1–7)-induced vasorelaxation. Here, we used two different angiotensin-(1–7) antagonists, A779 and d-Pro-angiotensin-(1–7), to address this question and also to study their influence on the vasodilatation induced by bradykinin. Isolated mesenteric microvessels from both wild-type and Mas-deficient C57Bl/6 mice were precontracted with noradrenaline, and vascular reactivity to angiotensin-(1–7) and bradykinin was subsequently studied using a small-vessel myograph. Furthermore, mechanisms for Mas effects were investigated in primary human umbilical vein endothelial cells. Both angiotensin-(1–7) and bradykinin triggered a concentration-dependent vasodilatation in wild-type microvessels, which was absent in the presence of a nitric oxide synthase inhibitor. In these vessels, the pre-incubation with the Mas antagonists A779 or d-Pro-angiotensin-(1–7) totally abolished the vasodilatory capacity of both angiotensin-(1–7) and bradykinin, which was nitric oxide mediated. Accordingly, Mas-deficient microvessels lacked the capacity to relax in response to either angiotensin-(1–7) or bradykinin. Pre-incubation of human umbilical vein endothelial cells with A779 prevented bradykinin-mediated NO generation and NO synthase phosphorylation at serine 1177. The angiotensin-(1–7) antagonists A779 and d-Pro-angiotensin-(1–7) equally block Mas, which completely controls the angiotensin-(1–7)-induced vasodilatation in mesenteric microvessels. Importantly, Mas also appears to be a critical player in NO-mediated vasodilatation induced by renin–angiotensin system-independent agonists by altering phosphorylation of NO synthase.
Key points
Two distinct angiotensin-(1–7) [Ang-(1–7)] receptor blockers, A779 and d-Pro-Ang-(1–7), can completely prevent Ang-(1–7)-induced vasorelaxation.
Genetic deficiency of Mas completely prevents vascular responses to Ang-(1–7).
Genetic deficiency of Mas completely prevents vascular responses to other NO-dependent vasorelaxants (bradykinin).
Mas plays a key role in NO-mediated vasodilatation by modulating vasorelaxant-mediated phosphorylation of endothelial nitric oxide synthase in endothelial cells.
Introduction
The renin–angiotensin system (RAS) was initially identified as an important regulator of arterial blood pressure, electrolyte homeostasis and water and sodium intake (Weir & Dzau, 1999; Wilson et al. 2005), but the RAS is also involved in the regulation of tissue regeneration, cellular proliferation and growth factor release (Unger et al. 1996; Takeda et al. 2004), as well as haematopoietic recovery after myelosuppression and progenitor engraftment (Rodgers et al. 2003; Ellefson et al. 2004). Renin metabolizes angiotensinogen to form the biologically inactive decapeptide, angiotensin I (Ang I). Angiotensin I is then cleaved by angiotensin-converting enzyme (ACE) to yield the main active peptide of the RAS, angiotensin II (Ang II). Angiotensin II is well known as a potent regulator of vasoconstriction, water intake and salt metabolism and is well known to worsen cardiovascular function under pathological conditions. Recently, a homologue of ACE, termed angiotensin-converting enzyme 2 (ACE2), has been identified (Donoghue et al. 2000; Tipnis et al. 2000). ACE2 cleaves a single peptide from either Ang I or the octapeptide Ang II to form angiotensin-(1–9) [Ang-(1–9)] or angiotensin-(1–7) [Ang-(1–7)], respectively (Vickers et al. 2002). In the last decade, Ang-(1–7) has become a peptide of interest, because it can counter-regulate adverse effects of Ang II (Freeman et al. 1996; Strawn et al. 1999; Zhu et al. 2002). We identified the G protein-coupled receptor Mas to be associated with Ang-(1–7)-induced signalling, demonstrating that genetic deletion of Mas, encoded by the Mas proto-oncogene, abolishes the binding of Ang-(1–7) to mouse kidney (Santos et al. 2003). This binding could be blocked by cotreatment with d-Ala7-angiotensin-(1–7) (A779), a specific Ang-(1–7) antagonist. Furthermore, Mas-deficient aortas lost their Ang-(1–7)-induced relaxation response (Santos et al. 2003).
We recently demonstrated that isolated microvessels lacking Mas showed significantly impaired dilatation not only in response to Ang-(1–7), but also in response to other endothelium-dependent vasodilators, including bradykinin (BK) and acetylcholine (Peiróet al. 2007). These results indicate that Mas deficiency may lead to microvascular endothelial dysfunction. We found that the concentration-dependent relaxation induced by Ang-(1–7) in vessels isolated from Mas-deficient mice was diminished by ∼40%. Similar to Mas-deficient vessels, the Ang-(1–7)-mediated relaxation was reduced by A779 in vessels from wild-type mice. Nevertheless, a residual relaxation in both wild-type vessels pretreated with A779 and Mas-deficient vessels was observed in response to Ang-(1–7) (Peiróet al. 2007). These data suggested that Ang-(1–7) may also interact with an additional specific receptor other than Mas, as speculated recently (Silva et al. 2007).
In order to investigate the presence of an additional receptor for Ang-(1–7) further, we examined in detail the response of isolated mesenteric vessels of Mas-deficient and wild-type mice to Ang-(1–7), BK and noradrenaline (NA), after a pretreatment with A779, d-Pro-Ang-(1–7), another specific Ang-(1–7) antagonist (Lemos et al. 2005), or the nitric oxide synthase (NOS) inhibitor l-NAME.
Methods
Animals
Six-month-old male C57Bl/6 mice or Mas-deficient mice on a C57Bl/6 background (Esteban et al. 2009) were used in the experiments. Animals were maintained under standardized conditions with an artificial 12 h–12 h dark–light cycle, with free access to food and water. All animal studies were performed according to national guidelines and were approved by the institutional animal care and ethics committees.
