

Abstract
The kinetic folding of ribonuclease H was studied by hydrogen exchange (HX) pulse labeling with analysis by an advanced fragment separation mass spectrometry technology. The results show that folding proceeds through distinct intermediates in a stepwise pathway that sequentially incorporates cooperative native-like structural elements to build the native protein. Each step is seen as a concerted transition of one or more segments from an HX-unprotected to an HX-protected state. Deconvolution of the data to near amino acid resolution shows that each step corresponds to the folding of a secondary structural element of the native protein, termed a “foldon.” Each folded segment is retained through subsequent steps of foldon addition, revealing a stepwise buildup of the native structure via a single dominant pathway. Analysis of the pertinent literature suggests that this model is consistent with experimental results for many proteins and some current theoretical results. Two biophysical principles appear to dictate this behavior. The principle of cooperativity determines the central role of native-like foldon units. An interaction principle termed “sequential stabilization” based on native-like interfoldon interactions orders the pathway.
Do proteins fold through varied and multiple tracks, or do they fold through predetermined intermediates according to understandable biophysical principles (1)? This question is fundamental for the interpretation of a large amount of biophysical and biological research. The question could be resolved if it were possible to define the intermediate structures and pathways that unfolded proteins move through on their way to the native state. Unfortunately, transient intermediates cannot be studied by the usual crystallographic and NMR methods. The range of kinetic and spectroscopic methods has been applied to many proteins, but these methods do not yield the necessary structural information.
We used a developing technology, hydrogen exchange pulse labeling measured by MS (HX MS), to study the folding of a cysteine-free variant of Escherichia coli ribonuclease H1 (RNase H), a mixed α/β protein that has served as a major protein-folding model (2–5). Previous studies showed that RNase H folds in a fast, unresolved burst phase (15 ms dead time) to an intermediate termed “Icore” and then much more slowly (in seconds) to the native state (3). HX pulse-labeling and equilibrium native-state HX experiments monitored by NMR showed that Icore comprises a continuous region of the protein between helix A and strand 5 and that β-strands 1, 2, and 3 and helix E acquire protection much later, consistent with mutational analysis (2–4). Single-molecule and mutational studies indicated that the intermediate is obligatory, on-pathway, and folds first even when Icore is not observably populated (6, 7).
The HX MS technique used here is able to follow the entire folding trajectory of RNase H in considerable structural and temporal detail. The analysis monitors every amide site, evaluates the folding cooperativity between them, and describes the separate folding steps. The results identify at near amino acid resolution the formation and stepwise incorporation of native-like foldon elements in four sequential events that progressively assemble the native structure. A comparison with other experimental and theoretical observations suggests that this pathway behavior is the prevalent mode for protein folding and that it is dictated by two straightforward biophysical principles.
Results
Folding by Spectrophotometry.
Fig. 1 shows results for RNase H folding monitored by circular dichroism under the conditions used in the HX MS studies (10 °C, pH 5). Folding is very similar to the observations in previous studies (25 °C, pH 5.5) but with slower final folding to the native state. The results fit to a three-state model [unfolded (U), intermediate (I), and native (N)] with the following parameters at 0 M urea. The free energy of unfolding for the intermediate (ΔGUI) is 4.1 kcal/mol, the free energy of global unfolding (ΔGUN) is 10.1 kcal/mol, and the rate constant for folding from the intermediate to the native state (kIN) is 0.07 s−1. These spectrophotometric data provide population-averaged kinetic and thermodynamic folding parameters with little structural detail or information about pathway steps.
Fig. 1.

The folding of RNase H monitored by circular dichroism. (A) A burst-phase intermediate (Icore) is formed within the dead time of the experiment (manual mixing) followed by slower folding (30 s) to the native state (0.6 M urea, 10 °C). (B) The equilibrium urea melt (black circles), the amplitude of the burst phase (diamonds), and the observable phase (white circles). (C) Folding and unfolding rates (chevron plot) at 10 °C (black trace) compared with the published fit at 25 °C (gray trace).
HX MS Experiment.
Fig. 2 diagrams the HX MS pulse-labeling experiment. To begin the folding process, denaturant-unfolded protein fully deuterated at amide sites in D2O is diluted into H2O folding buffer under conditions in which D-to-H back exchange is very slow (pH 5.0, 10 °C; ∼40-s HX time constant). After trial folding times, a brief labeling pulse to high pH is imposed (10 ms, pH 10, 10 °C), equivalent to ∼25 times the average HX time constant. When the protection factor (Pf) of an amide site at the time of the labeling pulse is very low (<10), it will appear unprotected and exchange to H. It will appear protected and remain almost fully D-labeled when Pf >100 or kop < 20 s−1, and it will be fractionally H/D labeled in the narrow range of intermediate Pf values. As a result, any amide position switches from fully unprotected when it is unfolded to almost fully protected when folding occurs. We associate the gain of HX protection with the formation of H-bonded secondary or tertiary structure (8, 9).
Fig. 2.

The HX MS pulse-labeling experiment. (A) Denaturant-unfolded fully deuterated protein is diluted into folding conditions (mixer 1). After some folding time, D-to-H exchange at still-exposed sites is induced by a brief high-pH pulse (mixer 2) and then is quenched to low pH where HX is very slow (mixer 3). Online proteolysis cuts the protein into many overlapping peptide fragments, and the peptides are separated by LC and MS. (B) The 228 unique peptides used in this work, identified and characterized in the MS data by the ExMS program (12), and plotted as a function of amino acid residue.
