

Abstract
Asthma affects nearly 300 million people worldwide. The majority respond to inhaled corticosteroid treatment with or without beta-adrenergic agonists. However, a subset of 5 to 10% with severe asthma do not respond optimally to these medications. Different phenotypes of asthma may explain why current therapies show limited benefits in subgroups of patients. Interleukin-13 is implicated as a central regulator in IgE synthesis, mucus hypersecretion, airway hyperresponsiveness, and fibrosis. Promising research suggests that the interleukin-13 pathway may be an important target in the treatment of the different asthma phenotypes.
Keywords: interleukin-13, asthma, airway hyper-reactivity, fibrosis, single nucleotide polymorphism
Asthma affects nearly 300 million people and one of every 250 deaths is attributed to this disease worldwide [1]. The cost of asthma hospitalizations, emergency room visits, lost school, and work days is significant. About 90% of patients respond to treatment regimens with inhaled corticosteroids with or without long-acting β2 agonists; how-ever, 5 to 10% seemingly do not. Treatment of this subgroup of patients accounts for greater than 50% of the total costs related to asthma [2,3].
Asthma is a reversible airway disease characterized by airway hyper-reactivity, inflammation and airway remodeling, and interleukin-13 (IL-13) is a recognized effector in these processes. For example, in a mouse model of asthma, IL-13 signaling results in mucin secretion, airway hyper-reactivity, fibrosis, and chitinase up-regulation [4].
Different phenotypes of asthma may explain why current therapies show limited benefits in subgroups of patients [5]. Patients with severe, poorly controlled asthma, who are insensitive to glucocorticoid treatment, highlight the need for new treatments. IL-13 signaling may be one pathway involved in the induction of corticosteroid-insensitive airway inflammation [6].
IL-13 Structure and Signaling
IL-13 has a mass of 13 kDa and folds into 4 alpha helical bundles, A, B, C, and D. It shares overlapping secondary structural features with interleukin-4 (IL-4); however, it has 25% sequence homology and is capable of IL-4 independent signaling [7,8]. IL-13 signals through a shared receptor with IL-4 via a heterodimer receptor complex comprised of IL-4 receptor alpha (IL-4Rα) and IL-13 receptor alpha 1 (IL-13Rα1) (also termed the type 2 interleukin 4 receptor). Signaling through this receptor is initiated with high affinity when IL-13 binds IL-13Rα1, leading to subsequent heterodimer formation with IL-4Rα. Crystal structure analysis shows the heterodimer receptor complex signals with different potencies in response to IL-4 versus IL-13. This suggests that the extracellular cytokine-receptor interactions modulate intracellular membrane-proximal signaling events [9]. Both intracellular subunit receptor tails interact with tyrosine kinases of the Janus family (JAK 1-3, TYK2) [10]. IL-4Rα associates with JAK1 whereas IL-13Rα1 interacts with either JAK2 or TYK2, but not JAK3. Once JAK1 is activated, the IL-4Rα tyrosine residues in the cytoplasmic domain are phosphorylated enabling the transcription factor STAT6 to dock. Once phosphorylated, 2 STAT6 molecules dimerize and translocate to the nucleus where the complex affects transcription of many IL-13 dependent genes.
IL-13 can also bind a high affinity receptor, IL-13 receptor alpha 2 (IL-13Rα2). In the mouse, this receptor has a soluble form and a membrane bound form resulting from alternative transcriptional splice variants. IL-13Rα2 in humans is primarily an intracellular rather than a membrane-bound molecule in both primary bronchial epithelial cells and fibroblasts and displays a diffuse granular cytoplasmic distribution in both cell types [11]. It is thought to act as a decoy receptor because of its short cytoplasmic tail. However, studies suggest that this receptor mediates IL-13 signaling and induces TGF-β production in both in vitro human and mouse and in vivo mouse experiments (Figure 1) [12,13].
Figure 1.
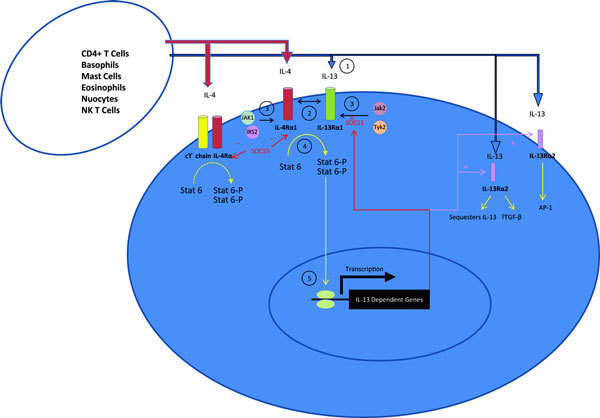
IL-13 signaling. Step 1, the binding of IL-13 to IL-13Rα1 leads to step 2, heterodimer formation with IL-4Rα1 and formation of the type 2 IL-4 receptor. Step 3 leads to Janus Kinase activation (JAK), followed by step 4, STAT6 phosphorylation, dimerization, and translocation to the nucleus. In step 5, Stat 6 heterodimers affect IL-13-dependent gene transcription. IL-13Rα2 is an IL-13 dependent gene. IL-13 can bind IL-13Rα2 that leads to sequestration of IL-13 or IL-13 signaling by TGF-β induction or AP-1 signaling. Mouse models suggest that suppressor of cytokine signaling (SOCS)1 is an IL-13 dependent gene that interacts with JAK2 to negatively regulate JAK2 association with IL-13Rα1 [14]. In a mouse model, SOCS5 targets IL-4Rα1 and impairs STAT6 signaling [15].
Extracellular regions of the IL-13Rα1 and 2 domains in mice are composed of 3 fibronectin type III domains D1, D2, and D3. The D2 and D3 domains have cytokine receptor homology modules (CRHs), which show similar structure to the class I cytokine receptor superfamily. Based on mutation studies, the IL-13Rα1 CRH module locations at leucine 319 and tyrosine 321 are important for binding to IL-13. So too, the IL-13Rα2 CRH module locations at tyrosine 207, aspartate 271, tyrosine 315, and aspartate 318 are important for binding to IL-13 [16].
Through DNA mutagenesis studies, it was shown that the alpha helices in IL-13 A, C, and D participate in interaction with IL-13Rα1 receptors [17,18]. Lysine residues 105, 106, and arginine 109 of the D-helix of IL-13 interact with IL-13Rα2. Glutamic acids at position 92 and 110 and leucine at position 104 are important for IL-13/IL-4 receptor stimulation [18].
IL-13 is produced by CD4+ T cells, NK T cells, mast cells, basophils, eosinophils, and nuocytes [17,21]. It is implicated as a central regulator in IgE synthesis, mucus hypersecretion, airway hyperresponsiveness (AHR), and fibrosis [4].
Through the use of the knockout mouse model, airway resistance, mucus production, and profibrogenic mediator induction are nearly totally dependent on IL-13Rα1, which serves as a signaling molecule for both IL-4 and IL-13 [4]. Allergen-induced TGF-β is completely dependent on IL-13Rα1.
IL-13 Role in Mucus Production
Goblet cell hyperplasia and mucus overproduction are features of asthma and chronic obstructive pulmonary disease and can lead to airway plugging, a pathologic feature of fatal asthma [21-23]. Animal models demonstrate that IL-13 induces goblet cell hyperplasia and mucus hypersecretion [24,25].
Human bronchial epithelial cells (HBEs), stimulated by IL-4 and IL-13, can also undergo changes from a fluid absorptive state to a hypersecretory state independent of goblet cell density changes [26,27].
Human in vitro studies demonstrate that IL-13 induces goblet cell hyperplasia, increases bronchial epithelial periodic acid schiff (PAS) cell staining, and MUC5AC expression. Experiments suggest that these effects are mediated by IL-13 signaling through IL-13Rα1 [28,29]. In these experiments, IL-13 also led to an increase in the soluble form of IL-13Rα2. IL-13Rα2 decreased the quantity of PAS+ cells, MUC5AC+ cells, goblet cells, and decreased both the mRNA expression and protein secretion of MUC5AC induced by IL-13 [29].
Antibody blockade of IL-13Rα2, in the presence of IL-13, on HBEs, led to increased PAS+ epithelial cells, goblet cells and MUC5AC+ cells and MUC5AC mRNA expression. This suggests that the soluble form of IL-13Rα2 may negatively modulate IL-13 signaling [29]. IL-4 was also able to increase the number of PAS+ cells, goblet cells and MUC5AC+ cells; however, IL-13Rα2 had no effects on the number of PAS+ cells, goblet cells and MUC5AC+ cells, MUC5AC mRNA expression or protein secretion induced by IL-4 [29].
