

Abstract
Perfluorinated Taddol-based phosphoramidite, CKphos, is a highly selective ligand for formation of the vinylogous amide cycloadduct in the Rh(I) catalyzed [2+2+2] cycloaddition of alkenyl isocyanates and alkynes. CKphos overrides substrate bias of product selectivity in the cycloaddition, providing indolizinones in excellent product and enantioselectivities. Excellent selectivities are attributed to a shortened Rh-P bond and coordination of one C6F5 to rhodium via a Z-type interaction, making the phosphoramidite a bidentate L,Z-ligand on rhodium. Evidence for the shortened Rh-P and C6F5 coordination is provided by X-ray, NMR and DFT computation analyses. Additionally, an anionic cobalt complex with CKphos was synthesized and two Co-C6F5 interactions are seen. Rh(C2H4)Cl•CKphos catalyst in the [2+2+2] cycloaddition of alkenyl isocyanates and alkynes represents a rare example of metal-C6F5 Z-type interaction affecting selectivity in transition metal catalysis.
Perfluoroaryl-Metal Interactions
Perfluoroaryls, such as C6F6, have a permanent quadrupole equal in magnitude and opposite in sign to C6H6 and have been demonstrated to interact with electron-rich Lewis bases via weak anion-π1 and lone pair-π2 interactions (Figure 1). The strength of anion-π interactions is comparable to a moderate to strong hydrogen bond (~20–50 kJ/mol). Theoretical studies revealed that anion-π interactions with π-acidic aromatics are dominated by electrostatic interactions.3 The importance of these counterintuitive,4 non-covalent attractive interactions is now being realized in chemical and biological processes.1b,c
Figure 1.

C6H6 and C6F6 electrostatic potential maps and depiction of metal-aryl interactions. Blue indicates net positive potential and net negative potential is red. The 0.004 au magnitude isovalue is plotted for the electron density. Mapping of the electrostatic potential ranges from −0.008 to 0.01 au.
Analogous metal-C6F5 interactions are rare. Only a handful of metal–C6Fn complexes have been reported: Rh,5 Ir,6 Ni,7 Re,8 Cr,9 W10 and Ru.11,12 DFT calculations reveal that electron density in perfluoroaryls localizes on the fluorines leaving a large, postive electrostatic potential on the π-system (Figure 1). Perfluoroaryls can be seen as acting as accepting electron density from electron-rich metal centers. C6Fn–metal interactions are markedly different from C6Hn–metal interactions, where an aryl donates electrons to a metal (L-type, Aryl→M).13 Lewis acidic metal-aryl interactions (Z-type, M→Aryl) where the metal donates electrons to an aryl acceptor are rare and remain unexplored.14
Recently, Lewis acidic, Z-type boron-metal interactions have been investigated using newly developed phosphinoborane ligands.15 Apart from phosphinoboranes, few ligand scaffolds exist to study Z-type metal interactions. Furthermore, the effect Z-type ligands have on transition metal catalysis has not been investigated. Herein, we report the development and analysis of a perfluorinated Taddol-based phosphoramidite,16 CKphos, that functions as a bidentate L,Z-ligand on Rh(I) through an L-type phosphoramidite and lewis acidic, Z-type, C6F5-metal interaction. Evidence of the Z-type interaction was provided by X-ray, NMR and DFT calculations. The bidentate CKphos ligand has a dramatic effect on product and enantioselectivity, due to the C6F5 aryls, in the Rh(C2H4)Cl•CKphos catalyzed [2+2+2] cycloaddition of alkenyl isocyanates and alkynes. To our knowledge, this is a rare example of an Lewis-acidic (Z-type) C6F5-metal interaction affecting selectivity in metal catalysis.
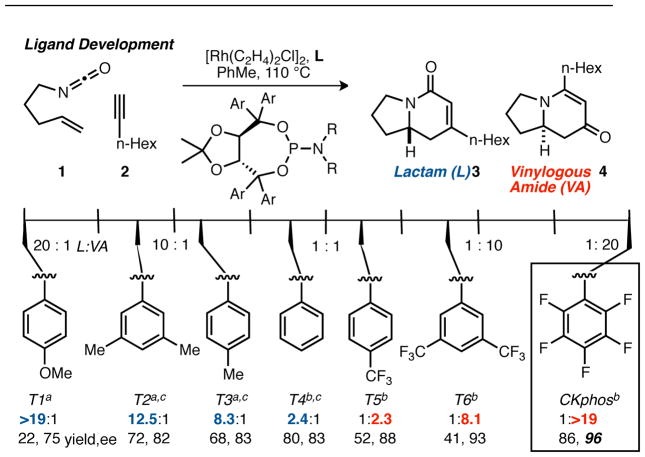
Perfluoroaryl Taddol phosphoramidite CKphos was designed to test our mechanistic hypothesis that phosphoramidite steric interactions in the Rh(I) catalyzed [2+2+2] cycloaddition17 of alkenyl isocyanates18 and alkynes controls product-, regio- and enantioselectivity (Table 1, eq 1).19 A long-standing goal of this project aimed to override substrate-based control of product selectivity through ligand development.20 Formation of vinylogous amide 4 with small, alkyl, or electron-deficient alkynes remained difficult because these substrates favor lactam 3.21 Due to the abundance (>200) of biologically active 5-alkyl substituted indolizidines, we sought to establish efficient, enantioselective methods of rapidly and efficiently synthesizing 5-alkyl indolizidine scaffolds.22
Table 1.