The subsequent HX MS analysis is designed to detect which residues are protected and which are not at each folding time. To preserve the H/D labeling reached during the labeling pulse, the protein sample is quenched immediately to a slow exchange condition (pH 2.5, 0 °C) and is proteolyzed quickly into many overlapping peptide fragments. The fragments then are separated and analyzed for carried deuterium by HPLC and mass spectrometry. Previous papers describe how to maximize the number of peptide fragments obtained (10), how to minimize the back exchange of D label during sample preparation (11), and a program (ExMS) for identifying and characterizing the many peptides in the final MS spectra (12). These methods provided 326 useful peptide fragments; 228 unique fragments for this 155-residue protein are indicated in Fig. 2B. Of these, 156 unique peptides passed the ExMS autocheck tests (no operator intervention) for all folding experiments out to 3 s and were used for the time-dependent analysis (shown in SI Appendix). For residue-resolved analysis, the entire peptide set was used.
Data Interpretation.
Each HX MS experiment at each folding time point records ∼1,000 mass spectra. The ExMS analysis separately identifies and characterizes each of the hundreds of peptide fragments detected. Each peptide monitors the folding history of the segment of the protein it represents. As folding proceeds and amides become protected at the time of the labeling pulse, each peptide converts from its unfolded, lighter state to a state that is heavier by a mass increment equal to the number of D-labeled sites protected.
Different folding scenarios will produce recognizably different results. At the individual peptide level, protection that occurs one or a few residues at a time will be seen as a continuous slide in time from an unprotected state to more protected states. If a folding step protects many amides in a given segment all together, the conversion will be seen as a bimodal envelope with occupancy shifting in time from the lighter to the heavier envelope.
For the whole protein, if folding is two-state, all segments will transition to native-like protection in the same concerted single-exponential step. In a multistep pathway, different regions fold at different times, and peptides that represent those regions will acquire protection accordingly. For example, in a classical stepwise pathway, 100% of any given segment will convert to its protected state in a concerted step, and other segments will convert in other steps, defining the pathway sequence. If a protein folds through several parallel pathways, any given segment will convert in fractional steps, earlier in some molecules of the protein population and later in others. If structure is formed and then lost, whether native-like or nonnative, the timed measurements of peptides that cover that region will reveal that behavior.
In summary, when many peptides are seen individually, their HX MS behavior can depict the spatially resolved and the time-resolved development of protection of segments throughout the protein. During data analysis, the many overlapping peptide fragments provide multiple internal consistency checks. In an ultimate step, multipeptide folding data can be deconvolved to near amino acid resolution. This information can specify the detailed protein folding behavior.
MS Results.
HX MS results for the 156 overlapping RNase H peptides measured as a function of folding time are given in SI Appendix. Four are shown in Fig. 3. The top frame shows the spectrum obtained for the unfolded protein (6 M urea, protonated, lower mass). The bottom frame shows the spectrum obtained from the native protein where the D label at H-bonded amide sites is protected and retained through the D-to-H labeling pulse. The time-dependent data provide a graphic snapshot of the fraction of the protein population that already is protected (heavier) and the fraction that is not yet protected (lighter) at each segmental position at the time of the labeling pulse.
Fig. 3.
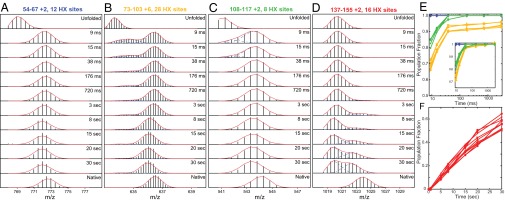
(A–D) Illustrative MS spectra versus folding time. Peptides shown (also see SI Appendix) cover each helical segment plus some neighboring sequence in the native protein. The top and bottom frames show control experiments in which the unfolded and native proteins were subjected to the same labeling pulse and analysis. Fitted envelopes separate the fractional populations of the unfolded, intermediate, and native state present at the time of the labeling pulse. Deuterons on side chains and the first two residues of each peptide are lost during sample preparation. The subpeaks within each isotopic envelope are caused by the natural abundance of 13C (∼1%) convolved with the carried number of deuterons. A leftward drift in folded peptide mass at long folding times (D) occurs because not-yet-protected sites are exposed to D-to-H exchange during the prepulse folding period (pH 5, 10 °C). (E and F) The time dependence for the formation of the protected state of different protein regions, color coded to match the RNase H foldon units in Fig. 5. (Inset) The unblocked folding phase of the yellow curves is renormalized to 100% to allow direct comparison with the folding time of the green segment. For this comparison, the experiment was replicated in triplicate, and only the highest-precision peptides were used. The green and yellow segments fold in detectably different phases. Peptides are identified in SI Appendix, Fig. S1.