In human airway epithelial cells, IL-13 is known to induce 15-Lipoxygenase-1 (15-LO1), an important enzyme in the arachidonic acid pathway, that forms stable 15-hydroxy-eicosatetraenoic acid (15-HETE) from metabolism of arachidonic acid. Human epithelial 15-LO1 expression is correlated with asthma severity [30]. IL-13 induction of 15-LO1 stimulates formation of 15-HETE that can be further metabolized through esterification to phosphatidylethanolamine (15-HETE-PE). IL-13 induction of 15-HETE-PE enhances MUC5AC expression in human airway epithelial cells [30].
IL-13 Role in Aspirin Exacerbated Respiratory Disease (AERD)
The triad of aspirin sensitivity, asthma, and nasal polyposis has been documented since at least 1922 [31]. About 10% of adults with asthma will experience a flare of their asthma accompanied by naso-ocular reactions after ingestion of aspirin or nonsteroidal anti-inflammatory drugs (NSAIDS) leading to intense eosinophilic inflammation of the nasobronchial tissues and cysteinyl-leukotriene (Cys-LTs) overproduction [33]. Polymorphisms within the IL-13 gene are associated with increased eotaxin-1 levels, increased eosinophil count, and the development of rhinosinusitis in patients with AERD [33].
IL-13 is implicated in regulating key arachidonic acid metabolic pathways including the prostaglandin pathway, the leukotriene pathway, and the lipoxin pathway (Figure 2).
Figure 2.
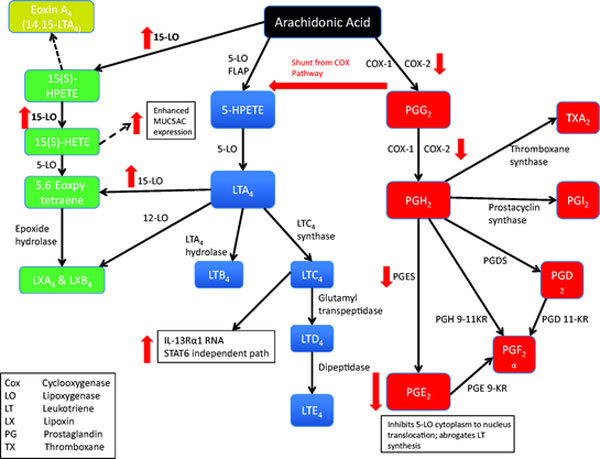
Arachidonic acid metabolic pathways and the role of IL-13. The prostaglandin pathway (red), the leukotriene pathway (blue), and the lipoxin pathway (green). PGE2 has inhibitory effects on leukotriene production including inhibition of 5-LO translocation from the cytoplasm to the nucleus and abrogation of leukotriene synthesis [24]. IL-13 down-regulates PGE2 both directly and indirectly through effects on PGES and COX-2. IL-13 also induces 15-LO that is correlated with asthma severity. IL-13 induction of 15-LO stimulates formation of 15-HETE and a metabolite of this enhances MUC5AC expression in human airway epithelial cells.
Interestingly, IL-4 increased LTC4 Synthase expression, a protein important in converting LTA4 to LTC4 that is most implicated in AERD; whereas IL-13 had no effect on LTC4 Synthase up-regulation [35]. LTC4 increased IL-13Rα1 RNA levels via a STAT6 independent pathway [36]. IL-13 appeared to have both upstream and downstream effects. In a mouse model, intranasal IL-13 induced an increase in Cys-LT receptors 1 and 2 RNA and decreased Cys-LT production at a dose of 0.035 μg [36]. At higher doses, intratracheal IL-13 induced an increase in 5-Lipoxygenase (5-LO) mRNA and increased BALF Cys-LTs (LTC4, D4, E4) within 15 minutes and LTB4 within 6 hours in BP2 mice [37].
IL-13 inhibits mPGES-1 (prostaglandin E synthase membrane-bound form) and COX-2 and up-regulates 15-prostaglandin-dehydrogenase (15-PGDH), the PGE-2 metabolizing enzyme, thereby leading to decreased PGE-2 levels [38]. PGE-2 regulates wound closure in airway epithelium, and exogenous application of PGE-2 stimulates wound closure, a vital process in airway epithelial repair after injury, maintenance of barrier function and limitation of airway hyperreactivity [39].
Lipoxins are important in inflammation resolution, acting locally at sites of inflammation. Administration of intrapleural lipoxin A4 (LXA4) to OVA sensitized rats, decreased allergen-induced pleural eosinophil and neutrophil infiltration [40]. In vitro treatment of the GM-CSF stimulated eosinophilic leukemia cell line (EoL-1) cells with LXA4 inhibited secretion of IL-8, IL-13, and eotaxin suggesting this molecule may have negative feedback implications on IL-13 mediated pathways [41].
Interleukin-13 in Pulmonary Fibrosis
Fibrosis is a major cause of morbidity and mortality with limited treatment options. Chronic inflammation induced by allergens, infections, toxins, autoimmune reactions, radiation, and mechanical injury can result in architectural lung changes, gas exchange impairment, and fibrosis with heterogeneous temporal development depending on the trigger. Experimental models identify IL-13 to be an important profibrotic mediator [25,42-50].
Transgenic mouse in vivo studies demonstrate that lung over-expression of IL-13 induces subepithelial airway fibrosis without additional stimulus [25]. Antibody blockade of IL-13 in mouse lungs challenged with Aspergillus fumigatus conidia, to simulate chronic allergic asthma, or bleomycin, to simulate toxin mediated fibrosis, led to decreased lung collagen deposition [42,43].
IL-13 induces CC-chemokines, including CCL3 (MIP-1α), CCL4 (MIP-1β), CCL20 (MIP-3 α), CCL2 (MCP-1), CCL11(eotaxin), CCL22 (MDC), and CCL6 (C10) [43,44]. Antibodies to CCL3, CCL2, and CCL6 profoundly abrogate lung remodeling responses in IL-13 transgenic mice and in bleomycin challenged mice, suggesting the pathogenesis of fibrosis has nonredundant mechanisms [43,44].
Both IL-4 and IL-13 have redundant signaling pathways with implications in pulmonary fibrosis. IL-4 and IL-13 are important in alternatively activated macrophage induction, which is believed to regulate fibrosis [45]. The use of knock-out mice deficient in IL-13R-1 implicates this receptor as the most important fibrosis signaling mechanism in fibrosis development [46]. Furthermore, several experimental models of fibrosis suggest that IL-13 is the dominant effector in toxin, [47,48] infection, [49] allergic, [42,50] and posttransplant bronchiolitis obliterans [51] models of fibrosis.
In addition to the redundancy of IL-4 and IL-13 signaling through IL-4Rα/Stat6, IL-13 may signal through a distinct pathway via the IL-13Rα2 driving TGFβ1 dependent pulmonary fibrosis [13]. IL-13 induces latent TGFβ1 production in macrophages. TGFβ1 forms homodimers that are noncovalently bound to a latency-associated protein (LAP) when stored in an inactive form in cells. IL-13 also indirectly activates TGFβ by up-regulating LAP cleavage proteins [52,53]. Additionally, IL-13 activates TGFβ through stimulation of matrix metalloproteinases and cathepsin-based proteolytic pathways [53,54]. In IL-13 transgenic mice, TGFβ1 plays a key role in subepithelial fibrosis evolution [52].
IL-13 in Airway Hyperresponsiveness
CD4+ T lymphocytes are important in the events leading to AHR in animal asthma models, via an IL-4R, STAT6 mechanism [55,56]. This process is independent of IL-4 and IL-5. Exogenous addition of IL-13 in T lymphocyte-deficient mice promotes AHR and airway inflammation [57].
IL-13 modulates Ca2+ responses in vitro in human airway smooth muscle through a STAT6/JAK-independent mechanism [58]. It signals through MAP kinases ERK and JNK effecting airway smooth muscle, leading some to hypothesize that STAT6 dependent pathways may be important in acute AHR, whereas STAT6-independent AHR and airway remodeling mechanisms may be important in chronic models [58-60].
Inflammation, remodeling, and AHR, all features of asthma, are induced by IL-13 overexpression [24,25]. Blockade of IL-13 by the soluble receptor-Fc fusion protein abrogates allergen-induced AHR [24,56].
IL-13 is overexpressed in sputum, bronchial submucosa, peripheral blood, and mast cells in the airway smooth muscle bundle in asthmatics further supporting its role in the pathogenesis of AHR [61-64]. Human IL-13 mRNA elevation in bronchial biopsy homogenates is seen in both atopic and nonatopic asthmatics versus nonasthmatics in RT-PCR experiments [65]. In situ hybridization studies of bronchial biopsy specimens from steroid responsive asthmatics versus steroid resistant asthmatics treated with one week of prednisolone demonstrated a decrease in IL-13 mRNA+ cells after treatment in the steroid responsive group that correlated with asthma clinical improvement in contrast to the steroid resistant group that maintained IL-13 mRNA+ cells and no clinical response [66]. Sputum IL-13 levels correspond to airway eosinophil percentage that is associated with airway inflammation in corticosteroid-naive subjects [67]. Furthermore, allergen challenged mild asthmatics have up-regulated IL-13 concentrations in bronchoalveolar lavage [68].
