

Abstract
Intrinsically disordered proteins (IDPs) are a unique class of proteins that do not require a stable structure for function. The importance of IDPs in many biological processes has been established but there remain unanswered questions about their evolution and conservation of their disordered state within a protein family. Our group has been studying the structural similarities among orthologous FlgM proteins, a model class of IDPs. We have previously shown that the FlgM protein from the thermophile Aquifex aeolicus has more structure at A. aeolicus' physiological temperature (85 °C) than is observed for the Salmonella typhimurium FlgM, suggesting that the disordered nature of FlgM varies among organisms and is not universally conserved. In this work, we extend these studies to the FlgM proteins from Escherichia coli, Pseudomonas aeruginosa, Proteus mirabilis, and Bacillus subtilis. We demonstrate that the B. subtilis, E. coli, and S. typhimurium FlgMs exist in a premolten globule-like conformation, though the B. subtilis FlgM is in a more compacted conformation than the other two. The P. aeruginosa and P. mirabilis FlgM proteins exist in a currently unknown conformation that is not either coil-like or premolten globule-like. The P. aeruginosa FlgM appears to contain more weak intramolecular contacts given its more compacted state than the P. mirabilis FlgM. These results provide experimental evidence that members of the same protein family can exhibit different degrees of disorder, though understanding how different disordered states evolve in the same protein family will require more study.
Keywords: FlgM, Intrinsically disordered protein, Circular dichroism, Molten globule
1. Introduction
It is well established that proteins with amino acid identities greater than 30% generally have nearly identical folds and function [1–4]. The relationship between amino acid sequence and protein fold exists because the amino acid sequence dictates the protein's final structure, which is critical for the protein's function [5]. The structure–function paradigm has guided protein structure/function studies for the last 50 years; however, this paradigm does not cover all proteins. A unique class of proteins was identified about 20 years ago that retain function but lack significant measurable secondary or tertiary structure in vitro [6–9]. These proteins are referred to by various names, including intrinsically disordered [10], natively denatured [11], natively unfolded [12], and intrinsically unstructured [9] but they all share the common characteristic of retaining function in the absence of well-ordered structure. The term intrinsically disordered protein (IDP) will be used to describe these proteins in this paper.
Over the past decade, computational and experimental studies together have highlighted the prevalence of IDPs, especially in higher eukaryotes [13]. IDPs are predicted to exist as a heterogeneous ensemble of structures, capable of assuming a wide range of possible conformations [14]. The ability to assume multiple conformations allows IDPs to interact with a variety of different proteins, thereby expanding their potential function. It has been hypothesized that this dynamic nature is as critical to the function of IDPs as a well-ordered structure is to enzyme function [9,15,16]. This hypothesis has been used to explain why IDPs are especially predominant among DNA binding proteins and proteins related to signaling pathways, which often bind multiple proteins [15,17–21].
IDPs have a lower number of hydrophobic residues and more polar or charged residues than compact, globular proteins [8,22]. This sequence relationship has allowed development of multiple algorithms for predicting disordered structure [23–28]. Because disorder can be maintained without formation of specific contacts, the high degree of sequence conservation and function in globular proteins does not appear to be maintained in IDPs. Rather, IDPs exhibit rapid evolution due to amino acid substitution, repeat expansions, insertions and deletions [29–31], leading to higher sequence divergence between homologous IDPs than what is observed in ordered, globular proteins. The consequence of this sequence divergence on conservation of the disordered state has not been well established.
One study examined conservation of disorder using the dynamic linker region in the replication protein A (RPA70) [14]. The backbone flexibility of homologous RPA70 linker regions from three kingdoms were measured by NMR spectroscopy and all five linkers showed similar flexibility, even in the absence of significant sequence similarity. This makes sense when one considers that disorder is generated by preventing positive associations between amino acids, while ordered protein structures are generated through specific amino acid interactions. Therefore, more amino acid variation is tolerated in disordered proteins, which helps explains the conclusion of the RPA70 study that disorder can be maintained in the absence of natural selection.
A logical candidate to extend these studies is the FlgM protein, which regulates flagella synthesis by binding to the transcription factor Sigma-28. FlgM is found in a wide range of Gram-positive and Gram-negative bacteria and acts as a regulator of the late stage genes involved in flagella synthesis [32–37]. Previous studies have identified 77 unique FlgM protein sequences and have shown that these orthologs form three phylogenetic groups that encompass a variety of characteristics, including classification (Gram-positive verse Gram-negative), pathogenicity, environment, and regulation [38]. Functional studies have been conducted on seven of these FlgM proteins elucidating aspects of FlgM function in regulating flagella synthesis [37,39–45] but structural studies have been limited to the Salmonella typhimurium and Aquifex aeolicus FlgM proteins [45–48].
Previous work with the S. typhimurium FlgM has demonstrated that this protein exhibits transient helical structure in the C-terminus but that the N-terminus remains disordered, even in the bound state [46,47]. In contrast, the A. aeolicus FlgM protein exhibits significant secondary structure at 20 °C, though the percentage of ordered structure decreases as the protein is heated to 85 °C, suggesting this FlgM protein undergoes cold-induced folding [49]. The observed differences in structure and the diversity of the FlgM family led us to ask whether the structural nature of FlgM changes as a function of organism. It is accepted that structure is generally conserved in orthologous globular proteins and as a result, family members are often not structurally characterized. However, there has not been extensive characterization of homologous IDPs and therefore, the principles governing the conservation of structural (or lack of structure) features within IDP families have not been established.
Results from our previous studies on the A. aeolicus FlgM protein suggested two possibilities [49]. One possibility is that bacterial IDPs exhibit more structural variability than either orthologous globular proteins or the RPA70 linker region, which would account for the structural variability observed between the A. aeolicus and S. typhimurium FlgM proteins. Alternatively, the measured structural differences between the A. aeolicus and S. typhimurium FlgM proteins could be due to the A. aeolicus FlgM coming from a thermophilic organism. In this work, we expand our studies to compare the degree of disorder exhibited by the FlgM proteins from Bacillus subtilis, Escherichia coli, Pseudomonas aeruginosa and Proteus mirabilis to distinguish between these possibilities. Our results demonstrate that the six FlgM proteins vary in the compactness of their structures and in how much transient helical structure appears to be present. These results show that the low degree of sequence conservation often observed in IDPs can result in altered structural features and highlights the importance of additional studies on orthologous IDPs.
2. Methods and materials
2.1. Materials
All materials were purchased from major chemical suppliers. Restriction enzymes for cloning were purchased from New England Biolabs. Pfu DNA polymerase was purchased from Stratagene. Primers were purchased from Integrated DNA Technologies. The expression clone for the A. aeolicus FlgM gene was obtained from Dr. Seth Darst [50]. The expression clone for the S. typhimurium FlgM gene was obtained from Dr. Gary Pielak [48].
2.2. Bioinformatics analysis
FlgM sequences were obtained from the protein sequence database at the National Center for Biotechnology Information (NCBI).
2.3. Sequence alignment
Protein sequence alignment was conducted using the ClustalW2 multiple sequence alignment program (http://www.ebi.ac.uk/Tools/clustalw2/index.html). Each alignment was completed using the full alignment parameter with scores reported as a percent identity. Alignments were conducted using the Gonnet matrix with a Gap Open Penalty of 10, Gap Extension Penalty of 0.2, Endgap of −1 and a Gap distance of 4.