Vascular reactivity experiments in mesenteric microvessels
Mice were anaesthetized with 70 mg kg−1 i.p. sodium pentobarbital and exsanguinated. Third-branch mesenteric arteries (mean internal diameter ranging between 150 and 200 μm) were mounted as ring preparations on a small-vessel myograph to measure isometric tension as described before (Vallejo et al. 2000; Peiróet al. 2007). Arteries were contracted with 10 μmol l−1 noradrenaline (NA; Sigma, St Louis, MO, USA), and then the vasoactive responses to Ang-(1–7) (Bachem, Bubendorf, Switzerland; 1 pmol l−1 to 1 μmol l−1) or bradykinin (BK; Sigma; 1 nmol l−1 to 30 μmol l−1) were tested by adding increasing concentrations of the drugs. In an acute experiment, the vasoactive response to a single concentration of Ang-(1–7) (1 μmol l−1) was investigated in both wild-type and Mas-deficient vessels. In some cases, the mesenteric segments were preincubated for 15 min with the Mas antagonists A779 (Bachem; 1 μmol l−1) or d-Pro-Ang-(1–7) (Biosyntan GmbH, Berlin, Germany; 1 μmol l−1) or with the NOS inhibitor l-NAME (100 μmol l−1) before addition of NA. In a parallel set of experiments, microvessels were preincubated with or without l-NAME (100 μmol l−1), contracted with NA and then maintained without any additional treatment throughout the duration of the experiment (time control). In another set of experiments, concentration-dependent curves to NA (10 pmol l−1 to 100 μmol l−1) were performed, either in control conditions or in the presence of l-NAME, A779, or both.
Cell culture
Human umbilical vein endothelial cells (HUVECs) were prepared essentially as described previously (Peiróet al. 2007). In brief, HUVECs were obtained by collagenase digestion from umbilical cords provided by Hospital Universitario de Getafe, as approved by the Hospital Ethics Committee. The HUVECs were cultured in M199 medium supplemented with 10% fetal calf serum, 25 mg ml−1 endothelial cell growth supplement and 100 mg ml−1 heparin. For experiments, cultures between passages one and five were used. The cells were pretreated with either A779 (1 μmol l−1) or saline for 15 min.
In order to investigate the impact of the pretreatment on NO generation and ligand–receptor-induced phosphorylation of endothelial nitric oxide synthase (eNOS), such pretreated cells were stimulated for 0, 1, 5, and 10 min with BK (10 μmol l−1).
Measurement of NO production in HUVECs
Intracellular NO was monitored with 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-2-FM diacetate; Molecular Probes, Invitrogen, Madrid, Spain), a fluorescence indicator of NO that emits fluorescence in response to a reaction with NO. To measure intracellular NO, HUVECs were seeded on 24-well plates. At 80% confluence, cells were loaded with 2.5 μmol l−1 DAF-2-FM in M199 without Phenol Red for 30 min, according to the manufacturer's instructions. After loading, cells were rinsed three times with culture medium and exposed to the desired treatments. To quantify the DAF-related fluorescence, the cells were observed under an inverted fluorescence Nikon Eclipse Ti-S microscope. Fluorescence from five different fields per well was measured (excitation wavelength 488 nm; emission wavelength 515 nm). Fluorescence signals were quantified using NIS-Elements 3.0 software (Nikon Izasa SA, Barcelona, Spain).
Western blotting
The activation of eNOS was assessed through phospho-eNOS (serine 1177) levels. The HUVECs were lysed in lysis buffer (10 mmol l−1 Tris, pH 7.4; 1% sodium dodecyl sulphate; 10 mmol l−1 sodium orthovanadate; 2 mmol l−1 phenylmethylsulphonyl fluoride; and 12.5 mg ml−1 aprotinin), and the protein concentration was determined with a BCA Protein Assay Kit (Perbio Science GmbH, Bonn, Germany) according to the manufacturer's instructions. Western blot was performed as described previously (Gembardt et al. 2005a). In brief, proteins and a prestained protein-weight marker (Amersham Biosciences GmbH, Freiburg, Germany) were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The blots were incubated with rabbit polyclonal antibodies against phospho-eNOS (serine 1177; dilution 1:1000; Cell Signalling, Cambridge, UK) and total eNOS (dilution 1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Subsequently, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (dilution 1:5000; Bio-Rad Laboratories, Hercules, CA, USA), and bands were detected by enhanced chemiluminescence (Amersham Biosciences GmbH, Freiburg, Germany). The bands on the X-ray film were quantified by densitometry scanning with free ImageJ software (National Institute of Health, http://rsb.info.nih.gov/ij/). Normalized results were expressed as the phospho-eNOS (Ser 1177) to total eNOS ratio.
Statistical analysis
Results were expressed as means ± SEM. Deviations from the mean regarding the curves to drugs were statistically analysed using factorial two-way ANOVA. Western blot data were analysed by unpaired Student's t test. Significance was considered from a value of P < 0.05.
Results
Mas deletion or pharmacological blockade blunts the vasorelaxation in response to Ang-(1–7) and BK in mesenteric microvessels
The vasodilator effects of Ang-(1–7) and BK were studied in murine mesenteric microvessels, precontracted with NA. The vasoconstrictor responses to NA were similar in wild-type and Mas-deficient microvessels (4.18 ± 0.23 and 4.14 ± 0.23 mN, respectively; results from 13–15 animals). In wild-type microvessels, Ang-(1–7) elicited a concentration-dependent relaxation; this response was significantly impaired in microvessels lacking the receptor Mas, although a residual relaxing effect was still observed (Fig. 1A). Likewise, Ang-(1–7)-induced vasorelaxation was impaired in wild-type microvessels pretreated with the Mas receptor antagonist A779 (Fig. 1A). The impairment observed in Mas-deficient and wild-type microvessels pretreated with A779 was statistically equivalent.