Sputum IL-13 concentration and the number of IL-13+ cells in the bronchial submucosa and airway smooth muscle bundle is increased in severe asthmatics [69]. Additionally, sputum IL-13 concentration is negatively associated with asthma control [69]. Steroid-responsive asthmatics, treated with oral steroids for 1 week, demonstrate decreased bronchial biopsy specimen IL-13 mRNA expression. However, patients clinically steroid nonresponsive, demonstrate persistent IL-13 mRNA expression [65]. This observation of persistent IL-13 sputum levels and bronchial biopsy specimen levels was observed in 2 cohorts of patients treated with intramuscular triamcinolone, to exclude noncompliance as a confounding variable [69].
IL-13 in Glucocorticoid Resistant Asthma
Glucocorticoids are anti-inflammatory medications of-ten used as maintenance therapy in acute and chronic asthma; however, some patients with severe asthma are steroid non-responsive. IL-13 remains elevated in glucocorticoid (GC) insensitive asthma, but not GC sensitive asthma [70,71].
GCs enter the cell by diffusion across the cell membrane with subsequent binding to the glucocorticoid receptor (GR), which dimerizes in the cytoplasm. The complex then translocates to the nucleus by nuclear transport proteins, whereby the complex modulates transcription of many genes pertaining to asthma through both activation and repression.
GCs have been shown to repress IL-13 transcription indirectly. In vitro experiments demonstrate that fluticasone can inhibit the IL-13 activating transcription factor GATA-3, from translocating into the nucleus through competition for the nuclear translocation protein importin α, in the T lymphocyte cell line HuT-78 and in peripheral blood mononuclear cells (PBMC) [72]. Additionally, fluticasone can induce expression of mitogen-activated protein kinase (MAPK) inhibitor phosphatase-1 (MKP-1), an inhibitor of p38 MAPK which is required for phosphorylation of GATA-3 before GATA-3 binding to importin α and transport into the cell nucleus [72].
GCs can also repress IL-13 transcription directly within the nucleus. The GC receptor has 2 isoforms designated GRα and GRβ based on alternative splice sites [73]. Both GRα and GRβ repress transcription of IL-13 [74]. GCs repress IL-13 gene transcription within the cell nucleus, in part, by competitively inhibiting activation mediated by NF-AT/AP-1 DNA binding sites in the proximal promoter [75]. GRβ mediates repressive function through the recruitment of histone deacetylase com-plexes [74]. Interestingly, higher numbers of GRβ immunoreactive cells in the bronchoalveolar lavage fluid and peripheral blood were identified in patients with GC-insensitive asthma [71]. Given that elevated levels of IL-13 are identified in GC-insensitive asthmatics, GRβ inability to down-regulate IL-13 may be responsible for this finding; however, no known studies have been performed to answer this question.
GC effects can be regulated at many steps within the cell. IL-2, IL-4, and IL-13 have all been found to be up-regulated in patients with steroid-resistant asthma [66,68,76]. Upon cell entry, endogenous GCs can be converted to an active or inactive form by the enzyme 11β-hydroxysteroid dehydrogenase (11β-HSD)-1 and 11β-HSD-2, respectively, thereby regulating the bioavailability of the GC substrate for the GR. IL-13 can up-regulate 11β-HSD-1 and this is thought to act as a negative feedback loop to curtail inflammation through the steroid anti-inflammatory effects previously mentioned [77]. It is unknown whether this pathway plays a role in steroid-resistant asthma.
In vitro, GC receptor function and binding affinity for GCs are theorized to be reduced by GR phosphorylation events within the cytoplasm. One such kinase thought to be important in GR phosphorylation is p38 MAPK, which is activated by IL-13, IL-2, and IL-4, since p38 MAPK inhibitor abrogates these effects [78].
IL-13 in Chitin Allergic Asthma
Chitin, the second most abundant biopolymer in nature, consists of N-acetyl-ß-D-glucosamine, and provides structural rigidity to fungi, crustaceans, helminthes, and insects [79]. Many chitin containing organisms have been implicated in allergy and asthma. Characterization of dust mite and cockroach anatomy, demonstrates chitin makes up a large component of the exoskeleton. Chitin can be introduced into the airway by inhalation of exoskeletons of dust mites, cockroaches, and by inhalation of fungi like Aspergillus and Alternaria.
Humans do not synthesize chitin, however, the human genome encodes for chitinases (enzymes that cleave chitin), and chitinase-like proteins (proteins that bind but do not cleave chitin) [80]. These proteins are hypothesized to counter parasitic infection [80]. The chitinase and chitinase-like proteins (C/CLP) are referred to as the 18-glycosyl hydrolase family [80]. In humans, the chitinase and chitinase-like proteins include Acid Mammalian Chitinase (AMCase), chitotriosidase, oviductin, HcGP-39/YKL-40 (chitinase 3-like-1), and YKL-39 (chitinase 3-like-2) [80].
In the mouse model, chitin challenge can induce accumulation of IL-4 expressing innate immune cells including eosinophils and basophils to the site of challenge, to the lungs or intraperitoneally, in a tissue nonspecific manner [81]. Mouse models of bronchial asthma demonstrate that AMCase is involved in the pathophysiology of asthma. AMCase has been shown to be induced by a T Helper-2 (TH2)-specific, IL-13-mediated pathway in epithelial cells and macrophages after ovalbumin sensitization and aerosol lung challenge in mice [82]. Additionally, AMCase mRNA was highly induced after A. fumigatus intranasal challenge in mice [83]. Inhibition of AMCase leads to abrogated TH2 inflammation, less bronchial hyper reactivity, and fewer eosinophils [84].
Through a series of experiments with knockout models of IL-13 signaling, including knockout mice lacking IL-13Rα1 or IL-4Rα, Munitz et al, demonstrated that key pathogenic molecules associated with asthma severity, such as chitinase, are entirely dependent on IL-13 signaling through IL-13Rα1, a component of the type II receptor.
In human lung autopsies, AMCase is expressed in exaggerated quantities in asthmatic lung epithelial cells and macrophages versus nonasthmatic lungs [82]. Interestingly, Siebold et al, reports both lower AMCase protein levels in BAL fluid and lower AMCase protein activity from asthmatic subjects with mild-to-moderate asthma compared with normal subjects suggesting a protective role of functional AMCase [85].
Investigation by a group at Yale, showed that asthma severity can be correlated with YKL-40 (chitinase 3-like-1) levels [86]. YKL-40 has been shown to be up-regulated by IL-13 [87].
IL-13 in Infectious Asthma
Infections are implicated in asthma flares [5]. Rhinovirus (RV), the virus responsible for the common cold, in children is a distinct risk factor for asthma exacerbations with an odds ratio of 6.8 [88]. A Finnish study compared cytokine responses in acute and convalescent rhinovirus versus respiratory syncytial virus (RSV) induced early wheezing in hospitalized, steroid-naive patients aged 3-35 months. Among the cytokines studied, IL-13 was the most increased in the RV group versus the RSV group with a 39-fold increase in the acute phase and 33-fold increase in the convalescent phase [89].
IL-13 is implicated in innate immune responses independent of IgE or T lymphocytes. In vitro, human mast cell activation with thymic stromal lymphopoietin (TSLP) or Toll-Like Receptor (TLR) 2 activated airway epithelial cells, in addition to IL-1, led to IL-5 and IL-13 production [90]. In mice, bone marrow derived mast cells stimulated with Staphylococcus aureus derived peptidoglycan, in a TLR2-dependent process, produced IL-13, IL-4, IL-5, IL-6, and TNF-α. Bone marrow derived mast cells stimulated with lipopolysaccharide derived from Escherichia coli, in a TLR4-dependent process, produced IL-13, TNF-α, IL-1β , and IL-6, but not IL-4 nor IL-5 [91]. In vitro HBE cells exposed to IL-4 and IL-13, mount decreased cellular antimicrobial activity and decreased mRNA levels of the antimicrobial human β-defensin 2 but not β-defensin 1 or LL-37 [92].
IL-13 Signaling Polymorphisms and Asthma
More than 10 papers report an association between single nucleotide polymorphisms (SNP) in IL-13 and the effects on asthma in adults and in children, in the context of infections, atopy, IgE levels, or risk for asthma (Table 1) [93-103]. Studies utilizing haplotype and multigene analysis have made similar associations of IL-13 SNPs and asthma (Table 2) [95,100,104].
Table 1.