Effect of aryl electronic modification on selectivity. [a] NR2 = piperidyl. [b] NR2 = pyrrolidyl. [c] Results from Ref. 21
|
Based on our proposed model,21 we hypothesized that we could favor vinylogous amide 4 by altering the Rh-P bond length and consequently the steric environment around Rh. We would control Rh–P bond length by manipulating the electronics of the phosphoramidite.23 Specifically, as the phosphoramidite was made increasingly electron-deficient the P–O σ* would lower in energy and backdonation from filled Rh d-orbitals to P–O σ* would increase, resulting in a shortened Rh–P bond length and increased selectivity for vinylogous amide.24
According to our hypothesis, relatively electron-rich phosphoramidites should have longer Rh–P bond lengths due to a higher lying P–O σ* and favor lactam 3, while electron-deficient phosphoramidites should have shorter Rh–P bond distances and favor vinylogous amide 4. Indeed, electron-rich, p-MeO T1 was synthesized and found to give excellent product selectivity for lactam 3 (Scheme 1). However, yield and enantioselectivity is low due to by-product formation and weak steric control from the ligand. On the other hand, electron-deficient p-CF3 T5 provides vinylogous amide 4 as the major product (1:2.3), which is a notable change in selectivity from that obtained with isosteric p-CH3 T3 (8.3:1). A further increase in electron-deficiency of the backbone aryls enhances selectivity for vinylogous amide 4. The most electron-deficient ligand, C6F5 CKphos, provides vinylogous amide 4 with excellent control of product-, regio- and enantioselectivity.
Scheme 1.

Mechanistic model for lactam and vinylogous amide formation.
X-ray Analysis of Rh(I)• CKphos Complexes
Perfluoroaryl Taddol-derived phosphoramidite, CKphos, was synthesized from the perfluorinated diol and dichloropyrridylphosphine. Treatment of [Rh(cod)Cl]2 with racemic CKphos ligand in the presence of DCM results in the Rh(cod)Cl•CKphos complex, which readily forms X-ray quality crystals after slow evaporation in DCM/Heptanes. X-ray analysis revealed a short, incomparison too ther Rh(cod)Cl•Phosphoramidite, Rh–P bond distance. This shortened bond distance is attributed to increased d→P–O σ* backdonation seen with the more electron-deficient C6F5 Taddol, CKphos, in comparison to the C6H5 Taddol T4 (Figure 2).
Figure 2.

X-ray structure of Rh(cod)Cl•CKphos. [a] ethylene bridge of cod omitted for clarity. [b] reaction of pentenyl isocyanate and 1-octyne. [c] Previously reported in ref. 21. Structures showing ellipsoids at 50% probability are included in the supplementary info.
Additionally, Rh(cod)Cl•CKphos shows a significant trans effect in comparison to the phenyl Taddol-derived phosphoramidite, T4, with C=C bond distances of 1.365 Å (CKPhos) and 1.378 Å (T4) seen. This is significantly shorter than C–C bond distance of 1.409 Å for T2 or 1.424 Å reported for [Rh(cod)Cl2]2 and closer to the bond distance of the free alkene, 1.337 Å.25 This suggests that the coordinated cyclooctadiene (cod) alkene trans to the phosphoramidite phosphorus has more Csp2 character, implying that there is less π-backbonding from rhodium to the alkene. Decreased π-backbonding to the alkene is likely the consequence of increased backdonation from filled rhodium d orbitals to lower lying P–O σ* of the electron-deficient phosphoramidite.23 Incorporation of the C6F5 aryls into the Taddol phosphoramidite serves to lower the P–O σ*, increase backdonation from rhodium and shorten the C=C bond trans to the phosphoramidite. Overall, the shortened Rh–P and C=C bond distances suggest that perfluoroaryl CKphos acts as a better π-acceptor ligand than phenyl-Taddol T4 on rhodium, decreasing the π-basicity of Rh(I).
Previously reported X-ray analyses of Rh(cod)Cl•Phosphoramidite complexes revealed that one Taddol-phosphoramidite aryl is situated above rhodium hindering one face of the rhodium square plane (Figure 2).21 In the Rh(cod)Cl•CKphos complex one aryl also sits directly above the rhodium square plane but it is significantly closer to rhodium than in Rh(cod)Cl•C6H5-T4. The Rh–C6F5 centroid bond distance in the Rh(cod)Cl•CKphos complex is 3.728 Å, which is 0.3 Å closer to rhodium than the 4.039 Å or 4.025 Å Rh–C6H5 centroid distance seen with T4 and T2. A search of the Cambridge Crystallographic Data Center (CCDC) found X-ray structures of C6H6–C6F626 and MenC6Hn–C6F627 complexes with intercentroidal distances between 3.5 and 3.7 Å. These distances suggest that the Taddol C6F5 is in close enough proximity to be interacting with rhodium in the ground state.
To determine if the close Rh–C6F5 centroid distance was an isolated occurrence, we examined Rh(cod)X•CKphos complexes with different counterions (Figure 3). X-ray analysis of Rh(cod)OTs•CKphos found that the Rh–C6F5 centroid distance with the tosylate counterion decreases from 3.728 Å (chloride) to 3.687 Å. Rh(cod)OTf•CKphos has a longer Rh–C6F5 distance and the C6F5 has shifted from being directly over rhodium, as seen with chloride and tosylate, to being situated over the oxygen of the triflate. Presumably this change is due to the highly ionic nature of the Rh–OTf bond, which is less tightly bound to rhodium than chloride or tosylate, and the anionic OTf–C6F5 interaction is more favorable than the Rh–C6F5 when the rhodium is less electron rich. This suggests that the perfluoroaryl is acting as a Lewis acid and for the interaction to occur the metal involved needs to be relatively Lewis basic.