Figure 1. Effect of genetic Mas deficiency or its pharmacological blockade on vascular reactivity.

Vasorelaxant responses to angiotensin-(1–7) [Ang-(1–7); 1 pmol l−1 to 1 mmol l−1; A] and bradykinin (BK; 1 nmol l−1 to 30 μmol l−1; B) in Mas-deficient or wild-type microvessels preincubated with the Mas antagonist A779 (1 μmol l−1). C, after the initial contraction with noradrenaline (NA), the isolated microvessels undergo a spontaneous loss of tension over time, which is similar between wild-type and Mas-deficient segments. Some of the segments used for time control were preincubated with l-NAME (100 μmol l−1). The vasoactive responses are expressed as a percentage of a previous contraction elicited by NA (10 μmol l−1). For each curve, results are shown as means ± SEM of 10–15 mesenteric microvessels from at least 5 different animals. D, acute vasorelaxant effect of a single concentration of Ang-(1–7) (1 μmol l−1) on wild-type and Mas-deficient mesenteric arteries, with and without A779 preincubation. *P < 0.05 vs. wild-type microvessels; †P < 0.05 vs. the respective time control without l-NAME.
Analogously, both Mas deficiency and pharmacological blockade with A779 resulted in an equal impairment of the concentration-dependent vasorelaxation triggered by the RAS-independent vasodilator BK (Fig. 1B). Again, a residual vasorelaxant response to BK after Mas deletion or blockade was still observed (Fig. 1B).
Murine mesenteric microvessels undergo a non-specific loss of tension mediated by NO
To understand the origin of the residual vasorelaxant responses to Ang-(1–7) or BK, parallel experiments were performed, in which arteries were contracted with NA and maintained without being subsequently exposed to Ang-(1–7) or BK. As shown in Fig. 1C, a spontaneous and gradual vasorelaxation was equally observed in both wild-type and Mas-deficient microvessels throughout the duration of the experiment. In fact, such spontaneous vasodilatation was not significantly different from the residual vasodilatation in response to Ang-(1–7) (Fig. 1A) and BK (Fig. 1B) observed after Mas deletion or blockade. The spontaneous loss of tension in both wild-type and Mas-deficient microvessels was dependent on NO release, because it was totally prevented by preincubation with the NOS-inhibitor l-NAME (Fig. 1C).
In order to provide further proof that there was no agonist-specific vasodilatation independent of Mas, we investigated the vasorelaxant effect of a single concentration of Ang-(1–7) (1 μmol l−1) on wild-type and Mas-deficient mesenteric arteries. The time course of this experiment did not allow for a significant spontaneous loss of tension to take place. While Ang-(1–7) induced a marked reduction of vascular tone in untreated wild-type mesenteric arteries (Fig. 1D), no significant vasoactive responses were observed in A779-pretreated or Mas-deficient vessels. Based on all of these observations, it appeared that the remaining vasorelaxant response to either Ang-(1–7) or BK observed in Mas-deficient or A779-pretreated wild-type microvessels in a concentration–response experiment (Fig. 1A and B) was unspecific and not mediated by the agonists themselves.
Effect of different Mas antagonists on Ang-(1–7)- and BK-induced relaxation in mesenteric microvessels
We next compared the ability of two different Mas antagonists, A779 and d-Pro-Ang-(1–7), to block the vasorelaxation specifically induced by Ang-(1–7) and BK. In both wild-type and Mas-deficient microvessels, the specific vasorelaxation was calculated as the difference between the total vasorelaxation observed in the presence of the agonist and the spontaneous vasorelaxation.
Figure 2A shows the effect of A779 and d-Pro-Ang-(1–7) on the vasodilatation induced by Ang-(1–7) in wild-type mesenteric microvessels. The pre-incubation with A779 completely blocked the vasorelaxation elicited by Ang-(1–7) at any concentration used. A similar effect was observed following pre-incubation with d-Pro-Ang-(1–7), either alone or in combination with A779 (Fig. 2A). The vasorelaxant response elicited by Ang-(1–7) in wild-type vessels was totally dependent on NO release, because it was blocked by pre-incubation with l-NAME (Fig. 2A). In microvessels lacking the receptor Mas, where Ang-(1–7) showed no vasorelaxant effects, vascular reactivity was not modified by l-NAME, A779 or d-Pro-Ang-(1–7) or by both antagonists in combination (Fig. 2B).
Figure 2. Effect of the Mas antagonists A779 and d-Pro-Ang-(1–7) on the vasodilatation elicited by Ang-(1–7).
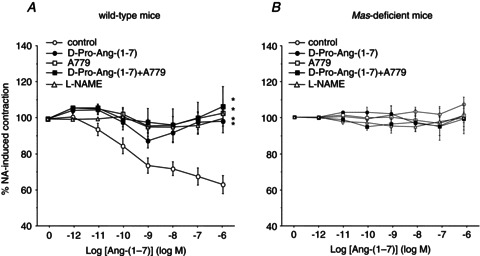
Both wild-type (A) and Mas-deficient microvessels (B) were exposed to Ang-(1–7) (1 pmol l−1 to 1 μmol l−1), after pre-incubation with or without A779 (1 μmol l−1), d-Pro-Ang-(1–7) (1 μmol l−1), both antagonists or l-NAME (100 μmol l−1). The vasoactive responses are expressed as a percentage of a previous contraction elicited by NA (10 μmol l−1). The curves are normalized to the loss of tension over time. For each curve, results are shown as means ± SEM of 10–15 mesenteric microvessels from at least 5 different animals. *P < 0.05 vs. wild-type microvessels.