Fig. 3A shows a peptide that monitors the C-terminal turn of helix A and most of β-strand 4 (blue in Fig. 5A). This segment forms structure that protects a sum of approximately eight deuterons before the first measured folding time point (9 ms), which appears as a shift of the peptide envelope to higher mass. The same state is adopted by 100% of the protein population; no lighter population exists. The segment remains folded at all later times in the folding process, and peptides that monitor other positions add on in subsequent folding steps. The same is true in all cases.
Fig. 5.

RNase H folding pathway. (A) RNase H foldon units. (B) The macroscopic folding reaction is well represented by a conventional free energy diagram.
The peptide in Fig. 3B monitors the kinked B/C helix plus the long connecting loop to helix D (yellow in Fig. 5A). The measured data for this peptide capture the last part of its transition to a folded and protected state. About 7% of the population remains unprotected until later in the folding process. This still unprotected portion could reflect an alternative pathway, or a barrier in a small population of the molecules as for a mis-isomerized proline, or the inherent dynamic EX1 behavior of this segment.
The peptide in Fig. 3C monitors most of helix D plus β-strand 5 (green in Fig. 5A). At the earliest measured time only a small population fraction is not yet folded, indicating a folding rate slightly faster than for the B/C region. The peptide in Fig. 3D monitors helix E and a long C-terminal protein segment (red in Fig. 5A). Protection develops with a halftime of ∼30 s Other peptides detect equally slow folding for the N-terminal β-strands 1, 2, and 3. This final folding step completes the native state.
The D-label protected in each earlier step does not yet match the native state, not because of the protection of fewer sites in the already-formed structure but because of less than complete protection at numerous positions, apparently because the folding intermediates, although “native-like” structurally, are not yet fully native and therefore are less stable and more dynamic. This reduced protection before the final folding step may relate to the proposed (un)locking step of a late dry molten globule folding intermediate (13).
Fig. 3 E and F plot the time dependence for folding of the different protein segments (see also SI Appendix, Fig. S1). The ordinate refers to the fraction of the protein population that has reached the protected state at specified regions of the protein. The Inset shows that the green segment folds detectably faster than the unblocked folding phase of the yellow segment.
These results identify four kinetically separable steps in RNase H folding.
Partially Folded Structure at High Resolution.
The HX MS data for the large data set of many overlapping peptides at various folding times can be deconvolved to near amino acid resolution (using the HDsite program). The results (Fig. 4) identify the structural regions that fold in each of the steps just described. They represent secondary structural elements of the native protein.
Fig. 4.

Protected D label at residue resolution. Site-resolved D occupancy was computed from MS data for many overlapping peptides after different folding times: at the earliest folding time in a competition folding versus labeling experiment (Top); after different prepulse folding times (Middle); and for a native protein control (Bottom). Dots through the native frame indicate amide H-bonds to main chain (open) and to side chains (filled) (PDB ID: 1F21). Individually resolved residues are shown in red. Switchable residues not distinguished because of inadequate peptide coverage are in blue and are connected by a dashed line. Small white spaces indicate absent amides because of proline or high probability protease cut sites. Regions in gray indicate segments that are in transition from unfolded to folded conformations and that produce bimodal MS spectra that cannot be analyzed for site resolution. Positions of helices A–E and β-strands 1–5 in the native protein are shown at the top, color coded to match their foldon identities (Fig. 5). Low D retention in the D-to-H labeling pulse indicates an equilibrium protection factor <10; full D retention indicates a protection factor >100 or kop <20 s−1. A nonzero D label in the absence of protection reflects the 6% D2O in the labeling pulse.
To guide the interpretation of residue-resolved pulse-labeling results, native RNase H was passed through the same pulse-labeling and analysis procedure. We expect that sites protected by H-bonding will resist exchange during the D-to-H labeling pulse, regardless of their exposure to solvent (8, 9). The results match this expectation.
To obtain the earliest possible kinetic information, a competition experiment (14, 15) was done in which unfolded deuterated protein was diluted into folding and labeling conditions in the same mixing step. In this experiment, protection competes kinetically with labeling. We find measurable protection in the helix A segment (∼50%) and more evanescent protection in segments in β-strand 4 and helices C and D but none in helix B and E or other β-strands (Fig. 4, Top). The protection develops as rapidly as the mixing process (perhaps 0.1 ms). Although helix A and strand 4 may fold on this time scale, helix C and D fold later, suggesting either some nascent prefolding protection when the still unfolded protein is placed into native conditions or their interaction with already formed helix A and strand 4 early in structure formation. The protection pattern does not correlate with intrinsic helix propensity, estimated by AGADIR (16) at 50% for helix E (see also ref. 17), 30% for helix A, and as negligible for helices B, C, and D. These observations may relate to current interest in initial protein collapse (18), the unfolded state structure (19), and its significance for subsequent protein folding (20).
By the 9-ms folding time point, the helix A and β-strand 4 segments (blue in Fig. 5A) have achieved protection in 100% of the refolding population. The residue-resolved pattern of protection is similar but not identical to the native protein. For example, the N terminus of helix A is less protected, as might be expected because of end fraying in the partly folded intermediate. The far N- and C-terminal segments remain wholly unprotected.
At this time point, peptides that monitor the segments shown in green and yellow in Fig. 5A exhibit bimodal envelopes (Fig. 3 and SI Appendix). The bimodal MS transition documents their concerted stepwise foldon behavior and measures the unfolded and folded fraction of the population at each time point. Bimodal data, noted in gray in Fig. 4, cannot be analyzed to site resolution but this limitation does not hinder the analysis of other peptides.