Figure 3.
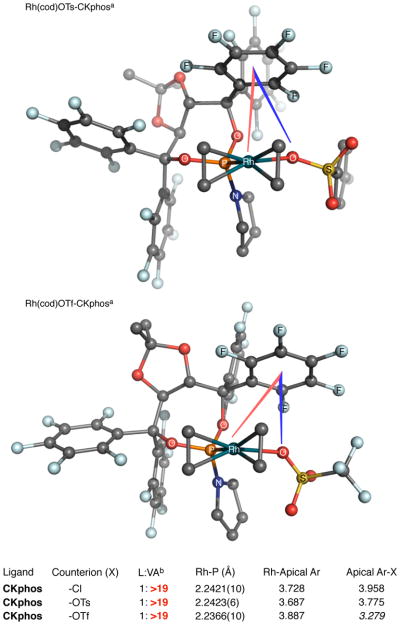
X-ray structures of Rh(cod)X•CKphos. [a] ethylene bridge of cod omitted for clarity. [b] reaction of pentenyl isocyanate and 1-octyne
DFT Studies
DFT computational analyses of Rh(cod)Cl•CKphos and Rh(cod)Cl•T4 were used to verify and better understand the Rh–C6F5 interaction implied by the ground state X-ray structure (Figure 4). Beginning with the X-ray structures, energy minimizations were performed, and significantly, the Rh–C6F5 interaction was found to be a persistent, favorable interaction in gas phase and not the result of crystal packing forces. Calculated bond distances were found to agree with those in the X-ray crystal data for both Rh(cod)Cl•CKphos and Rh(cod)Cl•T4. With three different basis sets the calculated Rh–C6F5 centroid bond distance is found to be shorter than that observed in the X-ray data by 0.311 to 0.516 Å. Agreement between computational and X-ray data provides further support for the Rh–C6F5 interaction.
Figure 4.
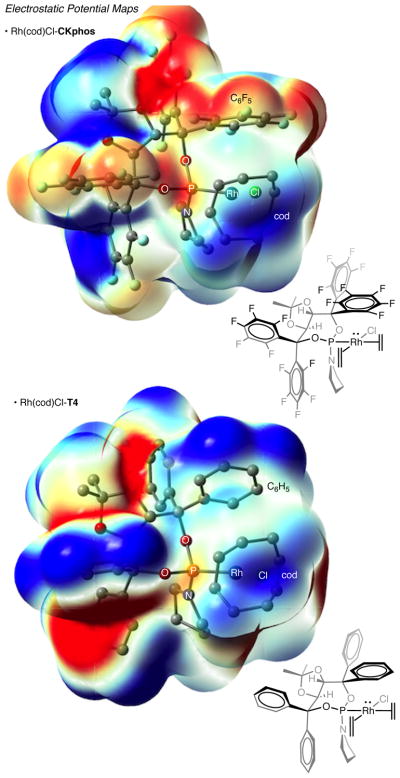
DFT calculations of the electrostatic potential maps for Rh(cod)Cl•CKphos and Rh(cod)Cl•T4. Blue indicates net positive potential; net negative potential is red. The 0.004 au magnitude isovalue is plotted for the electron density. Mapping of the electrostatic potential ranges from −0.008 to 0.01 au.
Electrostatic potential maps were generated from the DFT calculations of Rh(cod)Cl•CKphos and Rh(cod)Cl•T4. The maps show significant inversion of electrostatic potential between the perfluoroaryl and phenyl rhodium phosphoramidite complexes (Figure 4). In the Rh(cod)Cl•T4 potential map significant negative potential (red) is found above and below each of the aryl rings while positive potential (blue) gathers around the hydrogens of the phenyl. Rh(cod)Cl•CKphos map shows a clear inversion of electrostatic potential. Positive potential (blue) is now found above and below each perfluoroaryl and significant negative potential (red) is on the fluorines. An interesting observation is that the electrostatic potential of the C6F5 above rhodium, proposed to be interacting with it, is less electropositive (blue) and has more negative electrostatic potential (red) on its fluorines.
Furthermore, DFT calculations found that the HOMO for Rh(cod)Cl•CKphos is predominately on the rhodium dz2 orbital. As depicted in Figure 5, clearly the HOMO extends into the electropositive region below the highly electron deficient perfluoroaryl. This supports the notion that rhodium has significant electron density that is properly located to interact with the C6F5. Together these results support the proposal that perfluoroaryl C6F5 functions as a Lewis acid to accept electron density from electron rich rhodium as a Z-type ligand.
Figure 5.

HOMO of Rh(cod)Cl•CKPhos, depicted looking down the Rh-Cl bond with chlorine in the foreground.
NMR Studies
To investigate the Rh–C6F5 interaction in solution, we analyzed Rh(cod)Cl•phosphoramidite complexes by NMR spectroscopy. 31P-NMR shows coupling between phosphorus and rhodium (Figure 6). Rh(cod)Cl•T4 has a JRh–P coupling constant of 234 Hz and Rh(cod)Cl•CKphos has a JRh–P of 243 Hz. Roodt, Varshavsky and coworkers found a correlation between solid-state bond lengths from X-ray crystal data and coupling constants observed in solution by 31P-NMR.28 The difference in coupling constants between C6F5 and C6H5 may be correlated to the bond distance seen in the solid state and corresponds to the electronegativity of the aryl substituents on the Taddol phosphoramidite. Interestingly, 31P-NMR of Rh(cod)Cl•CKphos revealed phosphorus splitting into a doublet of doublet with J values of 243 Hz (JRh-P) and 5 Hz. A 19F-decoupled 31P-NMR study provided a broad doublet, suggesting that at least one fluorine on CKphos is coupling to phosphorus. Attempts to determine which fluorine is coupled to phosphorus by 31P-decoupled 19F-NMR have been unfruitful. As a result, we calculated JP-F coupling constants based on the Rh(cod)Cl•CKphos X-ray structure.