IL-13 Single-Nucleotide Polymorphisms (SNP) Associated With Asthma
| Gene | SNP(s)/SNP ID# | SNP Associated Phenotype | Race Background | P Value if Available | Reference |
|---|---|---|---|---|---|
| IL13 | Arg130Gln | Atopy | Multi-center | 0.01 | ([93]) |
| Total eosinophil, total serum IgE level | Childhood asthmatics with mild to moderate asthma | 0.0442 | |||
| Arg130Gln (= G4257A) | Atopic dermatitis | Japanese aged 11-61 | 0.043 | ([94]) | |
| Arg130Gln/IL4C-589T | Atopy | Canadian children with family | 0.006 | ([95]) | |
| Haplotype | history of asthma | ||||
| Arg130Gln | Atopy | Iceland chart review | 0.67 | ([96]) | |
| Arg130Gln | Late wheeze age 6 | Dutch children | 0.007 | ([97]) | |
| Arg130Gln | Asthma | British young | 0.017 B | ([98]) | |
| No association with IgE levels | Japanese young adults | 0.026 J | |||
| Arg130Gln | Total serum IgE | German children | 0.005 | ([99]) | |
| - 1112C/T | 0.0002 | ||||
| - 1112C/T rs1800925 | Severe RSV infection asthma | German children < 2 hospitalized for RSV | 0.026 | ([100]) | |
| - 1112C/T | Asthma | Dutch | .005 asthma | ([101]) | |
| BHR | .003 BHR | ||||
| + Skin test | .03 + ST | ||||
| - 646A/G | FEV1 postbronchodilator | African American | 0.009 | ([102]) | |
| IL4Rα | Gln551Arg | Atopic asthma | Meta analysis combo adult + children | OR 1.6; P = 0.004 | ([103]) |
BHR = Bronchial hyper-reactivity; B = British cohort; J = Japanese cohort.
Table 2.
Haplotype/Multi-Gene Analysis With Inclusion of the Arg130Gln IL-13 SNP and the Associated Haplotype Phenotype
| Gene | SNPs | rs Signatures | Haplotype Associated Phenotype | Race Background | P Value if Available | Reference |
|---|---|---|---|---|---|---|
| IL13 | - 1512A/C | rs1881457 | Severe RSV infection asthma | German children < 2 years old, | 0.009 (FASTEHPLUS) | ([100]) |
| - 1112C/T | rs1800925 | hospitalized for RSV | 0.01183 (FAMHAP) | |||
| Arg130Gln | rs20541 | |||||
| IL4 + IL13 | - 589C/T | rs2243250 | Severe RSV infection asthma | German children < 2 years old, | 0.0008 (FASTEHPLUS) | ([100]) |
| - 1512A/C | rs1881457 | hospitalized for RSV | 0.0011 (FAMHAP) | |||
| - 1112C/T | rs1800925 | |||||
| Arg130Gln | rs20541 | |||||
| IL4Rα + IL13 | Ile50Val Arg130Gln | Asthma + IgE levels | Chinese children 5 to 18 years of age | 0.013 (GMDR) | ([104]) | |
| IL4 + IL13 | - 589C/T | Atopic dermatitis | Canadian children with family | 0.006 | ([95]) | |
| Arg130Gln | Atopy (Asthma not investigated) | history of asthma or atopy | 0.009 |
GMDR = Generalized multifactor dimensionality reduction method.
Polymorphisms in the IL-13 gene associated with asthma are described at Arg130Gln (rs20541) (also described as Arg164Gln, Gln110Arg, +2044 NIaIV RFLP, and Arg144Gln based on IPI, Unit-ProtKB/Swiss-Prot, EMBL CDS databases) in a coding region of the IL-13 gene [19,97,105]. This leads to substitution of a positively charged arginine with a neutral glutamine and is the only nonsynonymous substitution present in all ethnic groups thus far studied (East and West Africa, Europe, China, and South America). Studies suggest that the Arg130Gln substitution results in decreased affinity of IL-13 for IL-13Rα2, increased expression of IL-13 and phosphorylation of STAT6 [16,106]. In a predominantly White cohort of 9960 people from Washington County, MD, the GG genotype frequency was 64%, the GA genotype frequency was 29%, the AA genotype frequency was 4%, with 3% samples missing. Frequency percentages were equivalent between male and female genders [107]. Furthermore, IL-13 Arg130Gln SNP did not show variation in allele frequency by race [108].
Polymorphisms are present in IL-13 and in the cognate receptors. A longitudinal Dutch family study population with asthma, followed an average of 26.5 years, with polymorphisms in both the IL-4Rα gene (S478P) and IL-13 at the-1112 promoter region (also described as - 1111,-1055) (rs1800925) demonstrate an odds ratio 4.87 times greater for the development of asthma versus individuals without these associated genotypes (P = 0.0004) [109]. The association of the C-1112T promoter polymorphism with asthma is reported in diverse ethnic populations with the allergic phenotype. Functional analysis of the promoter SNP identified a Yin-Yang 1 binding site activator that overlapped with a STAT motif repressor. The Yin-Yang 1 binding site is hypothesized to increase IL-13 transcription as opposed to STAT6 mediated repression of IL-13 transcription in TH2 cells [110]. However, these results did not carry over to nonpolarized CD4+ T cells. The type 2 interleukin 4 receptor is dose dependent in response patterns to IL-13 and IL-4 and transcriptional dysregulation at the promoter could have profound effects on IL-13 signaling.
A single nucleotide polymorphism in the IL-13 cognate receptor, IL4Rα, based on meta-analysis, identified the Q551R (+1652 A/G, rs1801275) IL4R variant to impart a combined OR, 1.6; P = 0.004 for risk of atopic asthma. The amino acid residue 55 is located in the cytoplasmic domain in the vicinity of the STAT6 binding site and is hypothesized to affect STAT6 signal transduction.
Clinical Trials Targeting IL-13/IL-4 Signaling
Clinical trials are currently underway to investigate blockade of IL-13/IL-13 signaling in the treatment of asthma (Table 3). A phase IIa clinical trial of a recombinant human IL-4 variant, pitrakinra AER 001 (AEROVANT, by Aerovance, Berkeley, CA) that competitively binds the IL-4Rα complex and interferes with IL-4 and IL-13 actions was published in 2007 [111]. The results demonstrate a reduction in the late phase response, after allergen challenge in allergic asthmatics, measured as a decreased fall in forced expiratory volume in 1 second (FEV1) and the area under the FEV1 time curve.
Table 3.
Clinical Trials Targeting IL-13/IL-4 Signaling
| Agent | Company | Mechanism | Status |
|---|---|---|---|
| CAT-354 | Cambridge Antibody Technology, Medimmune/AstraZeneca | Anti-IL-13 mAb | Phase IIa |
| QAX576 | Novartis | Anti-IL-13 mAb | Completed phase II |
| IMA-638 (Anrukinzumab) | Wyeth | Humanized Anti-IL-13 mAb | Completed phase II |
| IMA-026 | Wyeth | Anti-IL-13 mAb | Phase II |
| MILR1444A (Lebrikizumab) | Genentech | Humanized Anti-IL-13 mAb | Phase II |
| AIR645 | Altair Therapeutics | Anti-sense targeting mRNA for IL4Rα1 | Phase IIa |
| AER001 (Pitrakinra) | Aerovance | Human IL-4 mutein inhibits IL13Rα1 or IL2Rγ) from assembly into receptor complexes w/IL4Rα | Completed phase II |
| AMG317 | Amgen | Human Anti-IL-4Rα mAb | Completed phase II |
mAb = monoclonal antibody.
Anti-IL-13 monoclonal antibody CAT-354 (NCT00873860, by Cambridge Antibody Technology, 2006 acquired by Astra Zeneca, 2007 merged with MedImmune, Gaithersburg, MD) is in phase IIa investigation to study the efficacy and safety in adults with uncontrolled moderate-to-severe, persistent asthma.
Anti-IL-13 monoclonal antibody QAX576 (NCT00532233, by Novartis, Basel Switzerland) completed phase II clinical trials recruiting patients with idiopathic pulmonary fibrosis to treatment with single dose QAX576 to measure IL-13 production.
A 2'O-methoxyethyl second-generation antisense drug targeting IL4Rα1 mRNA AIR645 (NCT00941577, by Altair Therapeutics, Inc., San Diego, CA) is recruiting subjects with mild allergic asthma and is in phase IIa investigation.
A humanized mouse monoclonal antibody against human IL-13, IMA-638 (Wyeth, 2009 acquired by Pfizer, New York City, NY), completed 2 phase II investigations in subjects with persistent asthma (NCT00425061) and in subjects with mild atopic asthma (NCT00410280). Wyeth has another monoclonal antibody against human IL-13, IMA-026 that is in phase II clinical trials after completion of phase I investigation on allergen-induced late asthma responses in subjects with mild asthma (NCT00725582).
A humanized monoclonal antibody against IL-13, Lebrikizumab (NCT00930163, by Genentech, 2009 acquired by F. Hoffmann-La Roche Ltd., Basel, Switzerland), is in phase II investigation in asthmatic adults inadequately controlled with inhaled corticosteroids.
A human monoclonal antibody against IL4Rα, AMG317 (NCT00436670, by Amgen, Thousand Oaks, CA) has completed phase II investigation on safety and efficacy in subjects with moderate to severe asthma.