2.4. Disorder prediction
Predictions were conducted individually online using DisMeta prediction server (http://www-nmr.cabm.rutgers.edu/bioinformatics/disorder/), which is a web tool developed by the Northeast Structural Genomics Consortium as part of the Protein Structure Initiative.
2.5. Cloning of FlgM genes: A. aeolicus and S. typhimurium
FlgM clones were described previously [49]. B. subtilis, P. aeruginosa, P. mirabilis, and E. coli genomes were isolated using a Qiagen Genome Extraction kit. The FlgM gene in each genome was amplified using PCR. Forward and Reverse primers were designed using the gene sequences in the NCBI database. PCR products were digested using NdeI and BamHI and ligated into a similarly digested pET 14b vector using T4 ligase. Clones were selected from individual colonies and verified by complete gene sequencing.
2.6. Expression and purification of FlgM
All FlgM genes were expressed using the protocol developed for expression of the A. aeolicus FlgM gene [49]. Briefly, each clone was transformed into either BL21(DE3) or ROSETTA™ cells (Novagen) for expression. An overnight culture was used to inoculate expression cultures, which were induced at 18 °C overnight by addition of 1 mM IPTG. Expressed cells were collected by centrifugation and resuspended in Wash Buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 7.5), followed by addition of 1/10 volume Triton X-100 (10% solution), 10 μg/mL lysozyme, 5 μg/mL DNase, and 1 μg/mL MgCl2 to the resuspended cells. Cells were lysed using a combination of freeze/ thaw and sonication. Insoluble protein was removed by centrifugation and the resulting supernatant was passed over a HiTrap™ Chelating HP column (GE Healthcare). Bound protein (95% FlgM) was eluted using a 250 mM imidazole step gradient and the eluted protein was further purified using a Superdex™ 75 Prep column with Wash Buffer as the mobile phase. Purity was assessed by Coomassie staining as being >98% pure. Protein concentration was determined using a Bradford assay due to the low content of aromatic amino acids.
2.7. Circular dichroism (CD)
CD spectra were recorded at various temperatures using a Jasco J-815 CD spectrometer equipped with a Peltier temperature controller (Jasco, Easton, MD). 23 μM FlgM protein dialyzed into 10 mM phosphate was placed in a 1 mm quartz cell and spectra were obtained using a scanning rate of 100 nm/min and 0.1 nm wavelength steps from 190 to 260 nm. Spectra were averaged over 5 scans and background subtracted. The percent of α-helix was calculated using the formula (1) of Yang and Chen [51].
| (1) |
2.8. Gel filtration
Retention time of each FlgM protein was measured using an analytical Superdex-75™ gel filtration column. 100 μL samples of purified protein were separated and the retention time was determined to be the volume providing the maximum absorbance. Each sample was measured in triplicate and averaged.
2.9. FlgM crosslinking
1.0, 0.75, 0.5, and 0.25 mg/mL of purified FlgM protein were incubated with 1% glutaraldehyde. Alternatively, 0.5 mg/mL FlgM was incubated with 1%, 0.5%, 0.1%, 0.05% and 0.001% glutaraldehyde. Both sets of conditions were incubated at room temperature for 1 h. Cross-linked protein was separated on a 16% SDS–PAGE gel.
3. Results
3.1. Sequence similarity between FlgM genes
Previous phylogenetic analyses of the FlgM family identified three evolutionary groups of FlgM proteins [38]. We selected functionally characterized representatives from all three evolutionary groups including FlgM proteins from E. coli, P. aeruginosa, P. mirabilis and B. subtilis. The E. coli, S. typhimurium and P. mirabilis are from Group 1A. The P. aeruginosa FlgM is from Group 1B, the B. subtilis FlgM is from Group IIA and the A. aeolicus FlgM is from Group III [38]. All six FlgM proteins exhibit very low sequence identity, with the exception of the S. typhimurium and E. coli FlgM proteins, which come from evolutionarily similar bacteria (Fig. 1, Table 1). The sequence identity is so low that a BLAST search using the S. typhimurium FlgM protein sequence as a reference would identify only the E. coli FlgM protein and not any of the other four FlgM proteins studied.
Fig. 1.
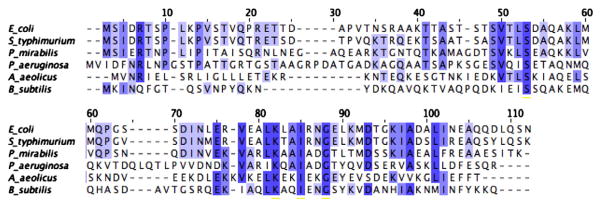
Sequence alignment shows low sequence identity. Aquifex aeolicus (AA), Bacillus subtilis (BS), Escherichia coli (EC), Pseudomonas aeruginosa (PA), Proteus mirabilis (PM), and Salmonella typhimurium (ST) FlgM genes were aligned by ClustlW. Conserved amino acids are shaded, with darker shading for more highly conserved amino acids.
Table 1.
Pairwise identity and similarities for FlgM proteins.
| Protein | Identical amino acids | % Identity | Similar amino acids | % Similarity |
|---|---|---|---|---|
| AA:BS | 17 | 19% | 24 | 27% |
| AA:EC | 19 | 21% | 30 | 32% |
| AA:PA | 21 | 22% | 34 | 35% |
| AA:PM | 16 | 17% | 27 | 29% |
| AA:ST | 20 | 22% | 30 | 32% |
| BS:EC | 19 | 21% | 27 | 29% |
| BS:PA | 17 | 17% | 32 | 33% |
| BS:PM | 17 | 18% | 23 | 25% |
| BS:ST | 17 | 18% | 23 | 25% |
| EC:PA | 20 | 20% | 35 | 34% |
| EC:PM | 42 | 43% | 56 | 60% |
| EC:ST | 77 | 79% | 81 | 84% |
| PA:PM | 31 | 30% | 39 | 38% |
| PA:ST | 23 | 23% | 36 | 35% |
| PM:ST | 42 | 43% | 55 | 56% |
AA: Aquifex aeolicus, BS: Bacillus subtilis, EC: Escherichia coli, PA: Pseudomonas aeruginosa, PM: Proteus mirabilis, ST: Salmonella typhimurium. Groups of similar amino acids: 1) Leu, Ile, Val; 2) Glu, Asp; 3) Lys, Arg; 4) Trp, Tyr, Phe; 5) Cys, Ser.
Conservation of function with low sequence conservation is a common feature of orthologous IDPs because the disordered state is a function of the chemical nature of a side chain rather than specific amino acid contacts. We therefore determined the percentage of sequence similarity by considering chemically similar amino acids as equivalent (Table 1). Similar amino acids were 5 to 17% higher than identical amino acids, with most FlgM pairs exhibiting over a 10% increase in similarity over identity. The high variation in sequence among IDPs can make the identification of orthologous IDPs difficult. Expanding alignment constraints to weight the chemical nature of a side chain more heavily may lead to algorithms more capable of accurately identifying orthologous IDPs in the future.