Figure 6.

31P-NMR of Rh(cod)Cl•Phosphoramidites. [a] ethylene bridge of cod omitted for clarity. [b] 31P-NMR shift of ligand.
DFT spin-spin calculations [B3LYP, 6–311(d,p) basis on P and F] on the optimized Rh(cod)Cl•CKphos structure identified three fluorines with JP-F coupling constants greater than 1 Hz (Figure 6). Each is an ortho fluorine, two (F1, F3) are within 5 Å of phosphorus and one (F2) is more than 5 Å. In the static DFT calculations, ortho fluorines F1, F2 and F3 appear to be coupling to phosphorus through long-range (5-bond) coupling,29 but in solution, aryl rotation likely disrupts long-range coupling for F1 and F2 because such coupling is sensitive to the molecule’s conformation.30 However, coordination of the C6F5 bearing F3 to rhodium will limit rotation of this C6F5 and rigidify the phosphoramidite overall. Rigidification of the phosphoramidite through Rh–C6F5 interaction could allow 31P–19F coupling to occur either through-bond or through-space. The magnitude of through-space coupling between NMR active nuclei diminishes as the sum of the Van-der Waals radii is exceeded.31 This would make through-space coupling unlikely; however, through-space 31P–19F coupling is reported on 3,3′-bistriflone-BINOL phosphoramidites and JP-F values of 5 Hz are seen between atoms 6 bonds apart.32
C6F5 Interactions with Co(-I)
To provide further evidence of the Metal–C6F5 interaction, we investigated CKphos as a ligand on highly Lewis basic, anionic cobalt. Co(CO)3nBu4N•CKphos was synthesized and analyzed by X-ray diffraction (Figure 7). Surprisingly, X-ray analysis revealed two Co–C6F5 interactions with Co–C6F5 centroid distances of 3.925 and 4.032 Å. These distances are longer than the Rh–centroid distance seen in Rh(cod)Cl•CKphos, a consequence of the smaller size of cobalt and its tetrahedral bonding environment. To accommodate a second Co–C6F5 interaction the phosphoramidite seven-membered ring changes its ring conformation in comparison to the ring conformation seen in Rh(cod)Cl•CKphos.
Figure 7.

X-ray structure of n-Bu4NCo(CO)3•CKphos, comparison of phosphoramidite ring conformations with rhodium and cobalt, and X-ray structure of Co(CO)3•PPh2(C6H4CH2NHMe2) [Ref. 34].
The geometry of cobalt in anionic tetracarbonyl cobaltate complexes with a wide range of non-coordinating cations is tetrahedral.33 Deviations from tetrahedral geometry are found when cobalt is strongly ligated by L-type ligands or through strong hydrogen bonding with large ammonium cations.34 Weak hydrogen bonds with large ammonium cations do not distort cobalt’s tetrahedral geometry.34c Co(CO)3nBu4N•CKphos adopts a distorted tetrahedral geometry at cobalt rather than a tetrahedral geometry, suggesting that the Co–C6F5 interaction may be similar to a strong hydrogen bond. Furthermore, Brammer and coworkers found that Co(CO)3PPh2(C6H4CH2NHMe2), a cobaltate complex with an intramolecular N+–H---Co− bond, adopts a similar distorted tetrahedral geometry.34c In comparison, one coordinating C6F5 of Co(CO)3nBu4•CKphos is in the same apical position above cobalt as the N–H hydrogen bond seen in Co(CO)3PPh2(C6H4CH2NHMe2). Also, in the Co(CO)3PPh2(C6H4CH2NHMe2) the distance between cobalt and the amine methyls is 4.075 and 4.124 Å, similar to the Co–C6F5 centroid distance (3.925, 4.032 Å) in Co(CO)3nBu4N•CKphos. Overall, the Co–C6F5 bond distances, change in phosphoramidite ring conformations and distorted tetrahedral geometry at cobalt provide reasonable evidence that anionic cobalt is interacting with two perfluoroaryls of CKphos as Lewis acidic, Z-type ligands.