Caveats in ANTI-IL-13 Therapeutics
CD4+ TH17 cells produce the cytokines IL-17A and IL-21, which when dysregulated, are implicated in inflammatory and autoimmune diseases. In a mouse model, IL-13 negatively regulates IL-17A and IL-21 production by downregulating expression of the transcription factor retinoic acidrelated γT (ROR-γt) [112]. Newcomb DC et al, speculate that blockade of IL-13 signaling could result in up-regulation of TH17 inflammation in disease states associated with altered TH17 signaling [112]. Evidence to support this hypothesis is demonstrated in double knock-out IL-4/IL-13 mice epicutaneously sensitized with OVA that resulted in increased systemic TH17 responses that affected the lungs after antigen challenge, and resulted in airway inflammation and airway hyperresponsiveness [113].
Further work is needed to define asthma phenotypes. Woodruff PG et al, reports IL-13 blockade may benefit only a subset of asthmatics [114]. In this asthma cohort, half of the asthmatics had IL-13 induced epithelial gene expression profiles in vitro and responses to inhaled corticosteroids, while the other half did not respond despite similar clinical phenotypes and the presence of atopy. Woodruff PG et al, speculate that airway barrier function defects, IL-17 induced inflammation, neutrophilic inflammation, and infection are important mechanisms that warrant further investigation.
Conclusions
IL-13 is an important cytokine in airway hyperresponsiveness, mucus production, airway remodeling, subepithelial airway fibrosis, infectious asthma, allergic asthma, and aspirin exacerbated respiratory disease. Research studies have implicated this cytokine in many of the pathologic events in different asthma phenotypes. As with many biologic events, often there are countervailing regulatory signals for activation and inactivation. It will be important to further understand both the mechanisms by which IL-13 is activated and inactivated and how these signaling pathways play into the bigger picture of cytokine networks, gene-gene interactions, and ultimate pathology.
Search Strategy and Selection Criteria
This Seminar is based on PubMed, ISI Web of Knowledge search for articles with "interleukin-13" in conjunction with "asthma," "airway hyperreactivity," "fibrosis," "single nucleotide polymorphism," "chitin," "aspirin exacerbated respiratory disease," "aspirin," "signaling," "mucus," glucocorticoid," "infection" in combination with "treatment" keywords, and other review articles and references from those review articles deemed relevant. Clinical trials were identified by using the keyword "interleukin-13" in clinicaltrials.gov. We prioritized more recent publications; however, there was no restriction on language or date of publication.
Notes
1. These amino acids are also enumerated as Lysine 137, 138; Arginine 141, Glutamic acids 124 and 142 in the International Protein Index (IPI), UniProtKB/Swiss-Prot, EMBL CDS protein sequence repositories [19].
End Note
Contributors: ELR contributed to design, content, literature review, and writing. RFL contributed to structure, editing, and content.
Acknowledgements
ELR acknowledges Thomas Wynn, PhD, at the National Institutes of Health/National Institutes of Allergy and Infectious Diseases and Daniel Hamilos, MD, at Harvard Medical School/Massachusetts General Hospital for their generous review of this paper and content suggestions.
References
- Masoli M, Fabian D, Holt S, Beasley R. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy. 2004;4:469–478. doi: 10.1111/j.1398-9995.2004.00526.x. [DOI] [PubMed] [Google Scholar]
- Bateman ED, Boushey HA, Bousquet J, Busse WW, Clark TJH. et al. Can guideline-defined asthma control be achieved? The Gaining Optimal Asthma Control Study. Am J Respir Crit Care Med. 2004;4:836–844. doi: 10.1164/rccm.200401-033OC. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Jonasson B, Klim JB. The costs of asthma. Eur Respir J. 1996;4:636–642. doi: 10.1183/09031936.96.09040636. [DOI] [PubMed] [Google Scholar]
- Munitz A, Brandt EB, Mingler M, Finkelman FD, Rothenberg. Distinct roles for IL-13 and IL-4 via IL-13 receptor α1 and the type II IL-4 receptor in asthma pathogenesis. Proceedings of the National Academy of Sciences. 2008;4:7240–7245. doi: 10.1073/pnas.0802465105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel SE. Asthma: defining of the persistent adult phenotypes. Lancet. 2006;4:804–813. doi: 10.1016/S0140-6736(06)69290-8. [DOI] [PubMed] [Google Scholar]
- Therien AG, Bernier V, Weicker S, Tawa P, Falgueyret JP. et al. Adenovirus IL-13-induced airway disease in mice: a corticosteroid-resistant model of severe asthma. Am J Respir Cell Mol Biol. 2008;4:26–35. doi: 10.1165/rcmb.2007-0240OC. [DOI] [PubMed] [Google Scholar]
- Moy FJ, Diblasio E, Wilhelm J, Powers R. Solution structure of human IL-13 and implication for receptor binding. J Mol Biol. 2001;4:219–230. doi: 10.1006/jmbi.2001.4764. [DOI] [PubMed] [Google Scholar]
- Zurawski SM, Vega F, Juyghe B, Zurawski G. Receptors for interleukin-13 and interleukin-4 are complex and share a novel component that functions in signal transduction. EMBO. 1993;4:2663–2670. doi: 10.1002/j.1460-2075.1993.tb05927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPorte SL, Juo ZS, Vaclavikova J, Colf LA, Qi X. et al. Molecular and structural receptor pleiotropy in the interleukin-4/13 system. Cell. 2008;4:259–272. doi: 10.1016/j.cell.2007.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly-Welch AE, Hanson EM, Boothby MR, Keegan AD. Interleukin-4 and interleukin-13 signaling connection maps. Science. 2003;4:1527–1528. doi: 10.1126/science.1085458. [DOI] [PubMed] [Google Scholar]
- Konstantinidis AK, Puddicombe SM, Mochizuki A, Sheth PD, Yang IA. et al. Cellular localization of interleukin 13 receptor α2 in human primary bronchial epithelial cells and fibroblasts. J Invest Allergol Clin Immunol. 2008;4(3):174–180. [PubMed] [Google Scholar]
- Fichtner-Feigl S, Fuss IJ, Young CA, Watanabe T, Geissler EK. et al. Introduction of IL-13 triggers TGF-beta 1-dependent tissue fibrosis in chronic 2,4,6-trinitrobenzene sulfonic acid colitis. J Immunol. 2007;4:5859–5870. doi: 10.4049/jimmunol.178.9.5859. [DOI] [PubMed] [Google Scholar]
- Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13 alpha 2 receptor is involved in induction of TGF-beta 1 production and fibrosis. Nat Med. 2006;4:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- Lee C, Kolesnik TB, Caminschi I, Chakravorty A, Carter W. et al. Suppressor of cytokine signaling (SOCS1) is a physiological regulator of the asthma response. Clin Exp Allergy. 2009;4:897–907. doi: 10.1111/j.1365-2222.2009.03217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki Y, Hayashi K, Matsumoto A, Seki N, Tsukada J. et al. Expression of the suppressor of cytokine signaling-5 (SOCS5) negatively regulates IL-4 dependent STAT6 activation and Th2 differentiation. Proc Natl Acad Sci USA. 2002;4:13003–13008. doi: 10.1073/pnas.202477099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima K, Sato K, Tanaka G, Kanaji S, Terada T. et al. Characterization of the interaction between interleukin-13 and interleukin-13 receptors. J Biol Chem. 2005;4:24915–24922. doi: 10.1074/jbc.M502571200. [DOI] [PubMed] [Google Scholar]
- Deng WP, Nickloff JA. Site-directed mutagenesis of virtually any plasmid by eliminating a unique site. Analytical Biochem. 1992;4:81–88. doi: 10.1016/0003-2697(92)90280-K. [DOI] [PubMed] [Google Scholar]
- Madhankumar AB, Mintz A, Debinski W. Alanine-scanning mutagenesis of α-helix D segment of interleukin-13 reveals new functionally important residues of the cytokine. J Biol Chem. 2002;4:43194–43205. doi: 10.1074/jbc.M205047200. [DOI] [PubMed] [Google Scholar]
- Jain E, Bairoch A, Duvaud S, Phan I, Redaschi N. et al. Infrastructure for the life sciences: design and implementation of the UniProt website. BMC Bioinformatics. 2009;4:136. doi: 10.1186/1471-2105-10-136. http://www.uniprot.org/uniparc/UPI000016ADBD Accessed December 1, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M. et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;4:1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy JV. Goblet cell and mucin gene abnormalities in asthma. Chest. 2002;4(Suppl):320S–326S. doi: 10.1378/chest.122.6_suppl.320s. [DOI] [PubMed] [Google Scholar]