3.2. Prediction of disorder
There are 17 disorder prediction algorithms listed on the DisProt website, though some of these predictors are related algorithms [52]. Each algorithm uses different relationships to predict which regions within a protein may be disordered, and there is not one ideal algorithm for all proteins. We analyzed the amino acid sequences of the six FlgM proteins in this study using the Disorder Prediction Meta-Server (DisMeta). DisMeta analyzes a protein sequence using 10 different disorder prediction algorithms (DISEMBL, DISOPRED2, DISpro, DRIPPRED, FoldIndex, FoldUnfold, GlobPlot2, IUPred, RONN, and VSL2); predictions from each algorithm provided separately by the server, along with a plot that shows how many of the algorithms predict a particular amino acid to be disordered. Protein sequences are also analyzed using seven different other sequence analysis tools (coils, ANCHOR, SignalP, TMHMM, SEG, PROFphd, and PSIPred) to provide secondary structure, binding site and complexity predictions. The results are presented in an easy to read graph that allows comparison of different algorithms simultaneously.
Two trends were apparent from the DisMeta analysis of the six FlgM proteins. First, the N-terminus of the FlgM proteins is predicted to be disordered by the majority of algorithms whereas the C-terminus is predicted to be ordered, consistent with experimental data. The exception to this was A. aeolicus FlgM, which is predicted to be predominantly ordered with a disordered region between amino acids 15 and 38, also consistent with experimental data. However, the specific regions and size of the predicted regions of disorder vary significantly throughout the six proteins. Second, the secondary structure prediction algorithms predict helical content in the C-terminus of all six FlgM sequences, though the predicted helical regions and the number of helices varied between the six sequences. Moreover, both the A. aeolicus and B. subtilis FlgM sequences are also predicted to contain helical content in their N-terminus. These results support the hypothesis that orthologous FlgM proteins exhibit different structural characteristics.
3.3. Size exclusion chromatography demonstrates different molecular weight predictions
An established characteristic of IDPs is an abnormally large Stokes radius due to their extended conformation. This correlates to higher predicted molecular weights, as determined by size exclusion chromatography (SEC), relative to similarly sized globular proteins. We analyzed the retention time of each FlgM protein using an analytical Superdex-75 gel filtration column. All six FlgM proteins eluted with a retention volume between 10 and 12 mL. The P. mirabilis FlgM had the shortest retention volume (10.6 mL) and theoretically the most extended structure, whereas the A. aeolicus FlgM had the longest retention volume (11.9 mL) and potentially the most compact structure. The long retention time of the A. aeolicus FlgM was not a surprise because our previous work had shown that the H1 and H2 helices are associated together at 20 °C, creating a more compact structure. The column was calibrated using a Low Molecular Weight calibration kit from GE Healthcare and the resulting calibration curve was used to calculate predicted molecular weights for each protein. As shown in Table 2, the molecular weights calculated by retention times are 2 to 4 times larger than the molecular weights calculated by amino acid sequence, suggesting that all six FlgM proteins are in some sort of extended conformation and not in a compacted, globular state. However, there are clear differences in how much the calculated molecular weight varies from the predicted molecular weight, providing the first experimental evidence that different FlgM proteins assume different conformations.
Table 2.
Predicted molecular weight from gel filtration retention times for FlgM proteins from different organisms.
| Organism | Retention time (mL) | Calculated molecular weight (kDa) | Predicted molecular weight (kDa) |
|---|---|---|---|
| Aquifex aeolicus | 11.9 | 24.4 | 10.2 |
| Bacillus subtilis | 11.8 | 25.4 | 10.0 |
| Escherichia coli | 11.0 | 35.9 | 10.3 |
| Pseudomonas aeruginosa | 11.1 | 35.7 | 11.3 |
| Proteus mirabilis | 10.6 | 43.0 | 10.7 |
| Salmonella typhimurium | 10.9 | 38.6 | 10.5 |
These results suggest that there are three types of conformation states: a compact state, a more extended conformation and a very extended conformation. Both the A. aeolicus and B. subtilis FlgM proteins are in the most compact state because their calculated molecular weight is only 2× the predicted molecular weight. The P. mirabilis FlgM appears to be in the most extended conformation because the calculated molecular weight is close to 4× the predicted molecular weight. The E. coli, P. aeruginosa and S. typhimurium FlgM proteins are potentially in a slightly more compacted state than the P. mirabilis FlgM because their calculated molecular weights are 3× the predicted molecular weight.
An alternative interpretation of these results is that the high molecular weight is due to oligomerization of FlgM. Previous work suggested that the B. subtilis FlgM exists as a dimer, though the experimental data supporting this conclusion could also be explained by the B. subtilis FlgM being in an extended, disordered conformation [39]. We used glutaraldehyde cross-linking to determine if we could observe FlgM oligomers to address this possibility. For each FlgM protein, we assessed potential oligomerization as a function of both protein concentration and glutaraldehyde concentration. Representative gels for the A. aeolicus and P. mirabilis FlgM proteins are shown in Fig. 2. As can be seen, the majority of the protein is monomeric, with only small amounts of dimer observed at very high protein concentrations, consistent with a concentration dependent oligomerization. If the high molecular weights observed in SEC were a function of oligomerization, then the majority of the FlgM protein should be in an oligomeric state (dimer in the case of A. aeolicus FlgM and tetra-mer in the case of P. mirabilis FlgM) and not monomeric.
Fig. 2.
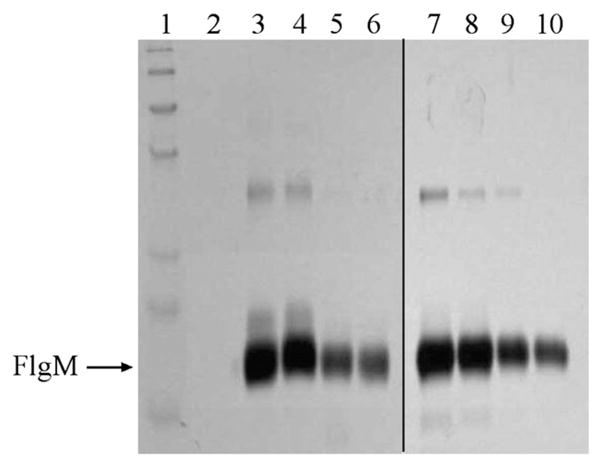
Cross-linking reveals concentration dependent oligomerization. Purified Aquifex aeolicus and Proteus mirabilis FlgM protein at varying concentrations was treated with 1% glutaraldehyde for 1 h at room temperature. Lane 1: marker, Lane 2: blank, Lanes 3–6: A. aeolicus FlgM at 1.0, 0.75, 0.5, 0.25 mg/mL respectively. Lanes 7–10: P. mirabilis FlgM at 1.0, 0.75, 0.5, 0.25 mg/mL respectively.
We further tested this hypothesis by measuring retention times at different concentrations of FlgM. If the high calculated molecular weights are due to a concentration-dependent oligomerization, then retention time should significantly change as a function of protein concentration. Retention times were measured at different protein concentrations and only minimal changes in retention time were observed (for example, 11.7 mL at 1 mg/mL compared to 11.9 mL at 0.1 mg/mL for the A. aeolicus FlgM). This further supports the model that the FlgM proteins are in extended, monomeric conformations and not forming an oligomeric species.
3.4. Orthologous FlgM proteins exhibit different circular dichroism spectra
The global secondary structure of the six FlgM proteins was measured at 20 °C using circular dichroism (CD) to characterize the conformational differences between FlgM orthologs. Representative average CD spectra of all six FlgM proteins are shown in Fig. 3. The A. aeolicus FlgM shows a distinctly helical spectrum at 20 °C, whereas all the mesophilic FlgM orthologs show the characteristic features of an unfolded polypeptide, namely a strong negative band at 200 nm and a weak negative signal around 220 nm. Deconvolution algorithms such as CDPro and K2D were not able to accurately fit the representative spectra so the percentage of helical content was determined using the formula developed by Yang and Chen [51] (Table 3). The A. aeolicus FlgM spectra has a high helical content (41%) consistent with the crystal structure and our previous studies [45,49]. All five of the mesophilic FlgM proteins are predicted to have helical contents between 10 and 21% but there is clear variation among the different proteins.