At the 720-ms folding time, helices A, B/C, and D, their local β-strand segments, and even the connecting C-to-D loop (blue, green, and yellow in Fig. 5A) have achieved protection patterns similar to the native protein. By 20 s, the extensive N- and C-terminal regions have begun to fold in a bimodal, concerted, foldon-dependent way.
Interestingly, the C-terminal Val155 amide remains deuterated in all cases. This apparent anomaly is caused by HX chemistry during the pulse, namely an expected 60-fold slowing factor for the C-terminal amide multiplied fivefold by the valine side-chain effect (21).
Anomalously Slow Exchange for β-Edge Amides.
The residue-level analysis at the early folding time points finds that some residues in strand 4 are moderately protected even when their native H-bond acceptors on strand 1 are not yet in place. Although this protection might be thought to indicate nonnative interactions, several amide hydrogens on the unprotected solvent-exposed edges of β-strands 3 and 5 in the native protein similarly avoid D-to-H exchange in the labeling pulse. Analogous behavior has been seen before (8, 9, 22).
Discussion
Pulse-labeling HX MS results obtained during the kinetic folding of the single-domain RNase H protein graphically display a time-resolved sequence of four concerted steps in structure formation. Each step corresponds to the folding of one or more secondary structural elements of the native protein. The residue-resolved pattern of HX protection mimics that seen for the native protein, implying native-like features for each added foldon unit and the intermediate states that they produce. Once formed, these units remain in place as subsequent units are added, demonstrating a sequential stepwise buildup of the native structure. Essentially the entire protein population joins in each step, indicating a single dominant route.
The helix A+ strand 4 segment (blue in Fig. 5A) folds within the first ms. Helix D and strand 5 (green in Fig. 5A) add on at ∼5 ms. In the native protein these two elements pack together to form a major hydrophobic core of the protein. Shortly thereafter (∼9 ms), the more tenuously associated yellow segment shown in yellow in Fig. 5A folds to complete the Icore intermediate previously identified by pulse-labeling HX NMR (3, 23). Finally, in a much slower reaction (∼30 s) the elements shown in red in Fig. 5A (strands 1, 2, and 3 and helix E) fold concertedly, even though they are drawn from the most distant protein termini.
It is hard to picture this sequence of segmental folding events as anything other than a classical pathway that constructs the native protein by the sequential incorporation of native-like foldon units. If any significant fraction of protein (>4%) formed a protecting structure that differed in any significant way, structurally or kinetically (e.g., an alternative parallel pathway), it would be seen clearly, as we see for the 7% yellow population. The folding behavior observed is well represented by the free energy diagram in Fig. 5B.
Experimental Work to the Contrary.
The folding model pictured in Fig. 5B is identical to the extensively worked out case of cytochrome c for which a series of HX NMR studies defined four native-like foldon-based intermediates in a well-ordered pathway (24–26). It also is consistent with the finding of individual native-like intermediates in many other proteins. However, other experimental studies have been interpreted in terms of more complex models. Those studies have depended mainly on spectroscopic measurements that provide little structural information and therefore are subject to ambiguity; prominent suggestions are that proteins may fold through many alternative intermediates, or through none at all, and that observed intermediates are nonnative and therefore nonproductive, or even obstructive.
In respect to the suggestion that proteins may fold through a number of alternative, independent, parallel routes, we have shown that the kinetic complexity observed in such spectroscopic studies can be explained with fewer fitting factors and equivalent or better χ2 fit by a single pathway in which a probabilistic barrier insertion affects some fraction of the population and not another (26, 27). Given only kinetic phase data and no structural information, it is not possible to distinguish a multiple pathway interpretation from a single pathway with an optional barrier that slows one population fraction and not another (26, 27).
Many small proteins are known to fold in an apparent two-state manner without observable intermediates. Folding intermediates, however, will be kinetically visible only under certain limited conditions. They must occupy a free-energy well that is relatively low compared with all prior wells and be blocked by a forward barrier that is sufficiently high relative to all prior barriers. Otherwise, intermediates will not accumulate noticeably even when they are present and important. In general, small proteins can be expected to fold in a two-state manner either because they tend to avoid barrier insertion or may be smaller than two foldons.
The requirement that the population of an intermediate depends on a subsequent large barrier has led to the idea that folding intermediates may be obstructive because visible intermediates and slowed folding go together. Rather it is the barrier, whether optional or intrinsic, that slows folding and correlates it with the population of an otherwise invisible intermediate. In the present experiments, the ability to distinguish and characterize RNase H folding intermediates depended on the presence of barriers between each of the folding steps. However, it seems clear that the sequential intermediates observed are on-pathway and constructive.
Do nonnative interactions imply that an intermediate is nonobligatory and off-pathway? Alternatively, the intermediate is on-pathway and productive but in addition has some nonnative character, which may or may not tend to slow folding. In a partly folded intermediate, energy-minimizing nonnative interactions can be expected (28) and have been seen in intermediates that are clearly on-pathway and constructive (29–33).
In summary, although a quantity of ambiguous protein folding data has led to different interpretations, it appears that the great majority of experimental protein-folding observations can be understood in terms of the reaction scheme in Fig. 5B.