- Shimura S, Andoh Y, Haraguchi M, Shirato K. Continuity of airway goblet cells and intraluminal mucus in the airways of patients with bronchial asthma. Eur Respir J. 1996;4:1395–1401. doi: 10.1183/09031936.96.09071395. [DOI] [PubMed] [Google Scholar]
- Aikawa T, Shimura S, Sasaki H, Ebina M, Takashima T. Marked goblet cell hyperplasia with mucus accumulation in the airways of patients who died of severe acute asthma attack. Chest. 1992;4:916–921. doi: 10.1378/chest.101.4.916. [DOI] [PubMed] [Google Scholar]
- Willis-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY. et al. Interleukin-13: central mediator of allergic asthma. Science. 1998;4:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP. et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;4:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danahay H, Atherton H, Jones G, Bridges RJ, Poll CT. Interleukin-13 induces a hypersecretory ion transport phenotype in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2002;4:L226–L236. doi: 10.1152/ajplung.00311.2001. [DOI] [PubMed] [Google Scholar]
- Galietta LJ, Pagesy P, Folli C, Caci E, Romio L. et al. IL-4 is a potent modulator of ion transport in the human bronchial epithelium in vitro. J Immunol. 2002;4:839–845. doi: 10.4049/jimmunol.168.2.839. [DOI] [PubMed] [Google Scholar]
- Yasuo M, Fujimoto K, Tanabe T, Yaegashi H, Tsushima K. et al. The relationship between calcium-activated chloride-channel 1 and MUC5AC in goblet cell hyperplasia induced by interleukin-13 in human bronchial epithelial cells. Respiration. 2006;4:347–359. doi: 10.1159/000091391. [DOI] [PubMed] [Google Scholar]
- Tanabe T, Fujimoto K, Yasuo M, Tsushima K, Yoshida K, Ise H, Yamaya M. Modulation of mucus production by interleukin-13 receptor a2 in the human airway epithelium. Clin Exp Allergy. 2007;4:122–134. doi: 10.1111/j.1365-2222.2007.02871.x. [DOI] [PubMed] [Google Scholar]
- Zhao J, Maskrey B, Balzar S, Chibana K, Mustovich A. et al. Interleukin-13-induced MUC5AC is regulated by 15-lipoxygenase 1 pathway in human bronchial epithelial cells. Am J Respir Crit Care Med. 2009;4:782–790. doi: 10.1164/rccm.200811-1744OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widal MF, Abrami P, Lenmoyez J. Anaphylaxie et idiosyncrasie. Presse Med. 1922;4:189–192. [Google Scholar]
- Szczeklik A, Stevenson DD. Aspirin-induced asthma: advances in pathogenesis and management. J Allergy Clin Immunol. 1999;4:5–13. doi: 10.1016/S0091-6749(99)70106-5. [DOI] [PubMed] [Google Scholar]
- Palikhe NS, Kim SH, Cho BY, Choi GS, Kim JH, Ye YM, Park HS. IL-13 gene polymorphisms are associated with rhinosinusitis and eosinophilic inflammation in aspirin intolerant asthma. Allergy Asthma Immunol Res. 2010;4:134–140. doi: 10.4168/aair.2010.2.2.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamand N, Surette ME, Picard S, Bourgoin S, Borgeat P. Cyclic AMP-mediated inhibition of 5-Lipoxygenase translocation and leukotriene biosynthesis in human neutrophils. Molec Pharmacol. 2002;4:250–256. doi: 10.1124/mol.62.2.250. [DOI] [PubMed] [Google Scholar]
- Hsieh FH, Lam BK, Penrose JF, Austen KF, Boyce JA. T helper cell type 2 cytokines coordinately regulate immunoglobulin E-dependent cysteinyl leukotriene production by human cord blood-derived mast cells: profound induction of leukotriene C4 synthase expression by interleukin-4. J Exp Med. 2001;4(1):123–133. doi: 10.1084/jem.193.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez J, Young HWJ, Corry DB, Lieberman MW. Interactions between leukotriene C4 and interleukin 13 signaling pathways in a mouse model of airway disease. Arch Pathol Lab Med. 2006;4:440–446. doi: 10.5858/2006-130-440-IBLCAI. [DOI] [PubMed] [Google Scholar]
- Vargaftig BB, Singer M. Leukotrienes mediate murine bronchopulmonary hyperreactivity, inflammation, and part of mucosal metaplasia and tissue injury induced by recombinant murine interleukin-13. Am J Respir Cell Mol Biol. 2003;4:410–419. doi: 10.1165/rcmb.2002-0032OC. [DOI] [PubMed] [Google Scholar]
- Trudeau J, Hu H, Chibana K, Chu HW, Westcott JY, Wenzel SE. Selective downregulation of prostaglandin E2-related pathways by the TH2 cytokine IL-13. J Allergy Clin Immunol. 2006. pp. 1446–1454. [DOI] [PubMed]
- Savla U, Appel HJ, Sporn PH, Waters CM. Prostaglandin E2 regulates wound closure in airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2001;4:L421–L431. doi: 10.1152/ajplung.2001.280.3.L421. [DOI] [PubMed] [Google Scholar]
- Bandeira-Melo C, Bozza PT, Diaz BL, Cordeiro RSB, Jose PJ, Martins MA, Serhan CN. Cutting edge: lipoxin (LX) A4 and aspirin-triggered 15-epi-LXA4 block allergen-induced eosinophil trafficking. J Immunol. 2000;4:2267–2271. doi: 10.4049/jimmunol.164.5.2267. [DOI] [PubMed] [Google Scholar]
- Starosta V, Pazdrak K, Boldogh I, Svider T, Kurosky A. Lipoxin A4 counterregulates GM-CSF signaling in eosinophilic granulocytes. J Immunol. 2008;4:8688–8699. doi: 10.4049/jimmunol.181.12.8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blease K, Jakubzick C, Westwick J, Lukacs N, Kunkel SL, Hogaboam CM. Therapeutic effect of IL-13 immunoneutralization during chronic experimental fungal asthma. J Immunol. 2001;4(8):5219–5224. doi: 10.4049/jimmunol.166.8.5219. [DOI] [PubMed] [Google Scholar]
- Belperio JA, Dy M, Burdick MD, Xue YY, Li K, Elias JA. et al. Interaction of IL-13 and C10 in the pathogenesis of bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2002;4(4):419–427. doi: 10.1165/rcmb.2002-0009OC. [DOI] [PubMed] [Google Scholar]
- Ma B, Zhu Z, Homer RJ, Gerard C, Strieter R, Elias JA. The C10/ CCL6 chemokine and CCR1 play critical roles in the pathogenesis of IL-13-induced inflammation and remodeling. J Immunol. 2004;4(3):1872–1881. doi: 10.4049/jimmunol.172.3.1872. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Fibrotic disease and the TH1/TH2 paradigm. Nat Rev Immunol. 2004;4:583–594. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam TR, Pesce JT, Sheikh F, Cheever AW, Mentink-Kane MM. et al. Unique functions of the type II interleukin 4 receptor identified in mice lacking the interleukin 13 receptor -1 chain. Nature Immunol. 2008;4(1):25–33. doi: 10.1038/ni1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliprantis AO, Wang J, Fathman JW, Lemaire R, Dorfman DM. et al. Transcription factor T-bet regulates skin sclerosis through its function in innate immunity and via IL-13. Proc Natl Acad Sci USA. 2007;4(8):2827–2830. doi: 10.1073/pnas.0700021104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodsick JE, Toews GB, Jakubzick C, Hogaboam C, Moore TA. et al. Protection from fluorescein isothiocyanate-induced fibrosis in IL-13-deficient, but not IL-4-deficient, mice results from impaired collagen synthesis by fibroblasts. J Immunol. 2004;4(7):4068–4076. doi: 10.4049/jimmunol.172.7.4068. [DOI] [PubMed] [Google Scholar]
- Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest. 1999;4(6):777–785. doi: 10.1172/JCI7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RK, Herbert C, Yang M, Koskinen AM, McKenzie AN, Foster PS. Role of interleukin-13 in eosinophil accumulation and airway remodeling in a mouse model of chronic asthma. Clin Exp Allergy. 2002;4(7):1104–1111. doi: 10.1046/j.1365-2222.2002.01420.x. [DOI] [PubMed] [Google Scholar]
- Keane MP, Gomperts BN, Weigt S, Xue YY, Burdick MD. et al. IL-13 is pivotal in the fibro-obliterative process of bronchiolitis obliterans syndrome. J Immunol. 2007;4(1):511–519. doi: 10.4049/jimmunol.178.1.511. [DOI] [PubMed] [Google Scholar]
- Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X. et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating trans-forming growth factor-B1. J Exp Med. 2001;4(6):809–821. doi: 10.1084/jem.194.6.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanone S, Zheng T, Zhu Z, Liu W, Lee CG. et al. Overlapping and enzyme-specific contributions of matrix metalloproteinase-9 and -12 in IL-13-induced inflammation and remodeling. J Clin Invest. 2002;4(4):463–474. doi: 10.1172/JCI14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant B, Chiaramonte MG, Cheever AW, Soloway PD, Wynn TA. Regulation of hepatic fibrosis and extracellular matrix genes by the Th response: new insight into the role of tissue inhibitors of matrix metalloproteinases. J Immunol. 2001;4(12):7017–7026. doi: 10.4049/jimmunol.167.12.7017. [DOI] [PubMed] [Google Scholar]