Fig. 3.
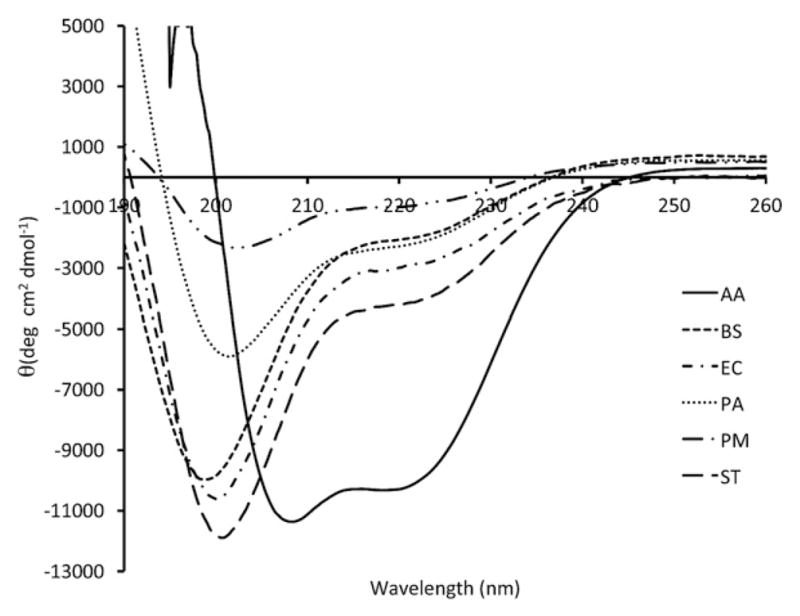
Homologous FlgM proteins exhibit different far-UV CD spectra. Far-UV spectra from different FlgM proteins exhibit different CD spectra. The Bacillus subtilis, Escherichia coli, and Salmonella typhimurium FlgM exhibit similar secondary structures. The Aquifex aeolicus FlgM exhibits the most ordered structure at 20 °C while the Proteus mirabilis and Pseudomonas aeruginosa FlgMs exhibit varying degrees of secondary structure. A. aeolicus (–), B. subtilis (- - - -), E. coli (-·-·-), P. aeruginosa (· · · · ·), P. mirabilis (–··–··–), S. typhimurium (– – –).
Table 3.
Percentage of α-helical content of FlgM proteins from different organisms in increasing amounts of TFE.
| 0% | 5% | 10% | 15% | 20% | 25% | |
|---|---|---|---|---|---|---|
| Aquifex aeolicus | 40.5±1.0% | N. D. | N. D. | N. D. | N. D. | N. D. |
| Bacillus subtilis | 14.3±0.8% | 17.3±0.4% | 21.1±0.4% | 25.0±0.4% | 29.7±0.7% | 33.3±1.0% |
| Escherichia coli | 17.2±0.7% | 20.1±1.6% | 21.8±0.4% | 26.1±1.9% | 28.8±0.8% | 34.7±4.4% |
| Pseudomonas aeruginosa | 14.9±0.7% | 13.8±0.1% | 15.0±0.5% | 17.1±0.4% | 18.7±0.3% | 20.7±0.7% |
| Proteus mirabilis | 10.8±0.9% | 13.0±0.3% | 13.5±0.9% | 14.3±1.0% | 15.6±0.8% | 17.2±0.7% |
| Salmonella typhimurium | 21.3±0.1% | 20.3±1.2% | 24.5±0.5% | 27.8±1.5% | 30.0±1.9% | 31.4±4.0% |
Spectra were collected at 20 °C from 3 independently prepared samples and the alpha-helical percentages for each sample were calculated. The percentages from each trial were averaged and the standard deviation for each set of samples was calculated.
An analysis developed by Uversky [53] was used to further characterize potential structural differences between the different orthologs. A study of the CD spectra of over 100 IDPs by Uversky has shown that a plot of [θ]222 verse [θ]200 can distinguish between two potential disordered protein structural states. IDPs with [θ]200 = −19,000± 2800° cm2 dmol−1 and [θ]222 = −1700±700° cm2 dmol−1 are in a coil-like state. The second state, a pre-molten globule like state, has [θ]200 = −10,700±1300° cm2 dmol−1 and [θ]222 = −3900± 1100° cm2 dmol−1. The pre-molten globule state was first defined in 1997 as a short-lived intermediate in the folding pathway of the trypto-phan synthase beta subunit [54]. One of the key structural differences between these two states is the polyprolineII (PII) helix content. The coil-like state has a PII content of ~30% while the pre-molten globule state has a PII content of ~10%.
The [θ]200 and [θ]222 values for the five mesophilic FlgMs were plotted to determine the state of the different orthologs (Fig. 4). The B. subtilis, E. coli, and S. typhimurium FlgM orthologs all have minima at 200 nm between −9000 and −12,000° cm2 dmol−1 and [θ]222 between −1900° cm2 dmol−1 and −3000° cm2 dmol−1, within or near the pre-molten globule range. This is consistent with previous studies on the S. typhimurium FlgM demonstrating that the C-terminus of this protein is composed of transiently formed helices. The [θ]200 and [θ]222 values for the P. aeruginosa and P. mirabilis FlgM proteins do not fit into either structural state, suggesting they are in unique conformations. There are two possible models that can explain these structural differences. The first is that the P. aeruginosa and P. mirabilis FlgM proteins spend more time in a non-helical conformation, reducing the PII signature even further (below 10%). A second possibility is that the transient helical structure is more stable; hence, the P. aeruginosa and the P. mirabilis FlgMs spend more time in this conformation. The slight red-shift of the 200 nm minima (Fig. 3) of the P. aeruginosa and the P. mirabilis FlgMs better support the second model but the weak overall CD signal better support the first.
Fig. 4.
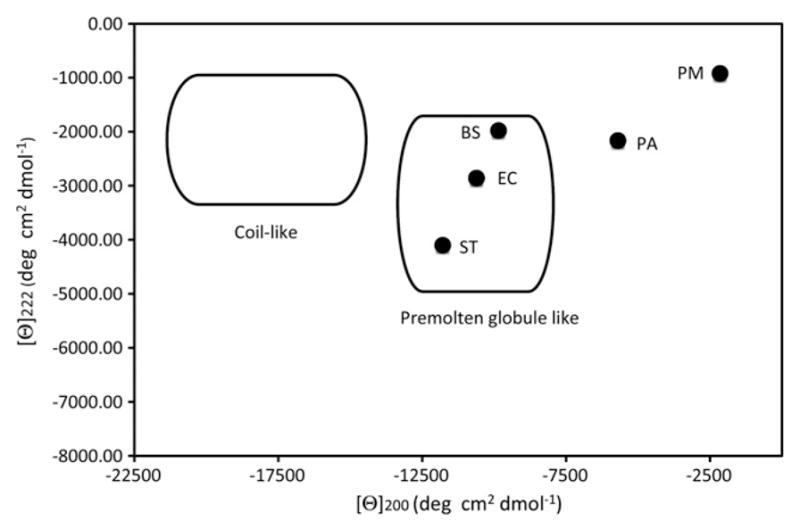
Plot of [θ]200 verse [θ]222 shows that the Bacillus subtilis, Escherichia coli and Salmonella typhimurium FlgM proteins are in a premolten globule state. The [θ]200 verse [θ]222 positions for the B. subtilis (BS), E. coli (EC), Proteus mirabilis (PA), Pseudomonas aeruginosa (PA), and S. typhimurium (ST) FlgM proteins were plotted together. The circle in the upper left of the figure represents the coil-like region and the circle in the center of the figure shows the region for the premolten globular state, as determined by Uversky [53].