Substrate Scope
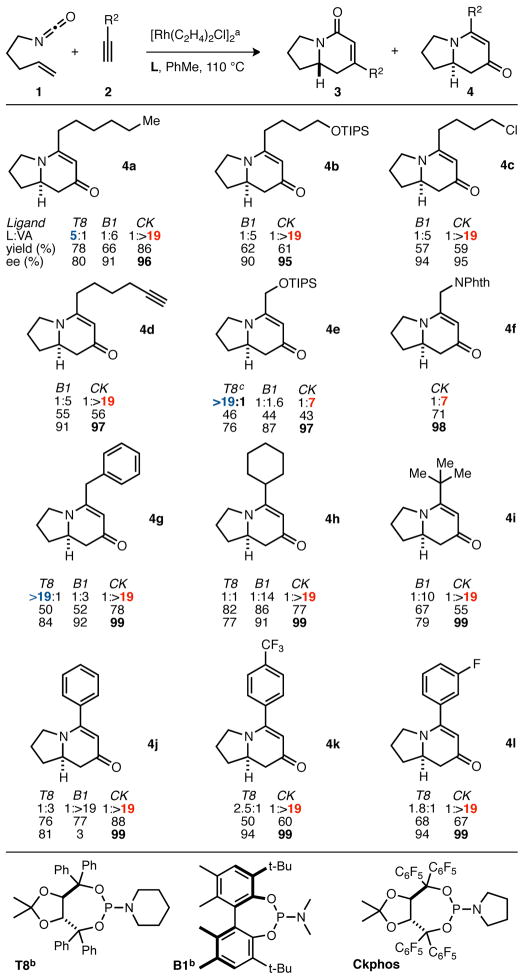
Perhaps the most striking discovery was that a simple change in aryl electronics of the Taddol phosphoramidite ligands had a profound effect on product and enantioselectivity in rhodium catalyzed [2+2+2] cycloadditions. Perfluoro Taddol phosphoramidite, CKphos, overrides substrate-based control of product selectivity for a wide range of alkynes, providing vinylogous amide indolizinones with excellent enantioselectivities (Table 2). Silyl-protected alcohols, alkyl chlorides, pendent alkynes and protected amines are well tolerated. Sterically encumbered alkynes participate well with increases in both product and enantioselectivity seen. Small, electron-deficient propargyl silyl ethers and protected amines, which were previously unselective, show good product and enantioselectivity (4e, 4f, 4g). Other problematic substrates, such as phenyl and electron-deficient aryl alkynes, suffer from either poor product selectivity (T8) or poor enantioselectivity (B1). CKphos provides excellent product and enantioselectivity for aryl substrates, even electron-deficient ones. The selective formation of 5-alkyl substituted vinylogous amide indolizinones is significant because there are approximately 200 known indolizidine alkaloids that contain 5-alkyl substituents.35
Table 2.
Substrate scope with Rh•CKphos catalyst. [a] 1, 2 (1.3 equiv), [Rh(C2H4)2Cl]2 2.5 mol %, L 5 mol % for 16 h. [b] Results previously reported, ref. 21.
|
Proposed Mechanism for Product Selectivity
The proposed mechanism for formation of lactam 3 and vinylogous amide 4 via the Rh(I) catalyzed [2+2+2] cycloaddition is illustrated in Scheme 1. From a single coordingation complex I both lactam and vinylogous amide products are formed; this depends on the direction of oxidative cyclization. The lactam product is formed from oxidative cyclization resulting in C-C bond formation to generate 5-membered rhodacycle IIa. Migratory insertion of the pendant alkene into IIa produces 7-membered rhodacycle IIIa; subsequent reductive elmination provides lactam 3. The vinylogous amide product occurs from oxidative cyclization leading to C-N bond formation, which results in 5-membered rhodacycle IIb. A strained geometry in the alkene coordination event caused by the alkene tether prohibits migratory insertion into IIb. For this reason, we propose a reversible CO extrusion-insertion occurs to access 5-membered rhodacycle IVb that the alkene inserts into forming 7-membered rhodacycle Vb. Reductive elmination of Vb provides vinylogous amide 4 and regenerates active catalyst.
Product distribution between lactam 3 and vinylogous amide 4 is determined from the direction of the irreversible oxidative cyclization of alkyne and isocyanate from coordination complex I, as previously reported.21 The phosphoramidite ligand environment organizes the coordination of alkyne and isocyanate such that smaller substituents (C–H, C=O) are on the hindered side of the rhodium square plane (Scheme 1). Oxidative cyclization with the small substituents cyclizing away from rhodium via TSa results in C–C bond formation and metallacycle IIa and leads to lactam 3. Cyclization with the large substituents moving away from rhodium via TSb results in C–N bond formation, metallacycle IIb and eventually vinylogous amide 4.
In the absence of ligand-substrate steric interactions, the LUMO coefficient of the isocyante controls the direction of oxidative cyclization in accord with the calculations of Stockis and Hoffman, who found that oxidative cyclization places the largest LUMO coefficients β to the metal.36 Electronic control of oxidative cyclization can be overriden by steric interactions during the oxidative cyclization event.37
We have found that manipulation of the aryl portion of Taddol phosphoramidites conclusively affects product selectivity with electron-rich aryls providing lactam 3 and electron-deficient giving vinylogous amide 4. X-ray analysis of Taddol phosphoramidites and CKphos reveal notable differences in Rh–P, C=C trans to phosphorus and Rh–aryl centroid distances. For example, the Rh–P difference between T2 and T4 is 0.015Å and corresponds to a 12.5:1 to 2.4:1 (L:VA) shift in product selectivity (Figure 2) while the Rh–P difference between T4 and CKphos is 0.012 Å and relates to a 2.4:1 to 1:>19 (L:VA) change in product selectivity. For the difference in Rh–P to account completely the difference would be expected to be greater than 0.015Å. A plot of Rh–P versus L:VA ratio shows a strong linear relationship with an R2 value of 0.90 (Figure 8). When CKphos is removed from this plot R2 increases to 0.95, suggesting another factor affects product selectivity other than Rh–P bond distance.38 Because there is a weak correlation between C=C trans to phosphorus and L:VA, and each of the other Rh(cod)Cl•Taddol phosphoramidites has approximately the same Rh–Aryl distance except CKphos, we suggest that the Rh–C6F5 interaction corroborated with the Rh-P interaction accounts for the increase in observed product selectivity.
Figure 8.

Graph of L:VA (3:4) ratio as a function of Rh-P bond distance. A better correlation is found when CKphos is excluded suggesting Rh-P bond distance is not the only factor responsible for product selectivity.