Theoretical Studies.
In 1992 Zwanzig et al. (34) showed that, in principle, protein folding need not require any particular pathway. A small energetic bias toward native-like interactions would allow the native state to be found quickly through multiple independent routes. The formulation was phrased in terms of native-centric selectivity at the amino acid level. Theoretical studies before and since, focused at the amino acid level, naturally emphasize the role of an ensemble of microscopically diverse structures, but this diversity is not at odds with higher-level, more deliberate pathway behavior. It now appears that distributed residue-level searching does not lead directly to the native protein but rather functions to construct distinct native-like foldon elements. Here the Levinthal paradox (35) is reduced to a manageable problem, because a random search for a small foldon can be accomplished rapidly. This microscopic behavior is out of the reach of macroscopic experiment. One must look forward to the promise of theoretical approaches for ultimately providing a deep understanding of protein folding from this basic level and up. The present work contributes to this effort by showing that a well-ordered pathway assembles native-like foldons to form structural intermediates and the native protein.
Physically based computational folding simulations can, in principle, discern folding mechanisms from the fundamental residue-level steps and up, but severe challenges exist. These include the immense computer power required to reach the time scale at which foldon behavior emerges, the great accuracy required for the force fields used, and the need for a method that can extract a descriptive trace of the progression of the folding reaction. Recent advances in molecular dynamics trajectory analysis have transcended these limitations in part, bridged the divide between the micro- and macroscopic views of protein folding, and demonstrated that thermally driven amino acid-level searching leads to the native structure through the formation of native-like structural elements and their sequential incorporation in repeatable folding pathways (36–38). A recent approach explicitly uses foldon conservation and sequential stabilization in an iterative search strategy (39). Some other theoretical efforts lead in this same direction (40). These scenarios closely resemble the reaction scheme in Fig. 5B. Other funnel and network models are conceptually different but are not inconsistent with this conclusion (41–46).
Principles of Protein Folding.
The results and considerations described here support a determinate mechanism for protein folding and its generality. Two straightforward biophysical principles appear to explain this behavior.
The first is the inherent cooperativity of protein structural units. The unfolding and refolding of cooperative foldon units and their role in forming intermediates and pathways has been detected, in greater or lesser detail, in many proteins by HX methods (23–25, 29, 47–55), by sulfhydryl labeling (56, 57), by relaxation dispersion NMR (58), and in theoretical simulations (36–38). The inherent cooperativity of native-like foldon units predisposes them rather than other fractional elements to account for the unit steps in protein folding and thus dictates the stepwise formation and native-like nature of pathway intermediates.
The second principle orders the pathway and determines its sequential nature. In the initial on-pathway step, a limited structural unit is assembled by a relatively unguided amino acid search (59), which makes the first on-pathway step the intrinsically slow step. In subsequent steps in which amino acid searching can be informed by existing structure, a faster assisted folding mode emerges called “sequential stabilization” (25, 60). Like the process of coupled folding upon binding (61–64), the prior structure will selectively guide and stabilize the units with which it interacts in the native protein. When prior structure can support more than one incoming foldon, the sequential stabilization principle can lead to pathway branching (65).
Simple considerations dictate that the ordered pathway buildup of native structure observed here must depend on the summed free energy of multiple native-like interactions acting together to select each correct step among the many competing alternatives. It seems likely that the concurrent interfoldon interactions produced by the two factors just considered could provide the required degree of selectivity, whereas individual residue-level interactions would not. When the bias toward native interactions is insufficient, probabilistic misfolding is likely to occur (25).
Materials and Methods
Protein.
The version of E. coli RNase H [Protein Data Bank (PDB) ID: 1F21] used here is the wild-type protein with its three cysteines changed to alanine. It was expressed and purified as described previously (66).
Kinetic Folding and Pulse Labeling.
Previously described methods (10–12) were used to obtain the many peptide fragments studied here. Of these, 156 unique peptides (203 with different charge states) were found to pass the ExMS autocheck tests (no operator intervention) and were used in the timed 9-ms to 3-s folding experiments. The HX MS experiment is diagrammed in Fig. 2 and detailed in SI Appendix.
Sample Analysis.
The selectively H/D-labeled reaction mixture flowed into a home-built cooled chamber containing an online flow analysis system (low pH, 0 °C) in which the protein sample was proteolyzed (immobilized tandem pepsin and fungal protease columns), caught on a trap column, washed, eluted, roughly separated (HPLC column, acetone, acetonitrile gradient), and continuously electrosprayed into the mass spectrometer for a second dimension of separation by mass (∼1,000 MS spectra). Previous papers describe methods for obtaining and analyzing many fragments (10) and for minimizing back exchange of the D-label during sample preparation (11).
Mass spectra were processed by the home-written ExMS program (12) to identify and characterize all of the individual peptide isotopic peaks and envelopes and to read out the placement of protecting structure and the time course for its formation during refolding. To quantify folded (HX-protected) and not-yet folded populations present during the H–D exchange-labeling pulse, bimodal MS envelopes were fit by a home-written program (HDpop), using binomial fitting with appropriate weighting. To obtain H–D labeling information at amino acid resolution, MS results for many overlapping peptides were analyzed using in-house software (HDsite).