To distinguish between the two possibilities, we measured the CD spectra in the presence of increasing amounts of 2,2,2-trifiuoroethanol (TFE). TFE is a highly polar molecule that helps reduce water's ability to solvate amide hydrogen bonds, promoting helix formation in transient and unstable helices [55]. TFE should have a greater stabilizing effect on the P. aeruginosa and P. mirabilis FlgMs if they contain more stable transient helical interactions than the other three mesophilic FlgM proteins. However, there should be a reduced increase in helical content if the P. aeruginosa and P. mirabilis FlgMs have less transient helical content. Far-UV CD spectra were measured for all five mesophilic FlgM proteins in the presence of 5–25% TFE. Representative spectra from the E. coli and P. mirabilis FlgMs are shown in Fig. 5 and the percent of α-helix is shown in Table 5. All five FlgM proteins exhibited a linear increase in helical content with increasing TFE, consistent with the presence of transient helical structure (Fig. 5B). The B. subtilis, E. coli and S. typhimurium FlgM proteins all exhibited a larger increase in helicity than the P. aeruginosa and P. mirabilis FlgMs. This was further shown by calculating the difference in helical content between 0 and 25% TFE (Table 3). These results support the model of the P. aeruginosa and P. mirabilis FlgMs having less transient structure than the other three mesophilic FlgM proteins.
Fig. 5.

TFE induces additional secondary structure in the more highly disordered FlgM proteins. (A) Representative far-UV CD spectra from Escherichia coli in increasing amounts of TFE. (B) Representative far-UV CD spectra from Proteus mirabilis in increasing amounts of TFE. (C) Plot of the %Helix verse %TFE. As can be seen, the helical percentage increases in a linear fashion. The Pseudomonas aeruginosa and P. mirabilis FlgM proteins show the smallest change in helical content. 0% TFE (–), 5% TFE (– –), 10% TFE (–··–··), 15% TFE (–·–·), 20% TFE (- - - -), 25% TFE (· · · · ·).
Table 5.
Amino acid composition of FlgM proteins from different organisms.
| Total number of amino acids | Hydrophobic amino acids | % Hydrophobic amino acids | Polar amino acids | % Polar amino acids | Charged amino acids | % Charged amino acids | |
|---|---|---|---|---|---|---|---|
| Bacillus subtilis | 88 | 35 | 40% | 32 | 36% | 21 | 24% |
| Escherichia coli | 97 | 41 | 42% | 34 | 35% | 22 | 23% |
| Pseudomonas aeruginosa | 107 | 48 | 45% | 34 | 32% | 25 | 23% |
| Proteus mirabilis | 99 | 44 | 44% | 29 | 29% | 26 | 26% |
| Salmonella typhimurium | 97 | 38 | 39% | 33 | 34% | 26 | 27% |
3.5. Temperature effects on mesophilic FlgM proteins
IDPs exhibit a unique thermal behavior relative to well-ordered globular proteins. Most ordered proteins exhibit a cooperative loss of CD signal as temperature increases as the protein unfolds. IDPs actually exhibit the formation of structure as temperature increases. This phenomenon is referred to as a “temperature-induced formation of secondary structure” and was first observed in α-synuclein but which has been observed in other IDPs as well [56,57]. It has been proposed that this occurs in IDPs because increased temperature will increase the strength of hydrophobic interactions, inducing a more ordered state [8]. This change in conformation is not a cooperative effect as the transition is a smooth, linear change and not a two-state transition characteristic of a cooperative folding event.
We investigated the thermal behavior of the five mesophilic FlgM proteins in the presence and absence of 25% TFE to compare the structural transitions between more ordered and less ordered forms of each protein. All five proteins exhibit non-cooperative transitions, both in the presence and absence of TFE, which was expected because even in the presence of TFE there is not significant structure. All but one of the mesophilic FlgMs exhibits similar “temperature-induced folding” as reported for α-synuclein but the magnitude of change varied (Table 4). The exception was the P. mirabilis FlgM, which did not exhibit any significant conformational changes between 15 and 85 °C. In the presence of TFE, all five proteins lost structure as temperature increased, though the magnitude of the change varied with S. typhimurium and B. subtilis FlgM exhibiting a 36–38% loss of helicity and the other three FlgMs exhibiting a 25–29% loss of helicity.
Table 4.
Change in helical percentage of FlgM proteins from different organisms with increasing temperature.
| No additives
|
25% TFE
|
|||||
|---|---|---|---|---|---|---|
| 15 °C | 85 °C | % change in helical content | 15 °C | 85 °C | % change in helical content | |
| Bacillus subtilis | 14.4±1.3% | 16.6±0.8% | 13% | 34.5±2.2% | 21.9±0.9% | −36.6% |
| Escherichia coli | 9.9±1.2% | 17.1±0.3% | 42% | 33.1±4.8% | 24.4±3.3% | −26.6% |
| Pseudomonas aeruginosa | 13.0±1.4% | 17.6±0.6% | 26% | 20.1±1.7% | 14.3±1.2% | −29.0% |
| Proteus mirabilis | 16.8±0.7% | 16.6±0.8% | −1% | 24.2±0.9% | 18.2±0.8% | −25.0% |
| Salmonella typhimurium | 20.8±1.0% | 22.2±1.1% | 6% | 28.5±4.2% | 17.7±2.3% | −38.0% |
Spectra were collected from 3 independently prepared samples and the alpha-helical percentages for each sample were calculated. The percentages from each trial were averaged and the standard deviation for each set of samples was calculated.
The difference in the thermal behavior of the five different orthologs was surprising because it was not consistent with our other results. We hypothesized that this might be a function of sequence and examined the distribution of amino acid types among the five FlgM proteins (Table 5). The B. subtilis and S. typhimurium FlgM proteins have a slightly higher percentage of polar and charged amino acids and a slightly lower percentage of hydrophobic amino acids compared to the E. coli, P. aeruginosa and P. mirabilis FlgMs, possibly explaining why these two FlgM proteins exhibit a larger decrease in helicity in the presence of TFE. It could also explain why these two proteins exhibit a lower degree of “temperature-induced structure.” However, the amino acid content does not explain why there is no “temperature-induced structure” in the P. mirabilis FlgM. The P. mirabilis FlgM is in the most extended conformation, as shown by SEC, and we propose that the sequence differences that cause the extended conformation also prevent any “temperature-induced structure.”
4. Discussion
IDPs are an interesting group of proteins due to their flexibility and potential for assuming a variety of conformations. These same characteristics, however, makes their true structural nature unclear. As a result, there are a number of unanswered questions regarding IDP structure and what truly constitutes disorder. One key question is how conserved the disorder character is among orthologous IDPs. As discussed above, the FlgM/Sigma-28 system provides a unique opportunity to explore this relationship as there are over 100 identified FlgM proteins.