Rh(cod)Cl•CKphos has the shortest Rh–P and Rh–aryl centroid distances of the phosphoramidite complexes studied; one result of this is an augmented steric environment on Rh(I) and an amplification of ligand-substrate steric interactions during coordination and oxidative cyclization, favoring vinylogous amide 4 via TSb (Scheme 1). Additionaly, a shortened C=C trans to phosphorus indicates that the π-basicity of Rh(I) is decreased, a result of increased rhodium backdonation to lower lying P–O σ* and a Lewis acidic, Z-type interaction between the π* of C6F5 and dz2 on Rh(I). Both serve to lower rhodium’s HOMO, which disfavors electronic control of cyclization by increasing the energy gap between the rhodium HOMO and isocyanate LUMO. In summary, CKphos favors vinylogous amide through enhanced ligand-substrate steric interactions seen in the ground state and felt in the transition state for oxidative cyclization caused by a shortened Rh–P and a Rh–C6F5 interaction. Steric effects are amplified by a lower rhodium HOMO that lessens electronic control of oxidative cyclization.
Conclusions
Perfluorinated Taddol phosphoramidite, CKphos, is a highly selective ligand for formation of vinylogous amide cycloadducts in Rh(I) catalyzed [2+2+2] cycloadditions of alkenyl isocyanates and alkynes. CKphos overrides substrate bias of product selectivity in the cycloaddition, providing indolizinones in excellent product and enantioselectivities. Excellent selectivities are attributed to a shortened Rh–P bond and coordination of one C6F5 to rhodium via a weak Lewic acidic, Z-type interaction, making the phosphoramidite a bidentate L,Z-ligand on rhodium. Evidence for the shortened Rh–P and C6F5 coordination is provided by X-ray, NMR and DFT computation analyses. Rh(C2H4)Cl•CKphos catalyst in the [2+2+2] cycloaddition of alkenyl isocyanates and alkynes represents a rare example of a L,Z-ligand affecting selectivity in transition metal catalysis.
Supplementary Material
Acknowledgments
We thank NIGMS (GM80442) for generous support. TR thanks Amgen and Roche for unrestricted support. DMD thanks the NIH Ruth Kirschstein predoctoral fellowship for support. We thank Dr. Chris Rithner (CSU) for assisting with NMR experiments. We thank Johnson Matthey for a generous gift of metal salts.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
Notes and references
- 1.For leading references on anion-π interactions, see: Quiñonero D, Garau C, Rotger C, Frontera A, Ballester P, Costa A, Deyà PM. Angew Chem Int Ed. 2002;41(18):3389–3392. doi: 10.1002/1521-3773(20020916)41:18<3389::AID-ANIE3389>3.0.CO;2-S.de Hoog P, Gamez P, Mutikainen I, Turpeinen U, Reedijk J. Angew Chem Int Ed. 2004;43:5815–581. doi: 10.1002/anie.200460486.Gamez P, Mooibroek TJ, Teat SJ, Reedijk J. Acc Chem Res. 2007;40:435–444. doi: 10.1021/ar7000099.Schottel BL, Chifotides HT, Dunbar KR. Chem Soc Rev. 2008;37:68–83. doi: 10.1039/b614208g.Frontera A, Gamez P, Mascal M, Mooibroek T, Reedijk J. Angew Chem Int Ed. 2011;50(41):9564–9583. doi: 10.1002/anie.201100208.Giese M, Albrecht M, Krappitz T, Peters M, Gossen V, Raabe G, Valkonen A, Rissanen K. Chem Commun. 2012;48:9983–9985. doi: 10.1039/c2cc34748b.
- 2.For leading references on lone pair-π interactions, see: Alkorta I, Rozas I, Elguero J. J Org Chem. 1997;62(14):4687–4691.Gallivan JP, Dougherty DA. Org Lett. 1999;1(1):103–105. doi: 10.1021/ol990577p.Egli M, Sarkhel S. Acc Chem Res. 2007;40:197–205. doi: 10.1021/ar068174u.Nelyubina YV, Barzilovich PY, Antipin MY, Aldoshin SM, Lyssenko KA. ChemPhysChem. 2011;12:2895–2898. doi: 10.1002/cphc.201100294.Amicangelo JC, Irwin DG, Lee CJ, Romano NC, Saxton NL. J Phys Chem A. 2012 doi: 10.1021/jp307984x.
- 3.Garau C, Frontera A, Quiñonero D, Ballester P, Costa A, Deyà PM. ChemPhysChem. 2003;4:1344. doi: 10.1002/cphc.200300886. [DOI] [PubMed] [Google Scholar]
- 4.The interaction between an anion and aromatic π-system is expected to be repulsive since aromatic systems typically act as electron donors. See ref. 1d for further explanation.