- Hogan SP, Matthaei KI, Young JM, Koskinen A, Young IG. et al. A novel T cell-regulated mechanism modulating allergen-induced airways hyperreactivity in BALB/c mice independently of IL-4 and IL-5. J Immunol. 1998;4:1501–1509. [PubMed] [Google Scholar]
- Venkayya R, Lam M, Willkom M, Grunig G, Corry DB. et al. The TH2 lymphocyte products IL-4 and IL-13 rapidly induce airway hyperresponsiveness through direct effects on resident airway cells. Am J Respir Cell Mol Biol. 2002;4:202–208. doi: 10.1165/ajrcmb.26.2.4600. [DOI] [PubMed] [Google Scholar]
- Grunig G, Warnock M, Wakil AE, Venkayya R, Brombacher F. et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;4:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynihan B, Tolloczko, Michoud MC, Tamaoka M, Ferraro P, Martin JG. MAP kinases mediate interleukin-13 effects on calcium signaling in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2008;4:L171–L177. doi: 10.1152/ajplung.00457.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Hogan SP, Henry PJ, Matthaei KI, McKenzie AN. et al. Interleukin-13 mediates airways hyperreactivity through the IL-4 receptor alpha chain and STAT-6 independently of IL-5 and eotaxin. Am J Respir Cell Mol Biol. 2001;4:522–530. doi: 10.1165/ajrcmb.25.4.4620. [DOI] [PubMed] [Google Scholar]
- Foster PS, Webb DC, Yang M, Herbert C, Kumar RK. Dissociation of T helper type 2 cytokine-dependent airway lesions from signal transducer and activator of transcription 6 signaling in experimental chronic asthma. Clin Exp Allergy. 2003;4:688–695. doi: 10.1046/j.1365-2222.2003.01647.x. [DOI] [PubMed] [Google Scholar]
- Berry MA, Parker D, Neale N, Woodman L, Morgan A. et al. Sputum and bronchial submucosal IL-13 expression in asthma and eosinophilic bronchitis. J Allergy Clin Immunol. 2004;4:1106–1109. doi: 10.1016/j.jaci.2004.08.032. [DOI] [PubMed] [Google Scholar]
- Komai-Koma M, McKay A, Thomson L, McSharry C, Chalmers GW. et al. Immuno-regulatory cytokines in asthma: IL-15 and IL-13 in induced sputum. Clin Exp Allergy. 2001;4:1441–1448. doi: 10.1046/j.1365-2222.2001.01174.x. [DOI] [PubMed] [Google Scholar]
- Park SW, Jangm HK, An MH, Min JW, Jang AS. et al. Interleukin-13 and interleukin-5 in induced sputum of eosinophilic bronchitis: comparison with asthma. Chest. 2005;4:1921–1927. doi: 10.1378/chest.128.4.1921. [DOI] [PubMed] [Google Scholar]
- Brightling CE, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID, Bradding P. Interleukin-4 and -13 expression is co-localized to mast cells within the airway smooth muscle in asthma. Clin Exp Allergy. 2003;4:1711–1716. doi: 10.1111/j.1365-2222.2003.01827.x. [DOI] [PubMed] [Google Scholar]
- Humbert M, Durham SR, Kimmitt P, Powell N, Assoufi B. et al. Elevated expression of messenger ribonucleic acid encoding IL-13 in the bronchial mucosa of atopic and nonatopic subjects with asthma. J Allergy Clin Immunol. 1997;4:657–665. doi: 10.1016/S0091-6749(97)70028-9. [DOI] [PubMed] [Google Scholar]
- Naseer T, Minshall EM, Leung DY, Laberge S, Ernst S. et al. Expression of IL-12 and IL-13 mRNA in asthma and their modulation in response to steroid therapy. Am J Respir Crit Car Med. 1997;4:845–851. doi: 10.1164/ajrccm.155.3.9117015. [DOI] [PubMed] [Google Scholar]
- Truyen E, Coteur L, Dilissen E, Overbergh L, Dupont LJ. et al. Evaluation of airway inflammation by quantitative TH1/ TH2 cytokine mRNA measurement in sputum of asthma patients. Thorax. 2006;4:202–208. doi: 10.1136/thx.2005.052399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SK, Xiao HQ, Kleine-Tebbe J, Paciotti G, Marsh DG. et al. IL-13 expression at the sites of allergen challenge in patients with asthma. J Immunol. 1995;4:2688–2694. [PubMed] [Google Scholar]
- Saha SK, Berry MA, Parker D, Siddiqui S, Morgan A. et al. Increased sputum and bronchial biopsy IL-13 expression in severe asthma. J Allergy Clin Immunol. 2008;4:685–691. doi: 10.1016/j.jaci.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Chung KF, Adcock IM. Update on glucocorticoid action and resistance. J Allergy Clin Immunol. 2006;4:522–543. doi: 10.1016/j.jaci.2006.01.032. [DOI] [PubMed] [Google Scholar]
- Hamid QA, Wenzel SE, Hauk PJ, Tsicopoulos A, Wallaert B. et al. Increased glucocorticoid receptor β in airway cells of glucocorticoidinsensitive asthma. Am J Respir Crit Care Med. 1999;4:1600–1604. doi: 10.1164/ajrccm.159.5.9804131. [DOI] [PubMed] [Google Scholar]
- Maneechotesuwan K, Yao X, Ito K, Jazrawi E, Usmani OS, Adcock IM, Barnes PJ. Suppression of GATA-3 nuclear import and phosphorylation: a novel mechanism of corticosteroid action in allergic disease. PLOS Med. 2009;4(5):1–12. doi: 10.1371/journal.pmed.1000076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encio IJ, Detera-Wadleigh SD. The genomic structure of the human glucocorticoid receptor. J Biol Chem. 1991;4:7182–7188. [PubMed] [Google Scholar]
- Kelly A, Bowen H, Jee YK, Mahfiche N, Soh C. et al. The glucocorticoid receptor b isoform can mediate transcriptional repression by recruiting histone deacetylases. J Allergy Clin Immunol. 2008;4:203–208. doi: 10.1016/j.jaci.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Jee YK, Gilmour J, Kelly A, Bowen H, Richards D. et al. Repression of interleukin-5 transcription by the glucocorticoid receptor targets GATA-3 signaling and involves histone deacetylase recruitment. J Biol Chem. 2005;4:23243–23250. doi: 10.1074/jbc.M503659200. [DOI] [PubMed] [Google Scholar]
- Leung DY, Martin RJ, Szefler SJ, Sher ER, Ying S, Kay AB, Hamid Q. Dysregulation of interleukin 4, interleukin 5, and interferon gamma gene expression in steroid-resistant asthma. J Exp Med. 1995;4:33–40. doi: 10.1084/jem.181.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu A, Fatma S, Cao J, Grunstein JS, Nino G, Grumbach Y, Grunstein MM. TH2 cytokine-induced upregulation of 11b-hydroxysteroid dehy-drogenase-1 facilitates glucocorticoid suppression of proasthmatic airway smooth muscle function. Am J Physiol Lung Cell Mol Physiol. 2009;4:L790–L803. doi: 10.1152/ajplung.90572.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irusen E, Matthews JG, Takahashi A, Barnes PJ, Chung KF, Adcock IM. p38 mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: role in steroid-insensitive asthma. J Allergy Clin Immunol. 2002;4:649–657. doi: 10.1067/mai.2002.122465. [DOI] [PubMed] [Google Scholar]
- Boot RG, Blommaart EFC, Swart E, Ghauharali-van der Vlugt K, Bijl N. et al. Identification of a novel acidic mammalian chitinase distinct from chitotriosidase. J Biol Chem. 2001;4:6770–6778. doi: 10.1074/jbc.M009886200. [DOI] [PubMed] [Google Scholar]
- Lee CG, Da Silva CA, Lee J-Y, Hartl D, Elias JA. Chitin regulation of immune responses: an old molecule with new roles. Current Opinion in Immunology. 2008;4:684–689. doi: 10.1016/j.coi.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM. Chitin induces accumulation in tissues of innate immune cells associated with allergy. Nature. 2007;4:92–96. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A, Zheng T, Homer RJ, Kim YK, Chen NY. et al. Acidic mammalian chitinase in asthmatic TH2 inflammation and IL-13 pathway activation. Science. 2004;4:1678–1682. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- Zimmermann N, Mishra A, King NE, Fulkerson PC, Doepker MP. et al. Transcript signatures in experimental asthma: identification of STAT6-dependent and-independent pathways. J Immunol. 2004;4:1815–1824. doi: 10.4049/jimmunol.172.3.1815. [DOI] [PubMed] [Google Scholar]
- Bierbaum S, Nickel R, Koch A, Lau S, Deichmann KA. et al. Polymorphisms and haplotypes of acid mammalian chitinase are associated with bronchial asthma. Am J Respir Crit Care Med. 2005;4:1505–1509. doi: 10.1164/rccm.200506-890OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibold MA, Donnelly S, Solon M, Innes A, Woodruff PC. et al. Chitotriosidase is the primary active chitinase in the human lung and is modulated by genotype and smoking habit. J Allergy Clin Immunol. 2008;4:944–950. doi: 10.1016/j.jaci.2008.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chupp GL, Lee CG, Jarjour N, Shim YM, Holm CT. et al. A chitinase-like protein in the lung and circulation of patients with severe asthma. N Engl J Med. 2007;4:2016–2027. doi: 10.1056/NEJMoa073600. [DOI] [PubMed] [Google Scholar]
- Stevens AL. Mechanical injury and inflammatory cytokines affect cartilage integrity and tissue homeostasis: a mass spectrometric analysis of proteins with relevance to arthritis. http://dspace.mit.edu/ handle/1721.1/37956 Accessed February 22, 2010.