Importantly, the relaxation induced by BK in wild-type microvessels was also completely abolished by pre-incubation with A779 and d-Pro-Ang-(1–7), either alone or in combination (Fig. 3A). As observed for Ang-(1–7), the vasodilatation induced by BK in wild-type microvessels was completely prevented by pre-incubation with l-NAME (Fig. 3A). Again, in the absence of a vasorelaxant response to BK due to Mas deficiency, neither A779 nor d-Pro-Ang-(1–7), alone or in combination, exerted any vasoactive effect (Fig. 3B). l-NAME did not modify vascular reactivity in Mas-deficient mice stimulated with BK (Fig. 3B).
Figure 3. Effect of the Mas antagonists A779 and d-Pro-Ang-(1–7) on BK-induced vasodilatation.
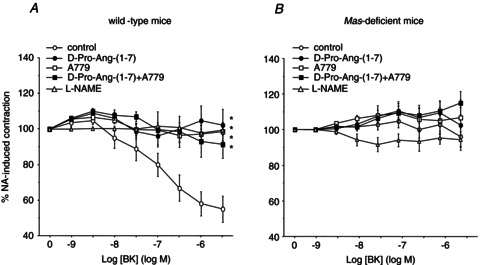
Both wild-type (A) and Mas-deficient microvessels (B) were preincubated with or without A779 (1 μmol l−1), d-Pro-Ang-(1–7) (1 μmol l−1), both antagonists or l-NAME (100 μmol l−1), and subsequently exposed to increasing concentrations of BK (from 1 pmol l−1 to 1 μmol l−1). The vasoactive responses are expressed as a percentage of a previous contraction elicited by NA (10 μmol l−1). The curves are normalized to the loss of tension over time. For each curve, results are shown as means ± SEM of 10–15 mesenteric microvessels from at least 5 different animals. *P < 0.05 vs. wild-type microvessels.
Effects of the Ang-(1–7) receptor antagonist A779 on NO production and eNOS phosphorylation in HUVECs
To evaluate whether the ex vivo findings correlate with in vitro effects, we investigated the impact of A779 on NO production in HUVECs. While A779 alone did not affect NO production over the 10 min of the experiments, BK increased the NO production in comparison to control cells. However, this significant increase was completely blunted by pretreatment with A779 (Fig. 4A). We then analysed eNOS phosphorylation in response to A779 treatment in HUVECs in order to characterize a potential mechanism responsible for both the blockade of BK-stimulated NO production in cell culture and the endothelial dysfunction observed in mice with genetic deficiency in Mas or after pharmacological Mas blockade. Treatment of primary HUVECs with A779 did not influence phosphorylation at serine 1177, used as a marker of eNOS activation. Notably, the phosphorylation pattern did not change for the control cells and A779-treated cells over the time of the experiment (data not shown). Congruent to NO production, BK stimulated the phosphorylation at serine 1177 as early as at 1 min. Pretreatment with A779 completely prevented the BK-mediated serine 1177 phosphorylation (Fig. 4B). None of the treatments changed the total eNOS levels over the 10 min of the experiment (data not shown).
Figure 4. Effect of the Mas antagonist A779 on NO production and endothelial nitric oxide synthase (eNOS)-activating phosphorylation in human umbilical vein endothelial cells (HUVECs).
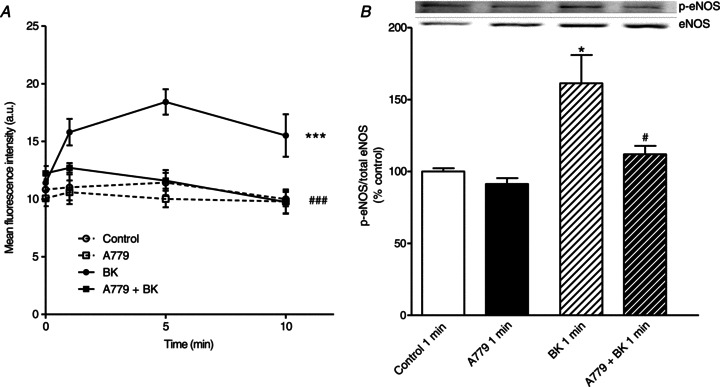
Cells were pretreated with or without A779 (1 μmol l−1) for 15 min. A, NO production in HUVECs is shown for different time points over the 10 min of stimulation. B, levels of eNOS phosphorylation at serine 1177. The inset shows a representative Western blot. *P < 0.05, ***P < 0.001 vs. saline-treated cells; #P < 0.05, ###P < 0.001 vs. BK-treated cells. Each experiment was repeated 6 times using cells from 4 independent isolations.
Response to NA in Mas-deficient and wild-type microvessels pretreated with Ang-(1–7) receptor antagonists
In order to investigate whether the reduced eNOS activity in response to A779 might have an influence not only on vasorelaxant agents, but also on vasoconstrictor substances due to less counteracting NO release, we analysed the impact of the Ang-(1–7) receptor antagonist A779 and the NOS inhibitor l-NAME on the vascular effects of NA. In microvessels from wild-type animals, NA led to a concentration-dependent constriction of the mesenteric arteries (Fig. 5A). After pretreatment with the Ang-(1–7) receptor antagonist A779 or l-NAME, the wild-type vessels showed a significantly increased sensitivity to NA stimulation (Fig. 5A), whereas none of the pretreatments could influence the response to NA in Mas-deficient vessels (Fig. 5B). This difference is caused by the fact that the Mas-deficient vessels already showed a significantly higher susceptibility to NA in baseline conditions (Fig. 5C).