- 5.(a) Belt ST, Duckett SB, Helliwell M, Perutz RN. J Chem Soc, Chem Comm. 1989;14:928–930. [Google Scholar]; (b) Lefort L, Crane TW, Farwell WD, Baruch DM, Kaeuper JA, Lachicotte RJ, Jones WD. Organometallics. 1998;17:3889–3899. [Google Scholar]
- 6.Bell TW, Helliwell M, Partridge MG, Perutz RN. Organometallics. 1992;11(5):1911–1918. [Google Scholar]
- 7.(a) Bach I, Porschke KR, Goddard R, Kopiske C, Kruger C, Rufinska A, Seevogel K. Organometallics. 1996;15:4959–4966. [Google Scholar]; (b) Johnson SA, Taylor ET, Cruise SJ. Organometallics. 2009;28:3842–3855. [Google Scholar]
- 8.Higgitt CL, Klahn A, Moore MH, Oelckers B, Partridge MG, Perutz RN. J Chem Soc, Dalton Trans. 1997;8:1269–1280. [Google Scholar]
- 9.Faggiani R, Hao N, Lock CJL, Sayer BG, McGlinchey MJ. Organometallics. 1983;2:96–100. [Google Scholar]
- 10.Barker JJ, Orpen AG, Seeley AJ, Timms PL. J Chem Soc, Dalton Trans. 1993;20:3097–3102. [Google Scholar]
- 11.Martin A, Orpen AG, Seeley AJ, Timms PL. J Chem Soc, Dalton Trans. 1994;15:2251–2255. [Google Scholar]
- 12.Koelle U, Hörnig A, Englert U. Organometallics. 1994;13:4064–4066. [Google Scholar]
- 13.For an explanation of L, X and Z-type ligands, see: Green MLH. J Organomet Chem. 1995;500:127–148.
- 14.A search of the Cambridge Crystallographic Database (CCDC) for “C6H5X[centroid]•Metal” yields 2074 structures while the same search for “C6F5X[centroid]•Metal” gives 9
- 15.For leading references on phosphino-borane, Z-type ligands see: Sircoglou M, Bontemps S, Bouhadir G, Saffon N, Miqueu K, Gu W, Mercy M, Chen C-H, Foxman BM, Maron L, Ozerov OV, Bourissou D. J Am Chem Soc. 2008;130(49):16729–16738. doi: 10.1021/ja8070072.Bontemps S, Bouhadir G, Weixing G, Mercy M, Chen C-H, Foxman BM, Maron L, Ozerov OV, Bourissou D. Angew Chem Int Ed. 2008;47:1481–1484. doi: 10.1002/anie.200705024.Moret M-E, Peters JC. Angew Chem Int Ed. 2011;50:2063–2067. doi: 10.1002/anie.201006918.
- 16.Teichert JF, Feringa BL. Angew Chem Int Ed. 2010;49:2486–2528. doi: 10.1002/anie.200904948. [DOI] [PubMed] [Google Scholar]
- 17.For reviews of metal-catalyzed [2+2+2] cycloadditions, see: Vollhardt KPC. Angew Chem Int Ed. 1984;23:539–556.Lautens M, Klute W, Tam W. Chem Rev. 1996;96:49–92. doi: 10.1021/cr950016l.Nakamura I, Yamamoto Y. Chem Rev. 2004;104:2127. doi: 10.1021/cr020095i.Gandon V, Aubert C, Malacria M. Curr Org Chem. 2005;9:1699–1712.Kotha S, Brahmachary E, Lahiri K. Eur J Org Chem. 2005;2005:4741–4767.Agenet N, Busine O, Slowinski F, Gandon V, Aubert C, Malacria M. Org React. 2007;68:1–302.Heller B, Hapke M. Chem Soc Rev. 2007;36:1085. doi: 10.1039/b607877j.Shibata TT, Tsuchikama KK. Org Biomol Chem. 2008;6:1317–1323. doi: 10.1039/b720031e.Galan BR, Rovis T. Angew Chem Int Ed. 2009;48(16):2830–2834. doi: 10.1002/anie.200804651.Shibata Y, Tanaka K. Synthesis. 2012;44:323–350.
- 18.For reviews of cycloadditions involving isocyanates, see: Varela JA, Saa C. Chem Rev. 2003;103:3787–3801. doi: 10.1021/cr030677f.Louie J. Curr Org Chem. 2005;9:605–623.Chopade PR, Louie J. Adv Synth Catal. 2006;348:2307–2327.D’Souza DM, Müller TJJ. Chem Soc Rev. 2007;36:1095–1108. doi: 10.1039/b608235c.Perreault S, Rovis T. Chem Soc Rev. 2009;38(11):3149–3159. doi: 10.1039/b816702h.Friedman RK, Oberg KM, Dalton DM, Rovis T. Pure Appl Chem. 2010;82(7):1353–1364. doi: 10.1351/PAC-CON-09-12-09.Domínguez GG, Pérez-Castells JJ. Chem Soc Rev. 2011;40:3430–3444. doi: 10.1039/c1cs15029d.Broere D, Ruijter E. Synthesis. 2012;44:2639–2672.
- 19.For our previous work in this area, see: Yu RT, Rovis T. J Am Chem Soc. 2006;128(9):2782–2783. doi: 10.1021/ja057803c.Yu RT, Rovis T. J Am Chem Soc. 2006;128(38):12370–12371. doi: 10.1021/ja064868m.Yu RT, Rovis T. J Am Chem Soc. 2008;130(11):3262–3263. doi: 10.1021/ja710065h.Lee EE, Rovis T. Org Lett. 2008;10(6):1231–1234. doi: 10.1021/ol800086s.Oinen ME, Yu RT, Rovis T. Org Lett. 2009;11(21):4934–4937. doi: 10.1021/ol9020805.Friedman RK, Rovis T. J Am Chem Soc. 2009;131(30):10775–10782. doi: 10.1021/ja903899c.Oberg KM, Lee EE, Rovis T. Tetrahedron. 2009;65(26):5056–5061. doi: 10.1016/j.tet.2009.02.021.