The RPA70 linker region has very low sequence identity; however, the disordered character of this region appears to be conserved [14]. This work demonstrates that all five FlgM proteins from mesophilic organisms exhibited different structural behaviors, showing that orthologous IDPs can have different structural characteristics. The most closely related FlgMs appear to be the E. coli and S. typhimurium FlgMs, which is consistent with their high sequence identity (79% identity: 84% similarity). However, this 20% difference in sequence is enough to alter the thermal behavior of the two proteins with the E. coli FlgM forming more “temperature-induced structure” than the S. typhimurium.
Aromatic amino acids can contribute to the CD signal at the 222 nm [58], which could impact the accuracy of any helical content predictions and the [θ]200 verse [θ]222 plot. There are no differences in phenylalanine or tryptophan content in any of the FlgM proteins but there are differences in tyrosine content: A. aeolicus, P. aeruginosa and S. typhimurium FlgMs each contain one tyrosine, E. coli and P. mirabilis FlgMs contain none, and B. subtilis FlgM contains three tyrosine residues. These differences might account for the spectral variations observed at 222 nm; however, the tyrosine content would not impact migration of these proteins during size exclusion chromatography, and the B. subtilis FlgM migrates differently than the other FlgM proteins. Moreover, the percentage of tyrosine in each protein is very low in all six proteins. We therefore argue that the structural differences are real rather than an artifact of differences in tyrosine content.
Our results support the existence of two structural types of FlgM proteins with some structural variation within each type (Table 6). The first structural group assumes a pre-molten globule conformation, as observed for the B. subtilis, E. coli and S. typhimurium FlgM proteins. The B. subtilis FlgM has a more compact structure than the other two FlgM proteins in this group, which may be unique to this particular FlgM or may indicate that there is a further classification than can be made. We propose that there are more transient interactions among different regions of the B. subtilis FlgM than in E. coli or S. typhimurium FlgM proteins, accounting for more compact structure. Note that the retention time that was measured for the B. subtilis FlgM is similar to the predicted molecular weight of a dimeric protein, which was the reported oligomeric state of the B. subtilis FlgM [39]. No evidence of a B. subtilis FlgM dimer was observed in our cross-linking experiments; hence perhaps the B. subtilis FlgM was classified as a dimer because the authors had not consider the possibility of an extended conformation. The initial characterization of the B. subtilis FlgM was conducted before the prevalence of IDPs was widely appreciated.
Table 6.
Summary of observed FlgM structural conformations.
| Organism | Conformation | Induction of helix by TFE | Folded state |
|---|---|---|---|
| Bacillus subtilis | Compacted | Strong | Pre-molten globule |
| Escherichia coli | Extended | Strong | Pre-molten globule |
| Pseudomonas aeruginosa | Extended | Weak | Unknown |
| Proteus mirabilis | Very extended | Weak | Unknown |
| Salmonella typhimurium | Extended | Strong | Pre-molten globule |
The second structural group encompasses the P. aeruginosa and P. mirabilis FlgM proteins. These two FlgM proteins exhibit characteristics that suggest they are structurally unique from each other as well as the other three FlgM proteins from mesophiles, although the evidence is not as strong. For example, they have similar 200/222 ratios, suggesting a similar conformation and both show similar effects in the presence of TFE. However, they migrate differently on SEC, suggesting different extended conformations. As there are only two examples of FlgM proteins that do not exhibit a pre-molten globule structure, it is premature to draw any conclusions about the characteristics of these two proteins without more studies. One possibility to consider is that observed structural variation is a function of these experiments being conducted in “dilute” solution conditions. It is known that the C-terminus of the S. typhimurium FlgM assumes a more ordered conformation inside the cell, presumably through stabilizing non-specific interactions with other proteins. The observed structural differences may represent variations in the weak stabilizing interactions each protein naturally contains but that the intracellular environment stabilized all five proteins so that they exist in a similar conformation. We currently do not have sufficient data to assess this possibility.
4.1. Relationship between structure and function with FlgM
Our results suggest that there are two different structural classes of FlgM proteins as well as some conserved structural elements across the FlgM proteins. First, all six proteins exist in an extended conformations. Second, all six proteins have helical content, though the degree of helical content varies. The primary difference is the stability of the helical regions of the protein. Four of the FlgM proteins have helical content that can be stabilized by TFE but the other two show only small increases in helical content, suggesting these helices are less stable. The impact of this difference in helix stability on function is still an open question.
Binding of FlgM to Sigma-28 has been structurally characterized in two systems. The S. typhimurium FlgM binds Sigma-28 through the α-helices at the C-terminus of the protein. These are transient helices in the free FlgM but the interaction with Sigma-28 stabilizes the C-terminus. The N-terminus of the protein remains in an extended and disordered state, even when bound. In contrast, the majority of the A. aeolicus FlgM protein exists in a structured state when bound to its complementary Sigma-28. It should be noted that this structure was determined at mesophilic temperatures. It is possible that the binding of the A. aeolicus FlgM N-terminus is a crystallographic artifact without any functional significance and the C-terminus of FlgM is the primary binding site for Sigma-28.
The proposed model is supported through two additional pieces of evidence. First, mutation studies on the E. coli FlgM have identified the C-terminus as the primary binding site [59]. In addition, the C-terminus of the FlgM protein is more highly conserved than the N-terminus. This was shown by the study conducted by Mons et al. [38] and we have expanded this study to include more FlgM protein sequences, with similar results. This makes sense since the main interaction between FlgM and Sigma-28 is through Region 4 of Sigma-28, which binds the −35 promoter sequence. The invariant nature of this promoter region provides a selective pressure on the Sigma-28 to conserve the promoter binding site, which in turn puts selective pressure onto FlgM to conserve the binding site with Sigma-28 even though it is evolving at a faster rate than Sigma-28 (unpublished data). This observation suggests that a common binding mechanism has been preserved.
The work presented here does not provide any insight into where the increased helical content is being generated in the presence of TFE. However, we propose that the helices being stabilized are in the C-terminal region of the protein. This would be consistent with the model of the binding site being in the C-terminus. It is possible that even the P. aeruginosa and P. mirabilis FlgM proteins assume a helical conformation upon binding to Sigma-28, similar to the S. typhimurium FlgM, though this remains to be shown experimentally. This raises some interesting possibilities regarding the binding of FlgM and IDP binding in general.
There are two models for binding; conformational selection and coupled binding-folding. The binding mechanism of FlgM to Sigma-28 has not been clearly established at this point. The transient structure in four of the six FlgM proteins would suggest that the conformational selection model might be the most consistent model for binding. However, the two FlgM proteins that exhibit little transient structure might bind through a coupled binding–folding mechanism. This would suggest that different members of the FlgM family might exhibit different binding mechanisms. It is more likely that there is a single binding mechanism but that leads to the question of how the different degrees of transient structure relate to the strength of the binding between the various FlgM and Sigma-28 combinations. The answers to these questions will provide insights beyond this system but will also provide insights into potential things to consider with other systems where there are multiple orthologous proteins.
The conservation of the C-terminus suggests that the binding mechanism may be similar for all FlgM proteins. We propose that there is a specific molecular recognition feature (MoRF) in FlgM that have been conserved and exist in all FlgM proteins. The existence of a MoRF would allow binding between FlgM and Sigma-28 regardless of the specific binding mechanism. There are four amino acids that are absolutely conserved between all six FlgM proteins and an additional eight amino acids that are similar. These sites are predominately located in the C-terminus of FlgM and several of them could contribute to form a conserved MoRF. If binding occurs through a MoRF, it would be predicted that FlgM proteins from different organisms would exhibit cross-species reactivity, which the FlgM from one species binding the Sigma-28 of another species. Future work will be necessary to identify if a MoRF exists and the specific sequence of that MoRF.