Figure 5. Concentration-dependent effects of NA in Mas-deficient and wild-type mesenteric arteries.
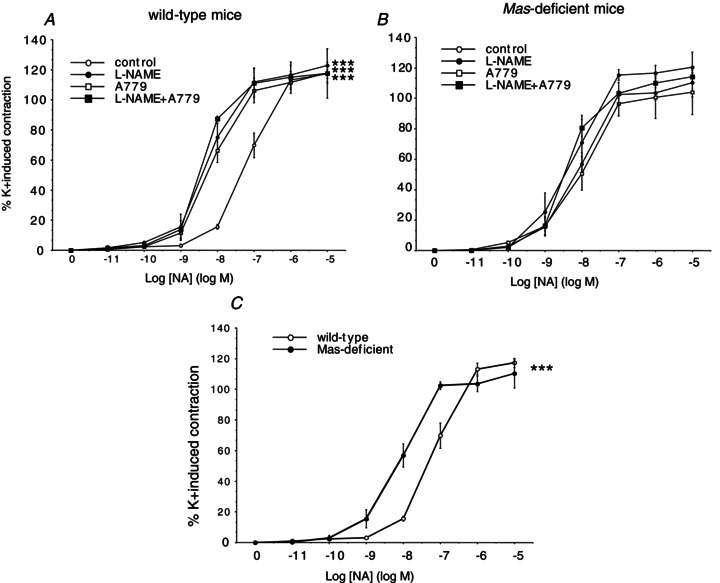
Vascular response to NA (10 pmol l−1 to 10 μmol l−1) in wild-type (A) and Mas-deficient microvessels (B) preincubated with the Mas antagonists A779 (1 μmol l−1) or d-Pro-Ang-(1–7), the eNOS inhibitor l-NAME, the combination of Mas antagonists with l-NAME, or saline. C, the vasoconstrictive response to NA is altered in Mas-deficient animals compared with their wild-type controls. For each curve, results are shown as means ± SEM of 10–15 mesenteric microvessels from at least 5 different animals. ***P < 0.0001 vs. wild-type microvessels.
Discussion
Previous studies led to a controversial discussion about the receptor(s) mediating Ang-(1–7) effects. Some publications suggested an involvement of the classical Ang II receptors, AT1 and AT2 (Gironacci et al. 1994, 2004; Handa et al. 1996), while other groups discussed a role for the BK B2 receptor in mediating Ang-(1–7)-stimulated effects (Heitsch et al. 2001). However, most reports indicated an independent Ang-(1–7) receptor, because defined actions of the heptapeptide could be completely blocked by A779, a specific Ang-(1–7) antagonist (Ambuhl et al. 1994; Santos et al. 1994). Our group identified the G protein-coupled receptor Mas to be associated with Ang-(1–7)-induced signalling, demonstrating that genetic deletion of Mas, encoded by the Mas proto-oncogene, abolished the binding of Ang-(1–7) to mouse kidney (Santos et al. 2003). Accordingly, Ang-(1–7) was able to bind to Mas-transfected cells and elicited arachidonic acid release, which was completely blocked by A779.
Previously, we generated data implying that part of the Ang-(1–7)-mediated vasorelaxant effects might be Mas independent (Peiróet al. 2007), because Ang-(1–7) induced a residual relaxation in both wild-type vessels pretreated with A779 and Mas-deficient vessels. These observations suggested that Ang-(1–7) may also interact with an additional receptor other than Mas to elicit vasodilatation, as speculated by others recently (Silva et al. 2007). In the present study, we wanted to investigate the mechanism of this Mas (A779)-independent vasorelaxant effect further. Interestingly, we have clearly demonstrated that the remaining vasorelaxant effect of Ang-(1–7) in Mas-deficient or A779-pretreated wild-type mesenteric arteries is due to a spontaneous loss of tension of the system over time that is completely independent of Ang-(1–7). Such relaxation, which is equivalent in both types of microvessels, depends on the release of NO and is not triggered by the heptapeptide. We could generate further proof that there is no remaining vasorelaxation that is independent of Mas (after subtracting the spontaneous loss of tension) by performing an acute experiment with a single concentration of Ang-(1–7) on wild-type and Mas-deficient mesenteric arteries. Given that no significant loss of tension occurred in this experimental setting, the marked reduction of vascular tone induced by a single concentration of Ang-(1–7) was completely blunted in A779-pretreated vessels or Mas-deficient vessels.
In a previous report, from a study performed in rat mesenteric microvessels, Neves et al. (2003) observed a similar spontaneous vasorelaxation over time, although the authors did not identify the vasodilator agent involved. Our finding that Ang-(1–7)-mediated vasorelaxation depends completely on the receptor Mas is congruent with data from our group, excluding an influence of the Ang II receptors (AT1 and AT2) on the Ang-(1–7)-stimulated vasorelaxation in an acute in vivo animal model (Gembardt et al. 2012).
In contrast to the work by Silva et al. (2007), we did not find different effects for A779 and d-Pro-Ang-(1–7), another specific Ang-(1–7) antagonist (Lemos et al. 2005). In our hands, both antagonists are equally potent, and they completely abolish the vasorelaxant effects specifically induced by the heptapeptide in our ex vivo experiments. The data suggest that both Ang-(1–7) antagonists target the same receptor, and thus indicate that there is only a single receptor (complex) triggering the Ang-(1–7)-induced vasorelaxant effects.