Supplementary Material
Acknowledgments
We thank Y. Bai, D. Barrick, N. R. Kallenbach, G. D. Rose, J. J. Skinner, T. R. Sosnick, and A. J. Wand for helpful suggestions on the manuscript. This work was supported by National Institutes of Health Grants RO1 GM031847 (to S.W.E.), R01 GM 50945 (to S.M.), National Science Foundation Grant MCB1020649 (to S.W.E.), the Mathers Charitable Foundation (S.W.E.), and National Institutes of Health Predoctoral Training Grants GM0827 (to B.T.W.) and GM008295 (to L.E.R.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305887110/-/DCSupplemental.
References
- 1.Sosnick TR, Barrick D. The folding of single domain proteins–have we reached a consensus? Curr Opin Struct Biol. 2011;21:12–24. doi: 10.1016/j.sbi.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chamberlain AK, Handel TM, Marqusee S. Detection of rare partially folded molecules in equilibrium with the native conformation of RNaseH. Nat Struct Biol. 1996;3(9):782–787. doi: 10.1038/nsb0996-782. [DOI] [PubMed] [Google Scholar]
- 3.Raschke TM, Marqusee S. The kinetic folding intermediate of ribonuclease H resembles the acid molten globule and partially unfolded molecules detected under native conditions. Nat Struct Biol. 1997;4(4):298–304. doi: 10.1038/nsb0497-298. [DOI] [PubMed] [Google Scholar]
- 4.Raschke TM, Kho J, Marqusee S. Confirmation of the hierarchical folding of RNase H: A protein engineering study. Nat Struct Biol. 1999;6(9):825–831. doi: 10.1038/12277. [DOI] [PubMed] [Google Scholar]
- 5.Cecconi C, Shank EA, Bustamante C, Marqusee S. Direct observation of the three-state folding of a single protein molecule. Science. 2005;309(5743):2057–2060. doi: 10.1126/science.1116702. [DOI] [PubMed] [Google Scholar]
- 6.Spudich GM, Miller EJ, Marqusee S. Destabilization of the Escherichia coli RNase H kinetic intermediate: Switching between a two-state and three-state folding mechanism. J Mol Biol. 2004;335(2):609–618. doi: 10.1016/j.jmb.2003.10.052. [DOI] [PubMed] [Google Scholar]
- 7.Connell KB, Miller EJ, Marqusee S. The folding trajectory of RNase H is dominated by its topology and not local stability: A protein engineering study of variants that fold via two-state and three-state mechanisms. J Mol Biol. 2009;391(2):450–460. doi: 10.1016/j.jmb.2009.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skinner JJ, Lim WK, Bédard S, Black BE, Englander SW. Protein hydrogen exchange: Testing current models. Protein Sci. 2012;21(7):987–995. doi: 10.1002/pro.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skinner JJ, Lim WK, Bédard S, Black BE, Englander SW. Protein dynamics viewed by hydrogen exchange. Protein Sci. 2012;21(7):996–1005. doi: 10.1002/pro.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayne L, et al. Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method. J Am Soc Mass Spectrom. 2011;22(11):1898–1905. doi: 10.1007/s13361-011-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walters BT, Ricciuti A, Mayne L, Englander SW. Minimizing back exchange in the hydrogen exchange-mass spectrometry experiment. J Am Soc Mass Spectrom. 2012;23(12):2132–2139. doi: 10.1007/s13361-012-0476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kan ZY, Mayne L, Chetty PS, Englander SW. ExMS: Data analysis for HX-MS experiments. J Am Soc Mass Spectrom. 2011;22(11):1906–1915. doi: 10.1007/s13361-011-0236-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baldwin RL, Frieden C, Rose GD. Dry molten globule intermediates and the mechanism of protein unfolding. Proteins. 2010;78(13):2725–2737. doi: 10.1002/prot.22803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roder H, Wüthrich K. Protein folding kinetics by combined use of rapid mixing techniques and NMR observation of individual amide protons. Proteins. 1986;1(1):34–42. doi: 10.1002/prot.340010107. [DOI] [PubMed] [Google Scholar]
- 15.Gladwin ST, Evans PA. Structure of very early protein folding intermediates: New insights through a variant of hydrogen exchange labelling. Fold Des. 1996;1(6):407–417. doi: 10.1016/S1359-0278(96)00057-0. [DOI] [PubMed] [Google Scholar]
- 16.Muñoz V, Serrano L. Development of the multiple sequence approximation within the AGADIR model of alpha-helix formation: Comparison with Zimm-Bragg and Lifson-Roig formalisms. Biopolymers. 1997;41(5):495–509. doi: 10.1002/(SICI)1097-0282(19970415)41:5<495::AID-BIP2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 17.Goedken ER, Raschke TM, Marqusee S. Importance of the C-terminal helix to the stability and enzymatic activity of Escherichia coli ribonuclease H. Biochemistry. 1997;36(23):7256–7263. doi: 10.1021/bi970060q. [DOI] [PubMed] [Google Scholar]