These results demonstrate that orthologous IDPs do not necessarily exhibit the same structural characteristics. These studies will have to be expanded to other IDP families to determine whether structural conservation is more prevalent or if structural diversity among IDPs is more common. Regardless of the ultimate outcome, it is clear that the principles regarding conservation of structure in IDPs are more complex than those for globular proteins.
Acknowledgments
The authors would like to thank Dr. Seth Darst for the clones of the A. aeolicus FlgM and σ28 genes and Dr. Gary Pielak for the clone of the S. typhimurium FlgM gene. We would also like to thank Dr. Robert Woody and Dr. Narasimha Sreerama for helpful discussions about the proper ways to analyze the CD data collected as part of this study. Funding for this study was provided by NIH 5U54CA096320-05.
Footnotes
This manuscript was prepared using Microsoft Word 2011 for Macintosh.
References
- 1.Al-Lazikani B, Jung J, Xiang Z, Honig B. Protein structure prediction. Curr Opin Chem Biol. 2001;5(1):51–56. doi: 10.1016/s1367-5931(00)00164-2. [DOI] [PubMed] [Google Scholar]
- 2.Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294(5540):93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 3.Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5(4):823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chothia C, Lesk AM. The evolution of protein structures. Cold Spring Harb Symp Quant Biol. 1987;52:399–405. doi: 10.1101/sqb.1987.052.01.046. [DOI] [PubMed] [Google Scholar]
- 5.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181(96):223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 6.Fink AL. Natively unfolded proteins. Curr Opin Struct Biol. 2005;15(1):35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27(10):527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 8.Uversky VN. What does it mean to be natively unfolded? Eur J Biochem. 2002;269(1):2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 9.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure–function paradigm. J Mol Biol. 1999;293(2):321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 10.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graph Model. 2001;19(1):26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 11.Schweers O, Schonbrunn-Hanebeck E, Marx A, Mandelkow E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem. 1994;269(39):24290–24297. [PubMed] [Google Scholar]
- 12.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35(43):13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 13.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337(3):635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Daughdrill GW, Narayanaswami P, Gilmore SH, Belczyk A, Brown CJ. Dynamic behavior of an intrinsically unstructured linker domain is conserved in the face of negligible amino acid sequence conservation. J Mol Evol. 2007;65(3):277–288. doi: 10.1007/s00239-007-9011-2. [DOI] [PubMed] [Google Scholar]
- 15.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6(3):197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 16.Vise P, Baral B, Stancik A, Lowry DF, Daughdrill GW. Identifying long-range structure in the intrinsically unstructured transactivation domain of p53. Proteins. 2007;67(3):526–530. doi: 10.1002/prot.21364. [DOI] [PubMed] [Google Scholar]
- 17.Bowman P, Galea CA, Lacy E, Kriwacki RW. Thermodynamic characterization of interactions between p27(Kip1) and activated and non-activated Cdk2: intrinsically unstructured proteins as thermodynamic tethers. Biochim Biophys Acta. 2006;1764(2):182–189. doi: 10.1016/j.bbapap.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 18.Campbell KM, Terrell AR, Laybourn PJ, Lumb KJ. Intrinsic structural disorder of the C-terminal activation domain from the bZIP transcription factor Fos. Biochemistry. 2000;39(10):2708–2713. doi: 10.1021/bi9923555. [DOI] [PubMed] [Google Scholar]
- 19.Lowry DF, Stancik A, Shrestha RM, Daughdrill GW. Modeling the accessible conformations of the intrinsically unstructured transactivation domain of p53. Proteins. 2008;71(2):587–598. doi: 10.1002/prot.21721. [DOI] [PubMed] [Google Scholar]
- 20.Sivakolundu SG, Bashford D, Kriwacki RW. Disordered p27Kip1 exhibits intrinsic structure resembling the Cdk2/cyclin A-bound conformation. J Mol Biol. 2005;353(5):1118–1128. doi: 10.1016/j.jmb.2005.08.074. [DOI] [PubMed] [Google Scholar]
- 21.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323(3):573–584. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 22.Williams RM, Obradovi Z, Mathura V, Braun W, Garner EC, Young J, Takayama S, Brown CJ, Dunker AK. The protein non-folding problem: amino acid determinants of intrinsic order and disorder. Pac Symp Biocomput. 2001:89–100. doi: 10.1142/9789814447362_0010. [DOI] [PubMed] [Google Scholar]
- 23.Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB. Protein disorder prediction: implications for structural proteomics. Structure. 2003;11(11):1453–1459. doi: 10.1016/j.str.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 24.Obradovic Z, Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK. Predicting intrinsic disorder from amino acid sequence. Proteins. 2003;53(Suppl 6):566–572. doi: 10.1002/prot.10532. [DOI] [PubMed] [Google Scholar]
- 25.Obradovic Z, Peng K, Vucetic S, Radivojac P, Dunker AK. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins. 2005;61(Suppl 7):176–182. doi: 10.1002/prot.20735. [DOI] [PubMed] [Google Scholar]
- 26.Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, Dunker AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry. 2005;44(6):1989–2000. doi: 10.1021/bi047993o. [DOI] [PubMed] [Google Scholar]
- 27.Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK, Obradovic Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J Bioinform Comput Biol. 2005;3(1):35–60. doi: 10.1142/s0219720005000886. [DOI] [PubMed] [Google Scholar]
- 28.Romero P, Obradovic Z, Dunker AK. Natively disordered proteins: functions and predictions. Appl Bioinformatics. 2004;3(2–3):105–113. doi: 10.2165/00822942-200403020-00005. [DOI] [PubMed] [Google Scholar]
- 29.Brown CJ, Takayama S, Campen AM, Vise P, Marshall TW, Oldfield CJ, Williams CJ, Dunker AK. Evolutionary rate heterogeneity in proteins with long disordered regions. J Mol Evol. 2002;55(1):104–110. doi: 10.1007/s00239-001-2309-6. [DOI] [PubMed] [Google Scholar]
- 30.Shaiu WL, Hu T, Hsieh TS. The hydrophilic, protease-sensitive terminal domains of eucaryotic DNA topoisomerases have essential intracellular functions. Pac Symp Biocomput. 1999:578–589. doi: 10.1142/9789814447300_0057. [DOI] [PubMed] [Google Scholar]
- 31.Tompa P. Intrinsically unstructured proteins evolve by repeat expansion. Bioessays. 2003;25(9):847–855. doi: 10.1002/bies.10324. [DOI] [PubMed] [Google Scholar]