In the present study, we have also shown that the blockade of Mas with either A779 or d-Pro-Ang-(1–7) not only abolished Ang-(1–7)-induced vasodilatation, but also completely prevented the vasodilatation induced by a RAS-independent vasodilator peptide, namely BK. However, microvessels genetically deficient in the receptor Mas lose the capacity to respond to BK. This observation, together with previous results from our group (Peiróet al. 2007), strongly suggests that Mas has a broader vascular action beyond mediating the actions of Ang-(1–7). Indeed, the presence of functional Mas appears to be a critical element for the microvessels to respond to a series of endothelium-dependent vasodilators, including Ang-(1–7), BK, and ACh. All three vasodilators trigger the release of endothelial NO to induce relaxation of the underlying smooth muscle. In the present study, we found that the effect of pharmacological blockade of Mas with either A779 or d-Pro-Ang-(1–7) on Ang-(1–7)-induced vasodilatation was equivalent to that obtained with the NO synthase inhibitor l-NAME. In accordance, Silva et al. (2007), have previously demonstrated that Ang-(1–7)-induced vasodilatation in rat aorta is completely NO dependent, with no involvement of cyclo-oxygenase-derived products, as stated by others (Benter et al. 1993; Machado et al. 2002). Likewise, NO accounted for the entire vasorelaxation response induced by BK in murine microvessels. These data suggest that Mas blockade may specifically interfere with the synthesis of endothelial NO. In accordance, we have previously reported that cultured HUVECs preincubated with A779 lose the ability to generate and release NO after stimulation with BK (Peiróet al. 2007). Thus, Mas does not appear to be a third receptor for BK, in addition to B1 and B2 receptors, as previously suggested, but rather plays a broader and central role in endothelial NO-mediated vasorelaxation.
This central role of Mas for endothelial function is further accentuated by the demonstration that A779 did abrogate eNOS activation by BK. These results clearly demonstrate that in the absence of functioning Mas, the signal pathways leading to eNOS stimulation are affected, rather than the baseline eNOS phosphorylation. Such alterations in eNOS phosphorylation were paralleled by a similar pattern of NO synthesis in endothelial cells. Also here, A779 prevented the effect of BK, without changing total eNOS levels. In contrast to the unaltered total quantities of eNOS in our experiments, Stegbauer et al. (2011) showed an effect of Ang-(1–7) on eNOS quantities. However, they demonstrated such regulation of the abundance of eNOS in pathological conditions after a long-lasting treatment, probably as an indirect result of a significant improvement of the pathophysiological condition in the investigated apoliprotein E-deficient mice. Given that we have used HUVECs from healthy donors and used the same short-term treatment with A779 as we used for our vessel preparations, we believe that the effects described by Stegbauer et al. (2011) are not related to our experimental settings, in which we explored an acute effect of Mas on eNOS phosphorylation, NO production and, consequently, vasorelaxation.
The spontaneous vasorelaxant effect in our microvessel preparation is not different between Mas-deficient and wild-type microvessels, probably because the baseline eNOS activity and NO release is less dependent on modulation of Mas than the ligand–receptor-mediated vasorelaxation. In fact, we have previously reported in HUVECs that Mas blockade reduces the agonist-induced but not the basal production of NO (Peiróet al. 2007). Furthermore, given that Mas has a central position in vascular function, the reactivity to vasoconstrictive agents stimulating both vascular smooth muscle cells and endothelial cells should be altered in Mas-deficient vessels. In the present study, indeed, we have shown that Mas deficiency and its pharmacological blockade generated an increased responsiveness to NA, while the inhibition of eNOS by l-NAME increased the vasoconstrictive potency of NA in the same manner. Therefore, it seems reasonable to suggest that the increased vasoconstrictive response to NA in Mas-deficient microvessels is caused by a reduced release of vasorelaxant NO in endothelial cells upon NA stimulation. Notably, at the NA concentration we used for preconstriction (10 μmol l−1) in our experiments, Mas-deficient and wild-type vessels showed the same maximal constriction.
Using sodium nitroprusside, we have previously shown that smooth muscle function is not impaired in Mas-deficient vessels or in vessels pretreated with A779 (Peiróet al. 2007). Taken together, such results and our present findings indicate that these effects are due to a reduced endothelial NO release and not to an increased constriction of the underlying smooth muscle. This clearly supports our hypothesis that the receptor Mas is a global regulator of endothelial function by regulating NO bioavailability and eNOS activity.
One additional possible mechanism for such a global position of Mas might be the peptide-independent intracellular signalling of the receptor. We and others previously described a constitutive activity of the receptor Mas (Gembardt et al. 2005b; Canals et al. 2006). The absence of this constitutive activity in vessels deficient for Mas might play a role in the lack of vasorelaxant capacity. However, this hypothesis requires further investigations to clarify the impact of this constitutive activity and to elucidate the possible cellular mechanisms involved.
In conclusion, we have shown that the Mas antagonists A779 and d-Pro-Ang-(1–7) equally block a single receptor, which mediates Ang-(1–7)-induced vasodilatation in mesenteric microvessels. Importantly, beyond mediating the vascular actions of Ang-(1–7), Mas appears to be a critical component required for NO-mediated vasodilatation induced by RAS-dependent and RAS-independent agonists. The receptor Mas therefore arises as a key pharmacological target to modulate endothelial function and might be an outstanding target for the treatment of diseases characterized by endothelial dysfunction.
Acknowledgments
We thank Elena Cercas for excellent technical assistance. The research was supported by grants from Plan Nacional de I+D (SAF2011-28011, SAF2011-24648 and Acciones Integradas HD2008-0056 and PRI-AIBDE-2011-0855), Instituto de Salud Carlos III (RETICEF RD06/0013, FIS PI10/00518 and Red Cardiovascular RD12/0042/0052), Sociedad Española de Farmacología – Almirall/Prodesfarma, and ‘Deutscher Akademische Austauschdiens’ (DAAD, Programm des Projektbezogenen Personenaustauschs – Acciones Integradas Hispano-Alemanas 5072341). E.P. is the recipient of a fellowship from Agencia Española de Cooperación Internacional. C.P. and C.F.S.-F. are engaged in the COST Action BM1005 ENOG.