- 18.Haran G. How, when and why proteins collapse: The relation to folding. Curr Opin Struct Biol. 2012;22(1):14–20. doi: 10.1016/j.sbi.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ratcliff K, Marqusee S. Identification of residual structure in the unfolded state of ribonuclease H1 from the moderately thermophilic Chlorobium tepidum: Comparison with thermophilic and mesophilic homologues. Biochemistry. 2010;49(25):5167–5175. doi: 10.1021/bi1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowler BE. Residual structure in unfolded proteins. Curr Opin Struct Biol. 2012;22(1):4–13. doi: 10.1016/j.sbi.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17(1):75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson JS, Hernández G, LeMaster DM. Backbone conformational dependence of peptide acidity. Biophys Chem. 2009;141(1):124–130. doi: 10.1016/j.bpc.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Raschke TM, Marqusee S. Hydrogen exchange studies of protein structure. Curr Opin Biotechnol. 1998;9(1):80–86. doi: 10.1016/s0958-1669(98)80088-8. [DOI] [PubMed] [Google Scholar]
- 24.Maity H, Maity M, Krishna MMG, Mayne L, Englander SW. Protein folding: The stepwise assembly of foldon units. Proc Natl Acad Sci USA. 2005;102(13):4741–4746. doi: 10.1073/pnas.0501043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Englander SW, Mayne L, Krishna MM. Protein folding and misfolding: Mechanism and principles. Q Rev Biophys. 2007;40(4):287–326. doi: 10.1017/S0033583508004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krishna MMG, Englander SW. A unified mechanism for protein folding: Predetermined pathways with optional errors. Protein Sci. 2007;16(3):449–464. doi: 10.1110/ps.062655907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bédard S, Krishna MMG, Mayne L, Englander SW. Protein folding: Independent unrelated pathways or predetermined pathway with optional errors. Proc Natl Acad Sci USA. 2008;105(20):7182–7187. doi: 10.1073/pnas.0801864105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng H, Takei J, Lipsitz R, Tjandra N, Bai Y. The high-resolution structure of a protein intermediate state: Implications for protein folding. Protein Sci. 2004;13(1):219–220. [Google Scholar]
- 29.Nishimura C, Dyson HJ, Wright PE. Identification of native and non-native structure in kinetic folding intermediates of apomyoglobin. J Mol Biol. 2006;355(1):139–156. doi: 10.1016/j.jmb.2005.10.047. [DOI] [PubMed] [Google Scholar]
- 30.Feng H, Zhou Z, Bai Y. A protein folding pathway with multiple folding intermediates at atomic resolution. Proc Natl Acad Sci USA. 2005;102(14):5026–5031. doi: 10.1073/pnas.0501372102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korzhnev DM, et al. Nonnative interactions in the FF domain folding pathway from an atomic resolution structure of a sparsely populated intermediate: An NMR relaxation dispersion study. J Am Chem Soc. 2011;133(28):10974–10982. doi: 10.1021/ja203686t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capaldi AP, Kleanthous C, Radford SE. Im7 folding mechanism: Misfolding on a path to the native state. Nat Struct Biol. 2002;9(3):209–216. doi: 10.1038/nsb757. [DOI] [PubMed] [Google Scholar]
- 33.Dyson HJ, Wright PE. Unfolded proteins and protein folding studied by NMR. Chem Rev. 2004;104(8):3607–3622. doi: 10.1021/cr030403s. [DOI] [PubMed] [Google Scholar]
- 34.Zwanzig R, Szabo A, Bagchi B. Levinthal’s paradox. Proc Natl Acad Sci USA. 1992;89(1):20–22. doi: 10.1073/pnas.89.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4(1):10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 36.Lindorff-Larsen K, Trbovic N, Maragakis P, Piana S, Shaw DE. Structure and dynamics of an unfolded protein examined by molecular dynamics simulation. J Am Chem Soc. 2012;134(8):3787–3791. doi: 10.1021/ja209931w. [DOI] [PubMed] [Google Scholar]
- 37.Piana S, Lindorff-Larsen K, Shaw DE. Protein folding kinetics and thermodynamics from atomistic simulation. Proc Natl Acad Sci USA. 2012;109(44):17845–17850. doi: 10.1073/pnas.1201811109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dror RO, Dirks RM, Grossman JP, Xu H, Shaw DE. Biomolecular simulation: A computational microscope for molecular biology. Annu Rev Biophys. 2012;41:429–452. doi: 10.1146/annurev-biophys-042910-155245. [DOI] [PubMed] [Google Scholar]
- 39.Adhikari AN, Freed KF, Sosnick TR. De novo prediction of protein folding pathways and structure using the principle of sequential stabilization. Proc Natl Acad Sci USA. 2012;109(43):17442–17447. doi: 10.1073/pnas.1209000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weinkam P, Zong C, Wolynes PG. A funneled energy landscape for cytochrome c directly predicts the sequential folding route inferred from hydrogen exchange experiments. Proc Natl Acad Sci USA. 2005;102(35):12401–12406. doi: 10.1073/pnas.0505274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lane TJ, Bowman GR, Beauchamp K, Voelz VA, Pande VS. Markov state model reveals folding and functional dynamics in ultra-long MD trajectories. J Am Chem Soc. 2011;133(45):18413–18419. doi: 10.1021/ja207470h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schafer NP, et al. Discrete kinetic models from funneled energy landscape simulations. PLoS ONE. 2012;7(12):e50635. doi: 10.1371/journal.pone.0050635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lane TJ, Shukla D, Beauchamp KA, Pande VS. To milliseconds and beyond: Challenges in the simulation of protein folding. Curr Opin Struct Biol. 2013;23(1):58–65. doi: 10.1016/j.sbi.