- 20.Yu RT, Lee EE, Malik G, Rovis T. Angew Chem Int Ed. 2009;48(13):2379–2382. doi: 10.1002/anie.200805455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dalton DM, Oberg KM, Yu RT, Lee EE, Perreault S, Oinen ME, Pease ML, Malik G, Rovis T. J Am Chem Soc. 2009;131(43):15717–15728. doi: 10.1021/ja905065j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Michael JP. Nat Prod Rep. 2001;18:520–542. doi: 10.1039/b005384h. [DOI] [PubMed] [Google Scholar]; (b) Michael JP. Nat Prod Rep. 2004;21:625–649. doi: 10.1039/b310689f. [DOI] [PubMed] [Google Scholar]; (c) Daly JW, Spande TF, Garraffo HM. J Nat Prod. 2005;68:1556–1575. doi: 10.1021/np0580560. [DOI] [PubMed] [Google Scholar]; (d) Michael JP. Nat Prod Rep. 2005;22:603–626. doi: 10.1039/b413748p. [DOI] [PubMed] [Google Scholar]; (e) Michael JP. Nat Prod Rep. 2007;24:191–222. doi: 10.1039/b509525p. [DOI] [PubMed] [Google Scholar]; (f) Michael JP. Beilstein J Org Chem. 2007;3:27. doi: 10.1186/1860-5397-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Michael JP. Nat Prod Rep. 2008;25:139–165. doi: 10.1039/b612166g. [DOI] [PubMed] [Google Scholar]; (h) Samoylenko V, Ashfaq MK, Jacob MR, Tekwani BL, Khan SI, Manly SP, Joshi VC, Walker LA, Muhammad I. J Nat Prod. 2009;72:92–98. doi: 10.1021/np800653z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Pivavarchyk M, Smith AM, Zhang Z, Zhou D, Wang X, Toyooka N, Tsuneki H, Sasaoka T, McIntosh JM, Crooks PA, Dwoskin LP. Eur J Pharmacol. 2011;658:132–139. doi: 10.1016/j.ejphar.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tolman CA. Chem Rev. 1977;77:313–348. (b) [Google Scholar]
- 24.(a) Orpen AG, Connelly NG. J Chem Soc, Chem Commun. 1985:1310–1311. [Google Scholar]; (b) Feringa BL. Acc Chem Res. 2000;33:346–353. doi: 10.1021/ar990084k. [DOI] [PubMed] [Google Scholar]
- 25.De Ridder DJA, Imhoff P. Acta Crystallogr E. 1994;50:1569–1572. [Google Scholar]
- 26.Williams JH, Cockcroft JK, Fitch AN. Angew Chem Int Ed. 1992;31:1655–1657. [Google Scholar]
- 27.(a) Dahl T. Acta Chem Scand. 1971;25:1031–1039. [Google Scholar]; (b) Dahl T. Acta Chem Scand. 1973;27:995–1003. [Google Scholar]; (c) Dahl T. Acta Chem Scand A. 1975;29:170–174. [Google Scholar]
- 28.Steyn GJJ, Roodt A, Poletaeva I, Varshavsky YS. J Organomet Chem. 1997;536–537:197–205. [Google Scholar]
- 29.Barfield M, Chakrabarti B. Chem Rev. 1969;69:757–778. [Google Scholar]
- 30.(a) Guenther H, Jikeli G. Chem Rev. 1977;77:599–637. [Google Scholar]; (b) Parr WJE, Schaefer T, Marat K. Can J Chem. 1977;55:3243–3247. [Google Scholar]
- 31.(a) Tuttle T, Gräfenstein J, Cremer D. Chem Phys Lett. 2004;394:5–13. doi: 10.1063/1.1711598. [DOI] [PubMed] [Google Scholar]; (b) Dra3ínský M, Jansa P, Bou3 P. Chem-Eur J. 2012;18:981–986. doi: 10.1002/chem.201102272. [DOI] [PubMed] [Google Scholar]
- 32.Kruck M, Muñoz MP, Bishop HL, Frost CG, Chapman CJ, Kociok-Köhn G, Butts CP, Lloyd-Jones GC. Chem-Eur J. 2008;14:7808–7812. doi: 10.1002/chem.200800825. [DOI] [PubMed] [Google Scholar]
- 33.(a) Schmidt JAR, Lobkovsky EB, Coates GW. J Am Chem Soc. 2005;127:11426–11435. doi: 10.1021/ja051874u. [DOI] [PubMed] [Google Scholar]; (b) Deng FG, Hu B, Sun W, Chen J, Xia CG. Dalton Trans. 2007;38:4262–4267. doi: 10.1039/b709800f. [DOI] [PubMed] [Google Scholar]
- 34.(a) Brammer L, McCann MC, Bullock RM, McMullan RK, Sherwood P. Organometallics. 1992;11:2339–2341. [Google Scholar]; (b) Brammer L, Mareque Rivas JC, Zhao D. Inorg Chem. 1998;37:5512–5518. doi: 10.1021/ic980317o. [DOI] [PubMed] [Google Scholar]; (c) Brammer L, Mareque Rivas JC, Spilling CD. J Organomet Chem. 2000;609:36–43. [Google Scholar]
- 35.Daly JW, Spande TF, Garraffo HM. J Nat Prod. 2005;68:1556–1575. doi: 10.1021/np0580560. [DOI] [PubMed] [Google Scholar]
- 36.Stockis A, Hoffmann R. J Am Chem Soc. 1980;102:2952–2962. [Google Scholar]
- 37.Wakatsuki Y, Nomura O, Kitaura K, Morokuma K, Yamazaki H. J Am Chem Soc. 1983;105:1907–1912. [Google Scholar]
- 38.Plots of the relationship between product selectivity and Rh–P, C=C trans to phosphorus and Rh–centroid are included in the supplementary information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.