Glossary
- A779
d-Ala7-angiotensin-(1–7)
- ACE
angiotensin-converting enzyme
- ACE2
angiotensin-converting enzyme 2
- Ang I
angiotensin I
- Ang II
angiotensin II
- Ang-(1–7)
angiotensin-(1–7)
- Ang-(1–9)
angiotensin-(1–9)
- AT1 receptor
angiotensin II type 1 receptor
- AT2 receptor
angiotensin II type 2 receptor
- BK
bradykinin
- eNOS
endothelial nitric oxide synthase
- HUVEC
human umbilical vein endothelial cell
- NA
noradrenaline
- NOS
nitric oxide synthase
- RAS
renin–angiotensin system
Author contributions
Conception and design of the experiments: C.P., L.R.-M., C.F.S.-F. and T.W. Collection, analysis and interpretation of data: C.P., S.V., F.G., E.P., S.N., V.A., C.H., C.F.S.-F. and T.W. Drafting the article: C.P., F.G., C.F.S.-F. and T.W. All authors have read the manuscript and approved its submission.
References
- Ambuhl P, Felix D, Khosla MC. [7-D-ALA]-angiotensin-(1–7): selective antagonism of angiotensin-(1–7) in the rat paraventricular nucleus. Brain Res Bull. 1994;35:289–291. doi: 10.1016/0361-9230(94)90103-1. [DOI] [PubMed] [Google Scholar]
- Benter IF, Diz DI, Ferrario CM. Cardiovascular actions of angiotensin(1–7) Peptides. 1993;14:679–684. doi: 10.1016/0196-9781(93)90097-z. [DOI] [PubMed] [Google Scholar]
- Canals M, Jenkins L, Kellett E, Milligan G. Up-regulation of the angiotensin II type 1 receptor by the MAS proto-oncogene is due to constitutive activation of Gq/G11 by MAS. J Biol Chem. 2006;281:16757–16767. doi: 10.1074/jbc.M601121200. [DOI] [PubMed] [Google Scholar]
- Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87:E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- Ellefson DD, diZerega GS, Espinoza T, Roda N, Maldonado S, Rodgers KE. Synergistic effects of co-administration of angiotensin 1–7 and Neupogen on hematopoietic recovery in mice. Cancer Chemother Pharmacol. 2004;53:15–24. doi: 10.1007/s00280-003-0710-0. [DOI] [PubMed] [Google Scholar]
- Esteban V, Heringer-Walther S, Sterner-Kock A, de Bruin R, van den Engel S, Wang Y, Mezzano S, Egido J, Schultheiss HP, Ruiz-Ortega M, Walther T. Angiotensin-(1–7) and the G protein-coupled receptor Mas are key players in renal inflammation. PLoS One. 2009;4:e5406. doi: 10.1371/journal.pone.0005406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman EJ, Chisolm GM, Ferrario CM, Tallant EA. Angiotensin-(1–7) inhibits vascular smooth muscle cell growth. Hypertension. 1996;28:104–108. doi: 10.1161/01.hyp.28.1.104. [DOI] [PubMed] [Google Scholar]
- Gembardt F, Sterner-Kock A, Imboden H, Spalteholz M, Reibitz F, Schultheiss HP, Siems WE, Walther T. Organ-specific distribution of ACE2 mRNA and correlating peptidase activity in rodents. Peptides. 2005a;26:1270–1277. doi: 10.1016/j.peptides.2005.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gembardt F, van Veghel R, Coffman TM, Schultheiss HP, Danser AHJ, Walther T. Hemodynamic effects of vasorelaxant compounds in mice lacking one, two or all three angiotensin II receptors. Hypertens Res. 2012;35:547–551. doi: 10.1038/hr.2012.5. [DOI] [PubMed] [Google Scholar]
- Gembardt F, Wieland T, Zhang J, Heiss A, Walther T. The G protein-coupled receptor Mas encoded by the Mas proto-oncogene shows mitogenic properties through a constitutive activation of rho. Naunyn Schmiedbergs Arch Pharmacol. 2005b;370:R19. [Google Scholar]
- Gironacci MM, Adler-Graschinsky E, Peña C, Enero MA. Effects of angiotensin II and angiotensin-(1-7) on the release of [3H]norepinephrine from rat atria. Hypertension. 1994;24:457–460. doi: 10.1161/01.hyp.24.4.457. [DOI] [PubMed] [Google Scholar]
- Gironacci MM, Valera MS, Yujnovsky I, Peña C. Angiotensin-(1–7) inhibitory mechanism of norepinephrine release in hypertensive rats. Hypertension. 2004;44:783–787. doi: 10.1161/01.HYP.0000143850.73831.9d. [DOI] [PubMed] [Google Scholar]
- Handa RK, Ferrario CM, Strandhoy JW. Renal actions of angiotensin-(1-7): in vivo and in vitro studies. Am J Physiol Renal Physiol. 1996;270:F141–F147. doi: 10.1152/ajprenal.1996.270.1.F141. [DOI] [PubMed] [Google Scholar]
- Heitsch H, Brovkovych S, Malinski T, Wiemer G. Angiotensin-(1-7)-stimulated nitric oxide and superoxide release from endothelial cells. Hypertension. 2001;37:72–76. doi: 10.1161/01.hyp.37.1.72. [DOI] [PubMed] [Google Scholar]
- Lemos VS, Silva DMR, Walther T, Alenina N, Bader M, Santos RAS. The endothelium-dependent vasodilator effect of the nonpeptide Ang(1-7) mimic AVE 0991 is abolished in the aorta of Mas-knockout mice. J Cardiovasc Pharmacol. 2005;46:274–279. doi: 10.1097/01.fjc.0000175237.41573.63. [DOI] [PubMed] [Google Scholar]