- Khetsuriani N, Kazerouni NN, Erdman DD, Lu X, Redd SC, Anderson LJ, Teague WG. Prevalence of viral respiratory tract infections in children with asthma. J Allergy Clin Immunol. 2007;4:314–321. doi: 10.1016/j.jaci.2006.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jartti T, Paul-Anttila M, Lehtinen P, Parikka V, Vuorinen T, Simell O, Ruuskanen O. Systemic T-helper and T-regulatory cell type cytokine responses in rhinovirus vs. respiratory syncytial virus induced early wheezing: an observational study. Respir Res. 2009;4(1):85–95. doi: 10.1186/1465-9921-10-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allakhverdi Z, Comeau MR, Jessup HK, Yoon BR, Brewer A. et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med. 2007;4:253–258. doi: 10.1084/jem.20062211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supajatura V, Ushio H, Nakao A, Akira S, Okumura K, Ra C, Ogawa H. Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. J Clin Invest. 2002;4:1351–1359. doi: 10.1172/JCI14704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisswenger C, Kandler K, Hess C, Garn H, Felgentreff K. et al. Allergic airway inflammation inhibits pulmonary antibacterial host defense. J Immunol. 2006;4:1833–1837. doi: 10.4049/jimmunol.177.3.1833. [DOI] [PubMed] [Google Scholar]
- DeMeo DL, Lange C, Silverman EK, Senter JM, Drazen JM. et al. Univariate and multivariate family-based association analysis of the IL-13 ARG130GLN polymorphism in the Childhood Asthma Management Program. Genetic Epidemiology. 2002;4:335–348. doi: 10.1002/gepi.10182. [DOI] [PubMed] [Google Scholar]
- Tsunemi Y, Saeki H, Nakamura K, Sekiya T, Hirai K. et al. Interleukin-13 gene polymorphism G4257A is associated with atopic dermatitis in Japanese patients. J Dermatol Sci. 2002;4:100–107. doi: 10.1016/S0923-1811(02)00065-8. [DOI] [PubMed] [Google Scholar]
- He JQ, Chan-Yeung M, Becker AB, Dimich-Ward H, Ferguson AC. et al. Genetic variants of the IL-13 and IL-4 genes and atopic diseases in at-risk children. Genes and Immunity. 2003;4:385–389. doi: 10.1038/sj.gene.6363985. [DOI] [PubMed] [Google Scholar]
- Hakonarson H, Bjornsdottir US, Ostermann E, Arnason T, Adalsteinsdottir AE. et al. Allelic frequencies and patterns of single-nucleotide polymorphisms in candidate genes for asthma and atopy in Iceland. Am J Respir Crit Care Med. 2001;4:2036–2044. doi: 10.1164/ajrccm.164.11.2101086. [DOI] [PubMed] [Google Scholar]
- Ermers MJJ, Hoebee B, Hodemaekers HM, Kimman TG, Kimpen JLL, Bont L. IL-13 genetic polymorphism identifies children with late wheezing after respiratory syncytial virus infection. J Allergy Clin Immunol. 2007;4:1086–1091. doi: 10.1016/j.jaci.2006.12.655. [DOI] [PubMed] [Google Scholar]
- Heinzmann A, Mao XQ, Akaiwa M, Kreomer RT, Gao PS. et al. Genetic variants of IL-13 signalling and human asthma and atopy. Human Molec Genetics. 2000;4:549–559. doi: 10.1093/hmg/9.4.549. [DOI] [PubMed] [Google Scholar]
- Liu X, Beaty TH, Deindl P, Huang SK, Lau S. et al. Associations between total serum IgE levels and the 6 potentially functional variants within the genes IL-4, IL-13, and IL-4RA in German children: The German Multicenter Atopy Study. J Allergy Clin Immunol. 2003;4:382–388. doi: 10.1067/mai.2003.1635. [DOI] [PubMed] [Google Scholar]
- Puthothu B, Krueger M, Forster J, Heinzmann A. Association between severe respiratory syncytial virus infection and IL-13/IL-4 haplotypes. J Infect Diseases. 2006;4:438–441. doi: 10.1086/499316. [DOI] [PubMed] [Google Scholar]
- Howard TD, Whittaker PA, Zaiman AL, Koppelman GH, Xu JF. et al. Identification and association of polymorphisms in the interleukin-13 gene with asthma and atopy in a Dutch population. Am J Respir Cell Molec Biol. 2001;4:377–384. doi: 10.1165/ajrcmb.25.3.4483. [DOI] [PubMed] [Google Scholar]
- Battle NC, Choudhry S, Tsai HJ, Eng C, Kumar G. et al. Ethnicity-specific gene-gene interaction between IL-13 and IL-4R alpha among African American with asthma. Am J Respir Crit Care Med. 2007;4:881–887. doi: 10.1164/rccm.200607-992OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loza MJ, Chang BL. Association between Q551R IL-4R genetic variants and atopic asthma risk demonstrated by meta-analysis. J Allergy Clin Immunol. 2007;4:578–585. doi: 10.1016/j.jaci.2007.05.019. [DOI] [PubMed] [Google Scholar]
- Chan HIS, Tang NLS, Leung TF, Huang W, Lam YYO. et al. Study of gene-gene interactions for endophenotypic quantitative traits in Chinese asthmatic children. Allergy. 2008;4:1031–1039. doi: 10.1111/j.1398-9995.2008.01639.x. [DOI] [PubMed] [Google Scholar]
- Tarazona-Santos E, Tishkoff SA. Divergent patterns of linkage disequilibrium and haplotype structure across global populations at the interleukin-13 (IL13) locus. Genes Immun. 2005;4:53–65. doi: 10.1038/sj.gene.6364149. [DOI] [PubMed] [Google Scholar]
- Vladich FD, Brazile SM, Stern D, Peck ML, Ghittoni R, Vercelli D. IL-13 R130Q, a common variant associated with allergy and asthma, enhances effector mechanisms essential for human allergic inflammation. J Clin Invest. 2005;4:747–754. doi: 10.1172/JCI22818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H-Y, Thuita L, Strickland P, Hoffman SC, Comstock GW, Helzlsouer KJ. Frequencies of single nucleotide polymorphisms in genes regulating inflammatory responses in a community-based population. BMC Genetics. 2007;4:7. doi: 10.1186/1471-2156-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyke AL, Cote ML, Wenzlaff AS, Land S, Schwartz AG. Cytokine SNPs: comparison of allele frequencies by race and implications for future studies. Cytokine. 2009;4:236–244. doi: 10.1016/j.cyto.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard TD, Koppelman GH, Hu JF, Zheng SQL, Postma DS, Meyers DA, Bleecker ER. Gene-gene interaction in asthma: IL4RA and IL13 in a Dutch population with asthma. Am J Hum Genetics. 2002;4:230–236. doi: 10.1086/338242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron L, Webster RB, Strempel JM, Kiesler P, Kabesch M. et al. TH2 cell-selective enhancement of humal IL13 transcription by IL13-1112C > T, a polymorphism associated with allergic inflammation. J Immunol. 2006;4:8633–8642. doi: 10.4049/jimmunol.177.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007;4:1422–1431. doi: 10.1016/S0140-6736(07)61600-6. [DOI] [PubMed] [Google Scholar]
- Newcomb DC, Zhou W, Moore ML, Goleniewska K, Hershey GKK, Kolls JK, Peebles RS. A functional IL-13 receptor is expressed on polarized murine CD4+ Th17 cells and IL-13 signaling attenuates Th17 cytokine production. J Immunol. 2009;4:5317–5321. doi: 10.4049/jimmunol.0803868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R, Kim HY, Yoon J, Oyoshi MK, MacGinnitie A. et al. Exaggerated IL-17 response to epicutaneous sensitization mediates airway inflammation in the absence of IL-4 and IL-13. J Allergy Clin Immunol. 2009;4:761–770. doi: 10.1016/j.jaci.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR. et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;4:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]