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lane TJ, Pande VS. A simple model predicts experimental folding rates and a hub-like topology. J Phys Chem B. 2012;116(23):6764–6774. doi: 10.1021/jp212332c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Onuchic JN, Wolynes PG. Theory of protein folding. Curr Opin Struct Biol. 2004;14(1):70–75. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 46.Bowman GR, Voelz VA, Pande VS. Taming the complexity of protein folding. Curr Opin Struct Biol. 2011;21(1):4–11. doi: 10.1016/j.sbi.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bai Y, Sosnick TR, Mayne L, Englander SW. Protein folding intermediates: Native-state hydrogen exchange. Science. 1995;269(5221):192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bollen YJM, Kamphuis MB, van Mierlo CPM. The folding energy landscape of apoflavodoxin is rugged: Hydrogen exchange reveals nonproductive misfolded intermediates. Proc Natl Acad Sci USA. 2006;103(11):4095–4100. doi: 10.1073/pnas.0509133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fuentes EJ, Wand AJ. Local dynamics and stability of apocytochrome b562 examined by hydrogen exchange. Biochemistry. 1998;37(11):3687–3698. doi: 10.1021/bi972579s. [DOI] [PubMed] [Google Scholar]
- 50.Chu R, Pei W, Takei J, Bai Y. Relationship between the native-state hydrogen exchange and folding pathways of a four-helix bundle protein. Biochemistry. 2002;41(25):7998–8003. doi: 10.1021/bi025872n. [DOI] [PubMed] [Google Scholar]
- 51.Gsponer J, et al. Determination of an ensemble of structures representing the intermediate state of the bacterial immunity protein Im7. Proc Natl Acad Sci USA. 2006;103(1):99–104. doi: 10.1073/pnas.0508667102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yan S, Kennedy SD, Koide S. Thermodynamic and kinetic exploration of the energy landscape of Borrelia burgdorferi OspA by native-state hydrogen exchange. J Mol Biol. 2002;323(2):363–375. doi: 10.1016/s0022-2836(02)00882-3. [DOI] [PubMed] [Google Scholar]
- 53.Pan J, Han J, Borchers CH, Konermann L. Characterizing short-lived protein folding intermediates by top-down hydrogen exchange mass spectrometry. Anal Chem. 2010;82(20):8591–8597. doi: 10.1021/ac101679j. [DOI] [PubMed] [Google Scholar]
- 54.Chamberlain AK, Marqusee S. Molten globule unfolding monitored by hydrogen exchange in urea. Biochemistry. 1998;37(7):1736–1742. doi: 10.1021/bi972692i. [DOI] [PubMed] [Google Scholar]
- 55.Chamberlain AK, Marqusee S. Touring the landscapes: Partially folded proteins examined by hydrogen exchange. Structure. 1997;5(7):859–863. doi: 10.1016/s0969-2126(97)00240-2. [DOI] [PubMed] [Google Scholar]
- 56.Silverman JA, Harbury PB. The equilibrium unfolding pathway of a (beta/alpha)8 barrel. J Mol Biol. 2002;324(5):1031–1040. doi: 10.1016/s0022-2836(02)01100-2. [DOI] [PubMed] [Google Scholar]
- 57.Stratton MM, Cutler TA, Ha JH, Loh SN. Probing local structural fluctuations in myoglobin by size-dependent thiol-disulfide exchange. Protein Sci. 2010;19(8):1587–1594. doi: 10.1002/pro.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Korzhnev DM, Kay LE. Probing invisible, low-populated States of protein molecules by relaxation dispersion NMR spectroscopy: An application to protein folding. Acc Chem Res. 2008;41(3):442–451. doi: 10.1021/ar700189y. [DOI] [PubMed] [Google Scholar]
- 59.Fersht AR. Nucleation mechanisms in protein folding. Curr Opin Struct Biol. 1997;7(1):3–9. doi: 10.1016/s0959-440x(97)80002-4. [DOI] [PubMed] [Google Scholar]
- 60.Rumbley J, Hoang L, Mayne LC, Englander SW. An amino acid code for protein folding. Proc Natl Acad Sci USA. 2001;98(1):105–112. doi: 10.1073/pnas.98.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kiefhaber T, Bachmann A, Jensen KS. Dynamics and mechanisms of coupled protein folding and binding reactions. Curr Opin Struct Biol. 2012;22(1):21–29. doi: 10.1016/j.sbi.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 62.Spolar RS, Record MT., Jr Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994;263(5148):777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- 63.Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12(1):54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 64.Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19(1):31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krishna MMG, Maity H, Rumbley JN, Englander SW. Branching in the sequential folding pathway of cytochrome c. Protein Sci. 2007;16(9):1946–1956. doi: 10.1110/ps.072922307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dabora JM, Marqusee S. Equilibrium unfolding of Escherichia coli ribonuclease H: Characterization of a partially folded state. Protein Sci. 1994;3(9):1401–1408. doi: 10.1002/pro.5560030906. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.