- Machado RDP, Ferreira MAND, Belo AV, Santos RAS, Andrade SP. Vasodilator effect of angiotensin-(1–7) in mature and sponge-induced neovasculature. Regul Pept. 2002;107:105–113. doi: 10.1016/s0167-0115(02)00070-8. [DOI] [PubMed] [Google Scholar]
- Neves LAA, Averill DB, Ferrario CM, Chappell MC, Aschner JL, Walkup MP, Brosnihan KB. Characterization of angiotensin-(1–7) receptor subtype in mesenteric arteries. Peptides. 2003;24:455–462. doi: 10.1016/s0196-9781(03)00062-7. [DOI] [PubMed] [Google Scholar]
- Peiró C, Vallejo S, Gembardt F, Azcutia V, Heringer-Walther S, Rodríguez-Mañas L, Schultheiss HP, Sánchez-Ferrer CF, Walther T. Endothelial dysfunction through genetic deletion or inhibition of the G protein-coupled receptor Mas: a new target to improve endothelial function. J Hypertens. 2007;25:2421–2425. doi: 10.1097/HJH.0b013e3282f0143c. [DOI] [PubMed] [Google Scholar]
- Rodgers K, Xiong S, DiZerega GS. Effect of angiotensin II and angiotensin(1–7) on hematopoietic recovery after intravenous chemotherapy. Cancer Chemother Pharmacol. 2003;51:97–106. doi: 10.1007/s00280-002-0509-4. [DOI] [PubMed] [Google Scholar]
- Santos RA, Campagnole-Santos MJ, Baracho NC, Fontes MA, Silva LC, Neves LA, Oliveira DR, Caligiorne SM, Rodrigues AR, Gropen Júnior C, Carvalho WS, Simoes e, Silva AC, Khosla MC. Characterization of a new angiotensin antagonist selective for angiotensin-(1–7): evidence that the actions of angiotensin-(1–7) are mediated by specific angiotensin receptors. Brain Res Bull. 1994;35:293–298. doi: 10.1016/0361-9230(94)90104-x. [DOI] [PubMed] [Google Scholar]
- Santos RA, Simoes e Silva AC, Maric C, Silva DMR, Machado RP, de Buhr I, Heringer-Walther S, Pinheiro SVB, Lopes MT, Bader M, Mendes EP, Lemos VS, Campagnole-Santos MJ, Schultheiss HP, Speth R, Walther T. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA. 2003;100:8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva DMR, Vianna HR, Cortes SF, Campagnole-Santos MJ, Santos RAS, Lemos VS. Evidence for a new angiotensin-(1–7) receptor subtype in the aorta of Sprague–Dawley rats. Peptides. 2007;28:702–707. doi: 10.1016/j.peptides.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Stegbauer J, Potthoff SA, Quack I, Mergia E, Clasen T, Friedrich S, Vonend O, Woznowski M, Königshausen E, Sellin L, Rump LC. Chronic treatment with angiotensin-(1–7) improves renal endothelial dysfunction in apolipoproteinE-deficient mice. Br J Pharmacol. 2011;163:974–983. doi: 10.1111/j.1476-5381.2011.01295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strawn WB, Ferrario CM, Tallant EA. Angiotensin-(1–7) reduces smooth muscle growth after vascular injury. Hypertension. 1999;33:207–211. doi: 10.1161/01.hyp.33.1.207. [DOI] [PubMed] [Google Scholar]
- Takeda H, Katagata Y, Hozumi Y, Kondo S. Effects of angiotensin II receptor signaling during skin wound healing. Am J Pathol. 2004;165:1653–1662. doi: 10.1016/S0002-9440(10)63422-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- Unger T, Chung O, Csikos T, Culman J, Gallinat S, Gohlke P, Höhle S, Meffert S, Stoll M, Stroth U, Zhu YZ. Angiotensin receptors. J Hypertens Suppl. 1996;14:S95–S103. [PubMed] [Google Scholar]
- Vallejo S, Angulo J, Peiró C, Nevado J, Sánchez-Ferrer A, Petidier R, Sánchez-Ferrer CF, Rodríguez-Mañas L. Highly glycated oxyhaemoglobin impairs nitric oxide relaxations in human mesenteric microvessels. Diabetologia. 2000;43:83–90. doi: 10.1007/s001250050011. [DOI] [PubMed] [Google Scholar]
- Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, Acton S, Patane M, Nichols A, Tummino P. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- Weir MR, Dzau VJ. The renin-angiotensin-aldosterone system: a specific target for hypertension management. Am J Hypertens. 1999;12:205S–213S. doi: 10.1016/s0895-7061(99)00103-x. [DOI] [PubMed] [Google Scholar]
- Wilson WL, Roques BP, Llorens-Cortes C, Speth RC, Harding JW, Wright JW. Roles of brain angiotensins II and III in thirst and sodium appetite. Brain Res. 2005;1060:108–117. doi: 10.1016/j.brainres.2005.08.032. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Zhong J, Zhu S, Liu D, Van Der Giet M, Tepel M. Angiotensin-(1–7) inhibits angiotensin II-induced signal transduction. J Cardiovasc Pharmacol. 2002;40:693–700. doi: 10.1097/00005344-200211000-00007. [DOI] [PubMed] [Google Scholar]