- 32.Gillen KL, Hughes KT. Molecular characterization of flgM, a gene encoding a negative regulator of flagellin synthesis in Salmonella typhimurium. J Bacteriol. 1991;173(20):6453–6459. doi: 10.1128/jb.173.20.6453-6459.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gillen KL, Hughes KT. Transcription from two promoters and autoregulation contribute to the control of expression of the Salmonella typhimurium flagellar regulatory gene flgM. J Bacteriol. 1993;175(21):7006–7015. doi: 10.1128/jb.175.21.7006-7015.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hughes KT, Gillen KL, Semon MJ, Karlinsey JE. Sensing structural intermediates in bacterial flagellar assembly by export of a negative regulator. Science. 1993;262(5137):1277–1280. doi: 10.1126/science.8235660. [DOI] [PubMed] [Google Scholar]
- 35.Kutsukake K. Excretion of the anti-sigma factor through a flagellar substructure couples flagellar gene expression with flagellar assembly in Salmonella typhimurium. Mol Gen Genet. 1994;243(6):605–612. doi: 10.1007/BF00279569. [DOI] [PubMed] [Google Scholar]
- 36.Ohnishi K, Kutsukake K, Suzuki H, Iino T. Gene fliA encodes an alternative sigma factor specific for flagellar operons in Salmonella typhimurium. Mol Gen Genet. 1990;221(2):139–147. doi: 10.1007/BF00261713. [DOI] [PubMed] [Google Scholar]
- 37.Ohnishi K, Kutsukake K, Suzuki H, Lino T. A novel transcriptional regulation mechanism in the flagellar regulon of Salmonella typhimurium: an antisigma factor inhibits the activity of the flagellum-specific sigma factor, sigma F. Mol Microbiol. 1992;6(21):3149–3157. doi: 10.1111/j.1365-2958.1992.tb01771.x. [DOI] [PubMed] [Google Scholar]
- 38.Pons T, Gonzalez B, Ceciliani F, Galizzi A. FlgM anti-sigma factors: identification of novel members of the family, evolutionary analysis, homology modeling, and analysis of sequence–structure–function relationships. J Mol Model. 2006;12(6):973–983. doi: 10.1007/s00894-005-0096-5. [DOI] [PubMed] [Google Scholar]
- 39.Bertero MG, Gonzales B, Tarricone C, Ceciliani F, Galizzi A. Overproduction and characterization of the Bacillus subtilis anti-sigma factor FlgM. J Biol Chem. 1999;274(17):12103–12107. doi: 10.1074/jbc.274.17.12103. [DOI] [PubMed] [Google Scholar]
- 40.Caramori T, Barilla D, Nessi C, Sacchi L, Galizzi A. Role of FlgM in sigma D-dependent gene expression in Bacillus subtilis. J Bacteriol. 1996;178(11):3113–3118. doi: 10.1128/jb.178.11.3113-3118.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chilcott GS, Hughes KT. Coupling of flagellar gene expression to flagellar assembly in Salmonella enterica serovar typhimurium and Escherichia coli. Microbiol Mol Biol Rev. 2000;64(4):694–708. doi: 10.1128/mmbr.64.4.694-708.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Correa NE, Barker JR, Klose KE. The Vibrio cholerae FlgM homologue is an anti-sigma28 factor that is secreted through the sheathed polar flagellum. J Bacteriol. 2004;186(14):4613–4619. doi: 10.1128/JB.186.14.4613-4619.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frisk A, Jyot J, Arora SK, Ramphal R. Identification and functional characterization of flgM, a gene encoding the anti-sigma 28 factor in Pseudomonas aeruginosa. J Bacteriol. 2002;184(6):1514–1521. doi: 10.1128/JB.184.6.1514-1521.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Josenhans C, Niehus E, Amersbach S, Horster A, Betz C, Drescher B, Hughes KT, Suerbaum S. Functional characterization of the antagonistic flagellar late regulators FliA and FlgM of Helicobacter pylori and their effects on the H. pylori transcriptome. Mol Microbiol. 2002;43(2):307–322. doi: 10.1046/j.1365-2958.2002.02765.x. [DOI] [PubMed] [Google Scholar]
- 45.Sorenson MK, Ray SS, Darst SA. Crystal structure of the flagellar sigma/ anti-sigma complex sigma(28)/FlgM reveals an intact sigma factor in an inactive conformation. Mol Cell. 2004;14(1):127–138. doi: 10.1016/s1097-2765(04)00150-9. [DOI] [PubMed] [Google Scholar]
- 46.Daughdrill GW, Chadsey MS, Karlinsey JE, Hughes KT, Dahlquist FW. The C-terminal half of the anti-sigma factor, FlgM, becomes structured when bound to its target, sigma 28. Nat Struct Biol. 1997;4(4):285–291. doi: 10.1038/nsb0497-285. [DOI] [PubMed] [Google Scholar]
- 47.Daughdrill GW, Hanely LJ, Dahlquist FW. The C-terminal half of the anti-sigma factor FlgM contains a dynamic equilibrium solution structure favoring helical conformations. Biochemistry. 1998;37(4):1076–1082. doi: 10.1021/bi971952t. [DOI] [PubMed] [Google Scholar]
- 48.Dedmon MM, Patel CN, Young GB, Pielak GJ. FlgM gains structure in living cells. Proc Natl Acad Sci U S A. 2002;99(20):12681–12684. doi: 10.1073/pnas.202331299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Molloy RG, Ma WK, Allen AC, Greenwood K, Bryan L, Sacora R, Williams L, Gage MJ. Aquifex aeolicus FlgM protein exhibits a temperature-dependent disordered nature. Biochim Biophys Acta. 2010;1804(7):1457–1466. doi: 10.1016/j.bbapap.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 50.Campbell EA, Darst SA. The anti-sigma factor SpoIIAB forms a 2:1 complex with sigma(F), contacting multiple conserved regions of the sigma factor. J Mol Biol. 2000;300(1):17–28. doi: 10.1006/jmbi.2000.3838. [DOI] [PubMed] [Google Scholar]
- 51.Chen YH, Yang JT. A new approach to the calculation of secondary structures of globular proteins by optical rotatory dispersion and circular dichroism. Biochem Biophys Res Commun. 1971;44(6):1285–1291. doi: 10.1016/s0006-291x(71)80225-5. [DOI] [PubMed] [Google Scholar]
- 52.Vucetic S, Obradovic Z, Vacic V, Radivojac P, Peng K, Iakoucheva LM, Cortese MS, Lawson JD, Brown CJ, Sikes JG, Newton CD, Dunker AK. DisProt: a database of protein disorder. Bioinformatics. 2005;21(1):137–140. doi: 10.1093/bioinformatics/bth476. [DOI] [PubMed] [Google Scholar]
- 53.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11(4):739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chaffotte AF, Guijarro JI, Guillou Y, Delepierre M, Goldberg ME. The “pre-molten globule,” a new intermediate in protein folding. J Protein Chem. 1997;16(5):433–439. doi: 10.1023/a:1026397008011. [DOI] [PubMed] [Google Scholar]
- 55.Myers JK, Pace CN, Scholtz JM. Trifluoroethanol effects on helix propensity and electrostatic interactions in the helical peptide from ribonuclease T1. Protein Sci. 1998;7(2):383–388. doi: 10.1002/pro.5560070219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. 2001;276(14):10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 57.Kim TD, Ryu HJ, Cho HI, Yang CH, Kim J. Thermal behavior of proteins: heat-resistant proteins and their heat-induced secondary structural changes. Biochemistry. 2000;39(48):14839–14846. doi: 10.1021/bi001441y. [DOI] [PubMed] [Google Scholar]
- 58.Woody RW. Aromatic side-chain contributions to far ultraviolet circular-dichroism of peptides and proteins. Biopolymers. 1978;17(6):1451–1467. [Google Scholar]
- 59.Chadsey MS, Hughes KT. A multipartite interaction between Salmonella transcription factor sigma28 and its anti-sigma factor FlgM: implications for sigma28 holoenzyme destabilization through stepwise binding. J Mol Biol. 2001;306(5):915–929. doi: 10.1006/jmbi.2001.4438. [DOI] [PubMed] [Google Scholar]